

Abstract
A small library of 7-pyrrolo[3,2-f]quinolinones was obtained by introducing benzoyl, sulfonyl and carbamoyl side chains at the 3-N position, and their cytotoxicity against a panel of leukemic and solid tumor cell lines was evaluated. Most of them showed high antiproliferative activity with GI50s ranging from micro-to sub-nanomolar values, and these values correlated well with the inhibitory activities of the compounds against tubulin polymerization. Based on a recently proposed colchicine bind site inhibitors (CBSIs) pharmacophore, the interactions of the novel 7-PPyQs at the colchicine domain were rationalized. The most active compounds (4a and 4b) did not induce significant cell death in normal human lymphocytes, suggesting that the compounds may be selective against cancer cells. In particular, 4a was a potent inducer of apoptosis in both the HeLa and Jurkat cell lines. On the other hand, the sulfonyl derivative 4b exhibited a lower potency in comparison with 4a. With both compounds, induction of apoptosis was associated with dissipation of the mitochondrial transmembrane potential and production of reactive oxygen species, suggesting that cells treated with the compounds followed the intrinsic pathway of apoptosis.
Keywords: Microtubules, Phenylpyrroloquinolinone, Tubulin, Apoptosis, Molecular docking, Structure-activity relationships
1. Introduction
Drugs interfering with microtubules (MTs) represent a class of compounds of great interest in the area of anticancer therapy. MTs are an essential component of the cellular cytoskeleton, as they regulate and participate in a variety of cellular functions that include motility, morphology, intracellular transport, signal transduction, and cell division [1]. MTs are composed of α/β tubulin heterodimers that have polymerized into cylindrical structures, and, therefore, both natural and synthetic agents able to interfere with tubulin polymerization or depolymerization, thereby altering MT dynamics, continue to attract considerable attention in the field of chemotherapeutic research [2]. Not only are there clinically used natural and semisynthetic antimitotics (such as, paclitaxel, other taxanes, eribulin, ixabepilone and vinca akaloids), but there is also a large number of structurally dissimilar small molecules with high affinity for the colchicine site on tubulin. These compounds are able to inhibit the proliferation of a wide variety of human cancer cells [3]. Moreover, these agents can also affect the tumor endothelial vasculature required for the growth of tumor mass. These types of tubulin inhibitors might provide new therapeutic approaches to treat cancers and overcome limitations of existing tubulin interactive drugs [4]. In the last decade, we have been developing phenylpyrroloquinolinone (PPyQ) derivatives that show interesting in vitro and in vivo antitumor activity. Both 2-PPyQs and 7-PPyQs act as tubulin polymerization inhibitors by binding at the colchicine site in β-tubulin [5,6]. Although less cytotoxic, the 2-PPyQ compounds were also found to exhibit interesting in vitro and in vivo antiangiogenic properties [7]. The more cytotoxic 7-PPyQ derivatives showed very remarkable in vitro biological properties and good antitumor activity in vivo. In particular, some 7-PPyQs, characterized by alkyl substitutions at the pyrrole nitrogen, showed increased cytotoxicity with nanomolar GI50 values, and these compounds overcame the resistance observed with the clinically used agents vincristine and taxol [8,9]. In the latter series, the 3N-cyclopropyl methyl 7-PPyQ derivative MG 2477 (Fig. 1, 10), was taken as lead compound due to its very strong cytotoxicity (nanomolar range GI50s) and its potent interaction with tubulin. Its activities as an inhibitor of tubulin polymerization and of colchicine binding to tubulin were similar to those of the reference compound combretastatin A-4: 0.90 μM assembly IC50 and 83% inhibition of colchicine binding for compound 10 versus values of 1.1 and 99%, respectively, for CA-4 [10,11]. Compound 10 was also demonstrated to induce autophagy in the A549 cell line [12]. Very recently, in an effort to produce additional highly active compounds, numerous related analogues were designed, synthesized and studied, resulting in the discovery of a potent 3N-acyl derivative MG 2603 (Fig. 1, 11) showing low nanomolar GI50 values. Compound 11, too, showed an anti-tubulin mechanism profile similar to that of 10, but it was also able to inhibit a number of kinases involved in tumor progression. Moreover, 11 showed reduced toxicity in non tumor cell lines and synergized with conventional chemotherapeutic agents in inhibiting leukemia cell proliferation [13].
Fig. 1.
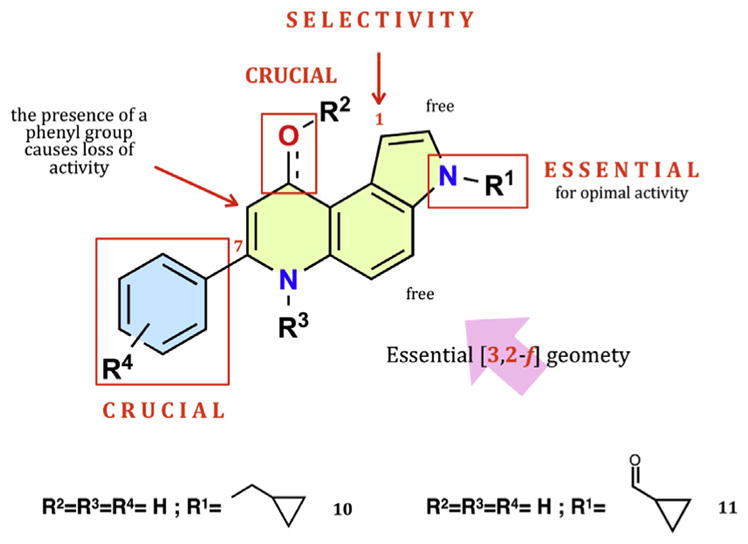
Structure-activity relationships of 7-phenyl-pyrrolo[3,2-f]quinolinones.
Driven by the SARs we have collected during the development of the PPyQs [14] (Fig. 1) and remembering the recent results with the 3N-acyl 7-PPyQ derivative, the aim of the present work was the design, synthesis and evaluation of novel analogues substituted at the pyrrole N with acyl, sulfonyl and carbamoyl side chains. In designing the novel derivatives, we preserved the structural elements crucial for the best antiproliferative activity, such as the [3,2-f] geometry of the pyrroloquinoline core, the un-substituted phenyl ring at the 7 and the carbonyl group at the 9 position, without any other substitutions except for the 3 position (Fig. 1). Biological investigations included cellular cytotoxicity, tubulin inhibition assays and an apoptosis assay, together with docking simulations in the colchicine site of β-tubulin. This allowed us to obtain more knowledge on the key substitutions at the pyrrole N for effective interactions at the colchicine site.
2. Results and discussion
2.1. Chemistry
Scheme 1 shows the route to 7-PPyQs bearing a side chain bound to the pyrrole N via a carbonyl group according to the previously reported general synthesis to 7-PPyQs [7]. The starting commercial 5-nitroindole was reacted with various acyl and sulfonyl chlorides in order to obtain directly the 3N-substituted indole derivatives 1a–d. Compound 1e was prepared by means of a one-pot procedure consisting first of activation of 5-nitroindole with p-nitrophenylchloroformate to give the reactive 3-p-nitrophenylcarbamate intermediate and then reaction with cyclopropylamine (19% yield). The next reduction step of 5-nitro- to 5-aminoindole intermediates was accomplished by a chemical procedure with , 37% HCl in methanol at reflux to give indole derivatives 2a–d. In the case of 1e, the reduction did not produce any corresponding amino compound. While, by a catalytic procedure with H2 and C/Pd 10% in EtOAc/EtOH at atmospheric pressure, indoline compounds 5a, b, d and e were obtained. Note that by the above chemical method aminoindole derivative 2a was obtained in poor yields due to the formation of a mixture of various chloro-derivatives (not shown). Therefore the synthesis to 4a did not proceed beyond the c step. Aminoindole derivatives 2b–d were then condensed with ethyl benzoyl acetate to provide the eneamine intermediates 3b–d to be then thermally cyclized into the final 7-PPyQs 4b–d. In the same way, indolines 5a, b, d and e gave eneamine derivatives 6a, b, d and e with benzoyl acetate. However, when these were submitted to thermal cyclization in diphenyl ether, 6a, b and d gave a mixture of isomeric compounds angular 7a, b, d and linear 8a, b, d, whereas derivative 6e did not react. It is worth emphasizing that, by the catalytic procedure at the reaction conditions used here, we never observed the formation either of aminoindolines or the subsequent cyclization to linear tricyclic compounds.
Scheme 1.

a) Benzoyl chloride, methansulfonyl chloride, p-methylbenzensulfonyl chloride, p-trifluoromethylbenzensulfonylchloride, NaH (60%), anhydrous DMF, rt, 2 h, 92%; p-nitrophenylchloroformate and cyclopropylamine, THF, 3 h, 87%; b) SnCl2 · 2H2O, HCl 37%, methanol, reflux, 36 h, 53%; c) ethylbenzoyl acetate, absolute ethanol, cat CH3COOH, drierite, reflux, 36 h, 60%; d) diphenyl ether, reflux, 15 min, 79%; e) H2, Pd/C 10%, EtOAc, atmospheric pressure, 50 °C, 24 h, 95%.
Scheme 2 shows an alternative method to obtain the final compounds 4a and 4e, which could not be prepared by the route shown in Scheme 1. The previously described 7-PPyQ 9 [8], available in our laboratory, was submitted to an acylation reaction with benzoyl chloride. After a laborious purification procedure, 7-PPyQ 4a was obtained in a 37% yield. Compound 4e was obtained by the same one pot procedure described above, consisting of the reaction of 7-PPyQ 9 to give the intermediate 3-p-nitrophenylcarbamate, which was not isolated, followed by reaction with cyclopropylamine (30% yield).
Scheme 2.

a) Benzoyl chloride, NaH (60%), anhydrous DMF, rt, 3 h, 37%; c) p-nitrophenylchloro formate; NaH (60%), cyclopropyl amine, THF, rt, 3 h, 30%.
2.2. Biological evaluation
2.2.1. In vitro antiproliferative activities and SAR analysis
On the basis of previous biological activity data on 7-PPyQs and docking simulations of 11 into the colchicine site of tubulin [13], the new compounds were designed to obtain additional SAR information by modifying the nature and size of substituents at the 3 position of the 7-PPyQ tricycle. Evaluation of antiproliferative activities of 4a–e, 7a,b, 7d, and 8a,b was performed with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay against a panel of 11 human tumor cell lines (CCRF-CEM, HL-60, RS4; 11, Jurkat, SEM, MV4; 11, THP-1, HeLa, A549, HT-29, MCF-7). GI50 values, the concentrations that inhibit cell growth by 50%, are presented in Table 1. Most of the novel 7-PPyQs possessed antiproliferative activity, inhibiting cell growth with nanomolar to micromolar GI50 values, except for the linear 1,2-dihydro 8b (GI50 > 10000 nM) having a methanesulfonyl group at the 3 position. In contrast, its angular isomer 7b showed GI50 values in a high nanomolar range, demonstrating that for partially hydrogenated pyrroloquinolinones the [3,2-f] geometry is also preferred for cytotoxic activity as it was for fully aromatic compounds. Comparable behavior was also observed for the benzamidic derivatives 7a and 8a, although not as dramatic: the linear 8a showed GI50 values in the micromolar, while the angular compound 7b had IC50 values in the sub-micromolar range. Moreover, from the data presented in Table 1, it is evident that the angular, fully aromatic 7-PPyQs 4a,b,d were more cytotoxic than the corresponding hydrogenated analogues 7a,b,d. This was most remarkable for the pair 4a and 7a, with the former having GI50 values in the 0.1–10 nM range and the latter GI50 values in the 250–2650 nM range. Of particular note were the low nanomolar and sub-nanomolar GI50 values obtained with the series 4a–4e, in both the leukemic and solid tumor cell lines. Overall, the most active of the new compounds was the benzamidic derivative 4a, with subnanomolar concentrations in five of the eleven cell lines, with slightly lower GI50 values in the solid tumor lines (GI50s 0.2, 0.1 and 0.2 nM in the HeLa, HT-29 and MCF-7 cells, respectively). Previously, we had observed that 7-PPyQs were more cytotoxic against leukemic cells. Thus, the preferential activity of 4a against solid tumor cell lines is worth emphasizing. The same relative activity against the solid tumor cell lines was also observed for sulfamidic derivatives 4b–d, but not for the ureidic derivative 4e. We also note that there did not appear to be a significant steric hindrance factor among the compounds evaluated here, with similar antiproliferative activities observed among the series 4a–4e and the previously evaluated compound 11.
Table 1.
In vitro cell growth inhibitory effects of compounds 4a-e, 7a,b,d, 8a-b, and 11.
| cmp | GI50 (nM)a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| CCRF-CEM | HL-60 | RS 4; 11 | Jurkat | SEM | MV 4; 11 | THP-1 | HeLa | A549 | HT-29 | MCF-7 | |
| 4a | 12 ± 0.2 | 2 ± 0.8 | 0.3 ± 0.1 | 16 ± 6 | 0.9 ± 0.1 | 2 ± 0.9 | 5 ± 1 | 0.2 ± 0.04 | 10 ± 6 | 0.1 ± 0.08 | 0.2 ± 0.1 |
| 4b | 42 ± 7 | 215 ± 81 | 18 ± 9 | 34 ± 4 | 23 ± 6 | 49 ± 16 | 92 ± 31 | 36 ± 9 | 3 ± 0.7 | 7 ± 2 | 152 ± 95 |
| 4c | 32 ± 1 | 335 ± 74 | 48 ± 16 | 55 ± 13 | 33 ± 15 | 65 ± 18 | 222 ± 71 | 26 ± 2 | 52 ± 5 | 5 ± 1 | 195 ± 51 |
| 4d | 32 ± 1 | 39 ± 16 | 147 ± 43 | 81 ± 3 | 33 ± 2 | 86 ± 22 | 42 ± 9 | 22 ± 2 | 63 ± 8 | 23 ± 3 | 5 ± 2 |
| 4e | 92 ± 14 | 155 ± 63 | 27 ± 3 | 145 ± 61 | 11 ± 4 | 85 ± 12 | 66 ± 19 | 212 ± 61 | 171 ± 32 | 255 ± 75 | 96 ± 31 |
| 7a | 1045 ± 212 | 2640 ± 309 | 256 ± 133 | 1345 ± 302 | 918 ± 211 | 1543 ± 223 | 2166 ± 614 | 331 ± 55 | 918 ± 268 | 1586 ± 145 | 1112 ± 178 |
| 7b | 235 ± 56 | 291 ± 13 | 35 ± 4 | 372 ± 26 | 271 ± 1 | 352 ± 36 | 71 ± 2 | 563 ± 65 | 451 ± 54 | 623 ± 59 | 521 ± 96 |
| 7d | 435 ± 085 | 1625 ± 35 | 211 ± 32 | 256 ± 21 | 336 ± 25 | 356 ± 63 | 846 ± 152 | 373 ± 25 | 541 ± 29 | 255 ± 62 | 475 ± 47 |
| 8a | 818 ± 154 | 3740 ± 233 | 132 ± 51 | 2316 ± 407 | 1656 ± 185 | 1978 ± 326 | 2323 ± 945 | 1447 ± 416 | 3562 ± 624 | 5436 ± 852 | 6323 ± 845 |
| 8b | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | 7638 ± 1126 | >10000 | >10000 | >10000 |
| 11b | 17 ± 4 | 2 ± 0.6 | 0.1 ± 0.05 | 0.3 ± 0.03 | 0.4 ± 0.01 | 19 ± 8 | 74 ± 25 | 1.6 ± 0.6 | 9 ± 0.4 | 1 ± 0.5 | 5 ± 1 |
IC50 = compound concentration required to inhibit tumor cell proliferation by 50%. Data are expressed as the mean ± SE from the dose-response curves of at least three independent experiments.
