

Abstract
Tetrandrine (Tet), a potent lysosomal inhibitor, blocks autophagic flux and induces cancer cell death. Previously, the present authors identified the prostate cancer cell line DU145 to exhibit high sensitivity towards Tet in 11 cancer cell lines. In the present study, autophagy in Tet-treated DU145 cells was investigated. Similar to other cell lines, such as PC-3 and 786-O cells, Tet neutralized the acidity of lysosome and blocked autophagy in DU145 cells. However, Tet failed to induce microtubule-associated protein 1 light chain 3 (LC3) conversion in DU145 cells. By contrast, it was observed by transmission electron microscopy that Tet induced an accumulation of autophagosomes in the cytoplasm. These contrasting results indicated that Tet triggered an LC3-independent autophagy in DU145 cells. Alkalizing lysosome with chloroquine enhanced Tet-induced cell death. The results of the present study indicated that detection of autophagy in tumor cells may assist in selecting lysosome inhibitors for chemotherapy treatment in prostate cancer.
Keywords: autophagy, tetrandrine, prostate cancer, DU145 cell line, autophagosome
Introduction
Tetrandrine (Tet), a natural product isolated from Stephania tetrandra, has been reported to be an anti-tumor and anti-inflammatory drug (1–8). Previously, the present authors have demonstrated that Tet induces cell death in 11 cancer cell lines (1). Tet directly neutralizes the lysosomal acidity, blocks the autophagic flux in the degradation step and causes energetic impairment, which eventually results in cell death (1). By contrast, Tet also acts as an autophagy agonist by inducing the synthesis of reactive oxygen species (3,4,9–11), indicating that Tet may function as a multi-target drug to regulate autophagy in cancer cells. Regardless of the Tet target, the present authors and other groups have previously consistently observed that Tet increases the number of autophagosomes, levels of microtubule-associated protein 1 light chain 3, type II (LC3-II) and the number of enhanced green fluorescent protein-LC3 puncta in cancer cells (1,3,9).
DU145, a commonly used prostate cancer cell line for research, is different from PC-3 and LNCaP cells in terms of autophagy regulation (12). As alternative transcripts of autophagy related 5 (ATG5) lack one or two exons in DU145 cells, and this causes the autophagy pathway to be genetically impaired (12). By contrast, DU145 cells exhibited the highest sensitivity to Tet in all of the 11 cancer cell lines that were previously tested by the authors of the present study (1), implying that impairment in autophagy may increase Tet-induced cell death. In order to investigate the role of autophagy on Tet-induced cell death in DU145 cells, the ultrastructural changes following Tet treatment were observed by transmission electron microscopy (TEM). Notably, Tet treatment triggered an accumulation of autophagosomes in this cell line, indicating that alternative autophagy was involved.
Materials and methods
Tetrandrine and other reagents
Tet (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) was dissolved as previously described (1). In brief, Tet powder was dissolved in one drop of 0.1 mM HCl and neutralized with NaHCO3 solution to 7.4 pH Tet was subsequently diluted with phosphate-buffered saline (PBS) for a final concentration of 10 mM. Chloroquine (CQ) was purchased from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany) and dissolved in sterilized deionized water for a final concentration of 100 mM. Rabbit anti-LC3B antibody was purchased from Sigma-Aldrich, Merck Millipore. Mouse anti-β-actin antibody was purchased from Santa Cruz Biotechnology Inc. Other primary (β-actin; cat. no., 8457; dilution, 1:2,000; p62, cat. no., 8025; dilution, 1:1,000) and anti-rabbit secondary antibody (cat. no., 7074; dilution, 1:5,000) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Lipofectamine 3000 reagent and LysoSensor Yellow/Blue DND-160 were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The EGFP-LC3 plasmid was obtained from Osaka University, Osaka, Japan.
Cell culture and transfection
DU145 and PC-3 cells (National Platform of Experimental Cell Resources for Sci-Tech Beijing, China) were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.), 100 units/ml penicillin and 0.1 mg/ml streptomycin (Thermo Fisher Scientific, Inc.), and maintained in an atmosphere of 5% CO2 at 37°C. The cells were digested with 0.25% of trypsin-EDTA solution for passage, and the cells were serum starved for 24 h prior to treatment.
