

Abstract
Channelrhodopsin-2 (ChR2)-based optogenetic technique has been increasingly applied to cardiovascular research. However, the potential effects of ChR2 protein overexpression on cardiomyocytes are not completely understood. The present work aimed to examine how the doxycycline-inducible lentiviral-mediated ChR2 expression may affect cell viability and electrophysiological property of neonatal rat ventricular myocyte (NRVM) cultures. Primary NVRMs were infected with lentivirus containing ChR2 or YFP gene and subjected to cytotoxicity analysis. ChR2-expressing cultures were then paced electrically or optically with a blue light-emitting diode, with activation spread recorded simultaneously using optical mapping. Results showed that ChR2 could be readily transduced to NRVMs by the doxycycline-inducible lentiviral system; however, high-level ChR2 (but not YFP) expression was associated with substantial cytotoxicity, which hindered optical pacing. Application of bromodeoxyuridine significantly reduced cell damage, allowing stimulation with light. Simultaneous optical Vm mapping showed that conduction velocity, action potential duration, and dVm/dtmax were similar in ChR2-expressing and control cultures. Finally, the ChR2-expressing cultures could be optically paced at multiple sites, with significantly reduced overall activation time. In summary, we demonstrated that inducible lentiviral-mediated ChR2 overexpression might cause cytotoxicity in NRVM cultures, which could be alleviated without impairing electrophysiological function, allowing simultaneous optical pacing and Vm mapping.
Introduction
The optogenetic technology, which allows remote control of electrical activity of cells of interest with high spatiotemporal resolution using light, has experienced explosive growth in basic and translational neuroscience in the last decade1–7. Recently, this innovative technique has been introduced to cardiac research both in vitro and in vivo to trigger8–14 or terminate electrical activity of cardiomyocytes15. For instance, Jia et al. described a “tandem cell unit” cell culture model that consisted of neonatal rat ventricular myocytes (NRVMs) co-cultured with HEK cells carrying the light-gated Channelrdoposin 2 (ChR2) gene10. They showed that illumination of the HEK cells led to depolarization and electrical excitation of NRVMs. Similarly, Nussinovitch et al. showed that optical illumination of fibroblasts expressing ChR2 or Archaerhodopsin-T can be used to stimulate or inhibit the electrical activity of co-cultured NRVMs11, 16. Those work proved the feasibility of indirect optogenetic control of cardiomyocytes in the dish. In addition to those in vitro studies, several animal studies have demonstrated that direct optogenetic cardiac pacing can be performed in vivo or ex vivo on transgenic mice8, zebrafish9 and rats17. Given those advancements, the application of optogenetic techniques in the treatment and diagnosis of cardiac diseases is still at the infant stage, partially due to the lack of well-characterized cardiac optogenetic models that contain the genetic encoded light-sensitive proteins. While transgenic animals are valuable and widely used for basic mechanistic studies, translational and clinical application of cardiac optogenetics would rather require using alternative methods for genetic manipulation, e.g., viral vector-based delivery of optogenetic constructs into cells or tissue of non-transgenic diseased hearts.
Lentiviruses have been extensively used as gene delivery vectors since the mid-1990s. Due to their ability to integrate into the genome of target cells, lentiviruses can infect both dividing and non-dividing cells and confer stable high-level expression18. This characteristic renders them of vital interest for gene therapy of cancer, cardiovascular disease, infectious disease, and neurodegenerative diseases. In optogenetic research, lentiviruses have achieved stable and effective rhodopsin proteins (e.g. ChR2) expression in a variety type of cells including neurons2, skeletal muscle cells19, and stem cells20. Recently, the use of lentiviruses for ChR2 delivery has been extended to cardiac optogenetic research, including neonatal cardiomyocytes21 and human pluripotent stem cells derived cardiomyocytes20, 22. An important question before cardiac optogenetic technique can be translated to clinical application is whether, and if yes, how the lentiviral-induced ChR2 expression affects cardiomyocyte viability and electrophysiological property. Viral-mediated ChR2 overexpression has been shown to induce cytotoxicity in neurons and glial cells23–25. In cardiac cells, however, this aspect has not yet been fully evaluated. In addition, the application of inducible optogenetic (ChR2) gene expression technique, which could benefit long-term gene therapy26, in cardiac optogenetics has not been examined.
Another critical element of cardiac optogenetic application is simultaneous and accurate recording of activation spread initiated by light stimulation. Optical mapping is a widely-used experimental technique for monitoring cardiac excitation27, which employs fluorescent dyes to image membrane potential (Vm) and other parameters such as intracellular Ca2+ transients at high spatial and temporal resolutions28–30. Since its introduction, optical mapping has revolutionized the study of impulse conduction and cardiac rhythm. Therefore, optical mapping could be a method of choice in cardiac optogenetics studies. However, as both methods employ visible light, their integration represents a challenge. Only very recently, Park et al. reported the first study of simultaneous optical control and Vm recordings in cardiomyocyte cultures21. However, their work primarily focused on the light-induced modulation of action potential duration (APD).
To address those issues, we developed and characterized an optogenetic NRVM culture model containing doxycycline (Doxy)-inducible lentiviral-mediated ChR2 expression. The results showed that the lentivirus-induced ChR2 protein overexpression caused substantial cytotoxicity in NRVM cultures, which could be alleviated by the application of bromodeoxyuridine (BrdU), allowing direct optical pacing and simultaneous Vm optical mapping. We also demonstrated that moderate ChR2 expression and light pacing did not influence the basic electrophysiological property of NRVMs. Preliminary results of this work have been published in abstract form31.
Results
Effects of viral transduction and ChR2 expression on NRVM cultures
We first examined the Doxy-activated lentivirus-mediated ChR2 expression in NVRMs and its effect on cell culture viability and electrical property. When the multiplicity of infection (MOI) was small (e.g. 10 or 20), the transduction efficiency (i.e. the percentage of ChR2-expressing positive cells) and the overall level of ChR2 expression were low; those monolayers could barely respond to (470 nm) LED light stimulation. Increasing MOI (e.g. to 40) led to high-level, homogeneous ChR2 expression throughout cell monolayer (Fig. 1A). However, severe cell damage was found in those cultures, including significantly lower (40.1 ± 4.0%, n = 10) cell density (Fig. 1B) as compared to controls (Fig. 1C) and adverse morphological changes such as smaller cell size, membrane blebbing, and strong ChR2 protein aggregation in the cytoplasm (Fig. 1D). Importantly, such monolayers usually developed rapid spontaneous beating and were unresponsive to external (optical or electrical) stimulation.
Figure 1.
