

Abstract
V-domain Immunoglobulin Suppressor of T cell Activation (VISTA) is an inhibitory immune-checkpoint molecule that suppresses CD4+ and CD8+ T cell activation when expressed on antigen-presenting cells. Vsir−/− mice developed loss of peripheral tolerance and multi-organ chronic inflammatory phenotypes. Vsir−/− CD4+ and CD8+ T cells were hyper-responsive towards self- and foreign antigens. Whether or not VISTA regulates innate immunity is unknown. Using a murine model of psoriasis induced by TLR7 agonist imiquimod (IMQ), we show that VISTA deficiency exacerbated psoriasiform inflammation. Enhanced TLR7 signaling in Vsir−/− dendritic cells (DCs) led to the hyper-activation of Erk1/2 and Jnk1/2, and augmented the production of IL-23. IL-23, in turn, promoted the expression of IL-17A in both TCRγδ+ T cells and CD4+ Th17 cells. Furthermore, VISTA regulates the peripheral homeostasis of CD27− γδ T cells and their activation upon TCR-mediated or cytokine-mediated stimulation. IL-17A-producing CD27− γδ T cells were expanded in the Vsir−/− mice and amplified the inflammatory cascade. In conclusion, this study has demonstrated that VISTA critically regulates the inflammatory responses mediated by DCs and IL-17-producing TCRγδ+ and CD4+ Th17 T cells following TLR7 stimulation. Our finding provides a rationale for therapeutically enhancing VISTA-mediated pathways to benefit the treatment of autoimmune and inflammatory disorders.
Introduction
V-domain Immunoglobulin Suppressor of T cell Activation (VISTA, gene name Vsir) is an inhibitory B7 family immune-checkpoint molecule1. Together with other T cell co-inhibitory receptors such as CTLA-4, PD-1, TIM3, and LAG3, these immune-checkpoint proteins play critical roles in maintaining peripheral tolerance and controlling immune responses against self and infectious agents, or cancer2,3.
The human and murine VISTA proteins share 90% identity and display similar expression patterns4. VISTA is constitutively expressed on CD11b+ myeloid dendritic cells (DCs), naïve CD4+ and CD8+ T cells, and Foxp3+CD4+ regulatory T cells. Similar to CTLA-4 and PD-1, VISTA controls peripheral tolerance and anti-tumor immunity1,3,5. VISTA expressed on APCs acts as a ligand to suppress the proliferation and cytokine production of both CD4+ and CD8+ T cells. VISTA expressed on CD4+ T cells also suppresses T cell activation in a T-cell autonomous manner6. Vsir knockout mice (Vsir−/−) developed loss of peripheral tolerance, manifested as spontaneous T cell activation, production of inflammatory cytokines and chemokines, and chronic multi-organ inflammation7. When bred onto an autoimmune-prone background, VISTA deficiency accelerated disease development in the experimental autoimmune encephalomyelitis (EAE) model8. Treatment with VISTA-blocking monoclonal antibody (mAb) enhanced T responses and anti-tumor immunity1,8. Our recent study has further demonstrated that VISTA and another B7 family immune-checkpoint PD-1 play non-redundant roles in controlling T cell responses9. Mice deficient for both genes developed the most severe inflammatory phenotypes, accompanied by spontaneous activation of CD4+ and CD8+ T cells. Combinational blockade of both VISTA and PD-1 proteins using blocking mAb led to synergized anti-tumor immune responses in murine models.
Irrespective of these findings, whether VISTA regulates innate immune responses is not known. To address this question, we have employed the imiquimod (IMQ)-induced murine model of psoriasis, where topical application of IMQ stimulates a network of innate immune cells, such as DCs and IL-17-producing γδ TCR+ T cells, leading to psoriasiform skin inflammation10. This murine model bears strong relevance to human psoriasis, which is also mediated by the IL-23/IL-17 inflammatory axis. Multiple studies have reported the presence of both IL-17-producing TCRγδ+ T cells (γδ T cells) and CD4+ Th17 cells in human psoriatic skin11–15. Treatment in human cancer patients with IMQ (Aldara®) has resulted in similar psoriasiform dermatitis, manifested as epidermal acanthosis and parakeratosis16–18.
In this study, using the IMQ-induced psoriasis model, we have demonstrated that VISTA plays a key role in suppressing the IL-23/IL-17-mediated inflammatory axis. VISTA inhibits the activation of DCs and the production of IL-23 following TLR7 stimulation. VISTA also regulates the activation of IL-17-producing γδ T cells and CD4+ Th17 T cells, as well as the peripheral homeostasis of CD27− γδ T cell subsets that are pre-committed to produce IL-17A. Consequently, VISTA deficiency exacerbated psoriasiform inflammation. Taken together its role in suppressing CD4+ and CD8+ T cell activation, our study indicates that VISTA is a unique immune-checkpoint that regulates both innate and adaptive immune responses.
Results
Vsir−/− mice developed exacerbated psoriasiform inflammation
To address the role of VISTA in regulating innate immunity, we examined IMQ-induced psoriasiform dermatitis in wild type (WT) and Vsir−/− mice that were topically treated with 3.5% IMQ on both ears. Skin inflammatory response was quantified by measuring ear thickness. Our previous study reported chronic inflammatory phenotypes in aged (>10 month of age) Vsir−/− mice7. We first examined untreated naïve Vsir−/− mice (7–8 weeks of age) but did not observe any spontaneous skin inflammation (unpublished data). IMQ treatment in the Vsir−/− mice resulted in more severe ear swelling when compared to WT mice (Fig. 1a). Histological analyses confirmed the development of severe epidermal acanthosis in the Vsir−/− ear skin (Fig. 1b) and increased epidermal thickness (Fig. 1c). Furthermore, Vsir−/− ears showed ~20 fold increase in the area of neutrophilic abscesses (Munro’s abscess), which is a histological hallmark in human psoriasis18 (Fig. 1d).
Figure 1.
