

Abstract
Familial platelet disorder with predisposition to acute myeloid leukaemia (FPD/AML) is characterized by germline RUNX1 mutations, thrombocytopaenia, platelet dysfunction and a risk of developing acute myeloid and in rare cases lymphoid T leukaemia. Here, we focus on a case of a man with a familial history of RUNX1 R174Q mutation who developed at the age of 42 years a T2‐ALL and, 2 years after remission, an AML‐M0. Both AML‐M0 and T2‐ALL blast populations demonstrated a loss of 1p36.32‐23 and 17q11.2 regions as well as other small deletions, clonal rearrangements of both TCRγ and TCRδ and a presence of 18 variants at a frequency of more than 40%. Additional variants were identified only in T2‐ALL or in AML‐M0 evoking the existence of a common original clone, which gave rise to subclonal populations. Next generation sequencing (NGS) performed on peripheral blood‐derived CD34+ cells 5 years prior to T2‐ALL development revealed only the missense TET2 P1962T mutation at a frequency of 1%, which increases to more than 40% in fully transformed leukaemic T2‐ALL and AML‐M0 clones. This result suggests that TET2 P1962T mutation in association with germline RUNX1 R174Q mutation leads to amplification of a haematopoietic clone susceptible to acquire other transforming alterations.
Keywords: FPD/AML, predisposition to leukaemia, RUNX1, TET2, AML‐M0, T2‐ALL
Introduction
Acquired genetic alterations in RUNX1 are frequently associated with numerous myeloid malignancies, especially acute myeloid leukaemia (AML) (40% of FAB M0 immature AMLs 1) and more rarely with T cell acute lymphoblastic leukaemia (T‐ALL) (25% of early thymic immature T‐ALL (ETP‐ALL) 2).
Germline RUNX1 mutations are found in FPD/AML characterized by thrombocytopaenia, and a 35% life‐time risk of developing myelodysplastic syndrome (MDS) and/or AML. 3 T‐ALL development has also been reported in rare cases 4, 5. In AML, contrary to T‐ALL, the leukaemic transformation is almost always associated with a somatic RUNX1 mutation on the second allele 4, 6, 7. Acquired mutations in CDC25C and GATA2 have also been identified at a high frequency but only in a Japanese cohort 4, 7, 8, 9, suggesting that environmental and/or ethnic factors may play an important role in leukaemia transformation. Somatic mutations in FLT3, PHF6, KIT, KRAS, RAD21, BCOR, BCORL1, CBL, CEBPA, MPL, TP53, WT1, SRSF2, DNMT3A, TET2 and ASXL1 were described in patients who developed AML/MDS and mutations in PHF6, WT1, NOTCH1, FLT3 and ASXL1 in patients with T‐ALL 4, 5, 7, 9.
Materials and methods
Patient samples
Biologic samples of one FPD/AML pedigree were collected between 2006 and 2013 after informed consent, in accordance with the Declaration of Helsinki. Genomic DNA was extracted from fibroblasts, CD34+ cells, peripheral blood mononuclear and bone marrow total cells using kit Qiagen (Les Ulis, France).
CGH arrays
Comparative genomic hybridization (CGH) arrays were performed on human CGH 2x400K (G4448A) (Agilent, Les Ulis, France) by hybridization of sample versus normal‐matched commercial reference. CGH data and protocols have been submitted to ArrayExpress at the EBI with the accession number E‐MTAB‐4623.
Exome capture and sequencing (v4+UTR, 70 Mb)
Library preparation, capture, sequencing and variant detection have been carried out by IntegraGen. Exons were captured from blood DNA using Agilent SureSelect Human All Exon v5–70 Mb kit and sequenced on IlluminaHiSEQ 2000 instrument as previously described 10. WES data and prodtocols have been submitted to ArrayExpress at the European Bioinformatics Institute (EBI) with the accession number E‐MTAB‐4679. Variants present in the internal control and in public databases at a frequency of >1% were excluded, and only non‐synonymous mutations predicted by PolyPhen2 to be probably or possibly deleterious were analysed.