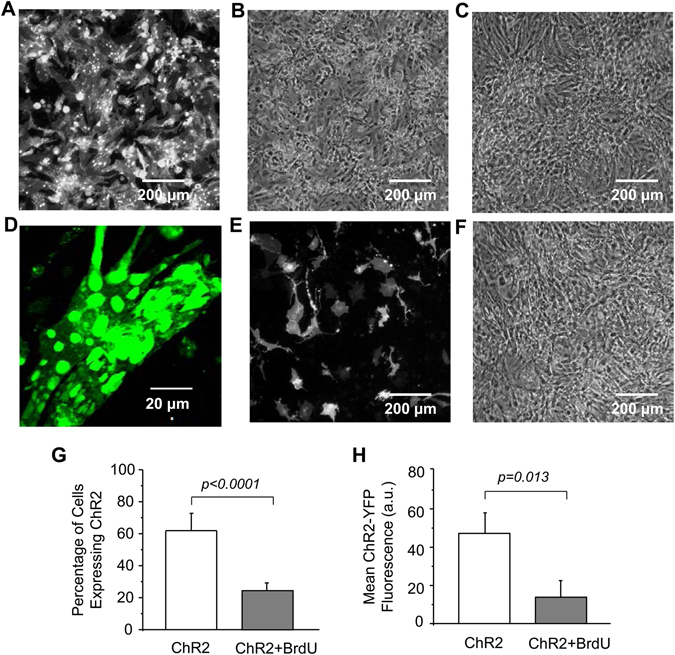
Effects of lentivirus transduction and doxycycline-induced ChR2 expression on NRVM cultures. (A and B) Fluorescent and phase-contrast images, respectively, of a ChR2-transduced monolayer; (C) Phase-contrast image of a control monolayer; (D) Confocal image of ChR2-expressing cardiomyocytes; (E and F) Fluorescent and phase-contrast images, respectively, of a ChR2-expressing monolayer pretreated with BrdU; (G) ChR2 transduction efficiency in cell cultures with or without BrdU pretreatment (two-tailed unpaired Student’s t test, n = 6); and (H) Effect of BrdU on ChR2-eYFP fluorescence intensity averaged over monolayer (two-tailed unpaired Student’s t test, n = 6).
To solve these problems, NRVM monolayers were treated with BrdU, a DNA replication inhibitor, prior to viral transduction. BrdU pretreatment led to a reduced and less uniform ChR2 expression (Fig. 1E) but alleviated cell damage. The cell morphology and density in the BrdU-treated ChR2 NRVM cultures (Fig. 1F) were similar to that in the control cultures (Fig. 1C). BrdU had no significant effect on the non-infected cell cultures (data not shown), but significantly reduced the viral transduction efficiency (Fig. 1G) and overall ChR2 expression (Fig. 1H) as compared to the non-treated cultures.
To examine the cause of cytotoxic effects observed in the ChR2-transduced cardiomyocytes, the gene transduction procedure was varied and cell viability was quantified using the trypan blue assay. Procedure modifications included application of ChR2 lentivirus at different MOIs, use of different virus batches, treatment with or without BrdU and Doxy, and use of YFP-only virus. Data showed that when the MOI was relatively low (e.g., 10 and 20) the cell viability of the ChR2-transduced cultures was similar to that of non-infected cultures (i.e. MOI = 0), regardless of whether cells were pretreated with BrdU or not (Fig. 2A). However, when the MOI was increased to 40, the cell viability declined dramatically in the ChR2-infected NRVM monolayers that were not treated with BrdU as compared with the control and low MOI-transduced cultures (Fig. 2A). BrdU pretreatment substantially reduced the high MOI infection-induced cell damage; the number of cells per well was not significantly different from that of control cultures treated with Doxy and BrdU only (Fig. 2A). The similar effects of MOI and BrdU treatment on ChR2-expressing cardiomyocytes have been observed in NRVM cultures infected with various batches of ChR2 lentivirus (see Figure S2 for an example). It is worth mentioning that neither BrdU nor Doxy treatment alone had significant effect on cell viability (Figure S3).
Figure 2.
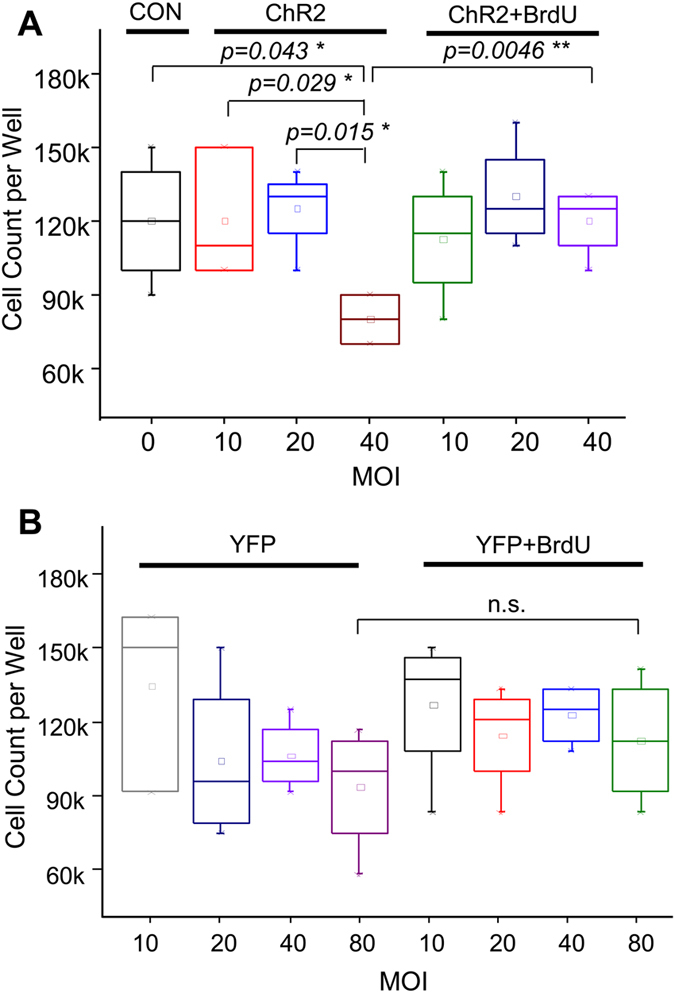
Effects of infection MOI and BrdU treatment on NRVM viability in ChR2 (A) or YFP (B) lentivirus-infected cultures. *: one-way ANOVA, **: two-way ANOVA, n = 3 or 4 per group.
To examine whether the cytotoxicity was caused by the viral-mediated ChR2 expression or the viral infection itself, NRVM cultures were infected with YFP-only lentivirus with various MOIs. Cell viability analysis shows that YFP lentiviral infection (and the mediated YFP expression) had no detrimental effect on NRVM cultures, regardless of the MOI or the BrdU treatment (Fig. 2B). In particular, no substantial cytotoxicity was detected in monolayers infected with YFP-only lentivirus, even with high MOIs (e.g. 80) and without the presence of BrdU. The effects of BrdU treatment on cell viability in YFP- or ChR2-transduced NRVM cultures with low (i.e. 10) and high (i.e. 40) MOIs are summarized in Figure S4.
Primary NRVM cultures often contain a certain number of non-myocyte cells, primarily fibroblasts32, which could be electrically coupled to cardiomyocytes33. If fibroblasts express ChR2, they may contribute to optogenetic stimulation of cardiac cells10. Therefore, we characterized the type of ChR2-expressing cells in the cultures using an anti-sarcomeric alpha-actinin antibody (a cardiomyocyte marker) and an anti-vimentin antibody (a fibroblast marker). The image analysis showed strong staining for alpha-actinin, which was largely overlapped with or in close proximity to ChR2-eYFP (Fig. 3A). On the contrary, NRVM cultures displayed much less staining for vimentin, which was barely colocalized with ChR2-eYFP (Fig. 3B). Qualitatively similar results were obtained in a total of eight cultures (n = 4 for each group). These results indicated that there were few fibroblasts in NRVM cultures and ChR2 was expressed mainly in cardiomyocytes.
Figure 3.