Data taken from ref.[13].
We conclude that the 7-PPyQ derivatives 4a–e, chemically modified at 3 position with substitutions such as carbonyl, sulfonyl and carbamoyl groups, maintained strong cytotoxicity against tumor cell lines. These substitutions were made to further explore the SARs, and they have confirmed what was observed previously with carbonyl compound 11. Substitutions with oxygenated groups, although of diverse nature and volume, confer very high antiproliferative activity on 7-PPyQs. Due to their broad spectrum of potent activity, 4a and 4b were selected for further biological investigations on mechanism of action.
2.2.2. Evaluation of cytotoxicity in human non-cancer cells
To obtain a preliminary indication of the cytotoxic potential of these derivatives in normal human cells, the two most active compounds (4a and 4b) were evaluated in vitro against peripheral blood lymphocytes (PBL) from healthy donors (Table 2). Compound 4a showed a GI50 of 28 μM in quiescent lymphocytes, while in the presence of the mitogenic stimulus phytohematoaglutinin (PHA), the GI50 decreased to about 15 μM. Notably, this value was almost 1000–2000 times higher than that observed against the lymphoblastic cell lines CCRF-CEM and Jurkat. These results indicate that 4a has a significant effect in rapidly proliferating cells but not in quiescent cells, as previously observed for other antimitotic derivatives developed by our group [13]. Compound 4b was completely inactive in both quiescent and proliferating lymphocytes.
Table 2.
Cytotoxicity of 4a-b for human peripheral blood lymphocytes (PBL).
Values are the mean ± SEM from three separate experiments.
Compound concentration required to reduce cell growth inhibition by 50%.
PBL not stimulated with PHA.
PBL stimulated with PHA.
2.2.3. Inhibition of tubulin polymerization and colchicine binding
To evaluate the tubulin interaction properties of compounds 4ae, we investigated their effects on inhibition of tubulin polymerization and the binding of [3H]colchicine to tubulin (Table 3) [15,16]. For comparison, CA-4 and 3c were examined in contemporaneous experiments as references compounds. Among the test compounds, 4a strongly inhibited tubulin assembly assay with an IC50 of 0.89 μM, a value that was lower than that obtained for the reference compound CA-4 (IC50 = 1.2 μM). Compounds 4b and 4c showed an IC50 similar to that of CA-4 while 4d and 4e were less effective than the reference compound (IC50 = 2.2–2.4 μM). These results correlate well with the growth inhibitory effects exhibited by the test compounds, indicating that their antiproliferative activity derives from an interaction with tubulin.
Table 3.
Inhibition of tubulin polymerization and colchicine binding by compounds 4a-e and CA-4.
| Compound | Tubulin assemblya IC50±S.D. (μM) | Colchicine bindingb % inhibition ±S.D. |
|---|---|---|
| 4a | 0.89 ± 0.04 | 70 ± 2 |
| 4b | 1.2 ± 0.01 | 42 ± 4 |
| 4c | 1.1 ± 0.04 | 37 ± 5 |
| 4d | 2.4 ± 0.2 | 29 ± 3 |
| 4e | 2.2 ± 0.3 | 18 ± 4 |
| CA-4 | 1.2 ± 0.1 | 98 ± 0.7 |
Inhibition of tubulin polymerization. Tubulin was at 10 μM.
Inhibition of [3H]colchicine binding. Tubulin and colchicine were at 1 and 5 μM concentrations, respectively.
In the colchicine studies, compound 4a was the most active inhibitor of the binding of [3H]colchicine to its domain on tubulin, with 70% inhibition occurring with this derivative at 5 μM. Nevertheless, 4a was less potent than CA-4 in this assay. In these experiments CA-4 inhibited colchicine binding by 98% at 5 μM. The other investigated compounds (4b–e) showed weaker inhibitory activity, with less than 50% inhibition of colchicine binding to tubulin.
2.2.4. Computational studies
Docking studies were carried out to investigate the binding mode of the novel inhibitors with the aim of interpreting experimental affinity data. A relevant number of experimentally derived complex structures of colchicine binding site inhibitors (CBSI) were recently deposited in the Protein Data Bank (PDB) [17]. Interestingly, the superposition of the different crystal structures reveals a significant variability in the sidechains of the residues belonging to the colchicine site depending on the chemical nature of the ligand, as shown in SI_Fig. 1 (see Supplementary Material). Moreover, the resolution of the crystal structures spanned a broad range (2.19–3.75 Å). As a consequence of this heterogeneity, we carried out a benchmark study, using the DockBench tool [18], to identify the most accurate docking model among 14 different ones and to determine which protein conformation was most appropriate to model our analogues. The benchmark study on the self-docking procedure was performed on 14 tubulin–CBSI complexes from the PDB as listed in table SI_Table 1. The benchmark results, summarized in SI_Fig. 2, revealed that several protocols showed a good ability to reproduce the experimental complex geometries for most of the experimental structures. Among them, GOLD software coupled to the PLP/goldscore/chemscore scoring function, gave the best results. In particular, GOLD protocols returned accurate predictions for the complete dataset. As a consequence of the overall good performance of the benchmark, we focused our attention on the identification of the most suitable crystallographic protein structure for the docking simulation of the PPyQ class of compounds. This step was crucial because of the variety of different sidechain orientations for certain residues in the colchicine site such as βGlu200, βCys239, βLeu248, and βLeu255, as shown in SI_Fig. 1. We have addressed this critical issue by comparing the shape similarity and the pharmacophoric determinants conservation between the ligands present in the complexes (summarized in SI_Table 1) and our representative compound, 4a. Plinabulin (PDB ID: 5C8Y) showed the highest shape similarity according Tversky coefficient (>0.7) and, more notably, the plinabulin key moieties for the tubulin interaction are nicely conserved as shown in Fig. 2, Panel A. Not surprisingly, all analogues showed a common binding mode similar to that of plinabulin, as shown by SI_Video 1. As depicted in Fig. 2 (Panel B), the diketopiperazinic core of plinabulin is mimicked by the pyrroloquinolinone core, maintaining the key hydrogen bond interaction with the backbone of βVal236 as anticipated by the shape-based superposition (plinabulin and 4a, Fig. 2, Panel A). In addition to the conserved hydrogen bond, the pyrroloquinolinone scaffold guarantees strong hydrophobic interactions with βLeu253, βAla314 and βIle368. The phenyl ring in position 7 reproduced the same scheme of interaction to the benzylidene moiety of plinabulin through hydrophobic interactions with residues βPhe167, βTyr200, and βLeu250. The substituents at the N-pyrrole were placed in the pocket formed by residues: βLys350, βThr351, βAla314, βAla352 and, for the more bulky substituents, also αThr179. Notably, this binding mode is compatible with a competitive mechanism of action at the colchicine site. The 1,2-dihydro derivatives showed minor differences in their orientation. The main difference is the reduction in the interaction strength with βVal236, in particular for the linear isomers (8a and 8b) (SI_Video 1).
Fig. 2.

Panel A. Superposition of 4a (green sticks, grey surface) and plinabulin (grey sticks) derived from the shape similarity calculations. Panel B. The energetically most favorable pose of 4a (in green) obtained by molecular docking simulation using the protein conformation of the plinabulin complex (PDB ID: 5C8Y). The ribbon as well as the residue atoms of the colchicine binding site are colored according the subunit to which they belong: white for β-tubulin and magenta for α-tubulin. Hydrogen atoms are not shown. Panel C. Per-residue analysis of the protein-ligand interaction for compound 4a (green) and plinabulin (red). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Supplementary video related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2016.10.026.
To evaluate if molecular docking was also able to explain the differences in the inhibition of colchicine binding by compounds 4a–e, we performed a per-residue analysis along the series, in which the contribution for each residue belonging to the binding site is computed for each synthesized compound (Fig. 2, Panel C). In particular, we measured the electrostatic interaction energy and score, taking into account hydrophobic interactions. In SI_Video 1 is reported the per-residue analysis for all the analogues in Table 1, including the reference compound 11. The resulting heatmap suggests a very similar pattern of interaction for all the compounds. In this context, the narrow differences in colchicine binding and tubulin assembly inhibition are difficult to rationalize. The higher inhibition in the colchicine binding of 4a among the 4a–4e derivatives could be ascribed to the orientation of the benzamidic moiety that is directed between the βLys350 and αThr179 residues, while in the 4b–4e analogues the N substituent assumes a slightly different orientation. The differences in the cell growth inhibition data are difficult to rationalize only with tubulin docking, possibly because of the involvement of other targets [13]. A clearer interpretation can be made for compound 8b, since its inhibition of tumor cell growth was negligible, and this analogue showed poor scores in molecular docking primarily because of a poor interaction with βVal236, as shown in the per residue analysis (SI_Video 1).
2.2.5. Compounds 4a and 4b induce mitotic arrest of the cell cycle
To investigate whether compounds 4a and 4b affected cell cycle progression, we evaluated by flow cytometry the effect of different concentrations of compounds after a 24 h of treatment of HeLa and Jurkat cells. As shown in Fig. 3, compound 4a caused a significant G2/M arrest in a concentration-dependent manner in both cell lines, with a rise in G2/M cells occurring at a concentration of 50 nM, while at the highest concentration (100 nM) more than 50% of the cells were arrested in G2/M. In the HeLa cells, the G2/M block was accompanied by a significant reduction of both G1 and S phase cells, suggesting that cell proliferation is impaired. For compound 4b, we observed a similar behavior but less marked as compared with 4a, in good agreement with the respective IC50 values found in the tubulin polymerization assay.
Fig. 3.

Percentage of cells in each phase of the cell cycle in HeLa (Panel A) and Jurkat cells (Panel B) treated with compounds 4a or 4b at the indicated concentrations for 24 h. Cells were fixed and labeled with PI and analyzed by flow cytometry as described in the Experimental Section. Data are presented as mean of two independent experiments with similar results.
2.2.6. Compounds 4a and 4b induce apoptosis through the mitochondrial death pathway
To evaluate the mode of cell death induced by 4a and 4b, we performed the annexin-V/propidium iodide (PI) assay by flow cytometry. Staining with annexin-V and with PI allows discrimination between live cells (annexin-V-/PI-), early apoptotic cells (annexin-V+/PI-), late apoptotic cells (annexin-V+/PI+) and necrotic cells (annexin-V-/PI+). The experiments were carried out both in Hela and Jurkat cells. As shown in Figs. 4 and 5, compounds 4a and 4b induce a significant time- and concentration-dependent increase of apoptotic cells in both cell lines. In particular, after 24 h we found a significant accumulation of early apoptotic (annexin-V+/PI-) cells starting from the lower concentrations and an increase of late apoptotic cells (annexin-V+/PI+) after 48 h treatments, indicating that the compounds trigger cells to a massive apoptotic cell death. In good agreement with the respective GI50 values (Table 1), compound 4a is the more potent inducer of apoptosis both in Hela and Jurkat cells at the 50 nM concentration.
Fig. 4.

Flow cytometric analysis of apoptotic cells after treatment of HeLa cells with 4a or 4b at the indicated concentrations after incubation for 24 or 48 h. The cells were harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. Data are presented as mean ± SEM of three independent experiments.
Fig. 5.

Flow cytometric analysis of apoptotic cells after treatment of Jurkat cells with 4a or 4b at the indicated concentrations after incubation for 24 or 48 h. The cells were harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. Data are presented as mean ± SEM of three independent experiments.
Loss of mitochondrial transmembrane potential (Δϕmt) and release of apoptogenic factor has been described as an early event in the apoptotic process [19,20]. Δϕmt was evaluated by flow cytometry using the fluorescence of the dye JC-1. In normal conditions (high Δϕmt), JC-1 displays a red fluorescence (590 nm), while mitochondrial depolarization is indicated by a shift to green fluorescence (525 nm).
In both Hela and Jurkat cells, treatment with 4a and 4b induced a marked increase in the percentage of cells with low Δϕmt (Fig. 6), and this occurred in a time- and concentration-dependent fashion. Depolarization of mitochondrial potential leads to the induction of the intrinsic pathway of apoptosis and is associated with the appearance of annexin-V positivity in the treated cells, as shown above and indicating the cells are in an early apoptotic stage. In fact, the disruption of Δϕmt and the intrinsic activation of apoptosis are characteristic of antimitotic drugs and have been observed with both microtubule stabilizing and destabilizing agents in different cell types. It is also well known that mitochondrial potential impairment and the resulting damage to mitochondrial function induce generation of reactive oxygen species (ROS) [21,22]. Superoxide anion is produced by mitochondria due to a shift from the normal 4-electron reduction of O2 to a 1-electron reduction when cytochrome c is released from mitochondria upon apoptosis [23,24].
Fig. 6.

Assessment of mitochondrial membrane potential (Δϕmt) after treatment of HeLa (Panel A) or Jurkat (Panels B) cells with the indicated compounds. Cells were treated with the indicated concentration of compound for 24 or 48 h and then stained with the fluorescent probe JC-1 for analysis of mitochondrial potential. Cells were then analyzed by flow cytometry as described in the Experimental Section. Data are presented as mean ± SEM of three independent experiments.
Using dichlorodihydrofluorescein diacetate (H2-DCFDA), which is oxidized to the fluorescent compound dichlorofluorescein (DCF) upon ROS induction [23], we measured ROS production after treatment with compounds 4a and 4b. As shown in Fig. 7 (Panels B and D), the two compounds induced the production of large amounts of ROS in comparison with control cells. This was observed in both the Jurkat and HeLa cells, in good agreement with the dissipation of Δϕmt described above.
Fig. 7.

Assessment of ROS production after treatment of HeLa (Panel A) or Jurkat (Panel B) cells with the indicated compounds. Cells were treated with the indicated concentration of compound for 24 or 48 h and then stained with H2-DCFDA for the evaluation of ROS levels. Cells were then analyzed by flow cytometry as described in the Experimental Section. Data are presented as mean ± SEM of three independent experiments.