Targeted NGS
The mutated regions were amplified by PCR using primers listed in Table S1. PCR products were end‐repaired, extended with an ‘A’ base on the 3′ end, ligated with indexed paired‐end adaptors (NEXTflex, Bioo Scientific, Saint Marcel, France) using the Bravo Platform (Agilent) and amplified by PCR for four cycles. Amplicon libraries were sequenced in an IlluminaMiSeq flow cell using the onboard cluster method, as paired‐end sequencing (2 × 250 bp reads) (Illumina, San Diego, CA, USA).
Results
We focus on a man with a familial history of RUNX1 R174Q mutation, previously referenced as AII‐1 11, 12, who developed at the age of 42 years an EGIL (European Group of Immunological Characterization of Leukemia) T2‐ALL with a loss of the 1p36 and 17q12 regions and the ASXL1 R693* mutation in the leukaemic clone. The patient was pre‐treated with corticosteroids and then treated according to the French GRAALL2003 protocol based on a polychemotherapy 13. As 15 days later the blasts were still present in the bone marrow, a second induction course with idarubicin and cytarabine was performed. The patient achieved complete remission 5.
Two years later, he was admitted to the hospital for relapse with 87% blasts in peripheral blood. The clinical course was aggressive and the patient died 1 month after hospitalization. The blasts exhibited basophilic cytoplasm and a lack of azurophilic granules (Figure S1A) and were characterized by immature marker expression (CD34+, CD13+, HLA‐DR+, CD33low, CD7+ and CD56low) and absence of cytoplasmic and surface expression of CD3 and cyMPO (Figure S1B, C). This phenotype was initially described by Suzuki as myeloid/NK‐cell precursor leukaemia 14 and represents a distinct subtype of AML‐M0 15. In contrast to previously described cases, we detected low expression of TdT, CD10, CD19, cyCD79a and CD5 (Figure S1B, C).
To investigate whether initial (T2‐ALL) and relapsed (AML‐M0) leukaemic blasts originated from the same clone, we performed first aCGH on both blast populations. In both samples, aCGH revealed loss of 1p36.32‐23 and of 17q11.2 and other small deletions (Table S2). One additional deletion on chromosome 11 and five duplicated regions on chromosome 14 were found exclusively in T2‐ALL (Table S2). In the AML‐M0 blasts, the loss of the 1p36.32‐23 region was associated with two large amplifications at the same break point (1p36.33‐32 and 1p36.23‐1p31.2) surrounding the deleted region (Figure S2). Other additional deletions and duplications were identified (Table S2). These results suggested that T2‐ALL and AML‐M0 blasts originated from the same clone, but harboured also specific features acquired during disease progression. Furthermore, identical clonal rearrangements of TCRγ (Vγ9‐Jγ1.1) and TCRδ (Dδ2‐Jδ1 and Dδ2‐Jδ3) (Figure S3 and data not shown) were identified in both T2‐ALL and AML‐M0 blasts strongly supporting their clonal affiliation. Finally, analysis of WES revealed 34 variants in the T2‐ALL and 49 in the AML‐M0. Interestingly, 20 variants were common between these two cell populations (Table 1). Taking into consideration that T2‐ALL blasts represented about 58% of the sequenced DNA and AML‐M0 blasts 87%, 18 genes were found to be mutated in the original clone at a frequency of more than 40% (or about 100% for X‐linked genes) (Table 1), suggesting that they were all present as heterozygous mutations. The results were confirmed by NGS (Table S3). Alterations in genes frequently associated with myeloid and/or lymphoid malignancies, such as PHF6, EZH2, ASXL1, JAK1, JAK3, TET2 and NOTCH1, were found. We also identified nine probably deleterious (CXCR4, IRS4, HCFC1, CTNND2, NRF1, PTPN13, PPP2R2B, MSRB2 and MYO1D) and two possibly deleterious de novo variants (EPHA10, PCNXL2) in genes that were also found with a frequency of >40% but had not previously been causally linked to AML/MDS. A second heterozygous NOTCH1 K1488N mutation occurred only in the T2‐ALL (Table 2), while the initial NOTCH1 K1607delinsNAK mutation became homozygous in AML‐M0 (Table 1). PRB4 mutated with an approximate 40% frequency only in T2‐ALL could be a passenger mutation. In the AML‐M0, only one variant was present at 50% but absent in T2‐ALL, SUDS3 R325H (Table 3). SUDS3 is a subunit of the histone deacetylase‐dependent SIN3A corepressor complex interacting with RUNX1 and its genetic alterations could lead to a deregulation of RUNX1 transcriptional program. No acquired RUNX1 mutation was found in T2‐ALL and AML‐M0.
