

Abstract
Glucocorticoid hormones (GC) are the main stress mediators associated with reproductive disorders. GC exert their effects through activation of the glucocorticoid receptor (GR) principally acting as a transcription factor. Beside well-established GR-mediated genomic actions, several lines of evidence suggest a role for rapid membrane-initiated GC signaling in gonadotrope cells triggered by a membrane-associated GR. Herein, we demonstrate the existence of a specific membrane-initiated GC signaling in LβT2 gonadotrope cells involving two related phosphoproteins: Ca2+/Calmodulin-dependent protein kinase II (CaMKII) and synapsin-I. Within 5 min, LβT2 cells treated with stress range of 10−7 M Corticosterone or a membrane impermeable-GC, BSA-conjugated corticosterone, exhibited a 2-fold increase in levels of phospho-CaMKII and phospho-synapsin-I. Biochemical approaches revealed that this rapid signaling is promoted by a palmitoylated GR. Importantly, GC significantly alter GnRH-induced CaMKII phosphorylation, consistent with a novel cross-talk between the GnRH receptor and GC. This negative effect of GC on GnRH signaling was further observed on LH release by mouse pituitary explants. Altogether, our work provides new findings in GC field by bringing novel understanding on how GR integrates plasma membrane, allowing GC membrane-initiated signaling that differs in presence of GnRH to disrupt GnRH-dependent signaling and LH secretion.
Introduction
Glucocorticoid hormones (GC) are the main mediators of stress and their increased secretion leads to a large variety of acute and long-term responses. Physical and emotional stressors have been reported to perturb female menstrual cycles1, 2, to alter the onset of puberty3, to be associated with a longer time-to-pregnancy and to increase the risk of infertility4. Psychosocial stress has been extensively studied in ewes5. It is now established that GC act at different levels of the reproductive axis: at the hypothalamus by reducing Gonadotropin-releasing-hormone (GnRH) pulse frequency6 and at the pituitary by rapidly decreasing responsiveness to GnRH to suppress pulsatile gonadotropins expression and secretion, notably LH7. GC responses are mediated by the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily, through two separate but inter-related mechanisms of actions8. After ligand binding, GC/GR complexes translocate into the nucleus to regulate target genes expression9. Several studies described a direct action of GC in pituitary gonadotrope cells to regulate gonadotropins and GnRH receptor (GnRH-R) expression10. In addition, signaling pathways mediating rapid non-genomic GC actions have been demonstrated in a number of studies; they involve various cytosolic proteins11 e.g Phospholipase C12, Mitogen-Activated Protein Kinases (MAPK)13, 14, Phosphatidylinositol 3-kinase15, Src kinase16, 17 or intracellular Ca2+ signaling18.
GC-induced rapid signaling has never been directly studied in gonadotrope cells yet, different arguments suggest their involvement in the rapid blunting of GnRH responsiveness during stress7. We speculate that GC could modulate signaling mechanisms downstream of the GnRH-R leading to a reduction of gonadotropin release. Such GC-induced rapid signaling should involve the activation of a putative membrane associated GR (MbGR). The existence of such a MbGR has been convincingly demonstrated in monocytes and B lymphocytes19, keratinocytes20, hippocampal neurons21 or in several cell lines16, 22, 23. Studies using classical GR antibodies24, 25 or using RNA interference-mediated GR reduction26 revealed that intracellular GR and MbGR may originate from the same gene. Membrane targeting of a steroid receptor has been already described for the estrogen receptor (ER) which undergoes membrane translocation after caveolin-1 association27 and protein palmitoylation28. Palmitoylation is a posttranslational process by which palmitate, a C16 fatty acid, is covalently linked to an internal cysteine via a thioester bond. As for ER, GR has been found associated with lipid rafts29 and caveolin-114, 30, 31. However, contrary to ER, Vernocchi et al., proposed that the membrane localization of GR is not totally dependent on caveolin-132. Posttranslational modifications that influence GR membrane localization33 have not been described yet.
In the present study, we used a murine gonadotrope cell line (LβT2) expressing GR and prone to release LH upon GnRH treatment, as well as mouse pituitary explants to better understand whether and how GC might rapidly interfere with GnRH signaling to prevent LH release during stress. Our data provide evidence, using a membrane-impermeable corticosterone conjugate, Cort-BSA (bovine serum albumin), and an inhibitor of protein palmitoylation (2-Br) that GC induce a novel rapid signaling in LβT2 cells through the activation of a palmitoylated membrane GR. We also demonstrate that GC interfere with GnRH induced Ca2+/Calmodulin-dependent kinase 2 (CaMKII) phosphorylation in LβT2 cells, ultimately decreasing LH release by pituitary explants.
Results
Specific membrane-initiated GC signaling in gonadotrope cells: role of CaMKII and synapsin-I
In ewes, GC have been described to reduce the LH response of pulse-like delivery of GnRH by 50% within 30 min, indicating rapid action of GC on pituitary7. To examine a potential relationship between rapid GC-induced signaling and LH release reduction, we investigated whether specific rapid GC signaling pathways were activated in gonadotrope cells. LβT2 cells were treated with a stress range of 10−7 M corticosterone (Cort) for up to 60 min and the level of phosphorylation of CaMKII (p-CaMKII) as well as one of its target, synapsin-I (Syn) were evaluated by Western blotting; signals were normalized to the level of total proteins and α-tubulin (Fig. 1a,b). Figure 1a shows that Cort induced a rapid and transient phosphorylation of CaMKII in LβT2 cells with a maximum (about 2.3-fold) observed after 15 min of treatment which then returned to basal value. Cort also stimulated the level of phospho-Syn (p-Syn) within 10 min (Fig. 1b) and such an effect was maintained up to 60 min of treatment. Cort treatment did not modify total levels of CaMKII and Syn. To bring support for a membrane GR-initiated effect, we used BSA-conjugated corticosterone (Cort-BSA), a membrane impermeable GC that allows discriminating specific activities of the MbGR from those of the intracellular GR. The ability of Cort-BSA to promote genomic effects was tested on the expression of a GC-induced gene Serum and Glucocorticoid regulated Kinase 1 (Sgk-1). As shown in Supplementary Fig. S1, Cort-BSA did not induce expression of Sgk-1 after 1 or 4 h of treatment, when compared to vehicle, in contrast to the expected effects of dexamethasone (Dex, a GR agonist) or Cort. These data confirm that Cort-BSA effects could only be attributed to non-genomic mechanisms in LβT2 cells. Exposure of LβT2 cells to Cort-BSA (10−7 M) for 5 min induced the same level (about 2-fold) of CaMKII phosphorylation as the one induced by Cort, but phosphorylation remained stable up to 60 min of treatment (Fig. 1c). A similar phosphorylation pattern was observed for p-Syn following Cort-BSA and Cort exposure, except an earlier return to basal level after Cort-BSA treatment (Fig. 1d). Total levels of CaMKII and Syn were unchanged after Cort-BSA treatment. Interestingly, Dex did not stimulate MAPK (Supplementary Fig. S2a) nor Src signaling pathways (Supplementary Fig. S2b) in contrast to their reported effect in most cellular models11 studied so far. The specificity of Cort/Cort-BSA rapid signaling was assessed using a GR antagonist (RU486) or a CaMKII inhibitor (KN93) (Fig. 1e). Co-incubation of Cort or Cort-BSA with these inhibitors prevented CaMKII phosphorylation measured after 5 min of treatment in LβT2 cells, without significantly affecting basal p-CaMKII. Interestingly, both inhibitors also affected the level of GC-induced p-Syn observed after 5 min of treatment without disturbing basal level of p-Syn (Fig. 1f). These data provide evidence that Cort or Cort-BSA rapidly promotes the phosphorylation of CaMKII and Syn in gonadotrope cells. This phosphorylation sequence therefore constitutes a new GC membrane-initiated signaling dependent on GR.
Figure 1.