2.2.7. Metabolic stability of 4a in human liver microsomes
Liver microsomal oxidation and hydrolysis represent major routes of drug metabolism in mammals, including humans [25]. In vitro studies were therefore carried out to get preliminary information on the stability of compound 4a to oxidative and hydrolytic metabolism by human liver microsomes. As shown in Fig. 8 (panel A), compound 4a (10 μM) was relatively stable in human liver microsomes (1 mg/mL) with more than 60% compound remaining after 60 min incubation at 37 °C. Interestingly, compound 4a disappearance was not influenced by the presence of NADPH (Fig. 8, panel A), a cofactor for both cytochrome P450- and flavin monooxygenase-mediated oxidations [25], and was accompanied by formation of a fluorescent metabolite whose retention time corresponded exactly to that of authentic compound 9 (panel B). Collectively, these findings indicate that compound 4a is partially susceptible to microsomal enzyme hydrolysis and that this catabolism produce compound 9 which retain a significant antiproliferative activity as previously demonstrated [8].
Fig. 8.

Assessment of metabolic stability of 4a in human liver microsomes. (A) 4a (10 μM) was incubated in the presence of human liver microsomes (1.0 mg/mL; HLMs;-△-), HLMs plus NADPH (1 mM; -●-), or buffer only (0.1 M KH2PO4, pH 7.4; –□–), for 0, 15, 30 or 60 min at 37 °C. The data are expressed as percent of parent compound (4a) remaining at each time compared with time 0 min, and represent the mean ± SD of n = 3 or 4 independent determinations. (B) Representative stacked HPLC-fluorescence traces of supernatants from mixtures containing 4a (10 μM) and HLMs (1.0 mg/mL), incubated for 0, 15, 30 or 60 min at 37 °C. M, 4a metabolite.
3. Conclusion
With the aim to further explore the SARs of the 7-PPyQ class of compounds, we examined the effects of various oxygenated functionalities at the 3 N position. A small series of novel derivatives was synthesized, and their antiproliferative activities and with their mechanism of action were investigated. The chemical series included both angular and linear compounds and fully aromatic and partially hydrogenated derivatives. Most compounds had significant antiproliferative activity, inhibiting cell growth with nanomolar to micromolar GI50 values, thus confirming that, in general, oxygenated substitutions at the 3 position improve cytotoxicity. In the series, the [3,2-f] angular geometry once again was required for obtaining potent cytotoxic compounds, and this [3,2-f] configuration was present both in the fully aromatic benzoyl 4a and the methanesulfonyl 4b. Compound 4a was the most active of the new agents, even having sub-nanomolar GI50s in some of the cell lines studied. Thus, compound 4a was even more cytotoxic than the previously described compound 11. Moreover, 4a had a significant effect only in rapidly proliferating cells but not in quiescent and proliferating lymphocytes. Investigations on the mechanism of action of 4a confirmed that it was a strong inhibitor of tubulin polymerization, as were previously described 7-PPyQ derivatives. Both in the Hela and Jurkat cell lines, 4a was more effective than 4b in blocking the cell cycle at the G2M phase, inducing apoptosis through the mitochondrial death pathway with production of ROS. In agreement with the experimental results obtained in this work, docking simulations suggested that the synthesized inhibitors had high affinity for the colchicine site of tubulin, with interactions with the binding site most similar to those observed with the known inhibitor plinabulin. The 1,2-dihydro derivative 8b was slightly penalized by affecting the geometry of the hydrogen bond with βVal236.
Additionally, in vitro metabolic stability studies indicated that a human liver microsomes esterases can catalyze the cleavage of the amide bond of 4a, leading to formation of an active metabolite, namely compound 9. These findings may be valuable for future in vivo studies.
4. Experimental section
4.1. Chemistry
Melting points were determined on a Buchi M – 560 capillary melting point apparatus and are uncorrected. Infrared spectra were recorded on a PerkinElmer 1760 FTIR spectrometer with KBr pressed disks and on a Varian ATR FTIR; all values are expressed in cm−1. UV–vis spectra were recorded on a Thermo Helyos α spectrometer. 1H NMR spectra were determined on Bruker 300 and 400 MHz spectrometers, with the solvents indicated; chemical shifts are reported in δ (ppm) downfield from tetramethylsilane as internal reference. Coupling constants are given in Hertz. In the case of multiplets, chemical shifts were measured starting from the approximate center. Integrals were satisfactorily in line with those expected on the basis of compound structure. Elemental analyses were performed in the Microanalytical Laboratory, Department of Pharmaceutical Sciences, University of Padova, on a PerkinElmer C, H, N elemental analyzer model 240B, and analyses indicated by the symbols of the elements were within ±0.4% of the theoretical values. Analytical data are presented in detail for each final compound in the Supporting Information. Mass spectra were obtained on a Mat 112 Varian Mat Bremen (70 eV) mass spectrometer and on an Applied Biosystems Mariner System 5220 LC/MS (nozzle potential 140 eV). Column flash chromatography was performed on Merck silica gel (250–400 mesh ASTM); chemical reactions were monitored by analytical thin-layer chromatography (TLC) on Merck silica gel 60 F-254 glass plates. Solutions were concentrated on a rotary evaporator under reduced pressure. Starting materials were purchased from Sigma-Aldrich and Alfa Aesar, and solvents were from Carlo Erba, Fluka and Lab-Scan. DMSO was obtained anhydrous by distillation under vacuum and stored on molecular sieves.
The purity of new tested compounds was checked by HPLC using the instrument HPLC VARIAN ProStar model 210, with detector DAD VARIAN ProStar 335. The analysis was performed with a flow of 1 mL/min, a C-18 column of dimensions 250 mm × 4.6 mm, a particle size of 5 μm, and a loop of 10 μL. The detector was set at 300 nm. The mobile phase consisted of phase A (Milli-Q H2O, 18.0 MΩ, TFA 0.05%) and phase B (95% MeCN, 5% phase A). Gradient elution was performed as reported: 0 min, % B = 10; 0–20 min, % B = 90; 25 min, % B = 90; 26 min, % B = 10; 31 min, % B = 10.
4.1.1. General procedure for the synthesis of 1N-substituted nitroindoles (1a–e)
As a typical procedure, the synthesis of 1-benzoyl-5-nitro-1H-indole 1a is described in detail. Into a two-necked 50 mL round-bottomed flask, 0.666 g (27.7 mmol, 3 eq.) of NaH, 60% dispersion in mineral oil, was placed and washed with toluene (3 × 10 mL). With stirring, a solution of commercial 5-nitroindole, 1.50 g (9.25 mmol, 1 eq.) in 5 mL of anhydrous DMF, was dropped into the flask, and the initial yellow color changed to red with the formation of H2 gas. After 30 min at room temperature, a solution of benzoyl chloride, 3.21 mL (27.7 mmol, d = 1.21 g/mL, 3 eq.) in 3 mL dry DMF, was added, and the reaction mixture was stirred for 2 h. The reaction was monitored by TLC analysis (eluent toluene/n-Hex/EtOAc, 1:1:1). At the end of the reaction, 25 mL of water was added, and the solvent was evaporated under reduced pressure, leaving a residue, which was extracted with EtOAc (3 × 50 mL). The organic phase, washed with water and dried over anhydrous Na2SO4, was concentrated under vacuum giving a crude yellow solid (2.446 g). This crude product was purified with a silica gel chromatographic column 3 × 35 cm, 230–400 mesh, eluent toluene/n-Hex/EtOAc, 1:1:1, yielding 2.345 g of a pure yellow solid.
4.1.1.1. 1-Benzoyl-5-nitro-1H-indole (1a)
Yield: 94.9%. Rf = 0.83 (eluent toluene/n-Hex/EtOAc, 1:1:1); 1H NMR (400 MHz, DMSO-d6) δ = 8.67 (d, J = 2.25 Hz, 1H, H-4), 8.43 (d, J = 9.12 Hz, 1H, H-7), 7.74 (m, 1H, H-4′), 8.28 (dd, J = 9.12 Hz and 2.37 Hz, 1H, H-6), 7.82 (m, 2H, H-2′ and -H-6′), 7.64 (m, 2H, H-3′ and H-5′), 7.62 (d, J = 3.64 Hz, 1H, H-2), 7.00 ppm (dd, J = 3.78 Hz and J = 0.60 Hz, 1H, H-3); 13C NMR (75 MHz, DMSO-d6) δ = 109.74 (C-3),113.19 (C-6), 117.05 (C-7), 120.55 (C-4), 129.69 (C-2′ and C-6′), 130.27 (C-3′ and C-5′), 131.56 (C-4′), 132.39 (C-2), 133.55 (C-3a), 133.94 (C-7a), 139.43 (C-1′), 144.66 (C-5), 169.24 ppm (C=O); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 267.0779; found, 267.0787.
4.1.1.2. 1-Methanesulfonyl-5-nitro-1H-indole (1b)
Compound 1b was prepared as for compound 1a by reacting 0.888 g of NaH 60% (37.03 mmol, 3 eq.) and 2 g (12.34mmol) of 5-nitroindole dissolved in 5 mL of DMF and 2.86 mL of methanesulfonyl chloride (37.03 mmol, d = 1.48 g/mL, 3 eq.). Reaction time: 2 h (TLC, eluent EtOAc/n-Hex, 2:1). 2.564 g of a solid bright yellow solid was obtained, and this was used in the next synthetic step without further purification. Yield: 91.9%. Rf = 0.54 (eluent EtOAc/n-Hex, 2:1); 1H NMR (400 MHz, DMSO-d6) δ = 8.67 (d, J = 2.20 Hz, 1H, H-4), 8.25 (dd, J = 9.14 Hz and J = 2.30 Hz, 1H, H-6), 8.05 (d, J = 9.16 Hz, 1H, H-7), 7.85 (d, J = 3.68 Hz, 1H, H-2), 7.08 (dd, J = 7.08 Hz and J = 0.72 Hz, 1H, H-3), 3.60 ppm (s, 3H, SO2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 42.07 (SO2CH3), 109.22 (C-3), 114.08 (C-7), 119.81 (C-6), 130.33 (C-2), 130.58 (C-3a), 137.71 (C-7a), 143.96 ppm (C-5); HRMS (ESIMS, 140 eV): m/z [M + H+] calculated for C9H9N2O4S+, 241.0283; found, 241.0236.
4.1.1.3. 1-(4-Methylbenzenesulfonyl)-5-nitro-1H-indole (1c)
Compound 1c was prepared as for compound 1a by reacting 0.444 g of NaH 60% (18.51 mmol, 3 eq.) and 1 g (6.17 mmol) of 5-nitroindole dissolved in 5 mL of DMF and 5.53 g of p-toluene-sulfonyl chloride (18.51 mmol, 3 eq.). Reaction time: 2 h (TLC, eluent n-Hex/EtOAc, 9:1). 1.786 g of a solid bright orange solid was obtained which was used in the next synthetic step without any additional further purification. Yield: 91.6%. Rf = 0.76 (eluent EtOAc/n-Hex, 9:1); 1H NMR (400 MHz, DMSO-d6) δ = 8.59 (d, J = 1.88 Hz, 1H, H-4), 8.21 (dd, J = 9.18 Hz and J = 2.26 Hz, 1H, H-6), 8.15 (dt, J = 9.18 Hz and J = 0.98 Hz, 1H, H-7), 8.07 (d, J = 3.72 Hz, 1H, H-2), 7.94 (m, AA’BB′, J = 8.44 Hz, J = 2.11 Hz and J = 1.83 Hz, 2H, H-2′ and H-6′), 7.43 (m, AA’BB′, J = 8.58 Hz and J = 0.62 Hz, 2H, H-3′ and H-5′), 7.07 (dd, J = 3.70 Hz and J = 0.66 Hz), 1H, H-3, 2.23 ppm (s, 3H, -CH3); 13C NMR (75 MHz, DMSO-d6) δ = 21.91 (-CH3), 110.93 (C-3), 114.54 (C-7), 118.85 (C-4), 120.55 (C-6), 127.77 (C-2′ and C-6′), 130.90 (C-2), 131.34 (C-3a), 131.37 (C-3′ and C-5′), 134.59 (C-4′), 137.81 (C-7a), 144.61 (C-5), 147.09 ppm (C-1′); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C15H13N2O4S+, 317.0596; found, 317.0585.
4.1.1.4. 5-Nitro-1-[4-(trifluoromethyl)benzenesulfonyl]-1H-indole (1d)
Compound 1d was prepared as described for compound 1a by reacting 0.670 g of NaH 60% (27.75 mmol, 3 eq.) and 1.5 g (9.25 mmol) of 5-nitroindole dissolved in 5 mL of DMF and 3.39 g of 4-(trifluoromethyl)benzenesulfonyl chloride (13.87 mmol, 1.5 eq.). Reaction time: 1 h (TLC, eluent n-Hex/EtOAc, 9:1). 2.845 g of a solid bright yellowish powdery solid was obtained, and this was used in the next synthetic step without further purification. Yield: 82.7%. Rf = 0.79 (eluent EtOAc/n-Hex/toluene, 1:1:1); 1H NMR (400 MHz, DMSO-d6) δ = 8.60 (d, J = 2.12 Hz, 1H, H-4), 8.29 (m, AA’BB′, J = 8.28 Hz, 2H, H-2′ and H-6′), 8.22 (dd, J = 9.20 Hz and J = 2.16 Hz, 1H, H-6), 8.18 (d, J = 9.16 Hz, 1H, H-7), 8.13 (d, J = 3.72 Hz, 1H, H-2), 8.01 (m, AA’BB′, J = 8.44 Hz, 2H, H-3′ and H-5′), 7.13 ppm (d, J = 3.72 Hz, 1H, H-1); 13C NMR (75 MHz, DMSO-d6) δ = 111.29 (C-3), 114.17 (C-7), 118.59 (C-4), 120.49 (C-6), 123.41 (q, J = 273.30 Hz, -CF3), 127.8250 (q, J = 3.65 Hz, C-3′ and C-5′), 128.46 (C-2′ and C-6′), 130.44 (C-2), 131.12 (C-3a), 134.84 (q, J = 32.69 Hz, C-4′), 137.42 (C-7a), 140.53 (C-1′), 144.46 ppm (C-5); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C15H10F3N2O4S+, 371.0313; found, 371.0309.