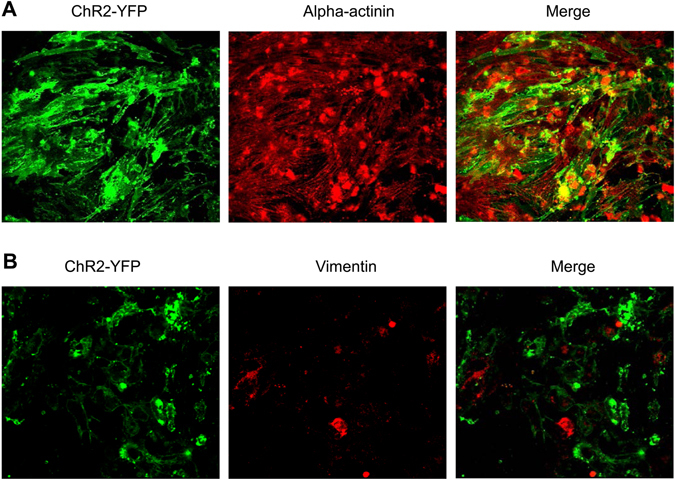
Characterization of lentivirus transduction-induced ChR2 expression in cardiomyocytes and fibroblasts. (A) NRVM culture stained with anti-sarcomeric alpha-actinin antibody; and (B) NRVM culture stained with anti-vimentin antibody. n = 4 per group.
Direct optical pacing of ChR2-expressing NRVM cultures
We next examined whether BrdU treatment enables direct optical pacing of cardiomyocytes containing Doxy-inducible ChR2 expression (hereinafter refers to NRVM cultures infected by ChR2 lentivirus at MOI of 40 and in the presence of BrdU and Doxy). As shown in Fig. 4A and Movie S1 (supplemental information), NRVMs beat spontaneously and irregularly prior to optical stimulation at a rate of ~0.3 Hz. Optical pacing with blue LED light at faster rates resulted in rhythm capture. In our hands, the slowest rate of light stimulation was about 0.5 Hz (Fig. 4B) although at such low pacing frequency the rhythm capture was incompletely as extra spontaneous cell contractions could be frequently observed (Fig. 4B, marked by the arrow). Further increasing pacing frequency (e.g. to 1 and 2 Hz) led to complete capture of cell culture rhythm by LED pulses (Fig. 4C and D), demonstrating the feasibility of direct optogenetic pacing of NRVM cultures. The highest frequency of optical pacing resulting in stable rhythm capture was 3.1 Hz (Movie S1). Similar results were obtained in a total amount of five monolayers.
Figure 4.
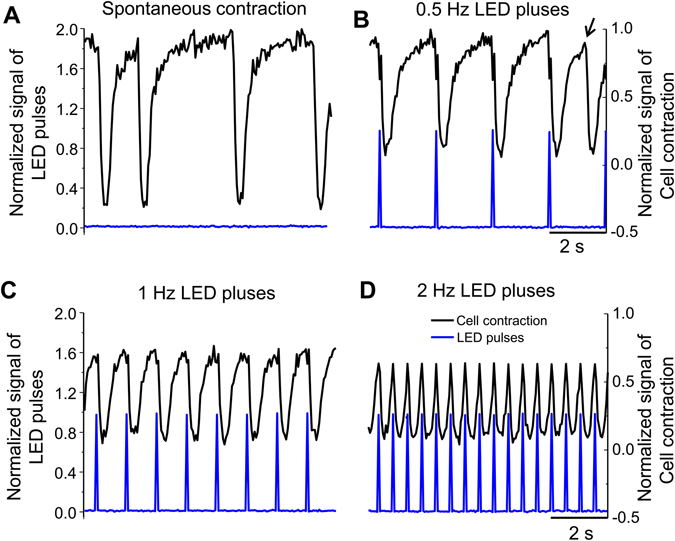
Direct optical pacing of ChR2-expressing NRVM cultures at varying light pulse frequencies. (A) Spontaneous contractions of a culture recorded in transmitted light; (B–D) Cell contractions triggered by light pacing at a frequency of 0.5, 1 and 2 Hz, respectively. The arrow marks an uncaptured contraction at the 0.5 Hz pacing rate.
Simultaneous optical pacing and Vm mapping of ChR2-expressing NRVM cultures
Normal AP propagation is critical for maintaining cardiac function. To examine the properties of light-triggered AP propagation in ChR2-expressing NRVM cultures, optical Vm mapping was performed during optical or electrical pacing (2 Hz). We first examined the patterns of electrical stimulation-induced AP propagation in control and ChR2-expressing NRVM monolayers. Optical mapping showed that activation spread had uniform patterns in ChR2-expressing monolayers (Fig. 5C and D), which were similar to those in the control cultures (Fig. 5A and B). The same ChR2-expressing monolayers were then subjected to stimulation by optical pulses and the AP propagation were recorded. Data showed that the activation patterns elicited by optical pulses (Fig. 5E and F) were very similar to those induced by electrical stimulus (Fig. 5C and D).
Figure 5.
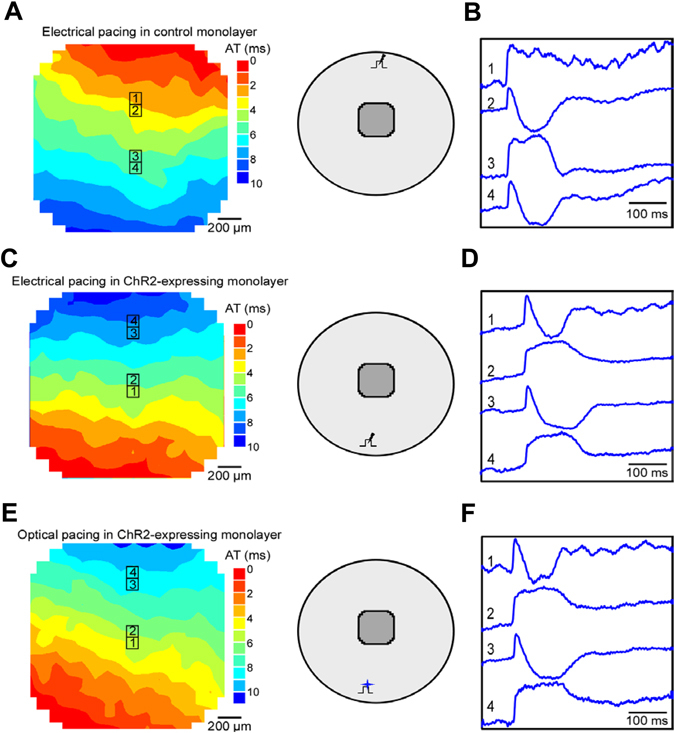
Isochronal map of activation spread and selected Vm traces in NRVM monolayers. (A and B) A control monolayer during electrical pacing; (C and D) A ChR2-expressing monolayer during electrical pacing; (E and F): A ChR2-expressing monolayer during optical pacing. The schematic insets show the corresponding pacing site and mapping area. The black lightning symbol in the inset represents electrical pacing and the blue star symbol represents optical stimulation.