GC rapidly stimulate CaMKII and synapsin-I phosphorylation in LβT2 cells. (a,b) LβT2 cells were treated with Cort (10−7 M) for 0 to 60 min. Cell lysates were subjected to western blotting (WB) with anti-CaMKII (a), anti-p-CaMKII (a), anti-synapsin (b), anti-p-synapsin (b) and anti-α-tubulin antibodies (a,b). Cropped blots are shown in the figure and are representative of 4 independent experiments. Results are expressed as means ± SEM fold stimulation when compared to time 0. One-way ANOVA with Dunnett’s posttest, *p < 0.05, **p < 0.01, and ***p < 0.001. (c,d) LβT2 were treated for 0 to 60 min with Cort-BSA (10−7 M). Cropped immunoblots are shown in the figure and are representative of 4 independent experiments. Results are expressed as means ± SEM fold stimulation when compared to time 0, one-way ANOVA with Dunnett’s posttest, *p < 0.05, **p < 0.01. (e,f) Phosphorylated CaMKII (e) and Syn (f) were significantly increased after 5 min treatment with either Cort or Cort-BSA. These events were abolished by co-incubation with RU486 (10−5 M) or KN93 (10−5 M). V: Vehicle. Statistical significance was assessed by one-way ANOVA with Newman-Keuls Multiple Comparison Test, n ≥ 3, ***p < 0.001 when compared to Vehicle, ### p < 0.001 within the same steroid treatment (Cort or Cort-BSA). Full length WBs are presented in Supplementary Fig. S4a–c. p-CaMKII: phospho-CaMKII; p-Syn: phospho-synaspin I.
We next examined GC effects in a more physiological context. To this aim, mouse pituitary explants were treated for 30 min with the GR agonist Dex. We found a significant GC-induced increase in levels of p-CaMKII (about 1.4-fold) as well as in p-Syn (about 2.2-fold) (Fig. 2). Dex treatment did not alter total CaMKII or total Syn contents. This effect was prevented by co-treatment with RU486 (not shown). CaMKII and Syn therefore constitute new key elements of GC membrane-initiated signaling in pituitary.
Figure 2.
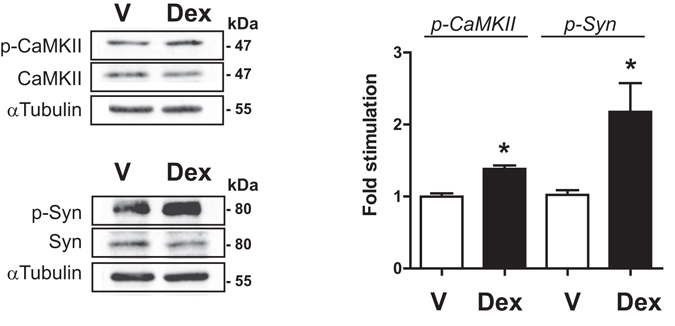
Dexamethasone stimulates phosphorylation of CaMKII and Syn in mouse pituitary explants. Explanted pituitaries were treated with Dex (10−7 M) or not (Vehicle, V) for 30 min. Lysates were subjected to immunoblotting with anti-p-CaMKII, anti-CaMKII, anti-Syn, anti-p-Syn and anti-α-tubulin antibodies. Dex induced significant increase in the phosphorylation state of CaMKII and Synapsin-I. Cropped blots (full length WBs are presented in Supplementary Fig. S4d) are shown in the figure and are representative image of 3 independent experiments. Results are expressed as means ± SEM fold stimulation when compared to vehicle. Statistical significance was assessed by One way ANOVA, Dunnett’s posttest, n = 3, *p ≤ 0.05. p-CaMKII: phospho-CaMKII; p-Syn: phospho-synaspin-I.
A palmitoylated membrane GR promotes CaMKII phosphorylation in LβT2 cells
This observed rapid membrane-initiated signaling strongly suggests the existence of a membrane form of GR (MbGR) in LβT2 gonadotrope cells. Therefore, we investigated the subcellular localization of endogenous GR in LβT2 cells by cell fractionation, each fraction being analyzed by Western blotting. As illustrated in Fig. 3a using classical GR antibodies (M20), a single band at approximately 100 kDa corresponding to GR was detected in total LβT2 cell lysates in the absence (Vehicle, V) or presence of Dex, as well as in all three subcellular compartments (Nuclei/Debris (N), Cytoplasmic (Cyto) and Membranous (Mb) fractions). Quantification of GR signal in each subcellular compartment in the vehicle condition further confirmed that GR was mainly located in the cytoplasm (ratio Cyto/Total = 2 ± 0.32). Noteworthy, our results demonstrate that a significant amount of GR was also recovered in the membranous fraction of LβT2 cells (ratio Mb/Total = 0.9 ± 0.2) (Fig. 3b). A membrane form of GR is therefore present in gonadotrope cells. Such a form could also be detected in various GC-sensitive cell lines originating from various tissues (Supplementary Fig. S3), clearly demonstrating that although the bulk of GR is in the cytoplasm and nucleus, a significant proportion of GR is tethered or anchored to cell membranes of GC-sensitive cells. To test whether subcellular GR localization could be modified in the presence of ligand, LβT2 cells were incubated with 10−7 M Dex for 30 min before cell fractionation. As expected, nuclear GR was significantly increased upon Dex treatment, while cytoplasmic signal was concomitantly reduced (Fig. 3b), in agreement with the hormone-dependent GR translocation from cytoplasm to the nucleus. Importantly, the presence of ligand did not modify the intensity of MbGR signal suggesting that acute Dex exposure does not affect MbGR level (Fig. 3b).
Figure 3.
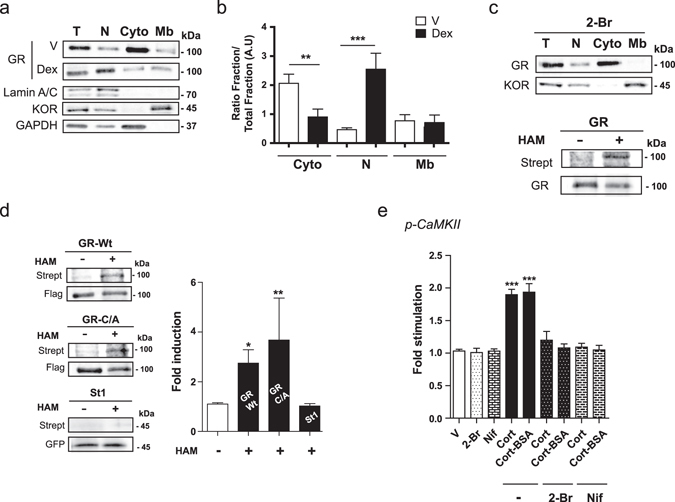
A palmitoylated membrane GR is involved in GC-induced rapid CaMKII phosphorylation in LβT2 cells. (a) LβT2 cells were treated or not (V) with Dex before subcellular fractionation to detect GR and specific markers by WB; lamin A/C, κ-opioid receptor (KOR) and GAPDH for nuclear/debris (N), membranous (Mb) and cytosolic (Cyto) fractions, respectively. T: total extract. (b) Quantification of GR in each fraction of LβT2 cells treated or not (V) with Dex, in comparison with T. Results are expressed as means ± SEM fold stimulation when compared to T, one-way ANOVA with Newman-Keuls Multiple Comparison Test, n = 4 experiments, **p < 0.01, and ***p < 0.001. (c) Upper panel, LβT2 cells were pre-incubated with 2-Br prior to cell fractionation. Equal amounts of proteins were analyzed by WB. n = 3 experiments. 2-Br: 2-bromopalmitate. Lower panel, LβT2 cell lysates were subjected to ABE assay after GR immunoprecipitation, followed by SDS-PAGE with the minus (−) and plus (+) HAM samples. Palmitoylation was detected by WB comparing streptavidin (Strept) signal to GR one. n = 3 experiments. (d) HEK293 cells were transfected with GRWt, GRC/A or stathmin 1-GFP (St1). Cell lysates were subjected to the ABE protocol followed by SDS-PAGE as described in (c). Strept signals detecting palmitoylation were normalized to the level of immunoprecipitated Flag-GR or GFP-St1 detected on the same membrane. Graph shows the average normalized densitometry data (Strept signal divided by anti-Flag or anti-GFP signals) displayed as fold induction of (+) HAM/(−) HAM control samples. One-way ANOVA with Dunnett’s posttest (n = 4 experiments, *p < 0.05, **p < 0.01). (e) LβT2 cells were pretreated with 2-Br or Nifedipine (Nif) respectively, and then treated with either Cort or Cort-BSA for 5 min. Cell lysates were subjected to WB with anti-CaMKII, anti-p-CaMKII, and anti-α-tubulin antibodies. Results are expressed as means ± SEM fold stimulation when compared to vehicle (V). One-way ANOVA with Dunnett’s post-test, n = 3 experiments, ***p < 0.001 when compared to V. Cropped blots are shown. Full length WBs are presented in Supplementary Fig. S4d,e.