4.1.1.5. N-cyclopropyl-5-nitro-1H-indole-1-carboxamide (1e)
To a stirred slurry of NaH (0.630 g of a 60% mineral oil dispersion, 26.27 mmol, 3 eq.) in THF (25 mL) at 0 °C, under N2, was cautiously added 5-nitroindole (1.42 g, 8.75mmol,1eq.) previously dissolved in THF (5 mL). The reaction mixture was stirred at 0 °C for 60 min, then transferred, via cannula, to a solution of 4-nitrophenyl chloroformate (2.11 g, 10.5 mmol, 1.2 eq.) in THF (8.5 mL). The resultant reaction mixture was stirred at ambient temperature for 15 h (TLC, eluent n-Hex/EtOAc, 2:1), prior to removal of the solvent by concentration in vacuo. The residue obtained was suspended in EtOAc (100 mL), then filtered and washed with EtOAc and Et2O to give 2.515 g (87.6%) of a pale yellow solid. The resulting activated carbamate 5-nitro-1-(4-nitrophenoxycarbonyl) indole was immediately used as follows: a 2.0 M solution of cyclopropylamine (0.382 mL, 5.48 mmol, d = 0.824 g/mL, 8 eq.) in THF (2.75 mL) was added to a solution of 5-nitro-1-(4-nitrophenoxycarbonyl)indole (0.245 g, 0.686 mmol, 1eq.) in THF (5 mL). The resultant reaction mixture was stirred at ambient temperature for 4 h and monitored by TLC (eluent n-Hex/EtOAc, 2:1), prior to removal of the solvent by concentration to dryness in vacuo. The residue obtained was partitioned between EtOAc (100 mL) and H2O (100 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 × 30 mL). The combined organic extracts were washed with sat’d NaHCO3 (100 mL), brine and finally dried over NaSO4 and concentrated in vacuo to give a yellow solid which was suspended in Et2O (35 mL), filtered and washed with Et2O (2 × 20 mL) to give 0.066 g of a pale yellow solid. Yield: 39.1%. Rf = 0.26 (eluent n-Hex/EtOAc, 2:1); 1H NMR (400 MHz, DMSO-d6) δ = 8.59 (d, J = 2.25 Hz, 1H, H-4), 8.50 (d, J = 2.31 Hz, 1H, NH), 8.39 (d, J = 9.18 Hz, 1H, H-7), 8.15 (dd, J = 9.21 Hz and J = 2.40 Hz, 1H, H-6), 8.02 (d, J = 3.89 Hz, 1H, H-2), 6.92 (dd, J = 3.69 Hz and J = 0.55, 1H, H-3), 2.80 (sex J = 3.12 Hz, 1H, NH-CH), 0.8–0.6 ppm (m, 4H, -CH2CH2-); 13C NMR (75 MHz, DMSOd6) δ = 6.75 (-CH2CH2-), 22.75 (NCH-CH2CH2-), 104.32 (C-3), 112.16 (C-7),116.74 (C-6),117.62 (C-4),127.30 (C-2),129.55 (C-3a),135.38 (C-7a), 140.95 (C-5), 160.11 ppm (C=O); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 246.0879; found, 246.0871.
4.1.2. General procedure for the synthesis of 1N-substituted aminoindoles 2a–d
As a typical procedure, the synthesis of 1-benzoyl-5-amino-1H-indole 2a is described in detail. Into a two-necked 50 mL round-bottomed flask, 3.241 g of 1-benzoyl-5-nitro-1H-indole (1a) (12.17 mmol, 1 eq.), 10.986 g of SnCl2·2H2O (48.68 mmol, 4 eq.), 2 mL of HCl 37% and 30 mL of methanol were added. The reaction mixture was refluxed for 3 h, and the reaction progress was monitored by TLC (n-Hex/EtOAc, 1:1). At the end, the solvent was evaporated, the residue was taken up with aqueous NaOH 20% (20 mL), and the resulting suspension was extracted with diethyl ether (4 × 50 mL). The combined extracts, washed with brine and treated with anhydrous Na2SO4, were evaporated to dryness on a rotary evaporator to yield 1.536 g of a semisolid yellow product, made up of three different reaction products. In order to obtain the desired pure 1-benzoyl-5-amino-1H-indole, the raw powder was purified in a silica gel chromatographic column 3 × 28 cm, 230–400 mesh, eluent n-Hex/EtOAc, 1:1, yielding 0.267 g of a pure yellow solid.
4.1.2.1. 1-Benzoyl-5-amino-1H-indole (2a)
Yield: 19.5%. Rf = 0.33 (eluent n-Hex/EtOAc, 1:1); 1H NMR (400 MHz, DMSO-d6) δ = 7.98 (d, J = 8.73 Hz, 1H, H-7), 7.70 (m, 2H, H-2′ and H-6′), 7.66 (m, 1H, H-4′), 7.59 (m, 2H, H-3′ and H-5′), 7.16 (d, J = 3.72 Hz, 1H, H-3), 6.75 (d, J = 2.07 Hz, 1H, H-4), 6.65 (dd, J = 8.74 Hz and J = 2.20 Hz, 1H, H-6), 6.51 (d, J = 3.42 Hz, 1H, H-2), 5.05 ppm (s, 2H, NH2); 13C NMR (75 MHz, DMSO-d6) δ = 101.99 (C-3), 113.14 (C-7), 115.82 (C-6), 118.34 (C-4), 128.44 (C-2′ and C-6′), 129.04 (C-3′ and C-5′), 129.47 (C-4′), 131.83 (C-1′), 133.84 (C-2), 134.72 (C-3a), 137.46 (C-7a), 144.04 (C-5), 172.01 ppm (C=O); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C15H13N2O+, 237.1028; found, 237.1031.
4.1.2.2. 1-Methanesulfonyl-5-amino-1H-indole (2b)
Compound 2b was prepared as described for compound 2a by reacting 1 g (4.16 mmol, 1 eq.) of the appropriate 5-nitroindole derivative 1b and 4.69 g of SnCl2·2H2O (20.80 mmol, 5 eq.), obtaining 0.963 g of a slightly brown solid. Yield: 80.6%; Rf = 0.37 (eluent n-Hex/EtOAc, 1:1); 1H NMR (400 MHz, DMSO-d6) δ = 7.48 (dt, J = 8.76 Hz and J = 0.66 Hz, 1H, H-7), 7.34 (d, J = 3.96 Hz, 1H, H-2), 6.75 (dd, J = 2.20 Hz and J = 0.50 Hz, 1H, H-4), 6.67 (dd, J = 8.76 Hz and J = 2.24 Hz, 1H, H-6), 6.59 (dd, J = 3.64 Hz and J = 0.75 Hz, 1H, H-3), 4.95 (bs, 2H, NH2), 3.24 ppm (s, 3H, SO2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 41.06 (SO2CH3), 105.02 (C-4), 108.96 (C-3), 113.92 (C-7), 114.21 (C-6), 127.28 (C-3a), 127.82 (C-2), 132.35 (C-7a), 145.95 ppm (C-5); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C9H11N2O2S+, 211.0541; found, 211.0542.
4.1.2.3. 1-[4-(Trifluoromethyl)benzenesulfonyl]-5-amino-1H-indole (2d)
Compound 2d was prepared as described for compound 2a by reacting 1.25 g (3.37 mmol, 1 eq.) of the appropriate 5-nitroindole derivative 1d and 3.80 g of SnCl2·2H2O (16.87 mmol, 5 eq.), obtaining 1.160 g of a slightly brown solid. Yield: 99.9%; Rf = 0.30 (eluent n-Hex/EtOAc/toluene, 1:1:1); 1H NMR (400 MHz, DMSOd6) = δ: 7.77 (m, J = 8.40 Hz, J = 1.98 and J = 1.76 Hz, 2H, H-3′ and H-5′), 7.67 (d, J = 8.76 Hz, 1H, H-7), 7.58 (d, J = 3.60 Hz, 1H, H-2), 7.30 (m, J = 8.12 Hz, 2H, H-2′ and H-6′), 6.77 (d, J = 2.04 Hz, 1H, H-4), 6.73 (dd, J = 8.72 Hz and J = 2.09 Hz, 1H, H-6), 6.61 (dd, J = 3.64 Hz and J = 0.60 Hz, 1H, H-3), 4.98 ppm (bs, 2H, -NH2); 13C NMR (101 MHz, DMSO-d6) δ: 105.92 (C-4), 110.02 (C-3), 114.18 (C-7), 114.51 (C-6), 124.55 (q, J = 245.01 Hz, CF3), 127.00 (C-3′ and C-5′), 127.50 (C-2), 127.81 (C-3a), 130.45 (C-2′ and C-6′), 133.83 (q, J = 33.01 Hz, C-4′), 134.69 (C-7a), 143.98 (C-5), 145.87 ppm (C-1′); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C15H12F3N2O2S+, 341.0572; found, 341.0569.
4.1.3. General procedure for the synthesis of 1N-substituted aminoindoles 2c, 5a, 5b, 5d, 5e
As a typical procedure, the synthesis of 1-(4-methylbenzenesulfonyl)-5-amino-1H-indole 2c is described in detail. Into a three-necked flask of 500 mL, previously dried in an oven, about 0.300 g of C/Pd 10% and approximately 60 mL of EtOAc were placed. After connecting the flask to an elastomer balloon containing H2 gas, the mixture was stirred at room temperature for 1 h in order to saturate the suspension of C/Pd with H2. Then, 1.9 g (6.00 mmol) of the appropriate 5-nitroindole derivative 1c in 15 mL of EtOAc was added dropwise to the suspension, and the mixture was stirred under H2 at atmospheric pressure and heated by means of an oil bath at 50–60 °C for 15 h, monitoring the progress of the reaction by TLC analysis (EtOAc/n-Hex, 9:1). At the end of the reaction, the mixture was filtered, and the solution was concentrated to dryness on a rotary evaporator to give 1.680 g of semisolid dark purple sticky product.
4.1.3.1. 1-(4-Methylbenzenesulfonyl)-5-amino-1H-indole (2c)
Yield: 97.8%. Rf = 0.76 (eluent n-Hex/EtOAc, 9:1); 1H NMR (400 MHz, DMSO-d6) δ = 7.77 (m, AA’BB′, J = 8.40 Hz, J = 1.98 Hz and J = 1.76 Hz, 2H, H-2′ and H-6′), 7.67 (d, J = 8.76 Hz, 1H, H-7), 7.58 (d, J = 3.60 Hz, 1H, H-2), 7.30 (m, AA’BB′, J = 8.12 Hz, 2H, H-3′ and H-5′), 6.77 (d, J = 2.04 Hz, 1H, H-4), 6.73 (dd, J = 8.72 Hz and J = 2.09 Hz, 1H, H-6), 6.61 (dd, J = 3.64 Hz and J = 0.60 Hz, 1H, H-3), 4.95 (bs, 2H, -NH2), 2.26 ppm (s, 3H, -CH3); 13C NMR (101 MHz, DMSO-d6) δ = 21.40 (-CH3), 105.92 (C-4), 110.02 (C-3), 114.18 (C-7), 114.51 (C-6), 127.00 (C-2′ and C-6′), 127.50 (C-2), 127.81 (C-3a), 130.45 (C-3′ and C-5′), 132.27 (C-4′), 134.69 (C-7a), 143.98 (C-5), 145.87 ppm (C-1′); HRMS (ESI-MS,140 eV):m/z [M + H+] calculated for C15H15N2O2S+, 287.0854; found, 287.0851.
4.1.3.2. 1-Benzoyl-2,3-dihydro-5-amino-1H-indole (5a)
Yield: 98.8%. Rf = 0.54 (eluent n-Hex/EtOAc, 5:4); 1H NMR (400 MHz, DMSO-d6) δ = 7.94 (d, J = 8.40, 1H, H-6), 7.49 (m, 6H, H-7, H-2′, H-3′, H-4′, H-5′ and H-6′), 6.48 (d, J = 1.89, 1H, H-4), 4.98 (bs, 2H, -NH2), 3.87 (t, J = 8.15 Hz, 2H, H2-2), 2.93 ppm (t, J = 8.14, 2H, H2-3); 13C NMR (101 MHz, DMSO-d6) δ = 29.27 (N-CH2CH2), 51.24 (N-CH2CH2), 110.98 (C-4), 113.94 (C-6), 115.98 (C-7), 128.24 (C-2′ and C-6′), 128.99 (C-3′ and C-5′), 129.32 (C-4′), 132.37 (C-3a), 134.23 (C-7a), 135.54 (C-1′), 143.82 (C-5), 166.83 ppm (C=O); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C15H15N2O+, 239.1184; found, 239.1179.
4.1.3.3. 1-Methanesulfonyl-2,3-dihydro-5-amino-1H-indole (5b)
Yield: 94.8%. Rf = 0.15 (eluent n-Hex/EtOAc, 2:1); 1H NMR (400 MHz, DMSO-d6) δ = 6.95 (d, J = 8.48 Hz, 1H, H-7), 6.50 (m, J = 2.37 Hz, 1H, H-4), 6.39 (dd, J = 8.50 Hz and J = 2.34 Hz, 1H, H-6), 4.91 (bs, 2H, -NH2), 3.82 (t, J = 8.24 Hz, 2H, N-CH2CH2), 2.96 (t, J = 8.20 Hz, 2H, N-CH2CH2), 2.81 ppm (s, 3H, SO2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 28.30 (N-CH2CH2), 33.48 (SO2CH3), 50.39 (N-CH2CH2), 111.26 (C-4), 112.92 (C-6), 115.57 (C-7), 131.88 (C-3a), 133.66 (C-7a), 146.17 ppm (C-5); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C9H13N2O2S+, 213.0698; found, 213.0663.
4.1.3.4. 1-[4-(Trifluoromethyl)benzenesulfonyl]-2,3-dihydro-5-amino-1H-indole (5d)
Yield: 98.2%. Rf = 0.51 (eluent n-Hex/EtOAc/toluene, 1:1:1); 1H-NMR (400 MHz, DMSO-d6) δ = 7.90 (m, J = 8.98 Hz, 2H, H-3′ and H-5′), 7.40 (m, J = 8.98 Hz, 2H, H-2′ and H-6′), 7.19 (d, J = 8.52 Hz, 1H, H-7), 6.41 (dd, J = 8.50 Hz and J = 2.26 Hz, 1H, H-6), 6.33 (d, J = 2.16 Hz, 1H, H-4), 4.99 (s, 2H, -NH2), 3.85 (t, J = 8.11 Hz, 2H, H2-3), 2.56 ppm (t, J = 8.10 Hz, 2H, H2-2); 13C NMR (101 MHz, DMSO-d6) δ = 28.17 (C-2), 50.69 (C-3), 111.00 (C-4), 113.13 (C-6), 116.93 (C-7), 126.27 (q, J = 3.80 Hz, C-3′ and C-5′), 127.35 (q, J = 269.56 Hz, CF3), 128.57 (C-2′ and C-6′), 132.44 (C-3a), 133.35 (q, J = 32.32 Hz, C-4′), 134.44 (C-7a), 140.59 (C-1′), 146.93 ppm (C-5).; HRMS (ESI-MS,140 eV):m/z [M + H+] calculated for C15H14F3N2O2S+, 341.0572; found, 341.0569.