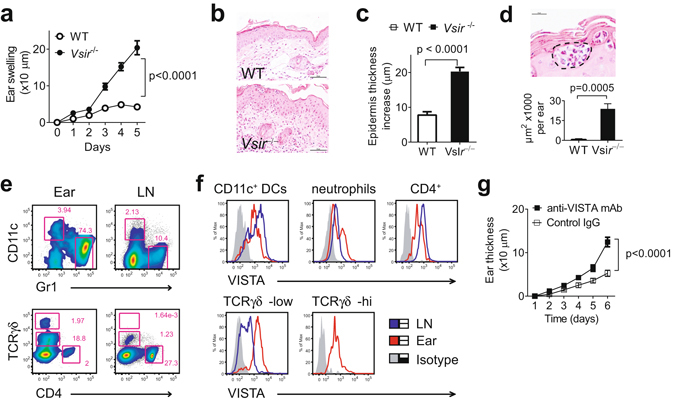
VISTA deficiency exacerbated the IMQ-induced psoriasiform inflammation. (a) WT and Vsir−/− mice were topically treated daily on each ear with 3.5% IMQ cream for 5 days. Ear thickness was measured daily. Ear swelling is shown as the increase of ear thickness when compared to day 0, and expressed as mean ± SEM (n = 18). On day 5, ears were harvested and processed for H&E staining. A representative image is shown (b). Scale bars: 50 μm. (c) The epidermal thickness was measured by examining at least 20 random fields throughout the cross section of ear tissues. The increase of epidermal thickness was calculated by subtracting the average value of naive ears. Data are pooled from 3 ears and shown as mean ± SEM. (d) A representative image of Munro’s abscess in epidermis is shown (within the dashed lines). Scale bar: 50 μm. Areas of Munro’s abscess were measured from the entire cross section of ear tissues. Data are pooled from 3 ears and shown as ± SEM. (e) The number of CD11c+ DC, Gr1+ neutrophils, γδ TCR+ and CD4+ T cells in IMQ-treated ears and ear-draining LN in WT mice was examined by flow cytometry. (f) Surface VISTA expression on these cell types was examined by flow cytometry. Representative data from at least three independent experiments are shown. (g) WT mice were treated with VISTA-specific mAb or control Ig (250 μg, on day 0, 2, and 4), in addition to the 3.5% IMQ cream. Ear swelling is shown as mean ± SEM (n = 10).
In addition to Gr1+ neutrophils, further examinations show that multiple other cell types were present in the WT psoriatic skin lesions, including CD11c+ DCs, CD4+ T cells, and γδ T cells (Fig. 1e). Similar lymphocyte populations were present in the ears of the Vsir−/− mice (data not shown). VISTA was highly expressed on these cells types (Fig. 1f), all of which are known to regulate the development of IMQ-induced psoriasiform inflammation17–20. Both γδlow and γδhigh T cells were present in the ear skin, whereas only γδlow T cells were present in the ear-draining lymph nodes (LN). VISTA expression on γδlow T cells was higher from inflamed ear skin than cells from the draining LN (Fig. 1f). To further determine whether the exacerbated psoriasiform inflammation was due to the pre-existing inflammatory environment in the Vsir−/− mice, we treated mice with a VISTA-specific mAb8. Consistent with results from the Vsir−/− mice, VISTA-specific mAb treatment significantly enhanced IMQ-induced psoriasiform inflammation (Fig. 1g), although the magnitude of disease was not as severe as those seen in the Vsir−/− mice.
Exaggerated neutrophil infiltration may potentially result from an augmented production of inflammatory cytokines and chemokines18. To investigate the inflammatory milieu, mRNA from IMQ-treated ear skin was harvested and examined by quantitative RT-PCR (Q-PCR). A panel of psoriasis-associated genes was induced in both WT and Vsir−/− skin following IMQ treatment (Fig. 2a). Vsir−/− skin showed significantly higher expression of inflammatory cytokine genes Il23p19, Il1β, Il6, Il17a, Il22, Tnfα, and Ifnγ, chemokine gene Cxcl2, and neutrophil chemotactic gene S100a9 (Fig. 2a). Serum protein levels of IL-1β, IL-6, IL-17A, IFN-γ, and CXCL2 were higher in the Vsir−/− mice (Fig. 2b). Serum levels of S100A9, TNF-α, IL-22, and IL23 were very low and were not reliably detected before or after IMQ treatment (unpublished results). Serum levels of IL-22 and TNF-α have been previously reported in mice following treatment with 5% IMQ cream on the back skin20. To further confirm that VISTA deficiency resulted in enhanced protein production of IL-22 and TNF-α, WT and Vsir−/− mice were treated with 3.5% IMQ cream on the shaved back skin. Serum was harvested six hours after the treatment, and the concentration of IL-22 and TNF-α in the serum was examined by ELISA. Consistently, IMQ treatment of back skin led to accumulation of higher levels of IL-22 and TNF-α in the serum of the Vsir−/− mice (Supplementary Fig. 1). Together, these results indicate that VISTA inhibits the expression of inflammatory cytokines and chemokines in response to IMQ.
Figure 2.
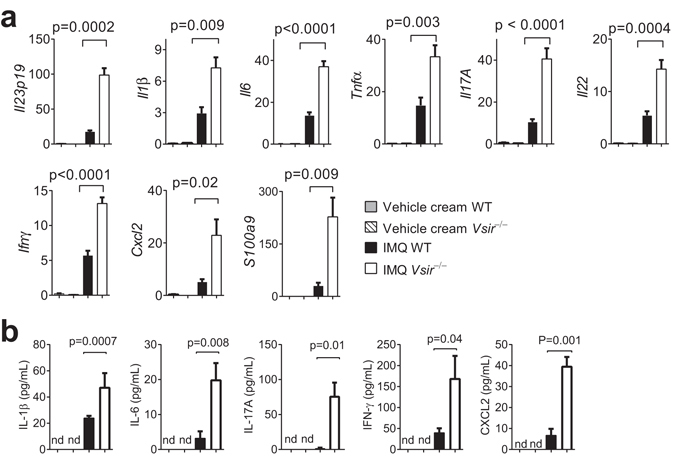
VISTA deficiency enhanced the production of inflammatory cytokines and chemokines. (a) WT and Vsir−/− mice were topically treated on both ears with 3.5% IMQ cream daily for 3 days. mRNA was isolated from ear tissues. Gene expression of inflammatory cytokines and chemokines (Il23p19, Il1b, Il6, Tnfa, Il17a, Il22, Ifng, Cxcl2, S100a9) was examined by quantitative RT-PCR. The relative mRNA abundance of each gene is normalized against the control gene Gapdh and shown as mean ± SEM (n = 4). Representative data from three independent experiments are shown. (b) Serum levels of IL-1β, IL-6, IL-17A, IFN-γ, and CXCL2 in WT and Vsir−/− mice before and after IMQ cream treatment were examined by ELISA. Data are shown as mean ± SEM (n = 4).