Since palmitoylation of ER was previously shown to be the major determinant for ER residence at the plasma membrane34, we evaluated the effect of the palmitoylation inhibitor 2-bromopalmitate (2-Br) on MbGR localization. As illustrated in Fig. 3c (upper panel), overnight incubation of LβT2 cells with 100 μM 2-Br totally abolished MbGR localization without modifying the presence of the membrane marker, the κ-opioid receptor (KOR), in the membranous fraction (ratio Mb/Total = 1.32 ± 0.2 vs 1.43 ± 0.3), strongly suggesting that endogenous GR is palmitoylated. To further confirm GR palmitoylation, we performed an ABE assay35 on LβT2 cells. During the ABE assay, unmodified free cysteines of immunoprecipitated GR were alkylated with NEM. Next, hydroxylamide (HAM) specifically cleaved palmitates from cysteines and then exposed these sites for biotinylation. Once the sites were labeled, palmitoylation was analyzed by SDS-PAGE and Western blotting using fluorophore-conjugated streptavidin (Strept); Strept signals correspond to protein biotinylation of palmitoylation sites. The same membrane was also used for Western blotting with an anti-GR antibody (Fig. 3c, lower panel). As shown in Fig. 3c (lower panel), Strept signals were only revealed after HAM treatment condition (+), despite an equivalent immunoprecipitation efficiency between the two conditions. This result confirms that GR is clearly palmitoylated in LβT2 cells and demonstrates that GR is addressed to the plasma membrane through palmitoylation. Next, we assessed whether the palmitoylation site of GR was conserved with the one described for ER, AR and PR28. We mutated the ER corresponding cysteine residue of GR (Cys671) into Alanine (C/A) on a Flag-form of GR and performed an ABE assay after overexpressing GRWt or GRC/A in HEK293 cells. A GFP-form of stathmin 1 (St1) known as a soluble non-palmitoylable protein36 was used as a negative control. Strept signal was normalized to the amount of tagged protein detected on the same blot (Fig. 3d). As shown in Fig. 3d, GRWt treated with HAM was significantly biotinylated and specifically labeled by Strept (about 2.7-fold induction compared to the absence of HAM), indicating that overexpressed GR is also palmitoylated in HEK293 cells. Importantly, mutation of Cys671 into Ala (GRC/A) led to a GR mutant that remained palmitoylated at an equivalent level as the GRWt (Strept/Flag signals between “−HAM” and “+HAM” increased by 3.6 ± 1.5 -fold vs 2.7 ± 1.2 -fold, respectively). Immunoprecipitated stathmin 1 (St1) was not labeled by Strept providing additional support for GR palmitoylation. Thus, in sharp contrast with ER, the putative palmitoylation consensus site Cys671 is not involved in the GR membrane targeting.
We also tested whether palmitoylated GR was responsible for rapid GC-induced signaling. As shown in Fig. 3e, overnight pretreatment of LβT2 cells with 100 µM 2-Br totally abolished both Cort- and Cort-BSA-induced CaMKII phosphorylation when compared to vehicle. Total CaMKII levels were not modified by 2-Br treatment (not shown). These data unambiguously demonstrate that the palmitoylated form of GR triggers GC rapid signaling on CaMKII phosphorylation.
Calmodulin-associated calcium promotes CaMKII autophosphorylation and subsequently CaMKII activation37. Therefore, we examined the mechanism by which GC would increase intracellular calcium concentration by following the effects of an L-type calcium channel blocker (Nifedipine) on GC induced CaMKII phosphorylation. As show in Fig. 3e, 1h pretreatment with 10 μM Nifedipine (Nif) prevented CaMKII phosphorylation measured after 5 min of Cort or Cort-BSA treatment. Nif did not modify total levels of CaMKII (not shown). Overall, GC rapidly promote membrane-initiated signaling involving a palmitoylated GR that elicits calcium entry through L-type calcium channel that participates to CaMKII phosphorylation and leads to synapsin phosphorylation.
GC affect GnRH signaling to reduce LH secretion
GnRH has been described to rapidly stimulate CaMKII phosphorylation (by about 2.5-fold) in rodent pituitary cells37, thus regulating gonadotropin subunit transcription. Therefore, we investigated whether GC could influence GnRH-induced CaMKII phosphorylation in LβT2 cells. Figure 4 shows that treatment with GnRH (10−9 M) for 5 min increased by about 2.3-fold p-CaMKII levels compared with vehicle. These phosphorylation levels were in the same range than those measured after Cort or Cort-BSA exposure alone (Fig. 4). Interestingly, co-treatment with GnRH and Cort or Cort-BSA for 5 min prevented GnRH-induced CaMKII phosphorylation. Total CaMKII levels remained unchanged after any of these hormonal stimulations. These data strongly indicate that GC affect GnRH signaling by preventing GnRH-dependent CaMKII phosphorylation in LβT2 cells.
Figure 4.
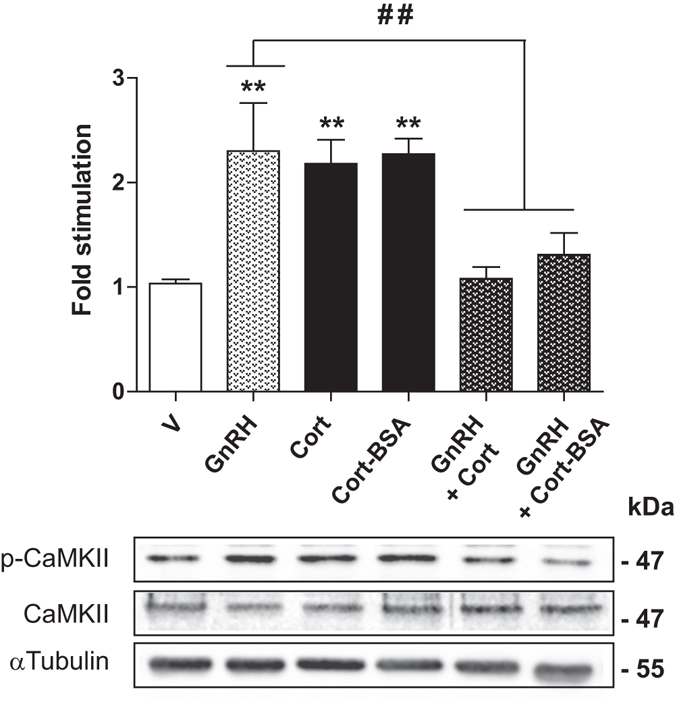
GC prevent GnRH-induced CaMKII phosphorylation. LβT2 cells were treated or not (V) with GnRH (10−8 M), Cort (10−7 M), Cort-BSA (10−7 M) or co-treated with either GnRH and Cort or GnRH and Cort-BSA for 5 min. Cell lysates were subjected to WB with anti-CaMKII, anti-p-CaMKII, and anti-α-tubulin antibodies. Cropped blots (full length WBs are presented in Supplementary Fig. S4e) are shown in the figure and are representative of 3 independent experiments. Results are expressed as means ± SEM fold stimulation when compared to vehicle. One-way ANOVA with Dunnett’s post-test, **p < 0.01. ## p < 0.01 comparing GnRH with both co-treatments.