Table 1.
Gene mutations common to both T2‐ALL and AML‐M0
| Gene, chr | Protein | Variant frequency in T2‐ALL (%) | Variant frequency in AML‐M0 (%) |
|---|---|---|---|
| PHF6, chrX | S247Y | 63 | 96 |
| IRS4, chrX | G1154V | 49 | 92 |
| HCFC1, chrX | R1982H | 55 | 89 |
| CXCR4, chr2 | V116Afs*55 | 36 | 80 |
| EZH2, chr7 | S669N | 34 | 44 |
| EPHA10, chr1 | R268H | 30 | 29† |
| CTNND2, chr5 | R82* | 28 | 48 |
| NRF1, chr7 | R206W | 27 | 50 |
| ASXL1, chr20 | R693* | 27 | 52 |
| PTPN13, chr4 | R1838Q | 25 | 47 |
| PPP2R2B, chr5 | K363Q | 25 | 51 |
| JAK1, chr1 | R879H | 24 | 54 |
| JAK3, chr19 | R657Q | 21 | 55 |
| TET2, chr4 | P1962T | 23 | 47 |
| MSRB2, chr10 | T59I | 23 | 42 |
| PCNXL2, chr1 | R1169W | 21 | 52† |
| MYO1D, chr17 | R560Q | 21 | 48 |
| NOTCH1, chr9 | K1607delinsNLQ | 29 | 97 |
| OPRD1, chr1 | I279N | 18 | 36 |
| RRP7A, chr22 | F241del | 11 | 20 |
*stop codon, †possibly damaging, in bold: mutation present at heterozygous state in T2‐ALL and at homozygous state in AML‐M0.
Table 2.
Genes mutated only in T2‐ALL
| Gene, chr | Protein | Variant frequency in T2‐ALL (%) |
|---|---|---|
| NOTCH1‐bis, chr9 | K1488N | 20 |
| PRB4, chr12 | G164R | 20 |
| ITGAX, chr16 | D810Y | 19 |
| AQP5, chr12 | L74M | 14† |
| GRIN3B, chr19 | L824M | 14 |
| CUBN, chr10 | A2914S | 13 |
| OBSCN, chr1 | A8630E | 12 |
| SCARF2, chr22 | R147S | 12 |
| DDX3X, chrX | G11W | 12 |
| PDZD2, chr5 | L34I | 11 |
| MCM3, chr6 | A461D | 11 |
| WNT3, chr17 | R60S | 11 |
| ZIK1, chr19 | A364delinsVLYF* | 11 |
| BCOR, chrX | L1203Sfs*33 | 11 |
*stop codon, †possibly damaging, in bold: mutation present at heterozygous state in T2‐ALL and at homozygous state in AML‐M0.
Table 3.