We next measured the electrophysiological parameters of AP propagation (i.e., APD, dV/dtmax, and conduction velocity) in the control and ChR2-expressing NRVM cultures paced electrically or ChR2-expressing NRVM cultures paced with light. Typically, APD measurements were hindered by cell motion artifact, which can be seen in Fig. 5 as deflections following AP upstrokes; such deflections often had opposite polarities at neighboring sites (e.g., sites 3 and 4 in Fig. 5A,C and E). Application of blebbistatin effectively suppressed motion artifact in AP recordings in control monolayers (Figure S5) and ChR2-expressing cell cultures (Figure S6) paced electrically. Blebbistatin had minimal effect on the activation map and conduction velocity in those monolayers (Figure S7A and B). Analysis of the Vm optical mapping data showed that the mean values of conduction velocity, APD80, and dV/dtmax were not significantly different between the ChR2-expressing (n = 6) and control cultures (n = 8) (Table 1), which is consistent with results reported by Park et al.21. Similarly, as in the monolayers paced electrically blebbistatin successfully suppressed the motion artifact but did not alter the light-induced AP propagation (Fig. 6) and conduction velocity (Figure S7C) in ChR2-expressing NRVM monolayers. In addition, there were no significant differences in the basic electrophysiological parameters in the ChR2-expressing cell cultures paced electrically or optically (Table 1), which are consistent with previous studies13.
Table 1.
Electrophysiological parameters (conduction velocity, APD80 and dV/dtma) measured in control and ChR2-expressing NRVM cultures.
| Conduction velocity (cm/s) | APD80 (ms) | dV/dtmax (V/s) | |
|---|---|---|---|
| Control cultures Electrical pacing (n = 8) | 19.3 ± 4.1 | 114.5 ± 29.8 | 32.3 ± 5.0 |
| ChR2-expressing Electrical pacing (n = 8) | 18.9 ± 1.0* (p = 0.44) | 124.7 ± 29.2* (p = 0.12) | 32.7 ± 2.9* (p = 0.87) |
| ChR2-expressing Optical pacing (n = 6) | 19.5 ± 1.4# (p = 0.557) | 133.8 ± 20.1# (p = 0.152) | 32.3 ± 6.5# (p = 0.387) |
Pacing frequency = 2 Hz, culture duration ranged from 6 to 8 days. *Non-significant vs. control cultures paced electrically (two-tailed unpaired Student’s t test). #Non-significant vs. ChR2-expressing cultures paced electrically (two-tailed unpaired Student’s t test).
Figure 6.
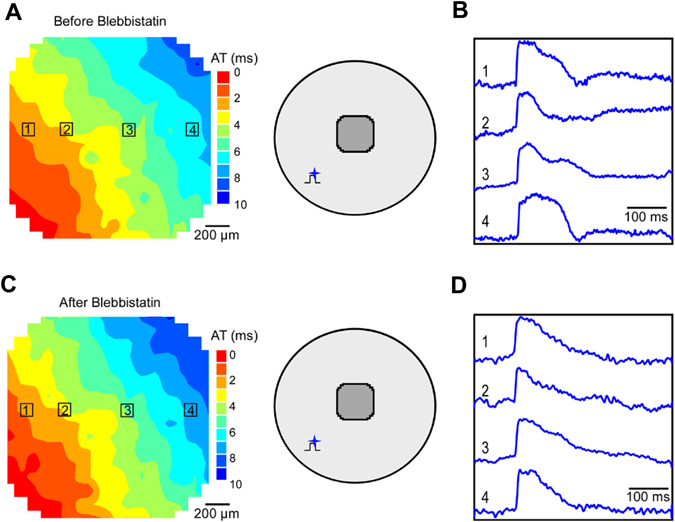
Suppression of motion artifact by blebbistatin. Activation maps and optical Vm recordings before (A and B) and after (C and D) blebbistatin application in a ChR2-expressing NRVM culture. The schematic insets show optical pacing site and mapping area.
Multisite optical pacing of NRVM cultures expressing ChR2
To examine the feasibility of multisite optogenetic pacing, we simultaneously illuminated a ChR2-expressing cell culture at two different sites and mapped the triggered AP propagation. Figure 7A and B show the AP propagation when the cell culture was paced optically at one site located either above or below the mapping area, respectively (sites 1 or 2 schematically shown in Fig. 7C). During single site optical pacing, the overall activation time across the mapping area was approximately 14 ms, which was not affected significantly by the location of pacing site. During dual-site pacing (Fig. 7D), two activation wavefronts were initiated at the top and bottom illumination sites simultaneously and travelled in the opposite direction colliding in the center of the monolayer. In this case, the overall activation time within the mapping area reduced to ~6 ms that is about half of that of single site pacing, suggesting that optogenetic pacing can be expanded to multiple sites.
Figure 7.
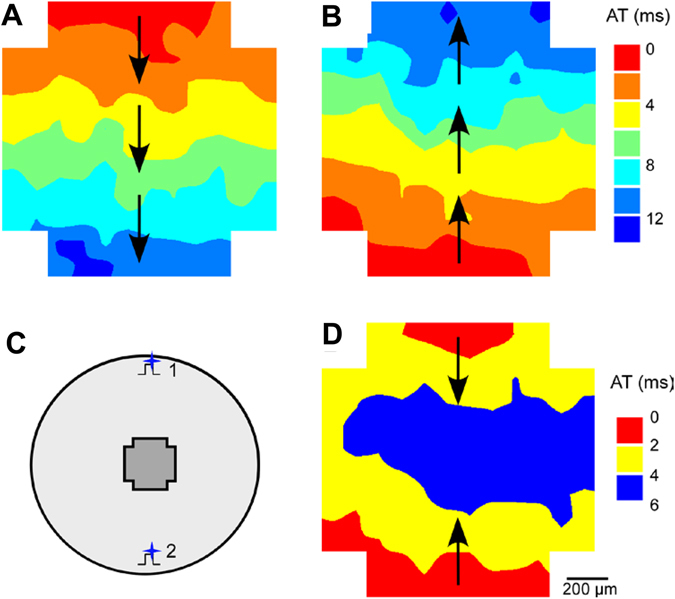
Multisite optical pacing of a ChR2-expressing NRVM culture. (A and B) Activation maps during pacing at a single site located on top or bottom of the cell culture, respectively; (C) A diagram schematically showing optical pacing sites and mapping area; (D) Activation maps of synchronous two-site pacing.
Discussion
In the present work, we developed and characterized an optogenetic NRVM culture model that is based on Doxy-inducible ChR2 expression. In particular, we examined the effect of lentivirus-mediated ChR2 expression on cell viability and electrophysiological property of NRVM cultures as well as the feasibility of direct optical pacing of ChR2-expressing cardiomyocytes. The main findings are: 1) infection of NRVM cultures with high MOI ChR2 lentivirus, but not YFP lentivirus, caused strong cardiomyocyte toxicity; 2) BrdU treatment alleviated cytotoxicity in ChR2-expressing cells; under such conditions, NRVM cultures could be stably paced using light pulses; and 3) optogenetic pacing had minor effects on the electrophysiological property of NRVM cultures.