Since CaMKII was also described to be involved in insulin secretion38, we wondered whether this kinase could also modulate LH secretion. Because detection of LH release in LβT2 culture media was not possible at early time points due to the low secretory activity of this cell line, we tested the effect of GC treatment on LH release from pituitary explants. The integrity of the pituitary explants was controlled at the end of the procedure by histological approaches (not shown). Mouse pituitary explants were pre-incubated for 30 min in DMEM at 37 °C before stimulation to measure basal level of LH content present in culture supernatants of each explant. Culture media was then replaced by fresh media containing either GnRH (10−8 M), Dex (10−7 M) or both hormones for 1 h. LH contents were subsequently assayed from culture supernatants. As anticipated, Fig. 5a shows that GnRH strongly increased (by about 9-fold) LH secretion. In contrast, Dex did not induce any significant LH release. Noteworthy, co-incubation of pituitary explants with both GnRH and Dex significantly reduced LH release by 1.5-fold when compared to GnRH alone (Fig. 5a).
Figure 5.
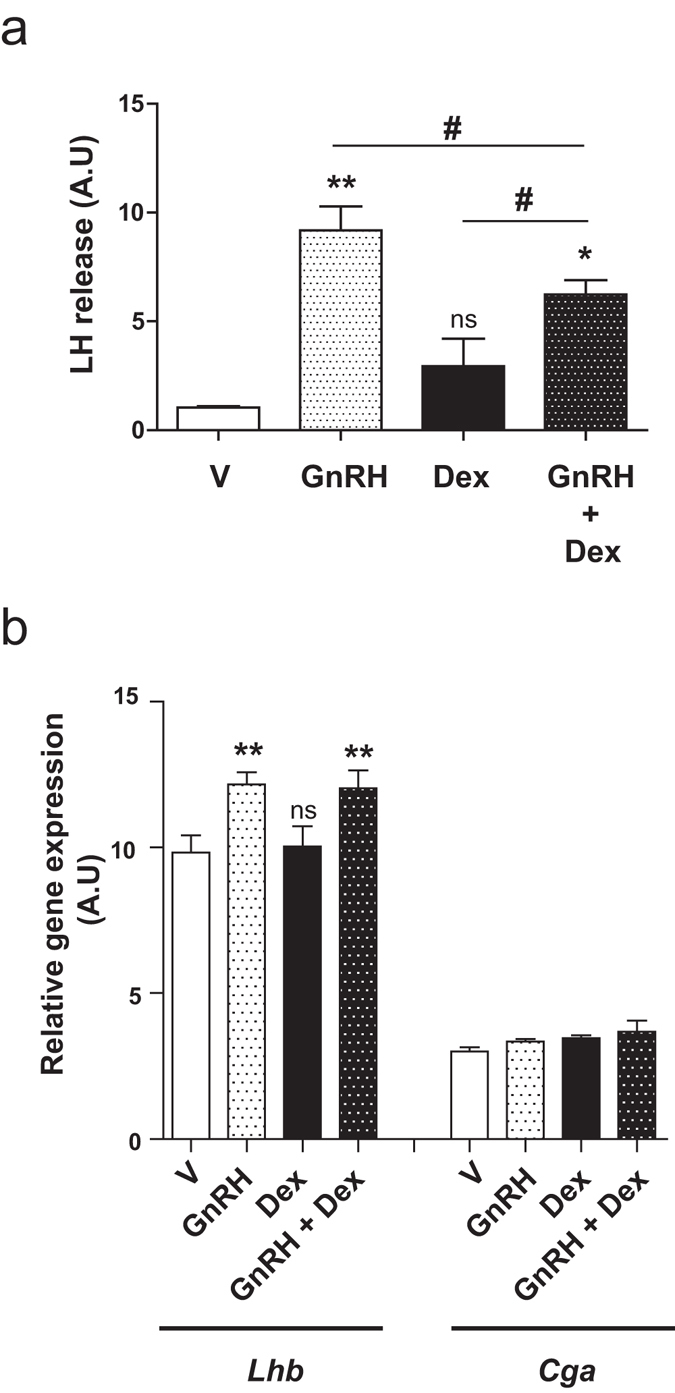
GC prevent GnRH-induced LH secretion in mice pituitary explants. (a) Pituitary explants were either treated with GnRH (10−8 M), Dex (10−7 M) or co-incubated with these two hormones for 1 h at 37 °C. LH concentrations were assayed in the culture supernatants as described in Methods. LH release after hormonal treatment was quantified (ng/ml) in comparison with LH amount measured in basal condition (pre-stimulation). GnRH significantly induced LH release when compared to vehicle. This effect significantly decreased after co-incubation of GnRH with Dex. One-way ANOVA with Dunnett’s post-test, *p < 0.05, **p < 0.01. # p < 0.05 comparing GnRH and Dex co-treatment with each individual treatment. (b) LβT2 were treated with GnRH (10−8 M), Dex (10−7 M) or co-incubated with these two hormones for 1 h at 37 °C before RT-qPCR to follow the expression of genes encoding the common α-glycoprotein subunit (Cga) and the β-subunit of LH (Lhb). Data show relative expression (arbitrary units) normalized with the reference gene 36b4. Within 1 h, GnRH significantly stimulated Lhb expression (by approximately 1.24-fold above basal), whereas Dex did not have any effects either alone or associated with GnRH. One-way ANOVA with Newman-Keuls Multiple Comparison posttest, n = 3, **p < 0.01.
Since Dex binds both membrane and non-membrane GR, one could expect Dex to exert genomic activities that could contribute to Dex inhibitory effect on GnRH-induced LH release. Therefore, to investigate a possible impact of Dex on the expression of genes encoding the common α-glycoprotein subunit (Cga) and the β-subunit of LH (Lhb), we performed RT-qPCR on total RNA extracted from LβT2 cells treated with GnRH (10−8 M), Dex (10−7 M) or both hormones for 1 h (Fig. 5b). GnRH treatment significantly increased Lhb expression (by about 1.24-fold) whereas Dex alone or associated with GnRH was ineffective on Lhb or Cga expression at this very early time point when compared to vehicle or GnRH respectively (Fig. 5b). In contrast and as shown in Supplementary Fig. S1, under the same experimental conditions, Dex was able to regulate Sgk1 expression; after 1 h treatment, Dex induced a 1.7-fold induction of Sgk-1 mRNA levels. These results confirm that Dex blockade of GnRH-induced LH secretion involves rapid non-genomic mechanisms.
Altogether, these results demonstrate that GC rapidly regulate GnRH-induced CaMKII phosphorylation as well as impact on GnRH-dependent LH secretion.
Discussion
Stress-induced disturbance of reproductive function is well described in many species including humans3, 5, 39, and is typically associated with an activation of the hypothalamic-pituitary adrenal axis, resulting in an increased secretion of GC. GC act directly via GR in the pituitary gland40 to elicit a rapid decrease (≤30 min) in responsiveness to GnRH, independent of changes in GnRH-R expression7, leading to a strong reduction of gonadotropin release41. Non-genomic mechanisms activated by a membrane anchored GR (MbGR) have been proposed to explain such rapid GC events.