Genes mutated only in AML‐M0
| Gene, chr | Protein | Variant frequency in AML‐M0 (%) |
|---|---|---|
| SUDS3, chr12 | R325H | 50 |
| SAP130, chr2 | G803‐G804insPTQN | 36 |
| GGT1, chr22 | R107C | 30 |
| LDB2, chr4 | K355N | 27 |
| ENPP2, chr8 | V785M | 26 |
| ATP8B1, chr18 | R941G, G940V | 23, 23 |
| LOC441155, chr6 | A81T | 22 |
| PVRIG, chr7 | C62F | 19 |
| MMEL1, chr1 | P743H | 18 |
| TMEM181, chr6 | E111* | 18 |
| RBFOX3, chr17 | E263* | 18 |
| GCGR, chr17 | E362* | 18 |
| CENPC, chr4 | E302* | 17 |
| PHACTR2, chr6 | G201C | 17 |
| MUC20, chr3 | T447M | 15 |
| FAM71F2, chr7 | P198H | 15 |
| WDR60, chr7 | G1009C | 15 |
| CSH2, chr17 | V27I | 15 |
| BAG6, chr6 | P252T | 14 |
| BCLAF1, chr6 | S38Yfs*38 | 14 |
| MYO9B, chr19 | A1659S | 14 |
| SLC17A7, chr19 | R314S | 14 |
| WDR52, chr3 | E1818* | 12 |
| SOX7, chr8 | P209T | 12 |
| C19orf26, chr19 | G349W | 12 |
| MXRA5, chrX | E937* | 12 |
| SOBP, chr6 | D569Y | 11 |
| HSPA12B, chr20 | R650S | 11 |
| SUV39H1, chrX | C41* | 11 |
*stop codon.
To examine whether some of the potentially deleterious variants accumulated before leukaemia development, we screened 20 variants found with a high penetrance in both T2‐ALL and AML‐M0 clones by NGS on CD34+ cells obtained from the patient's peripheral blood 5 years prior to T2‐ALL development. The only somatic mutation identified was a TET2 P1962T at a frequency of 1% (Table S3) reaching more than 40% at leukaemic stages. These results suggest that a missense TET2 P1962T mutation in association with germline RUNX1 R174Q mutation led to amplification of an haematopoietic clone susceptible to acquire other transforming alterations (Fig. 1).
Figure 1.
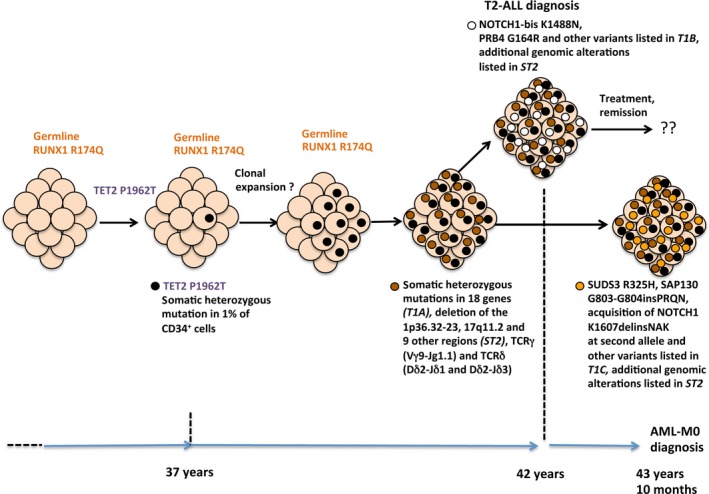
Clonal evolution in FPD/AML patient carrying germline RUNX1 R174Q mutation. The clonal evolution with the different mutations occurring at various stages of the disease (thrombocytopaenia, T2‐ALL, AML‐M0) is represented. Frequency of the variants is listed in the Tables 1, 3. The other deletions and amplifications are listed in Table S2.