For transfection, 1×105/ml cells were seeded in a 6-well plate and transiently transfected with EGFP-LC3 plasmid at a final concentration of 50 nM by using Lipofectamine 3000 reagent (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Following 6 h of incubation at 37°C, the supernatant was replaced with RPMI-1640 medium containing 1% FBS and incubated at 37°C for 48 h.
Cell viability measurement
Cells were digested and seeded (1×104/well) onto a 96-well plate. Cell viability was detected with Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc. Kumamoto, Japan). Briefly, cells were treated with Tet or CQ and incubated at 37°C in RPMI-1640 medium containing 10% (v/v) CCK-8 for 2 h. Optical densities were read at 450 nm by using a microplate reader (Thermo Fisher Scientific, Inc.).
Autophagy assays
DU145 cells were transfected with EGFP-LC3 plasmid and cultured at 37°C for 48 h. The EGFP-LC3 puncta formation in each group were observed and counted using fluorescence microscopy (Leica Microsystems GmbH, Wetzlar, Germany) (13,14).
TEM
TEM was performed as previously described (1). In brief, the cells were digested, washed with pre-cold PBS, centrifuged at 1,000 × g for 5 min at 4°C and fixed with 2% glutaraldehyde at 4°C overnight. The cell pellets were post-fixed with 1% osmium tetroxide for 1 h. The samples were subsequently dehydrated and cut into ultrathin 40-nm sections. The subcellular structures of cells in each group were observed with an electron microscope (JEOL Ltd., Tokyo, Japan).
Lysosome pH quantification
Lysosomal pH was quantified as previously described (1). A total of 1×105/ml cells were seeded in a 96-well plate, treated with Tet and washed with PBS. The cells were subsequently incubated in 5 µM of LysoSensor Yellow/Blue DND-160 (Thermo Fisher Scientific, Inc.) at room temperature for 5 min. The supernatants were removed and replaced with 100 µl RPMI-1640 medium. Cells used for the standard curve were incubated at room temperature with 10 µM monensin and 20 µM nigericin in 20 mM 2-(N-morpholino) ethanesulfonic acid, 110 mM KCl and 20 mM NaCl at pH 3.6 to 6.9 for 10 min. Fluorescence in each well was assessed based on the ratio of light excited at 329 nm vs. 384 nm.
Western blotting
Total protein from each group was extracted with radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 0.1% sodium dodecyl sulfate). The lysates were centrifuged at 8,000 × g at 4°C for 5 min and the supernatants were collected and subsequently quantified. The lysates in each group were diluted to the same concentration with RIPA lysis buffer, mixed with loading buffer and denatured at 95°C for 5 min. A total of 20 µg of the samples were loaded onto 12% SDS-PAGE and separated with electrophoresis. The proteins were then blotted onto a polyvinylidene difluoride membrane and blocked with 5% non-fat milk for 1 h at room temperature. The membrane was incubated with diluted primary antibodies at 4°C overnight. The membrane was washed with TBST and subsequently incubated with diluted secondary antibody for 1 h at room temperature. Following four washes, the blots were measured by using horseradish peroxidase substrate (Thermo Fisher Scientific, Inc.) and captured with a G:BOX Chemi Gel Documentation System (Syngene, Frederick, MD, USA). The relative density was calculated with Quantity One Software (v4.6.2, Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Student's t-test was performed to determine the statistical significance between two groups by using SPSS version 19.0 software (IBM SPSS, Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference. Data are presented as the mean ± standard deviation.