In the present study, we provide evidence that GC rapidly trigger CaMKII and Syn phosphorylation in gonadotrope LβT2 cells. We demonstrated that Cort elicited a rapid (significant up to 15 min) and transient effect on CaMKII phosphorylation whereas a significant induction of p-Syn was detected up to 60 min after GC exposure. Ex vivo, by using pituitary explants, we demonstrated that Dex also induces phosphorylation of CaMKII and Syn after 30 min treatment, likewise what observed in vitro, in gonadotrope LβT2 cells exposed to Cort. Interestingly, LβT2 cells treated with a membrane impermeable GC (Cort-BSA) induced similar phosphorylation patterns of p-CaMKII and p-Syn than those obtained with Cort. However, p-CaMKII rapidly returned to basal levels indicating dephophorylation processes after Cort treatment, as opposed to Cort-BSA. These data suggest a negative regulatory feedback thereby GC, acting genomically through unknown genomic events, might desensitize its activated-signaling pathway more rapidly and efficiently than Cort-BSA. In addition and contrary to Dex or Cort, no induction of Sgk-1 expression was observed after 1 h or 4 h of treatment with Cort-BSA, providing additional support that Cort-BSA effects are clearly only mediated by membrane-initiated non-genomic processes in LβT2 cells. Our results clearly differ from those reported in another study that showed genomic activities with Cort-BSA at a much higher concentrations (3 μg/ml vs 6.6 ng/ml in the present study) on human lymphoma cells42. In addition, contrary to other cell types14, 16, 17, 43, Dex was unable to induce phosphorylation of ERK1/2 or Src indicating that, depending on the cellular context and the nature of the associated signalosome, a wide variety of signal transduction pathways may be initiated. From plasma membrane of LβT2 cells, GC may promote L-type Calcium channel activation that triggers the calcium increase necessary for CaMKII phosphorylation and the resulting downstream signaling cascade.
To further understand this rapid signaling in gonadotrope LβT2 cells, we identified a MbGR and provided evidence that palmitoylation, a reversible posttranslational modification, promotes GR membrane targeting. We also demonstrated a direct involvement of palmitoylated form of GR in triggering CaMKII phosphorylation and activation, after Cort or Cort-BSA treatment in LβT2 cells. Contrary to others who used an hypothalamic cell line 4B33 and performed metabolic labeling presenting low sensitivity detection, we demonstrated that endogenous as well as overexpressed GR are truly palmitoylated. These data further support previous data performed in lymphocytes which showed that GR membrane targeting was prevented by a Golgi-disrupting agent (brefeldin A) treatment19, indicating that GR membrane attachment might take place in the Golgi where most of protein palmitoylation occurs44. Interestingly yet surprisingly, we demonstrated by site directed mutagenesis that ER and GR do not share the equivalent palmitoylation site, arguing against a consensus palmitoylation site among steroid receptors. Further studies are required to identify GR palmitoylation sites. Contrary to ER whose membrane transport relies on caveolin-1, two studies demonstrated that caveolin-1 is not the limiting factor for membrane transport of GR, without ruling out the possibility that it is a component of the transport machinery22, 42. A recent study using several protein structure algorithms predicted that some steroid receptor ligand binding domains contain one or more transmembrane helices and/or pore lining regions that may be essential for their translocation to the plasma membrane45. Herein, we also show as already described by others23 that Dex does not modify the level of MbGR in gonadotrope LβT2 cells. This finding contrasts though with the observation that Cort promoted MbGR level increase in hypothalamic cell line 4B16, and suggests that the regulation of MbGR might be dependent on the cell type.
In pituitary, GnRH-R is primary expressed in gonadotrope cells. Upon GnRH binding to GnRH-R, a wide range of intracellular signaling pathways including CaMKII phosphorylation is activated that ultimately regulates the synthesis and release of gonadotropins46. In the present study, we show that Cort or Cort-BSA rapidly induced phosphorylation of CaMKII and synapsin-I. However, in the presence of GnRH, GC rapidly prevented GnRH-induced CaMKII phosphorylation, by preventing calcium mobilization probably in localized sub-cellular regions, and efficiently reduced LH release by gonadotrope cells. Contrary to GnRH, Dex alone or associated with GnRH does not rapidly affect Lhb or Cga expression demonstrating that Dex inhibition of GnRH-induced LH secretion involves rapid non-genomic regulatory mechanisms.
These data suggest that during anestrus or in between GnRH pulses that could be widely spaced depending on the species, GC exert rapid non genomic signaling involving both CaMKII and synapsin-I, whereas during GnRH stimulation GC prevent GnRH-induced CaMKII phosphorylation that may account for the inhibitory effects of GC on LH secretion. Such paradoxical actions of GCs have already been described to depend on the timing, duration, and magnitude of GC exposure47. In the present situation, one could speculate that MbGR might interact with GnRH-R to regulate its associated signaling upon GnRH binding. GR and GnRH-R have been reported to co-localize with the lipid raft protein flotillin-1 at the plasma membrane, independently of the presence of ligands23. GnRH and GC were reported to synergistically inhibit cell proliferation via PKC activation and Sgk-1 up-regulation in LβT2 cells23. Rapid non-genomic as well as genomic cross-talk mechanisms between the GnRH-R and GR signaling pathways have already been described in LβT2 cells48. GnRH may rapidly activate the unliganded GR via GnRH-R dependent phosphorylation involving MAPKs, leading to nuclear translocation and GR transactivation via a glucocorticoid response element. GC and GnRH may also modulate GnRH-R mRNA levels via a genomic mechanism involving GR binding to an AP-1 motif of the murine GnRH-R promoter48. In the present study, we propose another level of cross-talk mechanisms in LβT2 cells in which GC may regulate the level of GnRH-induced phosphorylation of CaMKII and synapsin-I that ultimately regulate LH release. We showed that Dex inhibited GnRH-induced LH release by pituitary explants. It remains to determine whether this inhibition is triggered by MbGR. GC have already been described to rapidly inhibit ACTH release in pituitary AtT-20 cells49, 50 or Cl− secretion in human bronchial epithelial cells18. CaMKII is a multifunctional kinase that has been depicted to mediate insulin secretion51 by phosphorylating synapsin-I38, already known to control neurotransmitters release52. Synapsin-I has been described as an important molecular effector of the GR signaling pathway induced in hippocampus involved in increased stress-related memory53. We demonstrated in LβT2 cells that GC signaling pathways encompass both CaMKII and synapsin-I. However, in the presence of GnRH, GC do not trigger this signaling pathways any longer to ultimately prevent LH release. It remains to precisely decipher the molecular mechanisms by which GC and GnRH signaling cross-talk, even though such a task might be very difficult to explore in vivo given that both GC and GnRH signaling share several downstream effectors. In addition, owing to the fact that GR is expressed in almost all pituitary cells, GC action is not restricted to gonadotrope cells but also affect other cell lineage populations within pituitary.
Finally, our work brings new findings in GC field, unraveling novel understanding on how GR integrates plasma membrane allowing GC membrane-initiated signaling through L-type calcium channel activation, CaMKII and synapsin-I phosphorylation in gonadotrope cells. A novel conceptual advance in the cross-talk between GnRH and GC signaling pathways has been discovered that could ultimately regulate pituitary LH release. Future challenge should clarify the existence of other pathways between GnRH and GC signaling that could account for desensitization of gonadotrope cells to GnRH during stress.