VISTA regulates the production of IL17 by both γδ T cells and CD4+ Th17 cells
The IL-23/IL-17 inflammatory axis has a well-established role in the development of inflammatory and autoimmune diseases21,22. In psoriatic lesions, high levels of IL-17A induce the release of neutrophil chemoattractants from keratinocytes, thus amplifying the inflammatory cascade. IL-22, another key Th17 cytokine, promotes sustained epidermal acanthosis and parakeratosis18,23,24.
Both γδ T cells and CD4+ Th17 cells are IL-17-producing effector cells that drive human psoriasis and the IMQ-induced model of psoriasiform inflammation10,12–14. To investigate the contribution of γδ T cells and CD4+ Th17 cells during the development of psoriasiform inflammation in the Vsir−/− mice, we first examined their cell number in the ear and ear-draining LN. No significant difference was observed between WT and Vsir−/− mice (Fig. 3a). Since Il17a mRNA level was significantly augmented in the Vsir−/− skin (Fig. 2), we examined IL-17A protein expression in γδ T cells and CD4+ cells isolated from IMQ-treated WT and Vsir−/− mice. Vsir−/− γδ T cells in ear skin and draining LN produced significantly higher amount of IL-17A, but similar levels of IFN-γ and TNF-α when compared to WT cells (Fig. 3b and Supplementary Fig. 2). Vsir−/− CD4+ T cells also expressed higher amount of IL-17A than WT cells in the ear and ear-draining LN, though the percentage of IL-17A positive cells was much lower than γδ T cells (Fig. 3c). Thus, both γδ T cells and Th17 cells contributed to the higher IL-17A production in IMQ-treated Vsir−/− mice. In addition to γδ T cells and CD4+ T cells, CD11b+ myeloid cells expressed IFN-γ and TNF-α (Supplementary Fig. 3a). Higher numbers of IFN-γ and TNF-α-expressing CD11b+ cells were found to infiltrate IMQ-treated ears in the Vsir−/− mice, which may contribute to the higher levels of IFN-γ and TNF-α within the inflamed ear tissues (Supplementary Fig. 3b). Similar numbers of IFN-γ and TNF-α-expressing Cd11b+ cells were found in the ear-draining LN (Supplementary Fig. 3c).
Figure 3.
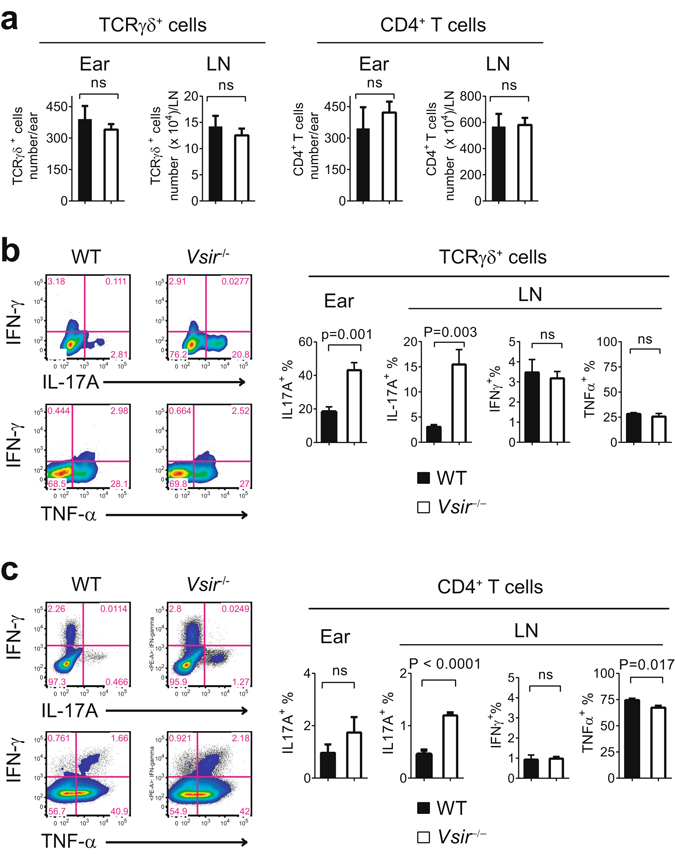
VISTA suppressed the production of IL-17A by γδ T cells and CD4+ Th17 cells following IMQ treatment. WT and Vsir−/− mice were topically treated on ears with 3.5% IMQ cream daily for 3 days. Cells from ear skin and draining LN were harvested and stimulated in vitro with PMA and Ionomycin. The number of γδ T cells and CD4+ T cells, and their expression of cytokines (IL-17A, IFN-γ, and TNF-α) were examined by surface and intracellular staining, and quantified by flow cytometry. Total cell number of recovered γδ T cells and CD4+ T cells from ear tissue and draining LN are shown (a). The percentages of γδ T cells (b) and CD4+ T cells (c) expressing cytokines IL-17A, IFN-γ, and TNF-α were examined by flow cytometry and are shown as mean ± SEM. Representative flow plots and representative data from three independent experiments are shown.
To further understand the mechanisms whereby VISTA regulates the homeostasis and activation of γδ T cells, we examined the subsets of γδ T cells in naïve WT and Vsir−/− mice25,26. The CD27+ and CD27− γδ T cell subsets are developed in thymus before exiting to the periphery25. The CD27− γδ T cells express higher levels of IL-1R and IL-23R, and are pre-committed to produce IL-17A upon TCR- or cytokine-mediated activation, whereas CD27+ γδ T cells predominantly produce IFN-γ25,27. To determine if loss of VISTA altered the development and peripheral homeostasis of γδ T cells, thymic and splenic γδ T cell subsets in WT and Vsir−/− mice were examined. Similar percentages of total γδ T cells and the Vγ4+ subset were observed in the spleen, indicating an overall normal development of γδ T cells in the absence of VISTA (unpublished data). On the other hand, higher percentage of the CD27− subset was present within the splenic but not thymic Vsir−/− γδ T cells (Fig. 4a). This result indicates that VISTA regulates the peripheral homeostasis of CD27− γδ T subsets.
Figure 4.