4.1.3.5. N-cyclopropyl-2,3-dihydro-5-amino-1H-indole-1-carboxamide (5e)
Yield: 91.2%. Rf = 0.23 (eluent n-Hex/EtOAc, 2:1); 1H NMR (400 MHz, DMSO-d6) δ = 7.56 (d, J = 8.43 Hz, 1H, H-7), 6.46 (m, 2H, H-4 and NH), 6.35 (dd, J = 8.44 Hz and J = 2.32 Hz, 1H, H-6), 4.63 (s, 2H, -NH2), 3.77 (t, J = 8.58 Hz, 2H, H2-2), 2.99 (t, J = 8.53 Hz, 2H, H2-3), 2.60 (sex, J = 3.46 Hz, 1H, NH-CH-CH2CH2-), 0.6–0.4 ppm (m, 4H, -CH2CH2-); 13C NMR (101 MHz, DMSO-d6) δ = 6.42 (-CH2CH2-), 23.47 (C-3), 27.89 (C-2), 46.89 (NH-CH-CH2CH2-), 111.27 (C-4), 112.39 (C-6), 115.04 ppm (C-7), 130.32 (C-3a), 133.83 (C-7a),143.82 (C-5),162.35 ppm (C=O); HRMS (ESI-MS,140 eV):m/z [M + H+] calculated for C12H16N3O+, 218.1293; found, 218.1245.
4.1.4. General procedure for the synthesis of acrylate derivatives 3b–d and 6a, 6b, 6d and 6e
As a typical procedure, the synthesis of (E,Z)-Ethyl 3-(1-(methanesulfonyl)-1H-indol-5-ylamino)-3-phenylacrylate 3b is described in detail. In a 100 mL round-bottomed flask, 1.4 g (6.66 mmol, 1 eq.) of 1-methanesulfonyl-5-amino-1H-indole 2a in 25 mL of absolute ethanol was condensed with 1.73 mL (9.99 mmol; d = 1.11 g/mL, 1.5 eq.) of commercial ethyl benzoylacetate and 0.5 mL of glacial acetic acid in the presence of 100 mg of Drierite (anhydrous CaSO4). The mixture was refluxed for about 24 h, the reaction being monitored by TLC analysis (eluent n-Hex/EtOAc, 2:1). Even though the reaction was not complete after 24 h, the mixture was cooled and filtered to remove the Drierite; the resulting solution was evaporated to dryness under vacuum and the residue (2.420 g) purified by silica gel chromatography (3 × 35 cm, 230–400 mesh, eluent n-Hex/EtOAc, 2:1) to yield 1.54 g of a deep yellow powdery solid.
4.1.4.1. (E,Z)-Ethyl 3-(1-(methanesulfonyl)-1H-indol-5-ylamino)-3-phenylacrylate (3b)
Yield: 60.2%. Rf = 0.65 (eluent n-Hex/EtOAc, 2:1); 1H NMR (400 MHz, DMSO-d6) δ = 10.25 (s, 1H, NH), 7.58 (d, J = 8.80 Hz, 1H, H-7), 7.49 (d, J = 3.68 Hz, 1H, H-2), 7.36 (m, 5H, 2′-,3′-,4′-,5′-,6′-H), 7.02 (d, J = 2.16 Hz, 1H, H-4), 6.84 (dd, J = 8.82 Hz and 2.18 Hz, 1H, H-6), 6.62 (dd, J = 3.68 Hz and J = 0.72 Hz, 1H, H-3), 4.94 (s, 1H, C=C-H), 4.15 (q, J = 7.09 Hz, 2H, OCH2CH3), 3.38 (s, 3H, SO2CH3),1.24 ppm (t, J = 7.10 Hz, 3H, OCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 14.76 (OCH2CH3), 41.35 (SO2CH3), 59.21 (OCH2CH3), 90.70 (C=C-H), 109.09 (C-3), 113.26 (C-7), 115.26 (C-4), 120.62 (C-6), 127.80 (C-2), 128.45 (C-2′ and C-6′), 128.94 (C-3′ and C-5′), 130.00 (C-4′), 130.77 (C-3a), 131.07 (C-1′), 135.80 (C-7a), 136.22 (C-5), 159.49 (C=C-H), 169.45 ppm (COO-CH2CH3); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C20H21N2O4S+, 385.1222; found, 385.1213.
4.1.4.2. (E,Z)-Ethyl 3-(1-(p-toluenesulfonyl)-1H-indol-5-ylamino)-3-phenylacrylate (3c)
Compound 3c was prepared as described for compound 3b by reacting 3.177 g (11.10 mmol) of the appropriate 5-aminoindole derivative 2c, obtaining after column chromatography 2.336 g of a brownish sticky semisolid product. Yield: 45.7%. Rf = 0.62 (eluent n-Hex/EtOAc, 2:1); 1H NMR (400 MHz, DMSO-d6) δ = 10.18 (s, 1H, NH), 7.79 (m, AA’BB′, J = 8.32 Hz, 2H, H-2′ and H-6′), 7.69 (d, J = 3.64 Hz, 1H, H-2), 7.65 (d, J = 8.84 Hz, 1H, H-7), 7.35 (m, AA’BB′, J = 8.61 Hz, 2H, H-3′ and H-5′), 7.31 (m, 5H, H-2″, H-3″,H-4″, H-5″ and H-6″), 6.92 (d, J = 1.88 Hz, 1H, H-4), 6.78 (dd, J = 8.61 Hz and J = 1.82 Hz, 1H, H-6), 6.61 (d, J = 3.68 Hz, 1H, H-3), 4.92 (s, 1H, C=C-H), 4.12 (q, J = 7.14 Hz, 2H, -OCH2CH3), 2.31 (s, 3H, -CH3), 1.22 ppm (t, J = 7.17 Hz, 3H, -OCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 14.85 (-OCH2CH3), 21.46 (-CH3), 59.34 (-OCH2CH3), 90.99 (C=C-H), 109.72 (C-3), 113.65 (C-7), 115.30 (C-4), 120.87 (C-6), 127.13 (C-2′ and C-6′), 128.24 (C-2), 128.53 (C-2″ and C-6″), 129.03 (C-3″ and C-5″), 130.11 (C-4″), 130.65 (C-3′ and C-5′), 130.91 (C-3a), 131.26 (C-1″), 134.53 (C-4′), 135.85 (C-7a), 136.69 (C-5), 145.95 (C-1′), 159.39 (C=C-H), 169.48 ppm (COOCH2CH3); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C26H25N2O4S+, 461.1535; found, 461.1539.
4.1.4.3. (E,Z)-Ethyl-3-(1-(4-(trifluoromethyl)benzenesulfonyl)-1Hindol-5-ylamino)-3-phenylacrylate (3d)
Compound 3c was prepared as described for compound 3b by reacting 1.160 g (3.41 mmol) of the appropriate 5-aminoindole derivative 2d, obtaining after column chromatography 1.020 g of a yellow powdery solid. Yield: 58.1%. Rf = 0.78 (eluent n-Hex/EtOAc, 8:2); 1H NMR (400 MHz, DMSO-d6) = δ: 10.18 (s, 1H, NH), 7.79 (m, AA’BB′, J = 8.32 Hz, 2H, H-2′ and H-6′), 7.69 (d, J = 3.64 Hz, 1H, H-2), 7.65 (d, J = 8.84 Hz, 1H, H-7), 7.35 (m, AA’BB′, J = 8.61 Hz, 2H, H-3′ and H-5′), 7.31 (m, 5H, H-2″, H-3″,H -4″, H-5″ and H-6″), 6.92 (d, J = 1.88 Hz, 1H, H-4), 6.78 (dd, J = 8.61 Hz and J = 1.82 Hz, 1H, H-6), 6.61 (d, J = 3.68 Hz, 1H, H-3), 4.92 (s, 1H, C=C-H), 4.12 (q, J = 7.14 Hz, 2H, -OCH2CH3), 1.22 ppm (t, J = 7.17 Hz, 3H, -OCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 14.84 (-OCH2CH3), 59.37 (-OCH2CH3), 90.90 (C=C-H), 109.71 (C-3), 113.64 (C-7), 115.36 (C-4), 120.89 (C-6), 127.11 (C-2′ and C-6′), 128.28 (C-2), 128.52 (C-2″ and C-6″), 128.99 (q, J = 269.28 Hz, CF3), 129.01 (q, J = 3.85 Hz, C-3″ and C-5″), 130.65 (C-3′ and C-5′), 130.91 (C-3a), 131.26 (C-1″), 133.28 (q, J = 32.35 Hz, C-4″) 134.53 (C-4′), 135.85 (C-7a), 136.69 (C-5), 145.95 (C-1′), 159.39 (C=C-H), 170.01 ppm (COOCH2CH3); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C26H22F3N2O4S+, 515.1252; found, 515.12.49.
4.1.4.4. (E,Z)-Ethyl 3-(1-(benzoyl)-2,3-dihydro-1H-indol-5-ylamino)-3-phenylacrylate (6a)
Compound 5a was prepared as described for compound 3b by reacting 1.96 g (8.22 mmol) of the appropriate 5-aminoindole derivative 5a, obtaining after column chromatography 0.940 g of a brown viscous oil. Yield: 27.7%. Rf = 0.70 (eluent n-Hex/EtOAc/toluene, 1:1:1); 1H NMR (400 MHz, DMSO-d6) δ = 10.14 (s, 1H, NH), 7.95 (m, 2H, H-2′ and H-6′), 7.68 (m, 1H, H-4′), 7.60 (m, 1H, H-4″), 7.60–7.32 (m, 8H, H-2″, -3″, -5″, -6″ and H-3′, -5′, and H-6, H-7), 6.72 (d, J = 1.32 Hz, 1H, H-4), 4.90 (s, 1H, C=CH-), 4.14 (q, J = 7.08 Hz, 2H, -OCH2CH3) 3.92 (t, J = 8.23 Hz, 2H, H-3), 2.90 (t, J = 8.32 Hz, 2H, H-2), 1.20 ppm (t, J = 7.09 Hz, 3H, -CH2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 15.24 (-OCH2CH3), 29.23 (C-3), 52.26 (C-2), 58.92 (-OCH2CH3), 91.07 (C=C-H), 114.23 (C-7), 116.32 (C-4), 119.28 (C-6), 127.11 (C-2′ and C-6′), 128.52 (C-2″ and C-6″), 129.91 (C-3″ and C-5″), 130.23 (C-3′ and C-5′), 130.91 (C-3a), 131.18 (C-1″), 133.84 (C-4″) 134.82 (C-4′), 135.75 (C-7a), 136.82 (C-5), 146.23 (C-1′), 158.83 (C=C-H), 165.24 (NC=O), 171.91 ppm (COOCH2CH3); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 413.1865; found, 413.1859.
4.1.4.5. (E,Z)-Ethyl 3-(1-(methanesulfonyl)-2,3-dihydro-1H-indol-5-ylamino)-3-phenylacrylate (6b)
Compound 6b was prepared as described for compound 3b by reacting 0.460 g (2.16 mmol) of the appropriate 5-aminoindole derivative 5b, obtaining after column chromatography 0.540 g of a brown sticky oil. Yield: 64.8%. Rf = 0.37 (eluent n-Hex/EtOAc, 2:1); 1H NMR (400 MHz, DMSO-d6) δ = 10.11 (s, 1H, NH), 7.38 (m, 1H, H-4′), 7.34 (m, 4H, H-2′, -3′, -5′ and 6′), 6.96 (d, J = 8.56 Hz, 1H, H-7), 6.72 (d, J = 2.20 Hz, 1H, H-4), 6.52 (dd, J = 8.72 Hz and J = 2.22 Hz, 1H, H-6), 4.90 (s, 1H, C=C-H), 4.31 (q, J = 7.09 Hz, 2H, OCH2CH3), 3.86 (t, J = 8.46 Hz, 2H, H-2), 2.93 (t, J = 8.44 Hz, 2H, H-3), 2.90 (s, 3H, SO2CH3), 1.23 ppm (t, J = 7.08 Hz, 3H, OCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 14.74 (OCH2CH3), 27.68 (C-3), 34.38 (SO2CH3), 50.31 (C-2), 59.21 (OCH2CH3), 90.63 (C=C-H), 113.64 (C-7), 120.18 (C-4), 122.20 (C-6), 128.38 (C-2′ and C-6′), 129.92 (C-3′ and C-5′), 130.03 (C-4′), 133.07 (C-3a), 135.75 (C-7a), 136.46 (C-1′), 138.14 (C-5), 159.19 (C=C-H), 169.39 ppm (COOCH2CH3); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C20H23N2O4S+, 387.1379; found, 387.1381.
4.1.4.6. (E,Z)-Ethyl 3-(1-(4-(trifluoromethyl)benzenesulfonyl)-2,3-dihydro-1H-indol-5-ylamino)-3-phenylacrylate (6d)
Compound 6d was prepared as described for compound 3b by reacting 1.560 g (4.55 mmol) of the appropriate 5-aminoindole derivative 5d, obtaining after column chromatography 1.29 g of a bright yellow powdery solid. Yield: 54.8%. Rf = 0.75 (eluent n-Hex/EtOAc, 8:2); 1H-NMR (400 MHz, DMSO-d6) δ = 10.15 (s, 1H, NH), 7.92 (m, J = 8.98 Hz, 2H, H-3″ and H-5″), 7.45 (m, J = 8.98 Hz, 2H, H-2″ and H-6″), 7.38 (m, 1H, H-4′), 7.34 (m, 4H, H-2′, -3′, -5′ and -6′), 6.96 (d, J = 8.56 Hz, 1H, H-7), 6.72 (d, J = 2.20 Hz, 1H, H-4), 6.52 (dd, J = 8.72 Hz and J = 2.22 Hz, 1H, H-6), 4.96 (s, 1H, C=C-H), 4.24 (q, J = 7.09 Hz, 2H, OCH2CH3), 3.96 (t, J = 8.46 Hz, 2H, H-2), 2.86 (t, J = 8.44 Hz, 2H, H-3), 1.15 ppm (t, J = 7.08 Hz, 3H, OCH2CH3); 13C-NMR (101 MHz, DMSO-d6) δ = 14.74 (OCH2CH3), 27.68 (C-3), 50.31 (C-2), 59.21 (OCH2CH3), 90.63 (C=C-H), 113.64 (C-7), 120.18 (C-4), 122.20 (C-6), 126.47 (q, J = 3.75 Hz, C-3″ and C-5″), 127, 98 (q, J = 265.67 Hz, CF3), 128.38 (2′- and 6′-C), 128.81 (C-2’‘and C-6″), 129.92 (3′- and 5′-C), 130.03 (C-4′), 133.07 (C-3a), 134.72 (q, J = 32.81 Hz, C-4″), 135.75 (C-7a), 136.46 (C-1′), 138.14 (C-5), 159.19 (C=C-H), 169.39 ppm (COOCH2CH3); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C26H24F3N2O4S+, 517.1409; found, 517.1401.