Results
DU145 cells are more sensitive to tetrandrine (Tet) compared with PC-3 cells
The present authors have previously observed that DU145 cells are more sensitive to Tet treatment than PC-3 cells at 48 h post-incubation (1). In the present study, the DU145 and PC-3 cells were incubated with a series of Tet concentrations for 12, 24 and 48 h, respectively. It was observed that Tet treatment inhibited cell viability in the two cell lines in a dose- and time-dependent manner (Fig. 1). The half maximal inhibitory concentration (IC50) values of Tet in DU145 cells were 24.0 (12 h), 10.2 (24 h) and 2.6 µM (48 h), whereas in PC-3 cells the values were 42.8 (12 h), 13.3 (24 h) and 9.5 µM (48 h), indicating that DU145 cells were more sensitive to Tet treatment compared with PC-3 cells (Fig. 1).
Figure 1.

IC50 values for DU145 and PC-3 cells. DU145 (A) and PC-3 (B) cells were treated with Tet as indicated. Cell viability was measured with cell-counting kit-8 assay. IC50 was defined as the concentration of an inhibitor whereby the response is reduced by half. Data are expressed as the mean ± standard deviation (n=6). **P<0.01 vs. the control (0 µM). Tet, tetrandrine; IC50, half maximal inhibitory concentration.
Tetrandrine fails to induce LC3 conversion in DU145 cells
Autophagy has an important role in Tet-induced cell death (1,3). In order to explore the role of autophagy in Tet-induced cell death of DU145 cells, the LC3 conversion was measured using western blotting. The DU145 cells were treated with 10 µM Tet for 12 h. At 10 µM, Tet was able to induce a significant increase in LC3-II and EGFP-LC3 puncta formation in PC-3 cells (1). It was observed that in DU145 cells LC3-II expression was undetectable in baseline and Tet treatment conditions (Fig. 2A). This observation was not unexpected as the lack of one or two exons in ATG5 interferes with the formation of LC3-II in DU145 cells (12). Consistently, EGFP-LC3 puncta was not be observed in Tet-treated DU145 cells (Fig. 2B).
Figure 2.

Tetrandrine fails to induce LC3 conversion in DU145 cells. (A) DU145 cells were treated with Tet (10 µM) for 12 h. The conversion of LC3 was detected using western blotting. β-actin was employed as a loading control. (B) DU145 cells were transfected with EGFP-LC3 plasmids and treated with 10 µM Tet for 12 h. The EGFP-LC3 puncta were observed under fluorescence microscopy (scale bar, 50 µM). (C) DU145 cells were treated with Tet or CQ for 1 h. Lysosomal pH values were measured by using LysoSensor Yellow/Blue DND-160. The values are shown as the mean ± standard deviation (n=6). (D and E) DU145 cells were treated with 10 µM of Tet for 12 h. The p62 levels were measured using (D) western blotting and (E) the p62/β-actin ratios are shown as the mean ± standard deviation (n=3). **P<0.01 vs. the ctrl. Ctrl, control; CQ, chloroquine; EGFP, enhanced green fluorescent protein; LC3, microtubule-associated protein 1 light chain 3, Tet, tetrandrine.
As autophagy mainly depends on the function of the lysosome, lysosomal pH of DU145 cells treated with a range of Tet concentrations (2.5 to 10 µM) was measured. The pH value increased in a concentration-dependent manner (Fig. 2C), indicating that Tet neutralized lysosomal acidity in DU145 cells.
De-acidulation of lysosomes results in the blockade of autophagic flux. As LC3 lipidation in DU145 cells is defective, assays based on LC3 turnover cannot be performed. Therefore, the expression of p62 (a protein that is degraded through autophagy and is upregulated when the autophagic flux is blocked) was analyzed. p62 protein in Tet-treated DU145 cells was upregulated, indicating that the autophagic flux may be blocked (Fig. 2D and E).
Tet triggers autophagosome accumulation in DU145 cells
In order to investigate the role of autophagy in DU145 cells, the ultrastructural changes of DU145 cells upon Tet treatment for 12 h were observed. Notably, Tet triggered a large amount of autophagosome accumulation in the cytoplasm (Fig. 3). There were no significant differences in the shapes of the autophagosomes in DU145 cells compared with autophagosomes in other cell lines induced by Tet treatment, indicating that Tet may have triggered an alternative autophagy in DU145 cells.