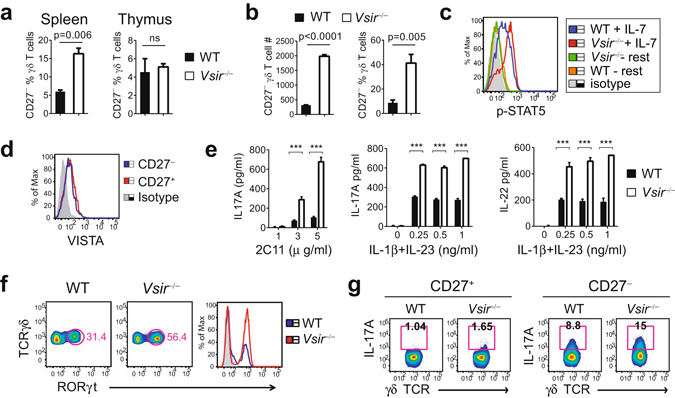
VISTA controls the peripheral homeostasis and activation of γδ T cells. (a) Naive γδ T cells were isolated from WT or Vsir−/− mice and their surface expression of CD27 was examined by flow cytometry (n = 4). The percentage of the CD27− subset is shown (mean ± SEM). (b and c) Naïve γδ T cells from WT and Vsir−/− mice (n = 4) were stimulated in vitro with IL-7 (10 ng/ml). The number and percentage of viable CD27− γδ T cells were quantified by flow cytometry after 4 days. Phosphorylated STAT5 was examined by intracellular staining and flow cytometry. (d) VISTA expression on CD27+ and CD27− naïve splenic γδ T cells was examined by flow cytometry. (e–g) Naïve splenic γδ T cells were purified from WT and Vsir−/− mice (cells pooled from 4 mice of each strain), and stimulated with either immobilized anti-CD3ε mAb (2C11) or cytokines IL-1β and IL-23. Culture supernatants were harvested after 24 hrs and the levels of IL-17A and IL-22 were examined by ELISA. Values from triplicated cultures are shown as mean ± SEM (e). Expression of RORγt at 24 hrs following IL-1β (0.25 ng/ml) and IL-23 (0.25 ng/ml) stimulation was examined by intracellular staining and flow cytometry (f). The percentage of IL-17A-producing CD27+ and CD27− γδ T subsets was examined by flow cytometry (g). ***P < 0.001. Representative results from three independent experiments are shown.
It has been shown that IL-7 preferentially expands the IL-17A-producing CD27− γδ T subset and promotes their peripheral homeostasis28,29. To determine whether the peripheral expansion of CD27− γδ subset in Vsir−/− mice resulted from unrestricted IL-7 receptor signaling, naive WT and Vsir−/− γδ T cells were stimulated in vitro with IL-7 for 4 days, and the number of CD27− γδ T cells was examined. IL-7 treatment expanded both WT and Vsir−/− CD27− γδ T subsets. However, Vsir−/− CD27− γδ T cells were hyper-proliferative than WT cells, resulting in a ~10 fold increase in viable cell number and a ~5 fold increase in percentage within the total expanded γδ T cell population (Fig. 4b). To determine how VISTA regulates the IL-7 receptor signaling, the phosphorylation status of STAT3 and STAT5 was examined, since both proteins are known to mediate IL-7 receptor signaling30. Our results show that higher level of phosphorylated STAT5 was induced in Vsir−/− γδ T cells than WT cells following IL-7 stimulation (Fig. 4c). On the other hand, similar level of phosphorylated STAT3 was observed (data not shown). These results indicate that VISTA restricts the activation of STAT5 but not STAT3 downstream of IL-7R signaling in γδ T cells.
γδ T cells are activated by both TCR-specific stimuli and inflammatory cytokines such as IL-1β and IL-2326. Since VISTA is expressed on γδ T cells (Figs 1f and 4d), it is possible that VISTA suppresses the activation of γδ T cells in an autonomous manner. To test this, naïve splenic WT and Vsir−/− γδ T cells were isolated and stimulated with either TCR crosslinking, or cytokines IL-1β and IL-23. Vsir−/− γδ T cells produced more IL-17A and IL-22 than WT cells (Fig. 4e). Activated Vsir−/− γδ T cells consistently expressed higher level of RORγt, a ROR family transcription factor that binds to and activates the Il-17a promoter (Fig. 4f)31,32. Both CD27+ and CD27− Vsir−/− γδ T subsets expressed more IL-17A than WT cells following IL-1β and IL-23 stimulation. These data indicate that in addition to regulating the peripheral homeostasis of CD27− γδ T cells, VISTA directly controls the activation of γδ T cells (Fig. 4g).
VISTA expression on dendritic cells suppresses IMQ-induced TLR7 signaling and IL-23 production
In both human psoriasis and murine models of psoriasiform inflammation, IL-23 is predominantly produced by myeloid DCs and promotes the expansion of pathogenic IL-17A-producing γδ T cells and CD4+ Th17 cells17,20,24. Since an elevated expression of IL23 gene was observed in IMQ-treated ear skin from Vsir−/− mice (Fig. 2), we hypothesize that VISTA expression on DCs suppresses IMQ/TLR7-induced IL-23 production. To test this hypothesis, WT and Vsir−/− mice were treated with 3.5% IMQ on the ears for 4 days. Ear tissues were harvested and the expression of IL-23p19 in ear CD11c+ DCs were examined by flow cytometry. Significantly higher percentages of Vsir−/− DCs expressed IL-23p19 protein than WT cells in ears and ear-draining LNs (Fig. 5a), whereas the number of DCs present in ears and the draining LNs was similar (Fig. 5b).
Figure 5.