Discussion
Here, we describe a unique case of one FPD/AML patient who developed first one T2‐ALL and less than 2 years later an AML‐M0. We show that these two leukaemic clones originate from the common subclone harbouring alterations in genes frequently mutated in both types of leukaemia. We cannot exclude that AML‐M0 was induced by the chemotherapy used for T2‐ALL treatment, as it involved anthracyclines and cyclophosphamide known to generate genomic instability. Interestingly, one of the variant TET2 P1962T was present at a frequency of 1% in haematopoietic progenitors of this patient 5 years prior to T2‐ALL development. A missense TET2 mutation at the same position (P1962L) has been described in AML 16 and T‐lymphoblastic lymphoma 17, and the role of TET2 in the clonal haematopoiesis preceding transformation in FPD/AML has been recently reported in another case of FPD/AML 18. However, this is the first report of a TET2P1962T mutation leading to the amplification of a leukaemic clone.
Our results suggest the following model of transformation: the germline RUNX1 mutation induces both alteration in haematopoiesis (more particularly increasing cycling) and a genetic instability 11. The acquisition of a somatic TET2 mutation enhances self‐renewal of haematopoietic stem/progenitor cells 17, 19, 20, leading to a clonal haematopoiesis. This is not a clonal haematopoiesis of undetermined potential (CHIP) in this germline context, but a true pre‐leukaemic state responsible for the emergence of progressively divergent haematological malignancies.
In conclusion, this report presents a FPD/AML patient with a germline RUNX1 mutation and an acquired TET2 mutation leading to clonal haematopoiesis. During a five‐year follow‐up, the haematopoietic clone acquired other genetic alterations including mutations in 18 genes. These subclonal alterations led first to T2‐ALL development, and later to a T/Myeloid phenotype with an AML‐M0 development (Fig. 1).
Clonal haematopoiesis in asymptomatic RUNX1 carriers under the age of 50 years was already reported 9 suggesting together with our results that the identification of clonal haematopoiesis before leukaemia development in FPD/AML patients could serve as a marker of pre‐leukaemic state. This finding might be helpful in patient care, especially if bone marrow transplantation is considered.
Conflict of interest disclosure
The authors declare no competing financial interests. The online version of the article contains a data supplement.
Author contribution
MVT, BH, ADI, AB, MG, DN and DMK performed genetic analysis and analysed mutational data. MVT, BH, VW, PI, ME, AV, FR, BV and RH conceptualized the idea, designed the research and analysed data. FR, PT, CTR and BV provided samples and data. MVT, BH, ADI, ME, AV, BV and RH wrote the manuscript, which was approved by all the authors.
Supporting information
Figure S1 Morphology and phenotype of AML‐M0 blasts.
Figure S2 Comparative genomic hybridization array on T2‐ALL and AML‐M0 blast populations.
Figure S3 TCRδ and TCRγ rearrangement analysis.
Table S1 List of primers used for NGS.
Table S2 Deletions and insertions found in AML‐M0 btasts and/or T2‐ALL blasts by CGH array.
Table S3 Variants found in AML‐M0 blasts, T2‐ALL blasts and CD34+ cells by NGS.
Acknowledgements
This work was supported by French grants from the Agence Nationale de la Recherche (ANR‐jeunes chercheurs, HR 2010), the ARC (projet libre, IP 2012). M.V.T. was supported by a doctoral fellowship from Société Francais d'Hématologie (SFH). I.A.D. was supported by a postdoctoral fellowship from ARC. The WES was cofunded by French Foundation for Rare Diseases. B.H. was supported by a doctoral fellowship from Ligue National Contre le Cancer. WES has been carried out by IntegraGen (France).