Inducible gene expression system is a valuable tool in cardiac optogenetic research and translational application as it allows for temporally and spatially controlled activation of light-sensitive proteins. For instance, tetracycline inducible lentiviral system can be used in animal model systems by expressing rtTA in specific heart cells (such as Purkinje cells or atrial cardiomyocytes) to allow optogenetic manipulation of desired cell types. Our results showed that ChR2 genes were readily transduced into NRVMs using a Doxy-inducible lentiviral vector, rendering homogeneous and high-level ChR2 protein expression. However, severe cell damage and detrimental electrophysiological alterations were often found in those NRVM cultures, which hindered optical pacing and rhythm capture. Similar optogenetic protein expression-induced cell malformation and toxicity have been previously reported in neurons and glial cells23–25. The observed cytotoxicity could be a major concern in translational and clinical applications of cardiac optogenetics, but the underlying mechanism is not entirely clear. One possibility is that the cell damage was caused by the lentivirus infection, as primary cells such as cardiomyocytes may be sensitive to high titers. However, the fact that YFP-only lentivirus infection did not cause toxic effect to NRVM cultures, even with higher MOIs, indicated that this was not the case. In addition, we found that BrdU treatment substantially reduced cytotoxicity in the ChR2 lentivirus-infected cell cultures, providing further support that the cytotoxic effect was not caused by viral infection because lentivirus doesn’t proliferate and therefore BrdU shouldn’t reduce the number of viral particles. Our data also showed that BrdU or Doxy treatment alone had no effect on NRVM monolayers. Taken together, it seemed like that the observed cytotoxicity was mainly caused by the Doxy-induced ChR2 overexpression.
While the possible effect of BrdU on protein expression was barely investigated, a study by Rauth et al. showed that BrdU treatment caused a marked decrease in the level of tyrosinase protein and mRNA expression34. In a following study, they reported that suppression is not due to incorporation of BrdU into the coding sequence of the tyrosinase or the upstream tyrosinase gene sequences in the plasmids35. Thus BrdU may not suppress ChR2 gene directly but instead inhibit its activator or regulatory genes, as suggested by a previous study36. Apparently, the exact mechanism by which BrdU prevents Doxy-inducible ChR2 overexpression requires further investigation. One downside of BrdU treatment was weaker and less uniform ChR2 expression, which may compromise the capability for precise spatial control of cells with light. In addition, as BrdU can replace thymidine during DNA replication and cause mutations, using it in vivo should be very careful. For instance, it has been reported that BrdU caused cardiovascular malformations in the chick embryo37. We also noticed that rather long duration of optical pulses (e.g. 20 ms) was required for reliable rhythm capture. Although such pulse durations were comparable to those used in previous cardiac optogenetics studies10, 11, 21, they are significantly longer than the durations used in electrical pacing (2–5 ms). The longer pulses required for optical pacing can be due to (i) relatively low level of ChR2 expression and (ii) relatively long time constant of ChR2 channel activation, which could be as long as 5 ms5, 38. Indeed, we have found that when the ChR2 transduction efficiency is low (e.g. <10%), the infected monolayers could barely be optically paced.
The high-level ChR2 protein expression-mediated cytotoxicity could happen via several mechanisms. One possible mechanism is related to intracellular acidification and Ca2+ overload. Although the largest portion of current flowing through the ChR2 channels is carried by Na+ ions, these channels are also permeable to H+ and Ca2+ ions5, 38–40. Thus, opening of ChR2 channels could produce significant proton and Ca2+ influxes, causing intracellular acidification and/or Ca2+ overload as reported in glial cells24 and oocytes39. In cardiac cells, intracellular acidification and Ca2+ overload can induce mitochondrial injury and activate Ca2+-dependent proteases, leading to cell damage and death41–44. However, this mechanism should not be the main cause responsible for the cell toxicity found in the present study because all NRVM cultures were kept in the dark most of the time whereas the signs of cell damage were noticeable from the start of experiments even before optical pacing.
Another possible mechanism underlying the cell injury may be related to the potential deleterious effects of ChR2 overexpression on the function of cell membrane. Previous studies in neurons and other types of cells have shown that overexpression of microbial opsins could alter the morphology and capacitance of the cell membrane, causing membrane damage and cell malformation25, 45–47. The resultant cell membrane dysfunction and cytotoxicity eventually led to cell death via a variety of signaling pathways including apoptosis. The considerable ChR2 protein aggregation, which was observed in some of the transduced NRVM cultures (Fig. 1A and D), could also contribute to the cell injury. It has been reported that the aggregation of halorhodopsin protein in the endoplasmic reticulum of neuron cells led to bleb formation, local swelling, and cell toxicity25, 45–48. The precise contribution of this mechanism to NRVM cytotoxicity could be explored in the future by reducing ChR2 aggregation via fusing an ER export signal peptide and/or plasma membrane-targeting signal peptide to the ChR2 sequence as suggested previously49.
An important aspect of the present work is the combination of optical stimulation delivered locally via a small optical fiber at one location with optical mapping of activation spread in a region spatially separated from the optical pacing site. A possible obstacle of optical mapping is that the light used for excitation of Vm-sensitive dye RH-237 fluorescence (in the wavelength range of 538–582 nm) could potentially cause partial activation of ChR2, membrane depolarization, and, therefore, change of electrophysiological properties at the mapping site. However, ChR2 absorption spectrum (peak absorption at 470 nm) has only a minor overlap with this wavelength range. Importantly, all measured electrophysiological parameters including conduction velocity, APD80 and upstroke rate of rise were similar in ChR2-expressing and control cultures. These data indicate that the effect of the excitation light on ChR2 activity was likely to be small and that optical mapping with RH-237 dye can be combined with optical pacing in cardiac rhythm studies. A definitive proof of this conclusion, however, requires verification using multielectrode electrical mapping of activation spread.
It should be noted that whereas conduction velocity and APD80 values measured in control and ChR2-expressing NRVM cultures were comparable to those ones measured previously in isotropic NRVM cultures50, 51, the dV/dtmax values were slightly lower than previously reported. This difference was likely caused by the lower optical resolution and stronger digital signal filtration (0.2 kHZ) used in the present study. We also showed that optogenetic pacing could be readily expanded to multiple sites. Therefore, optical stimulation may potentially achieve precise control of the location, size, shape and even sequence of illumination, making it a promising candidate for the development of novel cardiac resynchronization therapy device that may complement or replace the conventional biventricular electrical pacing52, 53.
A limitation of the present study is the inability to measure action potentials directly at the optical pacing site using current optical mapping approach. One reason for this limitation was the leakage of the blue LED light into the optical Vm recording channel. A similar light leakage was reported in a recent study21, in which Vm recordings were interrupted during optical pulses and the missing Vm signal portions were obtained by interpolation allowing examination of the effects of light pulses on APD. This light leakage could be eliminated by using the epi-fluorescent configuration for stimulation light path and better optical filtration (Figure S8). However, Vm measurements at the optical pacing site were still precluded due to excitation of the voltage-sensitive dye (RH-237) fluorescence by the blue LED light. Another limitation is the lack of functional analysis of the damaged ChR2-expressing cells such as patch clamp recordings of ionic currents and Ca2+ imaging, which could provide insights into more detailed mechanisms underlying the ChR2 expressing-mediated cytotoxicity (i.e. ChR2 functional expression or ChR2 overexpression). This is subject for future studies.
In summary, we developed and characterized a Doxy-inducible optogenetic NRVM culture model that allows direct optical control of electrical cardiomyocyte activity and simultaneous optical mapping of action potential propagation. This study expanded the utility of optogenetic tools in cardiac electrophysiological research.
Methods
All experimental protocols were approved by the University of Alabama at Birmingham institutional animal care and use committee and adhered to the National Institutes of Health’s guide for the care and use of laboratory animals (NIH Publication No. 85-23, revised 2011).