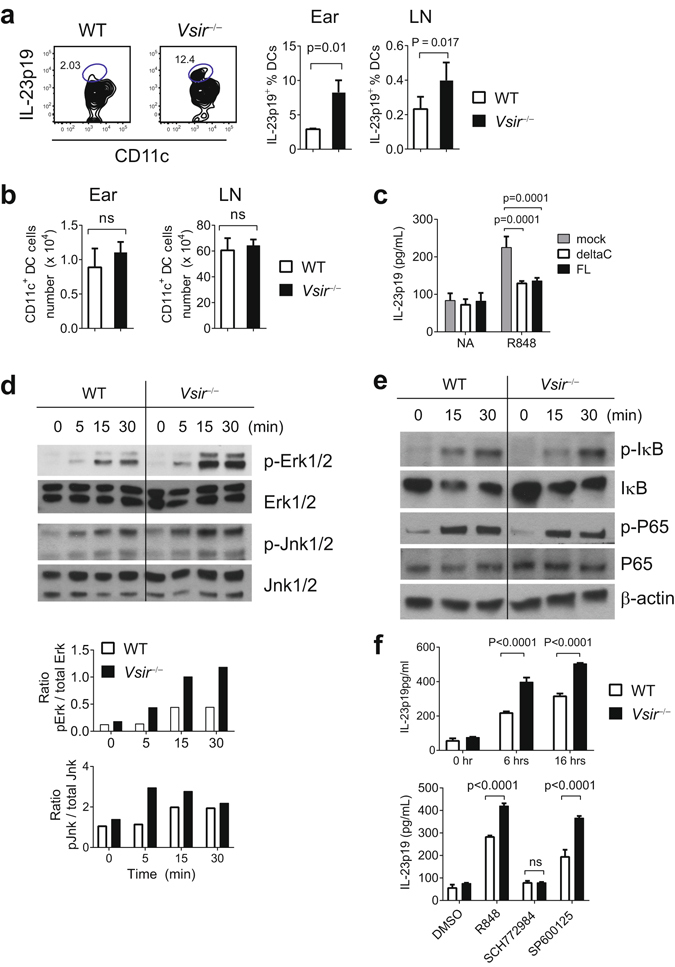
VISTA negatively regulates IMQ-induced activation of DCs and the production of IL-23. WT and Vsir−/− mice were treated on ears with 3.5% IMQ for 3 days. Cells from ear tissues and the ear-draining cervical LNs were harvested. Cells were stimulated with PMA and Ionomycin in vitro for 3 hrs. The expression of IL-23p19 in CD11c+ DCs was examined by flow cytometry. The percentages of IL-23p19-expressing DCs were quantified and shown as mean ± SEM (n = 6) in (a). The number of total CD11c+ DCs from ear tissue and draining LN is shown as mean ± SEM (n = 5) in (b). To determine whether ectopic expression of VISTA suppresses TLR7-induced IL-23 production, Vsir−/− BM-derived DC were transduced with lentivirus expressing full-length (FL), or mutant VISTA lacking the cytoplasmic tail (deltaC), or GFP control protein. After culture with GM-CSF (20 ng/ml) for 7 days, cells were stimulated with R848 (5 μg/ml) for 7 hrs. Culture supernatant was harvested and secreted IL-23p19/p40 was examined by ELISA (c). To examine TLR7 signaling in DCs, CD11c+ DCs were purified from the spleens of naïve Rag1−/− and Vsir−/−Rag1−/− mice, and stimulated with R848 (5 μg/ml) for indicated amount of time. Total cell lysates were prepared and examined for the levels of phosphorylated Erk1/2 and Jnk1/2 by western blotting (d). The ratio of phosphorylated versus total Erk1/2 and Jnk1/2 was calculated based on the total protein level from the same lysate run on a parallel gel (d). The activation of NF-κB signaling was examined by western blotting the level of phosphorylated and total IκB, as well as phosphorylated and total NF-κB p65 subunit (e). To determine whether Erk and Jnk were required for the production of IL-23, Splenic DCs were isolated from naïve WT and Vsir−/− mice, and stimulated with R848 (5 μg/ml) in the presence of Erk1/2 inhibitor (SCH772984, 10 μM), or Jnk1/2 inhibitor (SP600125, 10 μM), or DMSO solvent control for overnight. Culture supernatant was collected and secreted IL-23p19/p40 was quantified by ELISA. Values from triplicated cultures are shown as mean ± SEM in (f). Representative results from two to three independent experiments were shown.
Although there was similar expression of DC activation markers such as CD80, CD86, and CD40 on naive WT and Vsir−/− splenic DCs (Supplementary Fig. 4), it could not be formally excluded that an altered DC development in Vsir−/− mice may contribute to the hyper-response of DCs. To directly demonstrate the role of VISTA in suppressing DC cytokine production, we ectopically expressed either full-length VISTA, or a mutant VISTA lacking the cytoplasmic tail (deltaC), or GFP control protein in GM-CSF cultured Vsir−/− BM-derived DCs. Following stimulation with a TLR7/8 agonist R848, secreted IL-23 was examined by ELISA. Expression of both FL-VISTA and deltaC-VISTA significantly suppressed IL-23 production in Vsir−/− BMDCs (Fig. 5c). This result strongly supports the role of VISTA in inhibiting TLR7-mediated DCs activation and IL-23 expression. Furthermore, since the cytoplasmic tail is not required for the suppressive activity of VISTA, this result indicates that VISTA engages an unknown receptor, which in turn delivers an inhibitory signal.
The Il-23 promoter contains binding sites for AP-1 and NF-κB33. It has been shown that TLR4 stimulation in macrophages and DCs activates MAP kinases (Erk1/2, Jnk1/2, and p38), which are critical for the activation of transcription factor AP1 and the expression of Il-23p19 gene33,34. Furthermore, Erk1/2 inhibitor suppressed IL-23 production in DCs stimulated with TLR agonists34. To determine if VISTA regulates the activation of NF-κB and MAPK pathways, total cell lysates were prepared from WT and Vsir−/− splenic DCs that have been stimulated with R848 and examined by western blotting (Fig. 5d). R848 stimulation induced significantly higher levels of Erk1/2 phosphorylation and a moderately increased phosphorylation of Jnk1/2 in Vsir−/− DCs (Fig. 5d). On the contrary, similar levels of Iκ-B degradation and phosphorylation of NF-κB p65 were observed, indicating that the NF-κB pathway was not significantly altered in the absence of VISTA (Fig. 5e). Similar levels of phosphorylated p38 were present in lysates from WT and Vsir−/− BMDCs (unpublished data).
To further confirm the critical role of Erk1/2 in IL-23 production in DCs, WT and Vsir−/− DCs were purified from naïve Rag1−/− and Vsir−/−Rag1−/− mice and stimulated ex vivo with R848 in the presence of inhibitors of Erk1/2 or Jnk1/2, or solvent control (Fig. 5). Consistent with our hypothesis, Vsir−/− DCs produced higher level of IL-23p19/p40 than WT cells (Fig. 5f). Erk1/2 inhibitor completely abolished the ability of both WT and Vsir−/− DCs to produce IL-23, whereas Jnk1/2 inhibitor was moderately effective (Fig. 5f). Taken together, these results suggest that VISTA negatively regulates TLR7 signaling and inhibits the expression of IL-23 in DCs via suppressing the activation of Erk1/2.