4.1.4.7. (E,Z)-Ethyl 3-(1-(N-cyclopropyl-1-carboxamide)-2,3-dihydro-1H-indol-5-ylamino)-3-phenylacrylate (6e)
Compound 6e was prepared as described for compound 3b by reacting 0.128 g (0.589 mmol) of the appropriate 5-aminoindole derivative 5e, obtaining after column chromatography 0.139 g of a dark brown tarry oil. Yield: 60.4%. Rf = 0.56 (eluent CHCl3/MeOH, 95:5); 1H NMR (400 MHz, DMSO-d6) δ = 10.08 (s, 1H, NH), 7.52 (d, J = 8.55 Hz, 1H, H-7), 7.33 (m, 5H, H-2′, -3′, -4′, -5′ and -6′), 6.62 (m, 2H, H-4 and NH), 6.41 (dd, J = 8.55 Hz and J = 2.16 Hz, 1H, H-6), 4.83 (s, 1H, C= CH), 4.12 (q, J = 7.08 Hz, 2H, -OCH2CH3), 3.74 (t, J = 8.71 Hz, 2H, H2-2), 2.90 (t, J = 8.70 Hz, 2H, H2-3), 2.51 (m, 1H, -N-CH-CH2CH2-), 1.23 (t, J = 7.08 Hz, 3H, -CH3), 0.6–0.4 ppm (m, 4H, -CH2CH2-); 13C NMR (101 MHz, DMSO-d6) δ = 6.35 (-CH2CH2-), 14.96 (-CH3), 21.24 (C-3), 45.95 (NH-CH), 47.15 (-OCH2CH3), 59.06 (C-2), 90.24 (C=CH), 115.21 (C-7), 116.15 (C-6), 118.43 (C-4), 127.13 (C-3a), 127.45 (C-2′ and C-6′), 128.74 (C-3′ and C-5′), 130.14 (C-4′), 132.16 (C-7a), 132.74 (C-1′), 146.09 (C-5), 159.16 (C=CH), 161.45 (NC=ONH), 170.23 ppm (COOCH2CH3); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C23H26N3O3, 329.1974; found, 392.1971.
4.1.5. General procedure for the synthesis of phenylpyrroloquinolinones 4b–d, 7a, 7b, 7d and 8a, 8b, 8d
As a typical procedure, the synthesis of 3-methanesulfonyl-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one 4b is described in detail. In a two-necked round-bottomed flask, 20 mL of diphenyl ether was heated to boiling. To this 0.383 g (4.2 mmol) of the appropriate phenylacrylate derivative 3b was added portion wise, and the resulting mixture was refluxed for 15 min. After cooling to room temperature, 25 mL of diethyl ether was added, and the mixture was left for 12 h. The precipitate was collected by filtration and washed many times with diethyl ether. The product (0.437 g) was additionally purified by silica gel column chromatography (2.5 × 30 cm, 230–400 mesh, eluent CHCl3/MeOH, 95:5), obtaining 0.303 g of a slightly brown solid.
4.1.5.1. 3-Methanesulfonyl-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one (4b)
Yield: 89.9%; Rf = 0.16 (blue fluorescent spot, eluent CHCl3/MeOH, 95:5); mp: 321.5 °C (decomposition); UV-Vis (H2O/MeOH, 99:1): λmax (A) = 274 (A = 0.874), 347 nm (A = 0.432); fluorescence (H2O): λexc = 350.1 nm, λems = 488.8 nm; IR (KBr): ν = 3403.20 (NH), 3088 (C-H aromatic), 2958 (C-H aliphatic), 1610.20 (C=O), 1449.01 (C=C) 1170.20 cm−1 (SO2N); 1H NMR (400 MHz, DMSO-d6) δ = 11.87 (s, 1H, NH), 8.19 (d, J = 9.12 Hz, 1H, H-4), 7.92 (d, J = 3.52 Hz, 1H, H-1), 7.87 (m, J = 6.60 Hz and J = 4.20 Hz, 2H, H-2′ and H-6′), 7.82 (d, J = 9.08 Hz, 1H, H-5), 7.72 (d, J = 3.48 Hz, 1H, H-2), 7.62 (m, 1H, 4′-H), 7.60 (m, 2H, H-3′ and H-5′), 6.45 (d, J = 1.80 Hz, 1H, H-8), 3.50 ppm (s, 3H, SO2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 41.90 (SO2CH3), 109.08 (C-8), 109.78 (C-1), 116.19 (C-5), 117.30 (C-9a), 117.85 (C-4), 125.95 (C-9b), 127.56 (C-2), 127.80 (C-2′ and C-6′), 129.36 (C-3′ and C-5′), 129.56 (C-1′), 130.70 (C-4′), 133.82 (C-3a), 137.79 (C-5a), 148.37 (C-7), 177.48 ppm (C-9); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C18H15N2O3S+, 339.0803; found, 339.0798; RP-C18 HPLC: tR = 11.7 min, 97.5%.
4.1.5.2. 3-(p-Toluenesulfonyl)-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one (4c)
Compound 4c was prepared as described for compound 4b by reacting 2.336 g (5.07 mmol) of the appropriate phenylacrylate derivative 3c to yield 1.197 g of a slightly brown solid product. Yield: 56.9%; Rf = 0.64 (blue fluorescent spot, eluent CHCl3/MeOH, 9:1); mp: 277.6 °C (decomposition); UV-Vis (H2O/MeOH, 99:1): λmax (A) = 275 (A = 0.985), 347 nm (A = 0.569); fluorescence (H2O): λexc = 350.1 nm, λems = 489.1 nm; IR (KBr): ν = 3410.50 (NH), 3080 (C-H aromatic), 2965 (C-H aliphatic), 1611.40 (C=O), 1460.01 (C=C) 1170.17 cm−1 (SO2N); 1H NMR (400 MHz, DMSO-d6) δ = 11.85 (s, 1H, NH), 8.29 (d, J = 9.18 Hz, 1H, H-4), 7.90 (m, 1H, H-2), 7.89 (m, AA′BB′, J = 8.54 Hz, 2H, H-2′ and H-6′), 7.88 (m, 1H, H-1), 7.83 (m, 2H, H-2″ and H-6″), 7.79 (d, J = 9.18 Hz, 1H, H-5), 7.58 (m, 3H, H-3″, H-5″ and H-4″), 7.38 (m, 2H, H-3″ and H-5″), 6.40 (s, 1H, H-8), 2.30 ppm (s, 3H, -CH3); 13C NMR (75 MHz, DMSO-d6) δ = 21.91 (-CH3), 109.62 (C-8), 111.66 (C-1), 117.04 (C-5), 118.36 (C-9a), 118.51 (C-4), 127.54 (C-2′ and C-6′), 127.61 (C-9b), 128.29 (C-2″ and C-6″), 128.49 (C-2), 129.84 (C-3″ and C-5″), 130.59 (C-4″), 131.14 (C-3′ and C-5′), 131.20 (C-1″), 134.94 (C-4′), 135.08 (C-3a), 139.07 (C-5a), 146.49 (C-1′), 149.65 (C-7), 178.48 ppm (C-9); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C24H19N2O3S+, 415.1226; found, 415.1299; RP-C18 HPLC: tR = 14.7 min, 99.7%.
4.1.5.3. 3-((4-(Trifluoromethyl)benzene)sulfonyl)-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one (4d)
Compound 4d was prepared as described for compound 4b by reacting 1.555 g (3.02 mmol) of the appropriate phenylacrylate derivative 3d to yield 0.414 g of a brownish solid product. Yield: 29.3%; Rf = 0.65 (blue fluorescent spot, eluent CHCl3/MeOH, 9:1); mp: 341 °C (decomposition); UV-Vis (H2O/MeOH, 99:1): λmax (A) = 270 (A = 0.279), 345 nm (A = 0.169); fluorescence (H2O): λexc = 345.7 nm, λems = 491.1 nm; IR (KBr): ν = 3402.50 (NH), 3078 (C-H aromatic), 2969 (C-H aliphatic), 1608.30 (C=O), 1458.01 (C=C) 1169.37 (SO2N), 1325.12 cm−1 (C-F); 1H NMR (DMSO-d6): δ = 11.90 (s, 1H, NH), 8.32 (dd, J = 9.16 Hz and 0.68 Hz, 1H, H-4), 8.23 (m, J = 8.28 Hz, 2H, H-2′ and H-6′), 7.99 (m, 1H, H-2), 7.99 (m, 2H, H-3″ and H-5″), 7.93 (d, J = 3.32 Hz, 1H, H-1) 7.84 (m, 2H, H-2″ and H6″) 7.82 (d, J = 9.08 Hz, 1H, H-5), 7.59 (m, 1H, 4′), 7.59 (m, 2H, H-3′ and H-5′), 6.46 ppm (bs, 1H, H-8); 13C NMR (DMSO-d6): δ = 109.30 (C-8), 112.06 (C-1), 117.23 (C-5), 117.62 (C-9a), 118.03 (C-4), 126.41 (C-9b), 126.62 (q, J = 247.23, CF3), 127.29 (C-2), 127.65 (q, J = 32.40 Hz, C-3″ and C-5″), 127.94 (C-2″ and C-6″), 128.10 (C-2′ and C-6′), 129.48 (C-3′ and C-5′), 130.85 (C-4′), 131.24 (C-3a), 134.52 (q, J = 32.40, C-4″), 135.70 (C-1′), 139.53 (C-5a), 141.19 (C-1″), 155.26 (C-7), 180.72 ppm (C-9); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C24H16F3N2O3S+, 469.1027; found, 469.1040; RP-C18 HPLC: tR = 16.45 min, 98.5%.
4.1.5.4. 3-Benzoyl-1,2-dihydro-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one (7a) and 1n-benzoyl-2,3-dihydro-6-phenyl-5H-pyrrolo[2,3-g] quinolin-8-one (8a)
Compounds 7a and 8a were prepared as described for compound 4b by reacting 1.100 g (2.66 mmol) of the appropriate phenylacrylate derivative 6a to yield 0.443 g of a raw powdery solid consisting of the two isomers 7a and 8a. The two desired compounds were purified by liquid column chromatography (eluent CHCl3/MeOH, 95:5).
3-benzoyl-1,2-dihydro-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one (7a). 0.128 g were obtained. Yield: 15.3%; Rf = 0.49 (blue fluorescent spot, eluent CHCl3/MeOH, 9:1); mp: 322 °C (decomposition); UV–Vis (H2O/MeOH, 99:1): λmax (A) = 221 (0.717), 293 (0.670), 343 nm (0.320); λmin (A) = 258 (0.226), 318 nm (0.172); fluorescence (H2O): λexc = 300 nm, λems = 451 nm; IR (KBr): ν̃= 3180, 2950, 2880, 1615, 1490 cm−1; 1H NMR (400 MHz, DMSO)-d6) δ = 11.69 (s, 1H, NH), 7.82 (m, 2H, H-2″ and H-6″), 7.61 (m, 7H, H-3″, -4″, -5″, H-2′ and H-6′, H-4 and H-5), 7.52 (m, 3H, H-3′, -5′ and 4′), 6.24 (d, J = 1.82 Hz, 1H, H-8), 4.09 (t, J = 8.65 Hz, 2H, H-2), 3.68 ppm (t, J = 8.65 Hz, 2H, H-1); 13C NMR (DMSO-d6): δ = 25.34 (C-3), 50.17 (C-2),115.24 (C-4),109.45 (C-8),117.83 (C-9a),121.73 (C-5), 122.24 (C-9b), 127.12 (C-2′ and C-6′), 127.64 (C-2″ and C-6″), 128.41 (C-3″ and C-5″), 128.58 (C-3′ and C-5′), 129.45 (C-4′), 130.14 (C-4″), 132.13 (C-1′), 134.32 (C-3a), 135.23 (C-1″), 139.45 (C-5a), 154.13 (C-7), 165.34 (C=O), 178.34 ppm (C-9); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 367.1447; found, 367.1439; RP-C18 HPLC: tR = 12.38 min, 97.4%.
1N-benzoyl-2,3-dihydro-6-phenyl-5H-pyrrolo[2,3-g]quinolin-8-one (8a). 0.141 g were obtained. Yield: 16.8%; Rf = 0.37 (greenish fluorescent spot, eluent CHCl3/MeOH, 9:1); mp: 319 °C (decomposition); UV–Vis (H2O/MeOH, 99:1): λmax (A) = 222 (0.514), 284 (0.498), 350 nm (0.320); λmin (A) = 258 (0.226), 318 nm (0.172); fluorescence (H2O), λexc = 350 nm, λems = 468 nm; IR (KBr): ν̃= 3380, 3190, 2960, 1610, 1480 cm−1; 1H NMR (400 MHz, DMSO)-d6) δ = 11.72 (s, 1H, NH), 7.84 (m, 2H, H-2″ and H-6″), 7.72 (m, 7H, H-3″, -4″, -5″, H-2′ and H-6′, H-4 and H-9), 7.55 (m, 3H, H-3′, -5′ and 4′), 6.35 (s, 1H, H-7), 4.01 (t, J = 8.72 Hz, 2H, H-2), 3.28 ppm (t, J = 8.71 Hz, 2H, H-1); 13C NMR (DMSO-d6): δ = 26.28 (C-3), 49.24 (C-2), 116.16 (C-4), 108.24 (C-7), 118.13 (C-8a), 120.34 (C-9), 121.04 (C-3a), 126.92 (C-2′ and C-6′), 127.04 (C-2″ and C-6″), 127.97 (C-3″ and C-5″), 128.18 (C-3′ and C-5′), 129.05 (C-4′), 130.14 (C-4″), 131.14 (C-1′), 133.16 (C-9a), 136.14 (C-1″), 140.36 (C-4a), 153.16 (C-6), 166.18 (C=O), 177.90 ppm (C-8); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 367.1447; found, 367.1448; RP-C18 HPLC: tR = 12.02 min, 97.5%.