Methods
The antibodies and products used in the present study were purchased from the following sources:
Monoclonal anti–α tubulin (Sigma-Aldrich, St. Louis, MO, USA), Polyclonal anti-p-CaMKII (T286) (sc-12886-R, Santa Cruz Biotechnology, CA, USA), Polyclonal anti-CaMKII (H300) (sc-13082, Santa Cruz Biotechnology), Polyclonal anti-p-synapsin Ia/b (S603) (sc-135708, Santa Cruz Biotechnology), Polyclonal anti-synapsin Ia/b (H170) (sc-20780, Santa Cruz Biotechnology), Polyclonal anti-p-Src and anti-Src (Cell Signaling Technology, Danvers, MA, USA), Monoclonal anti-Flag M2 (Sigma-Aldrich), Polyclonal anti-GR M20 (Santa Cruz Biotechnology), Polyclonal anti-Lamin A/C (Cell Signaling Technology), Monoclonal anti-GAPDH (Sigma-Aldrich), monoclonal anti-GFP (Roche Diagnostics, Mannheim, Germany), Polyclonal anti-κ-Opioid Receptor (Abcam, Cambridge, UK), RU486 (mifepristone) (Sigma-Aldrich), 2-bromohexadecanoic acid (Sigma-Aldrich), Dexamethasone (Dex) (Sigma-Aldrich), Corticosterone (Sigma-Aldrich), Corticosterone-BSA conjugate (Cusabio Biotech Co., CliniSciences, Nanterre, France), KN93 (Sigma-Aldrich), DMEM-glutamax (Dulbecco’s Modified Minimum Essential Medium)-glucose 4.5 g/l (Life Technologies, Saint-Aubin, France), Fetal calf serum (FCS) (Biowest, Nuaillé, France), Lipofectamine 2000 (Life Technologies), OptiMEM (Life Technologies), Hydroxylamine (Sigma-Aldrich), EZ-link biotin-BMCC ((1-biotinamido-4-[4′-maleimidomethyl) cyclohexanecarboxamido] butane) (Thermo Fisher Scientific, Rockford, IL, USA), N-ethylmaleimide (NEM) (Merck Millipore, Darmstadt, Germany) and N-methylmaleimide (NMM) (Sigma-Aldrich).
Pituitary Culture
Adult male mice SV129 were decapitated and the whole pituitaries were rapidly collected and incubated in DMEM-glutamax medium at 37 °C in 5% CO2 for 2 h (equilibration) before hormonal treatment. This procedure was carried out in accordance with relevant guidelines and regulations following a protocol approved by the Animal Care Committee, Ministère de l’Agriculture, France (N°C94-043-12). All procedures were approved by the local ethics committee Consortium des Animaleries Paris Sud (CAPSud) (N°2012-021).
Cell line culture and transfection
Cell lines, LβT254, GT1-755 and HEK293 cells (obtained from American Type Culture Collection (ATCC; Manassas, VA, USA)) were cultured in DMEM containing 10% fetal calf serum (FCS) supplemented with 100 U/ml penicillin (Life technologies), 100 μg/ml streptomycin (Life technologies), at 37 °C in 5% CO2. KC3AC1 cells56 were seeded on collagen I-coated dishes and routinely cultured at 37 °C in 5% CO2 as previously described56. mhATF3F cells57 were cultured in DMEM/F12 (1:1) medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 10 nM insulin, 2 mM glutamine, 1 µM Dex, 30 nM sodium selenite, 1 µM 3,4,5-tri-iodo D-thyronine and 5% FCS. All cells were serum-starved for 24 h prior to drug treatment. HEK293 cells were transfected with Flag-GR using Lipofectamine 2000 according to the manufacturer’s instructions (Life Technologies). Twenty four hours after transfection, cells were used for ABE assays.
Construction of Flag-GR Wt or C/A mutant
Mouse GR cDNA was amplified by PCR using pSV2-GR plasmid, a generous gift of E. D. Sanchez (Dept Pharmacology, University of Toledo College of Medicine, Toledo OH 43614, US)58, and specific primers (Forward: 5′-GATCTATGGACTCCAAAGAATCC-3′, Reverse: 5′-CTAGATCATTTCTGATGAAACAGAAGC-3′) containing BgIII and Xbal restriction sites to facilitate cloning into the pFLAG-CMV2 vector (Sigma-Aldrich) leading to a N-terminal Flag form of GR. For Flag-GR C/A mutant, specific cysteine residue was mutated into alanine (Forward primer: 5′-GAAGAGTATCTCGCTATGAAAACCTTAC-3′; Reverse primer: 5′-CTGATTATTAATGAGCAGAGA-3′, mutated codons are underlined) by Quick change Site-Directed Mutagenesis Kit (Agilent Technologies). All plasmid sequences were verified by DNA sequencing.
Cell fractionation
Subcellular compartments of each cell line were separated according to specific protocols as previously described59. Cells were homogenized in a Dounce B homogenizer in buffer composed of 10 mM Hepes, 250 mM sucrose, 1 mM EDTA, pH 7.4, with an anti-protease cocktail (Sigma-Aldrich) at 4 °C and fractionated by differential centrifugation. The initial homogenate was first centrifuged at 850 × g for 5 minutes at 4 °C to obtain a pellet corresponding to nuclear (N) fraction that could also contain cell debris. Then, the post-nuclear supernatant was centrifuged at 10 000 × g for 10 minutes at 4 °C to remove mitochondria. The resulting supernatant was finally centrifuged at 400 000 × g for 6 minutes at 4 °C to obtain the soluble (supernatant, Cyto) and membrane (pellet, Mb) fractions. Each pellet was washed 3 times before resuspension in RIPA (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% Triton, protease inhibitors cocktail (Sigma-Aldrich).
Equal amounts (50 μg) of each fraction were analyzed by SDS–PAGE. The purity of each fraction was assessed by immunoblotting for specific protein markers: lamin A/C at approximately 70 kDa in the nuclear fraction, a 37 kDa band for glyceraldehyde 3-phosphate dehydrogenase (GAPDH, cytosol), and a band 45 kDa κ-opioid receptor (KOR, membranes). These analyses showed little contamination between compartment fractions indicating that the purity of the different fractions. Quantification of the membrane and the soluble pools of GR was analyzed and compared to the total extract (T) and expressed as a percentage of the total.
Protein extraction
After drug treatment, cells were scraped in RIPA (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% Triton, 10% Glycerol, protease inhibitors cocktail (Sigma-Aldrich) and phosphatase inhibitors (Sigma-Aldrich). Pituitaries were homogenized in RIPA with TissueLyser LT homogenizer (Qiagen, Les Ulis, France). After centrifugation (14,000 × g, 4 °C for 15 min) protein content was determined by bicinchoninic acid assay (Thermo Fisher Scientific).
Western Blot analysis
Equal protein amounts (50 μg) were resolved by either 7.5% or 10% SDS-PAGE and then transferred to nitrocellulose membrane (Whatman Schleicher & Schuell, Dassel, Germany). Blots were incubated for 1 h at room temperature (RT) in a blocking buffer (5% fat-free milk or 5% BSA in 0.2% Tween 20 phosphate-buffered saline (PBS-T) or in 0.2% Tween 20 Tris-buffered saline (TBS-T) depending on the nature of the first antibody, before an overnight incubation at 4 °C with primary monoclonal or polyclonal antibodies as indicated. After extensive washes with PBS-T or TBS-T, blots were incubated with an IRDye 800-conjugated affinity purified anti-rabbit IgG second antibody (Perbio Science, Brebières, France) and an IRDye 680- conjugated affinity purified anti-mouse IgG second antibody (Perbio Science) for 1 h at RT. After washes, proteins were visualized with an Odyssey-Fc apparatus (Li-Cor, Lincoln, NE, USA). Specific signals for different proteins were normalized by the infrared fluorescence of α-tubulin followed by total CaMKII or total Syn or total ERK or total Src signals as determined by densitometry using the Image Studio software (Li-Cor). All Western blots shown are representative of what was observed in at least three independent experiments, excepted in Supplementary Fig. S2 (2 independent experiments).
RT-qPCR
RT-qPCR analysis was undertaken using the Power SYBR Green PCR Master Mix (Life Technologies) with primers (Sgk-1 Forward: 5′-TCACTTCTCATTCCAGACCGC-3′, Reverse: 5′-ATAGCCCAAGGCACTGGCTA-3′; Lhb Forward: 5′-ATCACCTTCACCACCAGCAT-3′, Reverse: 5′ GACCCCCACAGTCAGAGCTA-3′; Cga Forward: 5′ GCTGTCATTCTGGTCATGCT-3′, Reverse: 5′-GAAGCAACAGCCCATACACT-3′; 36b4 Forward: 5′AGCGCGTCCTGGCATTGTCTGT-3′, Reverse: 5′-GGGCAGCAGTGGTGGCAGCAGC-3′) and a StepOne Real-Time PCR System (Life Technologies). Expression levels of Sgk-1, Lhb and Cga analyzed by qPCR were normalized relative to levels of 36b4 mRNA, Cga coding for the alpha subunit of LH and FSH.