ChR2 lentivirus plasmid construction and virus production
The ChR2(H134R)-eYFP insert was amplified from pAAV-CaMKIIa-hChR2(H134R)-eYFP plasmid (Addgene plasmid #26969) using the following PCR primers: GACGAATTCACCATGGACTATGGCGGCGCTTTGTC (forward) and GCGGAATTCTTTACTTGTACAGCTCGTCCATGCC (reverse). The eYFP insert was amplified from the same vector using the following PCR primers: GACGACGAATTCCACCATGGTGAGCAAGGGCG (forward) and GCGGAATTCTTTACTTGTACAGCTCGTCCATGCC (reverse). The TetO-FUW backbone, obtained from the EcoRI-digested TetO-FUW-OSKM (Addgene plasmid #20321), was ligated with EcoRI-digested ChR2-eYFP or eYFP PCR product to generate the inducible lentivirus plasmid TetO-FUW-ChR2-eYFP or TetO-FUW-eYFP (Figure S1A), respectively. The obtained plasmids were sequenced at University of Alabama at Birmingham Helflin Center for Genomic Science to confirm the constructions. To produce lentiviral particles, the TetO-FUW-ChR2-eYFP (or TetO-FUW-eYFP), psPAX2 (Addgene plasmid #12260) and pMD2.G (Addgene plasmid #12259) plasmids were co-transfected to 293T cells using calcium phosphate as described previously54. The packed replication-incompetent lentiviral particles were collected, purified, and concentrated on day 5 and stored at −80 °C properly.
The biological titration of lentivirus was performed with HeLa cells. Briefly, around 2.5 × 105 cells were plated in a 12-well plate. On day 2, cell number was counted from one of the wells. TetO-fuw-ChR2-eYFP or TetO-fuw-eYFP virus was thawed and diluted 10-fold serially ranging from 10−1 to 10−4. Each transduction was performed in duplicate in the presence of 8 μg/ml polybrene. Medium was replaced with addition of 2 μg/ml doxycycline 24 hours after the transduction. Cells were cultured for another two days and then trypsinzed from the plate. The trypsinzed cells were fixed with 1% formaldehyde in phosphate buffered saline (PBS) for 5 minutes, washed with PBS, and then resuspended with 150 μl PBS. eYPF expression were analyzed by FACS at University of Alabama at Birmingham CFAR flow cytometry core. The 1% to 20% eYFP positive dilution was used for titer calculation using the following formula: Number of target cells × (% of eYFP- positive cell/100)/volume of supernatant (in ml).
NRVM culture production and viral infection
NRVMs were obtained from 1–2 day old neonatal Sprague-Dawley rat hearts as previously described55, 56 with some modifications. In brief, hearts from 8–10 rats were excised, the ventricles minced and tissue pieces dissociated in Ca2+/Mg2+ free Hank’s balanced salt solution that contains 0.1% trypsin and 12 μg/mL pancreatin. The dispersed cells were re-suspended in Ultraculture serum-free medium (Fisher Scientific) supplemented with 2 mM L-glutamine, 2 μg/L vitamin B12, 20 U/mL penicillin and 20 μg/mL streptomycin. To reduce fibroblast content, cells were preplated in a 150-cm2 culture flask for 2 hours. After preplating, cells were re-suspended in the culture medium and counted. Approximately 2 × 105 cells were plated on collagen-coated cover glasses (diameter 15 mm) and incubated at 37 °C in a humidified atmosphere containing 5% CO2 with or without 0.1 mM of bromodeoxyuridine (BrdU).
Two days after cell plating, cultures were transduced using lentivirus containing ChR2-eYFP or eYFP gene with different MOIs (e.g., 10, 20, 40 or 80) in the presence of 8 μg/ml polybrene. Doxycycline (2 μg/ml) was added to the culture 24 hours after the infection to induce transgene expression. The expression of ChR2-eYFP or eYFP was monitored daily with a confocal microscope, which usually reached the peak level in the next 3–4 days and did not decrease significantly within the next week. The efficiency of lentiviral transduction (i.e. percentage of ChR2 positive cells in an infected monolayer) was calculated by dividing the number of YFP-expressing cells by the total number of NRVMs (with and without YFP expression), which were determined from fluorescent and phase contrast images, respectively. The mean fluorescence per monolayer was calculated by dividing the total fluorescence by the total number of cells. Five images were taken for each monolayer, and counts of cells were averaged within the same monolayer for statistical analysis. NRVM cultures were kept in dark and used for optogenetic studies 4–6 days after viral infection.
Immunocytochemistry
NRVMs cultured in 24-well plates were rinsed twice with PBS and fixed with 3% paraformaldehyde for 20 min at room temperature (RT). Permeabilization was performed by incubating the cultures with 0.1% TritonTMX-100 in PBS. After being blocked with 10% inactivated goat serum and 1% bovine serum albumin (BSA) in PBS, the preparations were incubated overnight at 4 °C with an anti-vimentin primary antibody (1:200, mouse monoclonal) or anti-sarcomeric-alpha-actinin primary antibody (1:800, mouse monoclonal). Thereafter, cultures were washed with 0.1% BSA in PBS, blocked again with blocking buffer (30 min at RT), and incubated with an Alexa Fluor 633 secondary antibody (1:800, goat anti-mouse), followed by washing and mounting. Images were acquired using a confocal microscope.
Cell death assay
The cell viability and cytotoxicity were quantitatively assessed using the trypan blue assay. Briefly, NRVMs in a 24 well plate were washed with PBS twice and incubated at 37 °C, 5% CO2 with 200 μL of 0.25% Trypsin-EDTA for 2 minutes. After trypsinization, 800 μL of cell culture medium containing 10% FBS was added to each monolayer. Cell suspensions were harvested and transferred to 15 mL tubes, and diluted with appropriated amount of culture medium. 10 μL cell suspension was then mixed with 10 μL 0.4% trypan blue solution for cell counting. Four areas of hemocytometer were counted and cell numbers were calculated according to the dilution factor and the total medium volume of each monolayer. Cells were counted three times by two individuals independently.
Confocal imaging of ChR2 expression and recording of light-induced cell contractions
A glass coverslip with NRVM culture was fitted in a perfusion chamber mounted on the stage of a confocal microscope. Cells were perfused with warm (36–37 °C) Hank’s solution. The 515-nm argon laser line of the confocal microscope was used to image YFP distribution. For optical stimulation, a 10x objective lens was coupled to a high-power 470-nm light emitting diode (LED) via an optical fiber (diameter 1000 µm) and positioned above the cell monolayer. Optical pulses with duration of 10–20 ms were delivered at a frequency varying between 0.5 and 3.1 Hz using a MyoPacer field stimulator. Cultures were illuminated with white light and changes in transmitted light intensity were video-recorded continuously at 30 fps using a CCD camera mounted on the confocal microscope. Images were analyzed offline using ImageJ software to determine light impulse-induced cell contractions, which were normalized to the displacement of a spot on the cell during spontaneous contraction.