Figure 3.
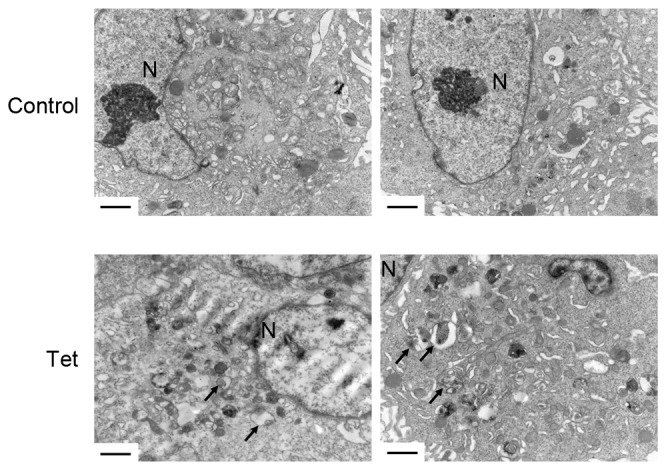
Tet triggers autophagosome accumulation in DU145 cells. DU145 cells were treated with 10 µM Tet for 12 h. The ultrastructure was observed using transmission electron microscopy. Scale bar, 2 µm. Black arrows, autophagosomes. N, nuclear; Tet, tetrandrine.
Lysosomal de-acidulation is required in tetrandrine-induced cell death
The present authors have previously reported that Tet is a potent lysosomal inhibitor in a number of cell lines, which is closely associated with autophagy and its anti-tumor effects (1). In the present study, whether Tet-induced lysosomal de-acidulation contributed to its anti-tumor effects in DU145 cells was investigated. To reduce Tet-induced cell death, DU145 cells were treated with 2.5 µM Tet for 12 h. At 2.5 µM, Tet exhibited mild inhibitory effects on DU145 cells (Fig. 1), and the lysosomal pH was increased to 5.64. DU145 cells were subsequently co-treated with 8 µM CQ to increase the lysosomal pH to 6.78 (Fig. 2C). Treatment with 8 µM CQ alone for 12 h did not result in a significant change in viability of DU145 cells (Fig. 4). However, the presence of CQ synergized Tet-induced cell death (Fig. 4). These results indicated that high lysosomal pH is required in Tet-induced cell death of DU145 cells.
Figure 4.
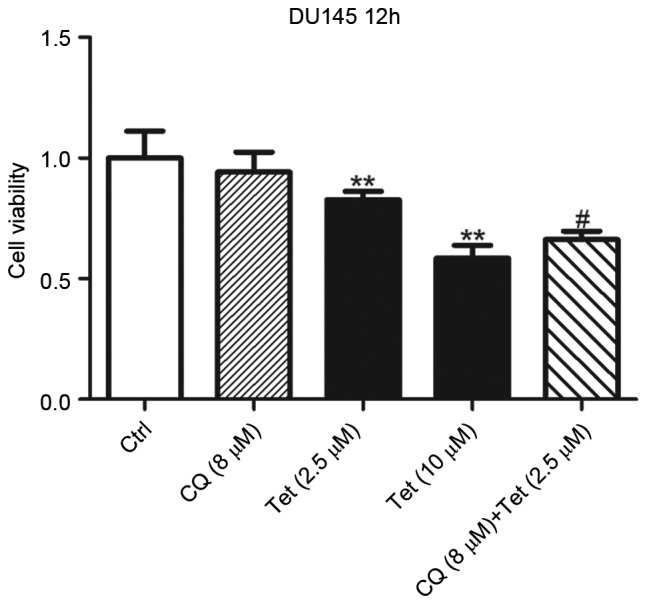
De-acidulation of the lysosome synergizes Tet-induced cell death in DU145 cells. DU145 cells were treated with Tet or CQ as indicated for 12 h. Cell viability was determined with cell counting kit-8 assay. Data are shown as the mean ± standard deviation (n=6). **P<0.01 vs. the ctrl and #P<0.01 vs. Tet (2.5 µM) group. Tet, tetrandrine; Ctrl, control; CQ, chloroquine.