4.1.5.5. 3-Methanesulfonyl-1,2-dihydro-7-phenyl-6H-pyrrolo[3,2-f] quinolin-9-one (7b) and 1N-methanesulfonyl-2,3-dihydro-6-phenyl-5H-pyrrolo[2,3-g]quinolin-8-one (8b)
Compounds 7b and 8b were prepared as described for compound 4b by reacting 0.488 g (1.27 mmol) of the appropriate phenylacrylate derivative 6b to yield 0.210 g of a raw powdery solid consisting of the two isomers 7b and 8b. The two desired compounds were purified by liquid column chromatography (eluent CHCl3/MeOH, 95:5)
4.1.5.5.1. 3-Methanesulfonyl-1,2-dihydro-7-phenyl-6H-pyrrolo [3,2-f]quinolin-9-one (7b)
0.084 g of a pale yellow solid were obtained. Yield: 19.4%; Rf = 0.21 (blue fluorescent spot, eluent CHCl3/MeOH, 95:5); mp: 311.4 °C (decomposition); UV–Vis (H2O/MeOH, 99:1): λmax (A) = 206 (1.435), 270 (1.341), 345 nm (0.642); λmin (A) = 195 (0.453), 247 (0.600), 314 nm (0.344); fluorescence (H2O): λexc = 350 nm, λems = 458 nm; IR (ATR ZnSe): ν = 3370, 3010, 2978, 1640, 1478, 1057 cm−1; 1H NMR (400 MHz, DMSO-d6) δ = 11.64 (s, 1H, NH), 7.82 (m, 2H, H-2′ and H-6′), 7.68 (d, J = 9.23 Hz, 1H, H-4), 7.63 (d, J = 8.87 Hz, 1H, H-5), 7.58 (m, 3H, H-3′, H-5′ and H-4′), 6.25 (sb, 1H, H-8), 4.02 (t, J = 8.60 Hz, 2H, H-2), 3.70 (t, J = 8.56 Hz, 2H, H-3), 2.97 ppm (s, 3H, SO2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 30.15 (C-1), 35.05 (SO2CH3), 51.63 (C-2), 108.77 (C-8), 119.32 (C-5), 119.58 (C-4), 119.79 (C-9a), 128.14 (C-2′ and C-6′), 129.84 (C-3′ and C-5′), 129.95 (C-4′), 131.00 (C-1′), 131.28 (C-9b), 134.88 (C-3a), 138.74 (C-5a), 150.45 (C-7), 178.97 ppm (C-9); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C18H17N2O3S+, 341.1211; found, 341.1250; RP-C18 HPLC: tR = 10.99 min, 96.8%.
4.1.5.5.2. 1N-methanesulfonyl-2,3-dihydro-6-phenyl-5H-pyrrolo [2,3-g]quinolin-8-one (8b)
0.068 g of a yellowish solid were obtained. Yield: 15.7; Rf = 0.10 (blue fluorescent spot, eluent CHCl3/MeOH, 95:5); mp: 317.7 °C (decomposition); UV–Vis (H2O/MeOH, 99:1): λmax (A) = 210 (1.137), 275 (0.875), 352 nm (0.447); λmin (A) = 190 (0.104), 247 (0.650), 314 nm (0.346); fluorescence (H2O): λexc = 350 nm, λems = 475 nm; IR (ATR ZnSe): ν = 3250, 2986, 1615, 1476, 1055 cm−1; 1H NMR (400 MHz, DMSO-d6) δ = 11.70 (s, 1H, NH), 7.91 (s, 1H, H-9), 7.81 (m, 2H, H-2′ and H-6′), 7.64 (s, 1H, H-4), 7.59 (m, 3H, H-3′, H-5′ and H-4′), 6.30 (bs, 1H, H-6), 4.01 (t, J = 8.26 Hz, 2H, H-2), 3.28 (t, J = 8.16 Hz, 2H, H-1), 3.03 ppm (s, 3H, SO2CH3); 13C NMR (101 MHz, DMSO-d6) δ = 27.91 (C-1), 34.40 (SO2CH3), 50.49 (C-2), 107.24 (C-7), 107.35 (C-9), 115.80 (C-4), 125.03 (C-9a), 127.80 (C-2′ and C-6′), 129.48 (C-3′ and C-5′), 130.84 (C-4′), 134.60 (C-1′), 137.83 (C-8a), 138.51 (C-3a), 139.011 (C-4a), 149.48 (C-7), 176.66 ppm (C-8); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C18H17N2O3S+, 341.1211; found, 341.1226; RP-C18 HPLC: tR = 10.52 min, 98.5%.
4.1.5.6. 3-((4-(Trifluoromethyl)benzene)sulfonyl)-1,2-dihydro-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one (7d) and 1N-((4-(trifluoromethyl) benzene)sulfonyl)-2,3-dihydro-6-phenyl-5H-pyrrolo [2,3-g]quinolin-8-one (8d)
Compounds 7d and 8d were prepared as described for compound 4b by reacting 1.100 g (2.12 mmol) of the appropriate phenylacrylate derivative 6d to yield 0.920 g of a raw sticky viscous tar. The tar was triturated with Et2O and purified by liquid column chromatography (eluent CHCl3/MeOH, 9:1) yielding 0.587 g of a powdery white solid consisting of an irresolvable mixture of the two isomers 7d and 8d. Yield: 58.6%; Rf = 0.61 (eluent CHCl3/MeOH, 9:1); mp: 301 °C (decomposition); UV-Vis (H2O/MeOH, 99:1): 275 nm (A = 0.337), 351 nm (A = 0.258); IR (KBr): ν = 3432.80 (NH), 3078 (C-H aromatic), 2980 (C-H aliphatic), 1600 (C=O), 1498 (C=C) 1171.51 (SO2N), 1323.63 cm−1 (C-F); 1H NMR (400 MHz, DMSO-d6): δ = 11.69 (s, 1H, NH), 11.67 (s, 1H, NH), 8.21 (m, J = 8.94 Hz, 2H), 8.14 (s, 1H), 8.02 (d, J = 9.17 Hz, 1H), 7.98 (m, 4H), 7.92 (d, J = 9.46 Hz, 1H), 7.80 (m, J = 8.22 Hz, 2H), 7.72 (d, J = 3.03 Hz, 1H), 7.69 (m, J = 8.23 Hz, 2H), 7.66 (d, J = 8.66 Hz, 2H), 7.61 (m, 2H), 7.41 (t, J = 2.05 Hz, 1H), 7.36 (s, 1H), 6.58 (s, 1H), 6.32 (s, 1H), 4.07 (t, J = 7.81 Hz, 4H), 3.54 (t, J = 7.83 Hz, 2H), 3.15 ppm (t, J = 8.23 Hz, 2H); 13C NMR (101 MHz, DMSO-d6): δ = 27.33, 29.03, 50.21, 50.87, 104.52, 105.27, 106.95, 112.24, 113.50, 118.56, 119.39, 119.65, 121.42, 121.55, 124.83 (q, J = 4.01 Hz), 126.29, 126.67 (q, J = 249.26 Hz), 126.88 (q, J = 247.28 Hz), 127.38, 127.59, 127.92,128.09,128.48,128.99,129.06,129.37,129.99,130.43,130.74, 131.60, 133.72 (q, J = 33.01 Hz), 134.02 (q, J = 32.72 Hz), 136.93, 137.25, 139.86, 150.67, 152.13, 179.12, 180.02 ppm; HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C24H18F3N2O3S+, 471.1234; found, 471.1221; RP-C18 HPLC: tR = 15.50 min, 82.0% and tR = 16.47 min, 17.4%. (NB: 32 H in NMR. If this represents both isomers, shouldn’t there be 34 H, unless some are silent?)
4.1.5.7. 3-Benzoyl-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one (4a)
Into a two-necked 50 mL round-bottomed flask, 0.041 g (1.7 mmol, 3 eq.) of NaH, 60% dispersion in mineral oil, was placed and washed with toluene (3 × 10 mL). With stirring, a solution of 7-phenyl-3H,6H-pyrrolo[3,2-f]quinolin-9-one (9, prepared as previously reported [8]), 0.150 g (0.57 mmol, 1 eq.) in 7 mL of anhydrous DMF, was dropped into the flask. After 30 min at room temperature, a solution of benzoyl chloride, 0.2 mL (1.7 mmol, d = 1.21 g/mL, 3 eq.) in 2 mL dry DMF, was added, and the reaction mixture was stirred for 2 h. The reaction was monitored by TLC analysis (eluent CHCl3/MeOH, 9:1). At the end of the reaction, 25 mL of water was added, and the solvent was evaporated under reduced pressure, leaving a residue, which was extracted with EtOAc (3 × 50 mL). The organic phase, washed with water, a 10% Na2CO3 solution, and brine was dried over anhydrous Na2SO4 and concentrated under vacuum to yield a crude yellow solid (0.171 g). This crude product was purified with a silica gel chromatographic column (3 × 35 cm, 230–400 mesh, CHCl3/MeOH, 9:1), yielding 0.057 g of a pure yellowish solid. Yield: 27.2%; Rf = 0.48 (blue fluorescent spot, eluent CHCl3/MeOH, 9:1); mp: 316.8 °C (decomposition); UV–Vis (H2O/MeOH, 99:1): λmax (A) = 204 (0.992), 279 (1.557), 352 nm (0.281); λmin (A) = 195 (0.322), 237 (0.443), 336 nm (0.225); fluorescence (H2O), λexc = 277 nm, λems = 460 nm; IR (KBr): ν = 3327 (NH), 1790 (C=O amidic), 1678 (C=O), 1660 cm−1 (C=C); 1H NMR (400 MHz, DMSOd6): δ = 11.87 (s, 1H, NH), 8.60 (d, J = 9.16 Hz, 1H, H-4), 7.88 (m, 2H, H-2′ and H-6′), 7.87 (d, J = 2.96 Hz, 1H, H-1), 7.82 (m, 2H, H-2″ and H-6″), 7.80 (d, J = 8.42 Hz, 1H, H-5), 7.73 (m, J = 7.51 Hz, J = 2.12 Hz and J = 1.22 Hz, 1H, H-4″), 7.65 (m, 2H, H-3″ and H-5″), 7.61 (m, 3H, H-3′, H-5′ and H-4′), 7.51 (d, J = 3.56 Hz, 1H, H-2), 6.44 ppm (d, J = 1.16 Hz, 1H, H-8); 13C NMR (101 MHz, DMSO-d6): δ = 109.13 (C-8), 110.23 (C-1), 116.18 (C-5), 117.21 (C-9a), 120.64 (C-4), 127.33 (C-9b), 127.93 (C-2′ and C-6′), 129.23 (C-3′ and C-5′), 129.37 (C-2), 129.46 (C-3′ and C-5′), 129.75 (C-2″ and C-6″), 130.79 (C-4′), 131.51 (C-1′), 132.79 (C-4″), 134.20 (C-1″), 137.00 (C-3a), 138.96 (C-5a), 149.20 (C-7), 168.99 (NC=O), 178.45 ppm (C-9); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 365.1385; found, 365.1382; RP-C18 HPLC: tR = 13.95 min, 96.5%.
4.1.5.8. N-cyclopropyl-7-phenyl-6H-pyrrolo[3,2-f]quinolin-9-one-3-carboxamide (4e)
To a stirred slurry of NaH (0.041 g of a 60% mineral oil dispersion, 1.73 mmol, 3 eq.) in THF (2 mL) at 0 °C, under N2, was cautiously added 7-phenyl-3H,6H-pyrrolo[3,2-f]quinolin-9-one (9, 0.15 g, 0.57 mmol, 1eq., prepared as reported [8]) previously dissolved in THF (7 mL). The reaction mixture was stirred at 0 °C for 60 min, then transferred, via cannula, to a solution of 4-nitrophenyl chloroformate (0.140 g, 0.69 mmol, 1.2 eq.) in THF (2 mL). The resultant reaction mixture was stirred at ambient temperature for 15 h (TLC, eluent n-Hex/EtOAc, 2:1), prior to removal of the solvent by concentration in vacuo. The residue obtained was suspended in EtOAc (100 mL), then filtered and washed with EtOAc and Et2O to yield 0.255 g (99.0%) of a pale yellow solid. The resulting activated N-(4-nitro)phenyl-7-phenyl-6H-pyrrolo [3,2-f]quinolin-9-one-3-carboxamide was immediately used as follows: a 2.0 M solution of cyclopropylamine (0.382 mL, 5.48 mmol, d = 0.824 g/mL, 8 eq.) in THF (2.75 mL) was added to a solution of the activated carbamate (0.255 g, 0.60 mmol, 1eq.) in THF (5 mL). The resultant reaction mixture was stirred at ambient temperature for 4 h and monitored by TLC (eluent CHCl3/MeOH, 85:15) prior to removal of the solvent by concentration to dryness, in vacuo. The residue obtained was partitioned between EtOAc (100 mL) and H2O (100 mL). The layers ware separated, and the aqueous phase was extracted with EtOAc (2 × 30 mL). The combined organic extract was washed with saturated NaHCO3 (100 mL) and brine, dried over NaSO4 and concentrated in vacuo to give a yellow solid that was suspended in Et2O (35 mL), filtered and washed with Et2O (2 × 20 mL) to give 0.062 g of a yellowish solid. Yield: 30.1%. Rf = 0.29 (eluent CHCl3/MeOH, 85:15); mp: 314 *C (decomposition); UV–Vis (H2O/MeOH, 99:1): λmax (A) = 338 (0.081), 270 (0.162), 204 nm (0.169); λmin (A) = 312 nm (0.046), 246 nm (0.079); fluorescence (H2O): λexc = 215.00 nm, λem = 430.00 nm; IR (KBr): ν = 3311.56 (NH), 1618 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ = 11.80 (s, 1H, NH), 8.56 (d, J = 9.12 Hz, 1H, H-4), 8.40 (d, J = 2.91 Hz, 1H, C=ONH), 7.91 (d, J = 3.54 Hz, 1H, H-2), 7.85 (m, 2H, H-2′ and H-6′), 7.75 (d, J = 2.67 Hz, 1H, H-1), 7.68 (d, J = 9.15 Hz, 1H, H-5), 7.58 (m, 3H, H-3′, H-5′ and H-4′), 6.41 (bs, 1H, H-8), 2.81 (m, J = 3.24 Hz, 1H, NH-CH), 0.80–0.60 ppm(m, 4H, -CH2CH2-); 13C NMR (101 MHz, DMSO-d6): δ = 6.31 (-CH2CH2-), 23.82 (NH-CH), 108.05 (C-1), 108.93 (C-8), 115.15 (C-5), 117.86 (C-9a), 120.14 (C-4), 124.15 (C-9b), 125.81 (C-2), 127.86 (C-2′ and C-6′), 129.43 (C-3′ and C-5′), 130.59 (C-4′), 130.85 (C-1′), 131.39 (C-3a), 134.85 (C-5a), 153.12 (C-7), 165.21 (NC=ONH), 178.43 ppm (C-9); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 344.1999; found, 344.1994; RP-C18 HPLC: tR = 10.93 min, 98.5%.