Discussion
The IL-23/IL-17-mediated inflammatory axis plays a critical role in many inflammatory disorders and autoimmune diseases such as psoriasis, rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease22. In the current study we have demonstrated a novel role of VISTA in regulating this inflammatory axis. In the IMQ-induced psoriasis model, VISTA deficiency augmented the inflammatory responses of DCs, γδ T cells, and Th17 cells, resulting in exacerbated psoriasiform dermatitis.
In both human psoriasis and murine model of psoriasiform inflammation, one of the main initial responders are IL-23-producing DCs17. IL-23 promotes the expansion and activation of IL-17-producing CD4+ Th17 cells and γδ T cells. This inflammatory milieu recruits and activates additional effector cells such as inflammatory monocytes and neutrophils, which amplify inflammation and drive epidermal hyperplasia. Our results indicate that VISTA controls the production of IL-23 in DCs via inhibiting the activation of Erk1/2. We predict that strategies that enhance VISTA-regulated inhibitory signaling will dampen IL-23-mediated inflammatory axis and benefit the treatment of not only human psoriasis, but also other inflammatory diseases driven by IL-23.
In addition to regulating the activation of DCs, VISTA negatively regulates IL-7-mediated homeostasis of CD27− γδ T cells, as well as the activation of γδ T cells in response to TCR-mediated or IL-23/IL-1β-mediated stimuli. These effects collectively contribute to the exaggerated psoriasiform inflammation in the Vsir−/− mice. It is noted that VISTA expression on γδ T cells was upregulated within the psoriatic skin when compared to the draining LN, indicating a potential feedback mechanism whereby inflammatory cytokines or other mediators may upregulate VISTA expression to dampen inflammation.
In addition to VISTA, other immune-checkpoint proteins including Programmed death-1 (PD-1) and B and T lymphocyte attenuator (BTLA) also regulate IL17 expression in γδ T cells35,36. Both receptors are expressed on γδ T cells and restrict their activation. PD-1 and BTLA knockout mice developed more severe psoriasiform dermatitis in the IMQ model35,36. These results warrant future efforts to determine whether these immune-checkpoint proteins act synergistically to regulate the function of γδ T cells.
In addition to psoriasis, the IL-23/IL-17 inflammatory axis regulates disease development in murine experimental autoimmune encephalomyelitis (EAE) and human autoimmune disease multiple sclerosis26,37,38. Previous studies have shown that VISTA genetic deletion or VISTA-blocking mAb treatment exacerbated disease in the EAE model1,7,9. Since both IL-17-producing γδ T cells and Th17 cells have been implicated as effector cells during EAE39,40, our current study provides additional mechanisms whereby VISTA regulates this disease.
In the context of cancer therapy, the IL-23/IL-17 inflammatory axis regulates the inflammatory tumor microenvironment (TME). Earlier studies have demonstrated the tumor-promoting role of IL-2341,42, whereas both tumor-promoting and tumor-inhibitory roles of IL-17 have been reported43–47. Results from this study indicate that blocking VISTA promotes the inflammatory responses mediated by IL-23/IL-17, particularly in the context of TLR stimulation. Our previous study has shown that VISTA-blocking mAb synergized with a tumor peptide vaccine and TLR agonists as adjuvants8. Future studies are warranted to determine whether the exacerbated IL23/IL17 inflammatory axis positively or negatively contributes to the anti-tumor immunity following VISTA blockade.
In conclusion, this study reveals a novel anti-inflammatory role of VISTA through regulating the IL-23/IL-17 inflammatory axis. Our findings distinguish VISTA from other immune-checkpoint proteins CTLA-4 and PD-1, and establish VISTA as a regulator of both innate and adaptive immunity1,4,6–9. Therapeutic agents have been developed to harness the immune-suppressive functions of immunecheckpoint proteins. For example, a fusion protein CTLA4-Ig (Abatacept) has been used in the clinic for treating autoimmune diseases such as rheumatoid arthritis48. Similarly, local overexpression of PD-L1-Ig or administration of purified PD-L1-Ig has been shown to promote allograft survival in murine models49–52. Our study indicates that enhancing the anti-inflammatory function of VISTA may benefit the treatment of a variety of inflammatory and autoimmune disorders.
Materials and Methods
Mice
C57BL/6 mice were purchased from Charles River Laboratories. Vsir−/− mice on a fully backcrossed C57BL/6 background were as described7,9. All animals were maintained in a pathogen-free facility at the Medical College of Wisconsin (Milwaukee, WI). All animal protocols were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. All methods were performed in accordance with the relevant guidelines and regulations.
Abs, cell lines, and reagent
Antibodies specific for γδ-TCR (GL3), CD27 (LG.3A10), IL-17A (eBio17B7), CD4 (GK1.5), CD8 (53-6.7), CD11b (M1/70), CD11c (N418), IFN-γ (XMG1.2), TNF-α (MP6-XT22), anti-CD3e (2C11) were purchased from BioLegend (San Diego, CA). Recombinant murine IL-1β and IL-23 were from Peprotech (Rocky Hill, NJ). Antibodies specific for p-STAT3, p-STAT5, p-Erk1/2 (Thr202/Tyr204), Erk1/2, p-Jnk1/2 (Thr183/Tyr185), Jnk1/2, and IκB were purchased from Cell Signaling Technology (Boston, MA). The VISTA-specific mAb was as described previously8. Erk1/2 inhibitor (SCH772984) was obtained from Selleckchem (Houston, TX) and Jnk1/2 inhibitor (SP600125)was obtained from Invivogen (San Diego, CA).
Imiquimod (IMQ)-induced psoriasiform inflammation model
WT and Vsir−/− mice were treated daily on both ears with 50 mg of 3.5% IMQ cream, which was prepared by diluting the 5% IMQ cream (Taro Pharmaceuticals, New York, NY) using the vehicle cream (Vanicream; Pharmaceutical Specialties, Cleveland, GA). Ear thickness was measured by using an Ozaki caliper (model G-A1-0.4 N) (Neill-Lavielle Supply, Louisville, KY). For histopathological analysis, H&E staining was performed on formaldehyde fixed, paraffin-embedded skin samples. Images were acquired using an INFINITY3-1C digital camera (Lumenera, Ottawa, Canada) attached to a Carl Zeiss microscope.