Simultaneous optical pacing and activation mapping
NRVM cultures were transferred to a recording chamber mounted on a Zeiss Axiovert inverted microscope and continuously perfused with warm Hank’s solution (36–37 °C). For electrical pacing, stimulation pulses were delivered using a bipolar electrode. For optical pacing, an optical fiber (diameter = 1000 µm) coupled to the 470-nm LED was positioned at a distance of ~1 mm from a cell monolayer and 20-ms light pulses were delivered at a rate of 2 Hz. At this distance, the measured diameter of the light spot was ~1.8 mm. The pacing light intensity was calculated as the light power (measured at the fiber exit using an optical power meter) divided by the area of the light spot. The threshold light intensity required for cell pacing was in the range of 1.0–1.3 mW/mm2 and the estimated energy was about 50 μJ. To map activation spread, cell cultures were stained with Vm-sensitive dye RH-237 (2.5 μmol/L) for 5 min. Fluorescence was excited using an Hg/Xe arc lamp and a 560/55-nm excitation filter and measured at >650 nm using a 16 × 16 photodiode array at spatial resolution of 110 µm per diode, as previously described56. The mapping area was located at a distance of 2–3 mm from the optical pacing site. The schematic diagram of the optical setup is shown in supplemental Figure S1B.
Optical signals were digitally filtered to increase the signal-to-noise ratio. Activation times were measured at the 50% level of action potential amplitude (APA) and used to construct the isochronal maps of activation spread. Conduction velocity was calculated at each recording site from local activation times and averaged across the whole map using custom-written data analysis software. The maximal upstroke rate of rise (dV/dtmax) was obtained by taking the first derivative of the optical signal that had been normalized assuming APA value of 100 mV. To measure the action potential duration (APD), an electro-mechanical uncoupler blebbistatin (2.5 μmol/L) was administered to eliminate the motion artifact in some cell cultures. APD80 was calculated as the interval between the activation and repolarization time measured at 80% level of the APA.
Materials
Plasmids for lentiviral vector construction were obtained from Addgene (Cambridge, MA). PCR primers were synthesized by Integrated DNA Technologies (Coralville, Iowa). The qPCR lentivirus titration kit was purchased from Applied Biological Material (Atlanta, GA). The PBS, TritonTMX-100, voltage-sensitive fluorescent dye RH-237, and Alexa Fluor 633 secondary antibody were purchased from Life Technologies (Carlsbad, CA). The anti-vimentin antibody, the anti-sarcomeric-α-actinin antibody, and all other reagents were from Sigma-Aldrich (St. Louis, MO).
Data were analyzed using OriginLab software. Comparisons between groups were performed using unpaired 2-tailed Student’s t-test (Figs 1, S3 and S7 and Table 1) or One-Way or Two-Way ANOVA test (Figs 2, S2 and S4). Data were considered significantly different at p < 0.05. Results are presented as bar (mean ± SEM) or box chart.
Electronic supplementary material
Acknowledgements
The authors thank Drs. Jing Qu and Wei Kong for assistance with lentiviral vector constructions and NRVM culture optical mapping, respectively. This work is supported by University of Alabama at Birmingham Comprehensive Cardiovascular Center Pilot Grant and National Institute of Health (NIH) R21 HL127599A1 and R01 HL121206A1 (to L.Z.), and NIH RO1 HL067748 (to V.G.F.).
Author Contributions
Li Q. performed experiments, analyzed data and prepared manuscript. Goh K.Y., Rong N., Hong H. and Rossi M. contributed to experiments, and Fast V.G. and Zhou L. designed experiments, analyzed data and wrote manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01723-2
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Vladimir G. Fast, Email: vfast@uab.edu
Lufang Zhou, Email: lfzhou@uab.edu.
References
- 1.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 2.Deisseroth K. Optogenetics. Nat Methods. 2011;8:26–29. doi: 10.1038/nmeth.f.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han X, et al. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front Syst Neurosci. 2011;5:18. doi: 10.3389/fnsys.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Husson SJ, et al. Microbial light-activatable proton pumps as neuronal inhibitors to functionally dissect neuronal networks in C. elegans. PLoS One. 2012;7:e40937. doi: 10.1371/journal.pone.0040937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagel G, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yawo H, Asano T, Sakai S, Ishizuka T. Optogenetic manipulation of neural and non-neural functions. Dev Growth Differ. 2013;55:474–490. doi: 10.1111/dgd.12053. [DOI] [PubMed] [Google Scholar]
- 7.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 8.Bruegmann T, et al. Optogenetic control of heart muscle in vitro and in vivo. Nat Methods. 2010;7:897–900. doi: 10.1038/nmeth.1512. [DOI] [PubMed] [Google Scholar]
- 9.Arrenberg AB, Stainier DY, Baier H, Huisken J. Optogenetic control of cardiac function. Science. 2010;330:971–974. doi: 10.1126/science.1195929. [DOI] [PubMed] [Google Scholar]
- 10.Jia Z, et al. Stimulating cardiac muscle by light: cardiac optogenetics by cell delivery. Circ Arrhythm Electrophysiol. 2011;4:753–760. doi: 10.1161/CIRCEP.111.964247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nussinovitch U, Shinnawi R, Gepstein L. Modulation of cardiac tissue electrophysiological properties with light-sensitive proteins. Cardiovasc Res. 2014;102:176–187. doi: 10.1093/cvr/cvu037. [DOI] [PubMed] [Google Scholar]
- 12.Burton RA, et al. Optical control of excitation waves in cardiac tissue. Nat Photonics. 2015;9:813–816. doi: 10.1038/nphoton.2015.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ambrosi CM, Boyle PM, Chen K, Trayanova NA, Entcheva E. Optogenetics-enabled assessment of viral gene and cell therapy for restoration of cardiac excitability. Sci Rep. 2015;5:17350. doi: 10.1038/srep17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu J, Chen K, Lucero RV, Ambrosi CM, Entcheva E. Cardiac Optogenetics: Enhancement by All-trans-Retinal. Sci Rep. 2015;5:16542. doi: 10.1038/srep16542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bingen BO, et al. Light-induced termination of spiral wave arrhythmias by optogenetic engineering of atrial cardiomyocytes. Cardiovasc Res. 2014;104:194–205. doi: 10.1093/cvr/cvu179. [DOI] [PubMed] [Google Scholar]
- 16.Nussinovitch U, Gepstein L. Optogenetics for suppression of cardiac electrical activity in human and rat cardiomyocyte cultures. Neurophotonics. 2015;2:031204. doi: 10.1117/1.NPh.2.3.031204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nussinovitch U, Gepstein L. Optogenetics for in vivo cardiac pacing and resynchronization therapies. Nat Biotechnol. 2015;33:750–754. doi: 10.1038/nbt.3268. [DOI] [PubMed] [Google Scholar]
- 18.Naldini L. Lentiviruses as gene transfer agents for delivery to non-dividing cells. Curr Opin Biotechnol. 1998;9:457–463. doi: 10.1016/S0958-1669(98)80029-3. [DOI] [PubMed] [Google Scholar]