ABE assay
LβT2 as well as HEK293 cells transfected with Flag-GR (GR) or Stathmin 1-GFP (St1) were lysed in presence of N-Ethylmaleimide (NEM). GR palmitoylation was assessed using the acyl biotin exchange (ABE) protocol as previously described35. The principle of this assay was to biotinylate palmitoylation sites after specific cleavage by hydroxylamide (HAM) on immunoprecipitated GR using either EZview Red anti-FLAG M2 agarose (Sigma-Aldrich) or anti-GR (M20) or GFP with anti-GFP associated with 50% slurry of protein G-coated sepharose beads (Roche Diagnostics). Once labeling sites was achieved, palmitoylation was analyzed by SDS-PAGE followed by Western blotting using Streptavidin-IRDye800CW conjugated (Rockland Immunochemicals, Gilbertsville, PA) as well as an anti-GR or anti-Flag or anti-GFP antibody. Streptavidin signals were normalized to the amount of Flag-GR protein present on the blot.
LH assay
Whole pituitaries were cultured in DMEM for 2 h at 37 °C for equilibration. The medium (200 μl) was replaced by fresh medium for 30 min at 37 °C and collected for LH assay (pre-stimulation). Medium containing ethanol (vehicle), GnRH (10−8 M), Dex (10−7 M) or both hormones were used to treat pituitaries for 1 h at 37 °C. Culture supernatants were then collected to measure LH content. LH concentration in culture media was determined using an ELISA method as previously described60 with reagents supplied by Dr Parlow (NHPP, Harbor-UCLA, CA, USA). Briefly, micro-titration plates (High binding, Greiner Bio-one) were coated overnight at 4 C with 10 ng of purified rat LH-I-10 diluted in carbonate buffer. Rat LH-RP3, used as standard, and media were incubated with anti-rat LH-S11 (dilution 1:4,000 overnight at 4 °C). Plates were then rinsed with PBS containing 1% BSA and 0.1% Tween20 for 1 h at RT and washed with PBS-0.1% Tween20 before addition of standards and samples for competition binding (2 h at 4 °C). After removal of unbound material, phosphatase alkaline-labeled secondary antibody (dilution 1: 2,000, Thermo Scientific, France) was added for 1 h at 4 °C and phosphatase alkaline activity was revealed with SigmaFast pNPP reagent. The minimum detectable LH concentration was 0.2 ng/ml and the inter-assay coefficient of variation were <10%. LH release after hormonal treatment was quantified in comparison with LH amount measured in basal condition (pre-stimulation).
Statistical Analysis
Means ± SEM of n experiments of measurements were analyzed for significant differences by one-way ANOVA using Prism 6 (GraphPad) software followed by appropriated posttests. Means are assumed to be significantly different if p < 0.05.
Electronic supplementary Material
Acknowledgements
We thank Drs S. Viengchareun for his help in performing luciferase assays and J. Fagart for discussion on GR structure. We thank N. Ba from Plateau de morphologie at Institut Biomedical de Bicêtre for preparing mouse pituitary slices and Dr C. Adam for the histological interpretations. We thank C. Denoyelle and F. Petit for their technical help as well as J.A. Girault who allowed us to use his ultracentrifuge (Beckman) at Institut du Fer à Moulin (Paris). This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and the University Paris Sud (Attractivity 2015 grant). M.A. is supported by a doctoral fellowship from the Ministère de l’Enseignement Supérieur et de la Recherche (MESR).
Author Contributions
M.A. designed and performed the experiments, analyzed primary data, aggregated data and contribute to the writing of the manuscript; V.S. performed L.H. assays and analyzed primary data; V.B. contributed to biochemical experiments; N.B. contributed to ex vivo experiments and to the conception of the project; J.C.-T. contributed to experimental design and analyzed the data; M.L. contributed to experimental design and analyzed the data; S.C. conceived and supervised the project, designed experiments, analyzed aggregated data and wrote the manuscript. All authors reviewed this manuscript and approved the submission.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01777-2
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Allsworth JE, et al. The influence of stress on the menstrual cycle among newly incarcerated women. Women’s health issues: official publication of the Jacobs Institute of Women’s Health. 2007;17:202–209. doi: 10.1016/j.whi.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo E, et al. Corticosterone Blocks Ovarian Cyclicity and the LH Surge via Decreased Kisspeptin Neuron Activation in Female Mice. Endocrinology. 2016;157:1187–1199. doi: 10.1210/en.2015-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whirledge S, Cidlowski JA. A role for glucocorticoids in stress-impaired reproduction: beyond the hypothalamus and pituitary. Endocrinology. 2013;154:4450–4468. doi: 10.1210/en.2013-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lynch CD, Sundaram R, Maisog JM, Sweeney AM, Buck Louis GM. Preconception stress increases the risk of infertility: results from a couple-based prospective cohort study–the LIFE study. Human reproduction. 2014;29:1067–1075. doi: 10.1093/humrep/deu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobson H, Fergani C, Routly JE, Smith RF. Effects of stress on reproduction in ewes. Anim Reprod Sci. 2012;130:135–140. doi: 10.1016/j.anireprosci.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Oakley AE, et al. Cortisol reduces gonadotropin-releasing hormone pulse frequency in follicular phase ewes: influence of ovarian steroids. Endocrinology. 2009;150:341–349. doi: 10.1210/en.2008-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breen KM, et al. Insight into the neuroendocrine site and cellular mechanism by which cortisol suppresses pituitary responsiveness to gonadotropin-releasing hormone. Endocrinology. 2008;149:767–773. doi: 10.1210/en.2007-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends in pharmacological sciences. 2013;34:518–530. doi: 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. The Journal of allergy and clinical immunology. 2013;132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breen KM, Mellon PL. Influence of stress-induced intermediates on gonadotropin gene expression in gonadotrope cells. Molecular and cellular endocrinology. 2014;385:71–77. doi: 10.1016/j.mce.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haller J, Mikics E, Makara GB. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Frontiers in neuroendocrinology. 2008;29:273–291. doi: 10.1016/j.yfrne.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Teng Z, Zhang M, Zhao M, Zhang W. Glucocorticoid exerts its non-genomic effect on IPSC by activation of a phospholipase C-dependent pathway in prefrontal cortex of rats. The Journal of physiology. 2013;591:3341–3353. doi: 10.1113/jphysiol.2013.254961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayroldi E, et al. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:4805–4820. doi: 10.1096/fj.12-216382. [DOI] [PubMed] [Google Scholar]
- 14.Samarasinghe RA, et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci USA. 2011;108:16657–16662. doi: 10.1073/pnas.1102821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solito E, et al. Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology. 2003;144:1164–1174. doi: 10.1210/en.2002-220592. [DOI] [PubMed] [Google Scholar]
- 16.Deng Q, et al. Rapid Glucocorticoid Feedback Inhibition of ACTH Secretion Involves Ligand-Dependent Membrane Association of Glucocorticoid Receptors. Endocrinology. 2015;156:3215–3227. doi: 10.1210/EN.2015-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchetti MC, Di Marco B, Cifone G, Migliorati G, Riccardi C. Dexamethasone-induced apoptosis of thymocytes: role of glucocorticoid receptor-associated Src kinase and caspase-8 activation. Blood. 2003;101:585–593. doi: 10.1182/blood-2002-06-1779. [DOI] [PubMed] [Google Scholar]