Discussion
LC3 conversion from type I to type II has been employed as a biomarker to evaluate the level of autophagy in previous studies (15,16). In the present study, a notable finding is that Tet triggered LC3-independent autophagy in ATG5 defective prostate cancer cells. This alternative autophagy was characterized as the accumulation of autophagosomes without affecting LC3 conversion. Therefore, evaluation of autophagy using LC3-based assays should be cautiously interpreted. Tet induced lysosomal de-acidulation in DU145 cells, which may result in autophagic flux inhibition and may contribute to its anti-tumor effects.
It has been reported that serum starvation, CQ treatment and rapamycin treatment all failed to induce LC3-I to LC3-II conversion in DU145 cells (12). Due to the lack of functional products of ATG5, the LC3-I to LC3-II conversion is blocked (12). Therefore, the EGFP-LC3 marked autophagosomes could not be observed in DU145 cells (Fig. 2B). By contrast, a recent study reported that sorafenib induces EGFP-LC3 foci formation in DU145 cells (17). The results of the present study indicated that Tet could not induce EGFP-LC3 puncta formation, but it increased the number of autophagosomes in DU145 cells. An alternative autophagy has been previously been discovered in ATG5 genetic knockout mouse embryonic fibroblasts (18). The results of the present study demonstrated that this type of autophagy may naturally exist in cancer cells. The present authors postulate that this alternative autophagy may universally exist in different types of cells. Therefore, only assessing autophagy by using experiments based on LC3 turnover may underestimate its actual level.
The present authors previously confirmed that Tet is a known lysosome inhibitor and is able to block autophagic flux (1). Like other cancer cell lines, it was observed in the present study that the lysosomal acidity was neutralized following Tet incubation in DU145 cells. Therefore, the present authors hypothesize that the autophagic flux may be blocked, although assays based on LC3 turnover or monomeric red fluorescent protein-EGFP-LC3 co-localization could not be performed in DU145 cells. The increased p62 levels induced by Tet supported this hypothesis.
The role of autophagy in chemotherapy remains unclear. It may either lead to cell death or maintain cell survival against chemotherapeutic drugs. In DU145 cells, sorafenib blocks the autophagic flux, whereas transiently re-expressing ATG5 in DU145 cells antagonizes sorafenib-induced cell death (17). Therefore, the present authors postulate that the autophagic flux may be relatively inadequate in DU145 cells. Blockade of autophagic flux with Tet resulted in cell death. This may explain why DU145 cells exhibited the highest sensitivity towards Tet in all of the 11 cell lines tested (1). It has been reported that CQ enhances the anti-tumor effects in cancer cells (19). The results of the present study indicated that lysosomal pH is able to affect Tet-induced cell death. Further alkalizing lysosomes with nontoxic concentrations of CQ enhanced Tet-induced cell death, indicating that low acidity of the lysosomal lumen is associated with Tet-induced cell death.
Collectively, the present study provided evidence that Tet induced alternative autophagy in DU145 cells. Detecting autophagy in tumor tissue may assist in selecting lysosome inhibitors for chemotherapy treatment in prostate cancer.
Acknowledgements
The authors would like to thank Dr Tamotsu Yoshimori at Osaka University (Osaka, Japan) for providing the EGFP-LC3 plasmid. The present study was supported by grants from the China Postdoctoral Science Foundation (No. 2016M601065).