- 19.Raman R, et al. Optogenetic skeletal muscle-powered adaptive biological machines. Proc Natl Acad Sci USA. 2016;113:3497–3502. doi: 10.1073/pnas.1516139113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuge Y, et al. Human pluripotent stem cell tools for cardiac optogenetics. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:6171–6174. doi: 10.1109/EMBC.2014.6945038. [DOI] [PubMed] [Google Scholar]
- 21.Park SA, Lee SR, Tung L, Yue DT. Optical mapping of optogenetically shaped cardiac action potentials. Sci Rep. 2014;4:6125. doi: 10.1038/srep06125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abilez OJ. Cardiac optogenetics. Conf Proc IEEE Eng Med Biol Soc. 2012;2012:1386–1389. doi: 10.1109/EMBC.2012.6346197. [DOI] [PubMed] [Google Scholar]
- 23.Zhao S, et al. Improved expression of halorhodopsin for light-induced silencing of neuronal activity. Brain Cell Biol. 2008;36:141–154. doi: 10.1007/s11068-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beppu K, et al. Optogenetic countering of glial acidosis suppresses glial glutamate release and ischemic brain damage. Neuron. 2014;81:314–320. doi: 10.1016/j.neuron.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 25.Miyashita T, Shao YR, Chung J, Pourzia O, Feldman DE. Long-term channelrhodopsin-2 (ChR2) expression can induce abnormal axonal morphology and targeting in cerebral cortex. Front Neural Circuits. 2013;7:8. doi: 10.3389/fncir.2013.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koponen JK, et al. Doxycycline-regulated lentiviral vector system with a novel reverse transactivator rtTA2S-M2 shows a tight control of gene expression in vitro and in vivo. Gene Ther. 2003;10:459–466. doi: 10.1038/sj.gt.3301889. [DOI] [PubMed] [Google Scholar]
- 27.Efimov IR, Nikolski VP, Salama G. Optical imaging of the heart. Circ Res. 2004;95:21–33. doi: 10.1161/01.RES.0000130529.18016.35. [DOI] [PubMed] [Google Scholar]
- 28.Fast VG, Cheek ER. Optical mapping of arrhythmias induced by strong electrical shocks in myocyte cultures. Circ Res. 2002;90:664–670. doi: 10.1161/01.RES.0000013403.24495.CC. [DOI] [PubMed] [Google Scholar]
- 29.Herron TJ, Lee P, Jalife J. Optical imaging of voltage and calcium in cardiac cells & tissues. Circ Res. 2012;110:609–623. doi: 10.1161/CIRCRESAHA.111.247494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radisic M, et al. Optical mapping of impulse propagation in engineered cardiac tissue. Tissue Eng Part A. 2009;15:851–860. doi: 10.1089/ten.tea.2008.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q, et al. Simultaneous Optical Pacing and Membrane Voltage Mapping in ChR2-Expressing Neonatal Rat Ventricular Myocyte Cultures. Biophys J. 2014;106:383a. doi: 10.1016/j.bpj.2013.11.2166. [DOI] [Google Scholar]
- 32.Golden, H. et al. In Cardiovascular Development Vol. 843 Methods in Molecular Biology (eds Peng, Xu & Antonyak, Marc) Ch. 20, 205–214 (Humana Press, 2012).
- 33.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 34.Rauth S, Hoganson GE, Davidson RL. Bromodeoxyuridine- and cyclic AMP-mediated regulation of tyrosinase in Syrian hamster melanoma cells. Somat Cell Mol Genet. 1990;16:583–592. doi: 10.1007/BF01233099. [DOI] [PubMed] [Google Scholar]
- 35.Rohr S, Fluckiger-Labrada R, Kucera JP. Photolithographically defined deposition of attachment factors as a versatile method for patterning the growth of different cell types in culture. Pflugers Arch. 2003;446:125–132. doi: 10.1007/s00424-002-1000-0. [DOI] [PubMed] [Google Scholar]
- 36.Tapscott SJ, Lassar AB, Davis RL, Weintraub H. 5-bromo-2′-deoxyuridine blocks myogenesis by extinguishing expression of MyoD1. Science. 1989;245:532–536. doi: 10.1126/science.2547249. [DOI] [PubMed] [Google Scholar]
- 37.Nishibatake M, Kargas SA, Bruyere HJ, Jr., Gilbert EF. Cardiovascular malformations induced by bromodeoxyuridine in the chick embryo. Teratology. 1987;36:125–132. doi: 10.1002/tera.1420360116. [DOI] [PubMed] [Google Scholar]
- 38.Entcheva E. Cardiac optogenetics. Am J Physiol Heart Circ Physiol. 2013;304:H1179–1191. doi: 10.1152/ajpheart.00432.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caldwell JH, et al. Increases in intracellular calcium triggered by channelrhodopsin-2 potentiate the response of metabotropic glutamate receptor mGluR7. J Biol Chem. 2008;283:24300–24307. doi: 10.1074/jbc.M802593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chater TE, Henley JM, Brown JT, Randall AD. Voltage- and temperature-dependent gating of heterologously expressed channelrhodopsin-2. J Neurosci Methods. 2010;193:7–13. doi: 10.1016/j.jneumeth.2010.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA. Calcium in cell injury and death. Annu Rev Pathol. 2006;1:405–434. doi: 10.1146/annurev.pathol.1.110304.100218. [DOI] [PubMed] [Google Scholar]
- 42.Piper HM. The calcium paradox revisited: an artefact of great heuristic value. Cardiovasc Res. 2000;45:123–127. doi: 10.1016/S0008-6363(99)00304-1. [DOI] [PubMed] [Google Scholar]
- 43.Oe H, et al. Calcium overload and cardiac myocyte cell damage induced by arachidonate lipoxygenation. Am J Physiol. 1994;267:H1396–1402. doi: 10.1152/ajpheart.1994.267.4.H1396. [DOI] [PubMed] [Google Scholar]
- 44.Tang J, et al. Cyclosporin A induces cardiomyocyte injury through calcium-sensing receptor-mediated calcium overload. Pharmazie. 2011;66:52–57. [PubMed] [Google Scholar]
- 45.Gradinaru V, et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27:14231–14238. doi: 10.1523/JNEUROSCI.3578-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmermann D, et al. Effects on capacitance by overexpression of membrane proteins. Biochem Biophys Res Commun. 2008;369:1022–1026. doi: 10.1016/j.bbrc.2008.02.153. [DOI] [PubMed] [Google Scholar]
- 48.Lin JY. A user’s guide to channelrhodopsin variants: features, limitations and future developments. Exp Physiol. 2011;96:19–25. doi: 10.1113/expphysiol.2009.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gradinaru V, Thompson KR, Deisseroth K. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008;36:129–139. doi: 10.1007/s11068-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fast VG, Kléber AG. Anisotropic conduction in monolayers of neonatal rat heart cells cultured on collagen substrate. Circ Res. 1994;75:591–595. doi: 10.1161/01.RES.75.3.591. [DOI] [PubMed] [Google Scholar]
- 51.Zlochiver S, et al. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bristow MR, et al. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med. 2004;350:2140–2150. doi: 10.1056/NEJMoa032423. [DOI] [PubMed] [Google Scholar]
- 53.Singh JP, Gras D. Biventricular pacing: current trends and future strategies. Eur Heart J. 2012;33:305–313. doi: 10.1093/eurheartj/ehr366. [DOI] [PubMed] [Google Scholar]
- 54.Carey BW, et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci USA. 2009;106:157–162. doi: 10.1073/pnas.0811426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohr S, Scholly DM, Kleber AG. Patterned growth of neonatal rat heart cells in culture. Morphological and electrophysiological characterization . Circ Res. 1991;68:114–130. doi: 10.1161/01.RES.68.1.114. [DOI] [PubMed] [Google Scholar]
- 56.Sowell B, Fast VG. Ionic mechanism of shock-induced arrhythmias: role of intracellular calcium. Heart Rhythm. 2012;9:96–104. doi: 10.1016/j.hrthm.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.