4.2. Biological assays
4.2.1. Cell growth conditions and antiproliferative assay
Human T-leukemia (CCRF-CEM and Jurkat), human B-leukemia (RS4; 11, SEM) cells and human myeloid leukemia (HL-60, THP-1, MV4; 11) cells, were grown in RPMI-1640 medium (Gibco, Milano, Italy). Breast adenocarcinoma (MCF-7), human cervix carcinoma (HeLa), non small cell lung adenocarcinoma (A549) and human colon adenocarcinoma (HT-29) cells were grown in DMEM medium (Gibco, Milano, Italy), all supplemented with 115 units/mL penicillin G (Gibco, Milano, Italy), 115 μg/mL streptomycin (Invitrogen, Milano, Italy), and 10% fetal bovine serum (Invitrogen, Milano, Italy). Stock solutions (10 mM) of the different compounds were obtained by dissolving them in DMSO. Individual wells of a 96-well tissue culture microtiter plate were inoculated with 100 μL of complete medium containing 8 × 103 cells. The plates were incubated at 37 °C in a humidified 5% CO2 incubator for 18 h prior to the experiments. After medium removal, 100 μL of fresh medium containing the test compound at different concentrations was added to each well in triplicate and incubated at 37 °C for 72 h. Cell viability was assayed by MTT test as previously described [26]. The GI50 was defined as the compound concentration required to inhibit cell proliferation by 50%. Peripheral blood lymphocytes (PBL) from healthy donors were obtained by separation on Lymphoprep (Fresenius KABI Norge AS) gradient. After extensive washing, cells were resuspended (1.0 × 106 cells/mL) in RPMI-1640 with 10% fetal bovine serum and incubated overnight. For cytotoxicity evaluations in proliferating PBL cultures, non-adherent cells were resuspended at 5 × 105 cells/mL in growth medium, containing 2.5 μg/mL PHA (Irvine Scientific). Different concentrations of the test compounds were added, and viability was determined 72 h later by the MTT test. For cytotoxicity evaluations in resting PBL cultures, non-adherent cells were resuspended (5 × 105 cells/mL) and treated for 72 h with the test compounds, as described above.
4.2.2. Effects on tubulin polymerization and on colchicine binding to tubulin
To evaluate the effect of the compounds on tubulin assembly in vitro [15], varying concentrations of compounds were pre-incubated with 10 μM bovine brain tubulin in glutamate buffer at 30 °C and then cooled to 0 °C. After addition of 0.4 mM GTP (final concentration), the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30 °C. Tubulin assembly was followed turbidimetrically at 350 nm. The IC50 was defined as the compound concentration that inhibited the extent of assembly by 50% after a 20 min incubation. The ability of the test compounds to inhibit colchicine binding to tubulin was measured as described [16], except that the reaction mixtures contained 1 μM tubulin, 5 μM [3H]colchicine and 5 μM test compound.
4.2.3. Molecular modeling
Compounds in Table 1 were built and their partial charges calculated after semi-empirical (PM6) energy minimization using the MOE2015 [27] program. Fourteen crystallographic structures were selected to perform docking studies (see SI_Table 1). Only the ligands occupying the colchicine binding site and the protein chain in the proximity of 4.5 Å were considered and subjected to the structure preparation tool of MOE 2015. Finally, Protonate 3D tool was used to assign the ionic state of each complex [28]. To identify the more appropriate docking protocol for the eleven complexes, we performed a self-docking benchmark using DockBench 1.01, a tool that compared the performance of 14 different posing/scoring protocols [29]. The active site was defined using a radius of 12 Å from the center of mass of the co-crystallized ligand. Each ligand was docked 20 times. All synthesized compounds were docked using GOLD using PLP [30], using the virtual screening tool of DockBench adopting the parameters already used in the benchmark study. Finally, the obtained conformations were rescored with the dock_pKi MOE function. The similarity studies were carried out with vROCS considering the Tversky coefficient [31].
To facilitate the visualization and analysis of data obtained from the docking simulations, we implemented an in-house tool, named MMsDocking video maker, for the automated production of a video that shows the most relevant docking data, such as docking poses, per residue IEhyd and IEele data, experimental binding data and scoring values. Videos were mounted using MEncoder [32], starting from images obtained with the following procedure: the heat maps in the background were drawn with GNUPLOT 4.6 [33] starting from per residue IEhyd and IEele data computed with MOE. Two dimensional depictions of compounds were generated using the open-source cheminformatics toolkit RDKit [34]. Representations of docking poses within the binding site were constructed using CHIMERA [35].
4.2.4. Flow cytometric analysis of cell cycle distribution
5 × 105 HeLa or Jurkat cells were treated with different concentrations of the test compounds for 24 h. After the incubation period, the cells were collected, centrifuged, and fixed with ice-cold ethanol (70%). The cells were then treated with lysis buffer containing RNase A and 0.1% Triton X-100 and stained with PI. Samples were analyzed on a Cytomic FC500 flow cytometer (Beckman Coulter). DNA histograms were analyzed using MultiCycle for Windows (Phoenix Flow Systems).
4.2.5. Apoptosis assay
Cell death was determined by flow cytometry of cells double stained with annexin V/FITC and PI. The Coulter Cytomics FC500 (Beckman Coulter) was used to measure the surface exposure of PS on apoptotic cells according to the manufacturer’s instructions (Annexin-V Fluos, Roche Diagnostics).
4.2.6. Analysis of mitochondrial potential and reactive oxygen species (ROS)
The mitochondrial membrane potential was measured with the lipophilic cation JC-1 (Molecular Probes, Eugene, OR, USA), while the production of ROS was followed by flow cytometry using the fluorescent dye H2DCFDA (Molecular Probes), as previously described [11].
4.2.7. Evaluation of the metabolic stability of compound 4a in human liver microsomes
4.2.7.1. Incubation procedure
Compound 4a (final concentration, 10 μM) was incubated in a medium (final volume, 0.2 mL) containing 0.1 M KH2PO4 (pH 7.4) and 1.0 mg/mL of pooled mixed-gender human liver microsomes (Xenotech LLC, Lenexa, USA; HLMs), in the absence or presence of 1 mM NADPH (Sigma-Aldrich). Control incubations were performed in the absence of both HLMs and NADPH (buffer only-incubations). The reactions were started by adding the microsomes following a 3-min thermal equilibration at 37 °C, conducted at 37 °C for different time periods (i.e. 0, 15, 30 and 60 min), and terminated by adding 0.1 mL of ice-cold acetonitrile. Samples were then centrifuged (4 °C) at 20,000g for 10 min, and aliquots of the supernatants were analyzed by HPLC with fluorescence detection, as described below
4.2.7.2. HPLC analysis
The chromatographic system consisted of a Hewlett-Packard 1100 HPLC system (Agilent Technologies Inc., formerly Hewlett-Packard, Palo Alto, USA) equipped with a degasser, a quaternary pump, an autosampler, a column oven, and a fluorescence detector; chromatographic data were collected and integrated using the Agilent ChemStation software. Chromatographic conditions were as follows: column, Agilent Zorbax SB C18 (4.6 × 75 mm, 3.5 μm); mobile phase, 0.1% HCOOH in H2O (solvent A) and 0.1% HCOOH in acetonitrile (solvent B); elution program, isocratic elution with 95% solvent A for 2 min, linear gradient from 5 to 40% solvent B in 8 min, followed by a further linear gradient from 40 to 60% solvent B in 2 min, and an isocratic elution with 60% solvent B for 7 min; post-run time, 5 min; flow rate, 1.0 mL/min; injection volume, 50 μL; column temperature, 30 °C; detection, fluorescence (excitation wavelength, 344 nm; emission wavelength, 493 nm). Under the above conditions, the retention time of 4a was 13.4 min. Metabolic stability of 4a, expressed as percent of compound remaining, was calculated by comparing the corresponding chromatographic peak area at each time point relative to that at time 0 min.
Supplementary Material
Acknowledgments
The computational work coordinated by S.M. has been supported with financial support from the University of Padova, Italy. MMS lab is also very grateful to Chemical Computing Group and OpenEye Scientific for the scientific and technical partnership. S.M. participates in the European COST Action CM1207 (GLISTEN). The work of M.S. has been supported by University of Padova, Italy (UNIPD, Progetto Giovani Studiosi 2013: Protocol number 79122). The content of this paper is solely the responsibility of the authors and does not necessarily reflect the official views of the Italian Ministry of Health.
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version, at http://dx.doi.org/10.1016/j.ejmech.2016.10.026. These data include MOL files and InChiKeys of the most important compounds described in this article.
Footnotes
Supporting Information file contains: the 14 tubulin-ligand complexes retrieved from the PDB, the superposition of the 14 tubulin-ligand complex used in the benchmark, docking benchmark results obtained with DockBench and HRMS spectra and HPLC traces of all the final tested compounds 4a-e, 7a,b,d and 8a,b,d. Movie. mp4 file contains an Animation of the energetically most favorable pose of all the final tested compounds and the gold standard 11 obtained by molecular docking simulation.
References
- 1.Islam MN, Iskander MN. Microtubulin binding sites as target for developing anticancer agents. Mini-Rev Med Chem. 2004;4:1077–1104. doi: 10.2174/1389557043402946. [DOI] [PubMed] [Google Scholar]
- 2.Desai A, Mitchison TJ. Microtubule polymerization dynamics. Ann Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 3.Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anti-Cancer Agents. 2002;2:1–17. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]
- 4.Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Vuuren RJ, Visagie MH, Theron AE, Joubert AM. Antimitotic drugs in the treatment of cancer. Cancer Chemother Pharmacol. 2015;76:1101–1112. doi: 10.1007/s00280-015-2903-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitika V, Kapil K. Microtubule targeting agents: a benchmark in cancer therapy. Curr Crug Ther. 2013;8:189–196. [Google Scholar]
- 7.Ferlin MG, Chiarelotto G, Gasparotto V, Dalla Via L, Pezzi V, Barzon L, Palu G, Castagliuolo I. Synthesis and in vitro and in vivo antitumor activity of 2-phenylpyrroloquinolin-4-ones. J Med Chem. 2005;48:3417–3427. doi: 10.1021/jm049387x. [DOI] [PubMed] [Google Scholar]
- 8.Gasparotto V, Castagliuolo I, Chiarelotto G, Pezzi V, Montanaro D, Brun P, Palù G, Viola G, Ferlin MG. Synthesis and biological activity of 7-phenyl-6,9-dihydro-3H-pyrrolo[3,2-f] quinolin-9-ones: a new class of antimitotic agents devoid of aromatase activity. J Med Chem. 2006;49:1910–1915. doi: 10.1021/jm0510676. [DOI] [PubMed] [Google Scholar]
- 9.Ferlin MG, Conconi MT, Urbani L, Oselladore B, Guidolin D, Di Liddo R, Parnigotto PP. Synthesis, in vitro and in vivo preliminary evaluation of antiangiogenic properties of some pyrroloazaflavones. Bioorg Med Chem. 2011;19:448–457. doi: 10.1016/j.bmc.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Gasparotto V, Castagliuolo I, Ferlin MG. 3-Substituted 7-phenyl-pyrroloquinolinones show potent cytotoxic activity in human cancer cell lines. J Med Chem. 2007;50:5509–5513. doi: 10.1021/jm070534b. [DOI] [PubMed] [Google Scholar]
- 11.Ferlin MG, Bortolozzi R, Brun P, Castagliuolo I, Hamel E, Basso G, Viola G. Synthesis and in vitro evaluation of 3H-pyrrolo[3,2-f]-quinolin-9-one derivatives that show potent and selective anti-leukemic activity. Chem-MedChem. 2010;5:1373–1385. doi: 10.1002/cmdc.201000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Viola G, Bortolozzi R, Hamel E, Moro S, Brun P, Castagliuolo I, Ferlin MG, Basso G. MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochem Pharmacol. 2012;83:16–26. doi: 10.1016/j.bcp.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carta D, Ferlin MG. An overview on 2-arylquinolin-4(1h)-ones and related structures as tubulin polymerisation inhibitors. Curr Top Med Chem. 2014;14:2322–2345. doi: 10.2174/1568026614666141127120421. [DOI] [PubMed] [Google Scholar]
- 14.Carta D, Bortolozzi R, Hamel E, Basso G, Moro S, Viola G, Ferlin MG. Novel 3-Substituted 7-Phenylpyrrolo[3,2-f]quinolin-9(6H)-ones as single entities with multitarget antiproliferative activity. J Med Chem. 2015;58:7991–8010. doi: 10.1021/acs.jmedchem.5b00805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 16.Verdier-Pinard P, Lai J, Yoo H, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Zhang H, Gigant B, Yu Y, Wu Y, Chen X, Lai Q, Yang Z, Chen Q, Yang J. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016;283:102–111. doi: 10.1111/febs.13555. [DOI] [PubMed] [Google Scholar]
- 18.C C G ( I. Molecular Operating Environment (MOE) (Internet) Available from: http://www.chemcomp.com.
- 19.Xiong S, Mu T, Wang G, Jiang X. Mitochondria-mediated apoptosis in mammals. Protein Cell. 2014;5:737–749. doi: 10.1007/s13238-014-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rovini A, Savry A, Braguer D, Carrè M. Microtubule-targeted agents: when mitochondria become essential to chemotherapy. Biochim Biophys Acta Bioenerg. 2011;1807:679–688. doi: 10.1016/j.bbabio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothe G, Valet G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2′,7′-dichlorofluorescin. J Leukoc Biol. 1990;47:440–448. [PubMed] [Google Scholar]
- 23.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 24.Nohl H, Gille L, Staniek K. Intracellular generation of reactive oxygen species by mitochondria. Biochem Pharmacol. 2005;69:719–723. doi: 10.1016/j.bcp.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Parkinson A, Ogilvie BW. Biotransformation of xenobiotics, in casarett & Doull’s toxicology. In: Klaassen CD, editor. The Basic Science of Poisons. 7. McGraw-Hill; New York: 2008. pp. 161–304. [Google Scholar]
- 26.Romagnoli R, Baraldi PG, Lopez-Cara C, Preti D, Aghazadeh Tabrizi M, Balzarini J, Bassetto M, Brancale A, Fu X, Gao Y, Li J, Zhang S, Hamel E, Bortolozzi R, Basso G, Viola G. Concise synthesis and biological evaluation of 2-Aroyl-5-Amino benzo[ b ]thiophene derivatives as a novel class of potent antimitotic agents. J Med Chem. 2013;56:9296–9309. doi: 10.1021/jm4013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart JJP. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model. 2007;13:1173–1213. doi: 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins Struct Funct Bioinforma. 2009;75:187–205. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuzzolin A, Sturlese M, Malvacio I, Ciancetta A, Moro S. DockBench: an integrated informatic platform bridging the gap between the robust validation of docking protocols and virtual screening simulations. Molecules. 2015;20:9977–9993. doi: 10.3390/molecules20069977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cambridge Crystallographic Data Centre: 12 Union Road. Cambridge CB2 1EZ; UK: GOLD suite, version 5.2 (Internet) Available from: http://www.ccdc.cam.ac.uk. [Google Scholar]
- 31.(Interne). Santa Fe, NM, USA, OpenEye Scientific Software Inc. OEChem, 2016 (cited 2016 May 24). Available from: http://www.eyesopen.com/.
- 32.MEncoder. Available from: http://www.mplayerhq.hu/design7/projects.html.
- 33.Gnuplot. Available from: http://www.gnuplot.info/index.html.
- 34.RDKit: Cheminformatics and Machine Learning Software. Available from: http://www.rdkit.org.
- 35.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.