To quantify the amount of Munro’s abscess, the entire cross section of the ear tissue was examined. Munro’s abscess was identified as areas within the epidermis that were occupied with aggregated neutrophils. The areas of Munro’s abscess were quantified by manually defining the boundaries and measuring the area using the Image J software.
The thickness of the epidermis was also quantified using the Image J software. More than 20 random fields were measured throughout the entire cross section of the ear tissue. The increase of epidermal thickness was calculated by subtracting the average epidermal thickness of naïve ear tissues.
Quantitative real-time PCR (RT-PCR)
Total RNA of ear skin was prepared using an RNeasy Fibrous Tissue Kit (Qiagen, Hilden, Germany). Quantitative RT-PCR was performed via StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA).
Primer sequences are described in the following: Il23-p19 (forward: CCAGCAGCTCTCTCGGAATC; reverse: TCATATGTCCCGCTGGTGC); Il1b (forward: CGCAGCAGCACATCAACAAGAGC; reverse: TGTCCTCATCCTGGAAGGTCCACG); Il6 (forward: GCAGAAAAAGGCAAAGAATC; reverse: CTACATTTGCCGAAGAGC); Tnfa (forward: AGGCAGTCAGATCATCTTC; reverse: TTATCTCTCAGCTCCACG); Il17A (forward: GAGCTTCCCAGATCACAGAG; reverse: AGACTACCTCAACCGTTCCA); Il22 (forward: CTG CTT CTC ATT GCC CTG TG; reverse: AGC ATA AAG GTG CGG TTG AC); Ifnγ (forward: GTTACTGCCACGGCACAGTCATTG; reverse: ACCATCCTTTTGCCAGTTCCTCCAG); Cxcl2 (forward: GAAGTCATAGCCACTCTCAAGG; reverse: CTTCCGTTGAGGGACAGC); S100a9 (forward:ATACTCTAGGAAGGAAGGACACC; reverse: TCCATGATGTCATTTATGAGGGC); gapdh (forward: GTGGAGTCATACTGGAACATGTAG; reverse: AATGGTGAAGGTCGGTGTG).
Generation of BM-derived DC and lentiviral transduction
Bone marrow (BM) cells were harvested from the femur and tibia from naive Vsir−/− mice, and cultured in GM-CSF (20 ng/ml). On day 3, cells were infected with lentivirus expressing full-length (FL), or mutant VISTA lacking the cytoplasmic tail (deltaC), or GFP control protein. Infected cells were selected in puromycin (5 μg/mL) for additional 4 days. On day 7, cells were stimulated with R848 (5 μg/ml) for 7 hrs. Culture supernatant was harvested and secreted IL-23p19/p40 was examined by ELISA (Biolegend Inc, San Diego, CA).
Flow Cytometry and data analysis
CD11c+ DCs and γδ T cells were purified from spleens of naïve WT and Vsir−/− mice using MACS Microbead kits (Miltenyi Biotech, San Diego, CA). DCs were positively selected using the Cd11c Microbeads (130-108-338). γδ T cells were purified using the TCRγδ+ T Cell Isolation Kit (130-092-125). Purity was examined by flow cytometry and was typically >90%.
Cells from ear skin were harvested following digestion at 37 °C for 45 min with Liberase TL (Roche, Pleasanton, CA) and Dnase (Sigma, St Louis) to obtain single cell suspensions. To detect intracellular cytokine expression, cells were stimulated for 4 hrs in complete RPMI medium containing PMA(50 μg/ml), ionomycin (1 μg/ml), 10% FBS, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, 1% penicillin-streptavidin, 1x monensin, 1x Brefeldin A (BioLegend, San Diego, CA). Cells were then fixed with 1% paraformaldehyde, permeabilized with 0.5% saponin, stained for intracellular cytokines, and analyzed by flow cytometry.
Flow cytometry was performed using an Acuri C6 or LSR II (BD Biosciences, San Jose, CA). Data were analyzed with FlowJo version 10.0.7 analysis software (Tree Star, San Carlos, CA).
Graphs and Statistical analysis
All graphs and statistical analysis were generated using Prism 6 (GraphPad Software, Inc., San Diego, CA). Student’s t test (two tailed) or ANOVA was used for data analyses. A P-value less than 0.05 is considered as statistically significant.
Electronic supplementary material
Acknowledgements
This study was supported by research funding from NCI R01 CA164225 (L.W.), Advancing A Healthier Wisconsin Research and Education Program (AHW REP) fund (L.W.), Ann’s Hope Foundation from the Medical College of Wisconsin Cancer Center (L.W.), the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Cancer Research Program under Award No. W81XWH-14-1-0587 (L.W.), Worldwide Cancer Research Foundation (UK) research grant 16-1161 (L.W.). This work is also supported by the Uehara Foundation (Y.I.), NIH R01 AI102893 (S.M.), NCI R01 CA179363 (S.M.), Nicholas Family Foundation (S.M.), and Gardetto Family (S.M.), NIH R01 AI089805 (Y.H.H.), NIH R01AR063091 NIAMS (S.T.H.), NIH R01 CA120777 (M.J.T.).
Author Contributions
L.W. designed and supervised this research project, analyzed data and wrote the manuscript. S.M. and Y.H.H. provided reagents and protocols, and edited the manuscript. S.T.H., and M.J.T. provided consultation. N.L., Y.Y., W.X., N.A., Y.I., X.W., Y.F., H.M., and M.O. performed experiments and analyzed data.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Na Li and Wenwen Xu contributed equally to this work.