References
- 1.Qiu W, Su M, Xie F, Ai J, Ren Y, Zhang J, Guan R, He W, Gong Y, Guo Y. Tetrandrine blocks autophagic flux and induces apoptosis via energetic impairment in cancer cells. Cell Death Dis. 2014;5:e1123. doi: 10.1038/cddis.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma JW, Zhang Y, Li R, Ye JC, Li HY, Zhang YK, Ma ZL, Li JY, Zhong XY, Yang X. Tetrandrine suppresses human glioma growth by inhibiting cell survival, proliferation and tumour angiogenesis through attenuating STAT3 phosphorylation. Eur J Pharmacol. 2015;764:228–239. doi: 10.1016/j.ejphar.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 3.Wang H, Liu T, Li L, Wang Q, Yu C, Liu X, Li W. Tetrandrine is a potent cell autophagy agonist via activated intracellular reactive oxygen species. Cell Biosci. 2015;5:4. doi: 10.1186/2045-3701-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu FS, Yu CS, Chen JC, Yang JL, Lu HF, Chang SJ, Lin MW, Chung JG. Tetrandrine induces apoptosis via caspase-8, −9, and −3 and poly (ADP ribose) polymerase dependent pathways and autophagy through beclin-1/LC3-I, II signaling pathways in human oral cancer HSC-3 cells. Environ Toxicol. 2016;31:395–406. doi: 10.1002/tox.22053. [DOI] [PubMed] [Google Scholar]
- 5.Chen JC, Hwang JH, Chiu WH, Chan YC. Tetrandrine and caffeine modulated cell cycle and increased glioma cell death via caspase-dependent and caspase-independent apoptosis pathways. Nutr Cancer. 2014;66:700–706. doi: 10.1080/01635581.2014.902974. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Li P, Yang S, Tong N, Zhang J, Zhao X. Tetrandrine enhances the anticancer effects of arsenic trioxide in vitro. Int J Clin Pharmacol Ther. 2014;52:416–424. doi: 10.5414/CP201939. [DOI] [PubMed] [Google Scholar]
- 7.Qin R, Shen H, Cao Y, Fang Y, Li H, Chen Q, Xu W. Tetrandrine induces mitochondria-mediated apoptosis in human gastric cancer BGC-823 cells. PLoS One. 2013;8:e76486. doi: 10.1371/journal.pone.0076486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu SJ, Ng LT. Tetrandrine inhibits proinflammatory cytokines, iNOS and COX-2 expression in human monocytic cells. Biol Pharm Bull. 2007;30:59–62. doi: 10.1248/bpb.30.59. [DOI] [PubMed] [Google Scholar]
- 9.Gong K, Chen C, Zhan Y, Chen Y, Huang Z, Li W. Autophagy-related gene 7 (ATG7) and reactive oxygen species/extracellular signal-regulated kinase regulate tetrandrine-induced autophagy in human hepatocellular carcinoma. J Biol Chem. 2012;287:35576–35588. doi: 10.1074/jbc.M112.370585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang AC, Lien JC, Lin MW, Yang JS, Wu PP, Chang SJ, Lai TY. Tetrandrine induces cell death in SAS human oral cancer cells through caspase activation-dependent apoptosis and LC3-I and LC3-II activation-dependent autophagy. Int J Oncol. 2013;43:485–494. doi: 10.3892/ijo.2013.1952. [DOI] [PubMed] [Google Scholar]
- 11.Liu T, Men Q, Wu G, Yu C, Huang Z, Liu X, Li W. Tetrandrine induces autophagy and differentiation by activating ROS and Notch1 signaling in leukemia cells. Oncotarget. 2015;6:7992–8006. doi: 10.18632/oncotarget.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouyang DY, Xu LH, He XH, Zhang YT, Zeng LH, Cai JY, Ren S. Autophagy is differentially induced in prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing profiles of ATG5. Autophagy. 2013;9:20–32. doi: 10.4161/auto.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 15.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kharaziha P, Chioureas D, Baltatzis G, Fonseca P, Rodriguez P, Gogvadze V, Lennartsson L, Björklund AC, Zhivotovsky B, Grandér D, et al. Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget. 2015;6:37066–37082. doi: 10.18632/oncotarget.5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 19.Mei L, Chen Y, Wang Z, Wang J, Wan J, Yu C, Liu X, Li W. Synergistic anti-tumour effects of tetrandrine and chloroquine combination therapy in human cancer: A potential antagonistic role for p21. Br J Pharmacol. 2015;172:2232–2245. doi: 10.1111/bph.13045. [DOI] [PMC free article] [PubMed] [Google Scholar]