References
- 1. Ichikawa M, Yoshimi A, Nakagawa M, et al A role for RUNX1 in hematopoiesis and myeloid leukemia. Int J Hematol. 2013; 97: 726–34. [DOI] [PubMed] [Google Scholar]
- 2. Grossmann V, Kern W, Harbich S, et al Prognostic relevance of RUNX1 mutations in T‐cell acute lymphoblastic leukemia. Haematologica. 2011; 96: 1874–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Owen CJ, Toze CL, Koochin A, et al Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood. 2008; 112: 4639–45. [DOI] [PubMed] [Google Scholar]
- 4. Antony‐Debre I, Duployez N, Bucci M, et al Somatic mutations associated with leukemic progression of familial platelet disorder with predisposition to acute myeloid leukemia. Leukemia. 2015; 30: 999–1002. [DOI] [PubMed] [Google Scholar]
- 5. Prebet T, Carbuccia N, Raslova H, et al Concomitant germ‐line RUNX1 and acquired ASXL1 mutations in a T‐cell acute lymphoblastic leukemia. Eur J Haematol. 2013; 91: 277–9. [DOI] [PubMed] [Google Scholar]
- 6. Preudhomme C, Renneville A, Bourdon V, et al High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood. 2009; 113: 5583–7. [DOI] [PubMed] [Google Scholar]
- 7. Haslam K, Langabeer SE, Hayat A, et al Targeted next‐generation sequencing of familial platelet disorder with predisposition to acute myeloid leukaemia. Br J Haematol. 2015; 175: 161–3. [DOI] [PubMed] [Google Scholar]
- 8. Yoshimi A, Toya T, Kawazu M, et al Recurrent CDC25C mutations drive malignant transformation in FPD/AML. Nat Commun. 2014; 5: 4770. [DOI] [PubMed] [Google Scholar]
- 9. Churpek JE, Pyrtel K, Kanchi KL, et al Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015; 126: 2484–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gnirke A, Melnikov A, Maguire J, et al Solution hybrid selection with ultra‐long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009; 27: 182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Antony‐Debre I, Manchev VT, Balayn N, et al Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood. 2015; 125: 930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bluteau D, Gilles L, Hilpert M, et al Down‐regulation of the RUNX1‐target gene NR4A3 contributes to hematopoiesis deregulation in familial platelet disorder/acute myelogenous leukemia. Blood. 2011; 118: 6310–20. [DOI] [PubMed] [Google Scholar]
- 13. Huguet F, Leguay T, Raffoux E, et al Pediatric‐inspire therapy in adults with Philadelphia chromosome‐negative acute lymphoblastic leukemia: the GRAALL‐2003 study. J Clin Oncol. 2009; 27: 911–8. [DOI] [PubMed] [Google Scholar]
- 14. Suzuki R, Yamamoto K, Seto M, et al CD7+ and CD56+ myeloid/natural killer cell precursor acute leukemia: a distinct hematolymphoid disease entity. Blood. 1997; 90: 2417–28. [PubMed] [Google Scholar]
- 15. Oshimi K. Progress in understanding and managing natural killer‐cell malignancies. Br J Haematol. 2007; 139: 532–44. [DOI] [PubMed] [Google Scholar]
- 16. Weissmann S, Alpermann T, Grossmann V, et al Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012; 26: 934–42. [DOI] [PubMed] [Google Scholar]
- 17. Quivoron C, Couronne L, Della Valle V, et al TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011; 20: 25–38. [DOI] [PubMed] [Google Scholar]
- 18. Sakurai M, Kasahara H, Yoshida K, et al Genetic basis of myeloid transformation in familial platelet disorder/acute myeloid leukemia patients with haploinsufficient RUNX1 allele. Blood Cancer J. 2016; 6: e392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kunimoto H, Fukuchi Y, Sakurai M, et al Tet2 disruption leads to enhanced self‐renewal and altered differentiation of fetal liver hematopoietic stem cells. Sci Rep. 2012; 2: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moran‐Crusio K, Reavie L, Shih A, et al Tet2 loss leads to increased hematopoietic stem cell self‐renewal and myeloid transformation. Cancer Cell. 2011; 20: 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Morphology and phenotype of AML‐M0 blasts.
Figure S2 Comparative genomic hybridization array on T2‐ALL and AML‐M0 blast populations.
Figure S3 TCRδ and TCRγ rearrangement analysis.
Table S1 List of primers used for NGS.
Table S2 Deletions and insertions found in AML‐M0 btasts and/or T2‐ALL blasts by CGH array.
Table S3 Variants found in AML‐M0 blasts, T2‐ALL blasts and CD34+ cells by NGS.