A correction to this article is available online at https://doi.org/10.1038/s41598-018-22151-w.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01411-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wang L, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lines JL, et al. VISTA Is an Immune Checkpoint Molecule for Human T Cells. Cancer Res. 2014;74:1924–1932. doi: 10.1158/0008-5472.CAN-13-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flies DB, Wang S, Xu H, Chen L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol. 2011;187:1537–1541. doi: 10.4049/jimmunol.1100660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flies, D. B. et al. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J Clin Invest (2014). [DOI] [PMC free article] [PubMed]
- 7.Wang L, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci USA. 2014;111:14846–14851. doi: 10.1073/pnas.1407447111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Mercier I, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014;74:1933–1944. doi: 10.1158/0008-5472.CAN-13-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu, J. et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci USA (2015). [DOI] [PMC free article] [PubMed]
- 10.van der Fits L, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182:5836–5845. doi: 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- 11.Gray EE, Suzuki K, Cyster JG. Cutting edge: Identification of a motile IL-17-producing gammadelta T cell population in the dermis. J Immunol. 2011;186:6091–6095. doi: 10.4049/jimmunol.1100427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai Y, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laggner U, et al. Identification of a novel proinflammatory human skin-homing Vgamma9Vdelta2 T cell subset with a potential role in psoriasis. J Immunol. 2011;187:2783–2793. doi: 10.4049/jimmunol.1100804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JH, et al. Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1-high T cells. J Allergy Clin Immunol. 2016;137:1466–1476 e1463. doi: 10.1016/j.jaci.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 15.Cibrian D, et al. CD69 controls the uptake of L-tryptophan through LAT1-CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat Immunol. 2016;17:985–996. doi: 10.1038/ni.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajan N, Langtry JA. Generalized exacerbation of psoriasis associated with imiquimod cream treatment of superficial basal cell carcinomas. Clin Exp Dermatol. 2006;31:140–141. doi: 10.1111/j.1365-2230.2005.01938.x. [DOI] [PubMed] [Google Scholar]
- 17.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339–1350. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 18.Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–255. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pantelyushin S, et al. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest. 2012;122:2252–2256. doi: 10.1172/JCI61862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wohn C, et al. Langerin(neg) conventional dendritic cells produce IL-23 to drive psoriatic plaque formation in mice. Proc Natl Acad Sci USA. 2013;110:10723–10728. doi: 10.1073/pnas.1307569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Croxford AL, Kulig P, Becher B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. 2014;25:415–421. doi: 10.1016/j.cytogfr.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. 2014;13:21–38. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- 24.Sutton CE, et al. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 25.Ribot JC, Silva-Santos B. Differentiation and activation of gammadelta T Lymphocytes: Focus on CD27 and CD28 costimulatory receptors. Adv Exp Med Biol. 2013;785:95–105. doi: 10.1007/978-1-4614-6217-0_11. [DOI] [PubMed] [Google Scholar]
- 26.Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ribot JC, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol. 2009;10:427–436. doi: 10.1038/ni.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baccala R, et al. Gamma delta T cell homeostasis is controlled by IL-7 and IL-15 together with subset-specific factors. J Immunol. 2005;174:4606–4612. doi: 10.4049/jimmunol.174.8.4606. [DOI] [PubMed] [Google Scholar]
- 29.Michel ML, et al. Interleukin 7 (IL-7) selectively promotes mouse and human IL-17-producing gammadelta cells. Proc Natl Acad Sci USA. 2012;109:17549–17554. doi: 10.1073/pnas.1204327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin JX, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2:331–339. doi: 10.1016/1074-7613(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 31.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 32.Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 33.Liu W, et al. AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem. 2009;284:24006–24016. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brereton CF, Sutton CE, Lalor SJ, Lavelle EC, Mills KH. Inhibition of ERK MAPK suppresses IL-23- and IL-1-driven IL-17 production and attenuates autoimmune disease. J Immunol. 2009;183:1715–1723. doi: 10.4049/jimmunol.0803851. [DOI] [PubMed] [Google Scholar]
- 35.Bekiaris V, Sedy JR, Macauley MG, Rhode-Kurnow A, Ware CF. The inhibitory receptor BTLA controls gammadelta T cell homeostasis and inflammatory responses. Immunity. 2014;39:1082–1094. doi: 10.1016/j.immuni.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imai Y, et al. Cutting Edge: PD-1 Regulates Imiquimod-Induced Psoriasiform Dermatitis through Inhibition of IL-17A Expression by Innate gammadelta-Low T Cells. J Immunol. 2015;195:421–425. doi: 10.4049/jimmunol.1500448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 38.Blink SE, Miller SD. The contribution of gammadelta T cells to the pathogenesis of EAE and MS. Curr Mol Med. 2009;9:15–22. doi: 10.2174/156652409787314516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malik S, Want MY, Awasthi A. The Emerging Roles of Gamma-Delta T Cells in Tissue Inflammation in Experimental Autoimmune Encephalomyelitis. Front Immunol. 2016;7:14. doi: 10.3389/fimmu.2016.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teng MW, et al. IL-23 suppresses innate immune response independently of IL-17A during carcinogenesis and metastasis. Proc Natl Acad Sci USA. 2010;107:8328–8333. doi: 10.1073/pnas.1003251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smyth MJ, Teng MW. Targeting the IL-12/IL-23 axis: An alternative approach to removing tumor induced immune suppression. Oncoimmunology. 2014;3:e28964. doi: 10.4161/onci.28964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muranski P, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Y, et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208:491–503. doi: 10.1084/jem.20100269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–256. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maniati E, Soper R, Hagemann T. Up for Mischief? IL-17/Th17 in the tumour microenvironment. Oncogene. 2010;29:5653–5662. doi: 10.1038/onc.2010.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silva-Santos B, Serre K, Norell H. gammadelta T cells in cancer. Nat Rev Immunol. 2015;15:683–691. doi: 10.1038/nri3904. [DOI] [PubMed] [Google Scholar]
- 48.Fiocco, U. et al. Co-stimulatory modulation in rheumatoid arthritis: The role of (CTLA4-Ig) abatacept. Autoimmun Rev (2008). [DOI] [PubMed]
- 49.Ozkaynak E, et al. Programmed death-1 targeting can promote allograft survival. J Immunol. 2002;169:6546–6553. doi: 10.4049/jimmunol.169.11.6546. [DOI] [PubMed] [Google Scholar]
- 50.Dudler J, et al. Gene transfer of programmed death ligand-1.Ig prolongs cardiac allograft survival. Transplantation. 2006;82:1733–1737. doi: 10.1097/01.tp.0000250757.69384.79. [DOI] [PubMed] [Google Scholar]
- 51.Watson MP, George AJ, Larkin DF. Differential effects of costimulatory pathway modulation on corneal allograft survival. Invest Ophthalmol Vis Sci. 2006;47:3417–3422. doi: 10.1167/iovs.05-1597. [DOI] [PubMed] [Google Scholar]
- 52.Nosov M, et al. Role of lentivirus-mediated overexpression of programmed death-ligand 1 on corneal allograft survival. Am J Transplant. 2012;12:1313–1322. doi: 10.1111/j.1600-6143.2011.03948.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.