- 18.Urbach V, Walsh DE, Mainprice B, Bousquet J, Harvey BJ. Rapid non-genomic inhibition of ATP-induced Cl- secretion by dexamethasone in human bronchial epithelium. The Journal of physiology. 2002;545:869–878. doi: 10.1113/jphysiol.2002.028183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartholome B, et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18:70–80. doi: 10.1096/fj.03-0328com. [DOI] [PubMed] [Google Scholar]
- 20.Stojadinovic O, Sawaya A, Pastar I, Tomic-Canic M. Glucocorticoid receptor localizes to adherens junctions at the plasma membrane of keratinocytes. PloS one. 2013;8:e63453. doi: 10.1371/journal.pone.0063453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komatsuzaki Y, et al. Rapid spinogenesis of pyramidal neurons induced by activation of glucocorticoid receptors in adult male rat hippocampus. Biochemical and biophysical research communications. 2005;335:1002–1007. doi: 10.1016/j.bbrc.2005.07.173. [DOI] [PubMed] [Google Scholar]
- 22.Spies CM, et al. Membrane glucocorticoid receptors are down regulated by glucocorticoids in patients with systemic lupus erythematosus and use a caveolin-1-independent expression pathway. Annals of the rheumatic diseases. 2006;65:1139–1146. doi: 10.1136/ard.2005.048272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wehmeyer L, Du Toit A, Lang DM, Hapgood JP. Lipid raft- and protein kinase C-mediated synergism between glucocorticoid- and gonadotropin-releasing hormone signaling results in decreased cell proliferation. J Biol Chem. 2014;289:10235–10251. doi: 10.1074/jbc.M113.544742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maier C, et al. G-protein-coupled glucocorticoid receptors on the pituitary cell membrane. J Cell Sci. 2005;118:3353–3361. doi: 10.1242/jcs.02462. [DOI] [PubMed] [Google Scholar]
- 25.Nahar J, Rainville JR, Dohanich GP, Tasker JG. Further evidence for a membrane receptor that binds glucocorticoids in the rodent hypothalamus. Steroids. 2016 doi: 10.1016/j.steroids.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strehl C, et al. Origin and functional activity of the membrane-bound glucocorticoid receptor. Arthritis and rheumatism. 2011;63:3779–3788. doi: 10.1002/art.30637. [DOI] [PubMed] [Google Scholar]
- 27.Acconcia F, et al. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol Biol Cell. 2005;16:231–237. doi: 10.1091/mbc.E04-07-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedram A, et al. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282:22278–22288. doi: 10.1074/jbc.M611877200. [DOI] [PubMed] [Google Scholar]
- 29.Jain S, Li Y, Kumar A, Sehgal PB. Transcriptional signaling from membrane raft-associated glucocorticoid receptor. Biochemical and biophysical research communications. 2005;336:3–8. doi: 10.1016/j.bbrc.2005.08.057. [DOI] [PubMed] [Google Scholar]
- 30.Matthews L, et al. Caveolin mediates rapid glucocorticoid effects and couples glucocorticoid action to the antiproliferative program. Molecular endocrinology. 2008;22:1320–1330. doi: 10.1210/me.2007-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peffer ME, et al. Caveolin-1 regulates genomic action of the glucocorticoid receptor in neural stem cells. Mol Cell Biol. 2014;34:2611–2623. doi: 10.1128/MCB.01121-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vernocchi S, et al. Membrane glucocorticoid receptor activation induces proteomic changes aligning with classical glucocorticoid effects. Molecular & cellular proteomics: MCP. 2013;12:1764–1779. doi: 10.1074/mcp.M112.022947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng Q, Waxse B, Riquelme D, Zhang J, Aguilera G. Helix 8 of the ligand binding domain of the glucocorticoid receptor (GR) is essential for ligand binding. Molecular and cellular endocrinology. 2015;408:23–32. doi: 10.1016/j.mce.2015.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochemical and biophysical research communications. 2004;316:878–883. doi: 10.1016/j.bbrc.2004.02.129. [DOI] [PubMed] [Google Scholar]
- 35.Brigidi, G. S. & Bamji, S. X. Detection of protein palmitoylation in cultured hippocampal neurons by immunoprecipitation and acyl-biotin exchange (ABE). Journal of visualized experiments: JoVE, doi:10.3791/50031 (2013). [DOI] [PMC free article] [PubMed]
- 36.Chauvin S, Sobel A. Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Progress in neurobiology. 2015;126:1–18. doi: 10.1016/j.pneurobio.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Haisenleder DJ, Burger LL, Aylor KW, Dalkin AC, Marshall JC. Gonadotropin-releasing hormone stimulation of gonadotropin subunit transcription: evidence for the involvement of calcium/calmodulin-dependent kinase II (Ca/CAMK II) activation in rat pituitaries. Endocrinology. 2003;144:2768–2774. doi: 10.1210/en.2002-0168. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto H, Matsumoto K, Araki E, Miyamoto E. New aspects of neurotransmitter release and exocytosis: involvement of Ca2+/calmodulin-dependent phosphorylation of synapsin I in insulin exocytosis. Journal of pharmacological sciences. 2003;93:30–34. doi: 10.1254/jphs.93.30. [DOI] [PubMed] [Google Scholar]
- 39.Breen KM, et al. Stress levels of glucocorticoids inhibit LHbeta-subunit gene expression in gonadotrope cells. Molecular endocrinology. 2012;26:1716–1731. doi: 10.1210/me.2011-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breen KM, et al. Does cortisol acting via the type II glucocorticoid receptor mediate suppression of pulsatile luteinizing hormone secretion in response to psychosocial stress? Endocrinology. 2007;148:1882–1890. doi: 10.1210/en.2006-0973. [DOI] [PubMed] [Google Scholar]
- 41.Collu R, Tache Y, Ducharme JR. Hormonal modifications induced by chronic stress in rats. Journal of steroid biochemistry. 1979;11:989–1000. doi: 10.1016/0022-4731(79)90042-6. [DOI] [PubMed] [Google Scholar]
- 42.Vernocchi S, et al. Membrane Glucocorticoid Receptor activation induces proteomic changes aligning withclassical glucocorticoid effects. Molecular & cellular proteomics: MCP. 2013 doi: 10.1074/mcp.M112.022947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutierrez-Mecinas M, et al. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc Natl Acad Sci USA. 2011;108:13806–13811. doi: 10.1073/pnas.1104383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greaves J, Chamberlain LH. DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends in biochemical sciences. 2011;36:245–253. doi: 10.1016/j.tibs.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Morrill GA, Kostellow AB, Gupta RK. Transmembrane helices in “classical” nuclear reproductive steroid receptors: a perspective. Nuclear receptor signaling. 2015;13:e003. doi: 10.1621/nrs.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duran-Pasten ML, Fiordelisio T. GnRH-Induced Ca Signaling Patterns and Gonadotropin Secretion in Pituitary Gonadotrophs. Functional Adaptations to Both Ordinary and Extraordinary Physiological Demands. Frontiers in endocrinology. 2013;4:127. doi: 10.3389/fendo.2013.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bellavance MA, Rivest S. The HPA - Immune Axis and the Immunomodulatory Actions of Glucocorticoids in the Brain. Front Immunol. 2014;5:136. doi: 10.3389/fimmu.2014.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kotitschke A, Sadie-Van Gijsen H, Avenant C, Fernandes S, Hapgood JP. Genomic and nongenomic cross talk between the gonadotropin-releasing hormone receptor and glucocorticoid receptor signaling pathways. Molecular endocrinology. 2009;23:1726–1745. doi: 10.1210/me.2008-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loechner KJ, Knox RJ, McLaughlin JT, Dunlap K. Dexamethasone-mediated inhibition of calcium transients and ACTH release in a pituitary cell line (AtT-20) Steroids. 1999;64:404–412. doi: 10.1016/S0039-128X(98)00121-4. [DOI] [PubMed] [Google Scholar]
- 50.Castellino F, Heuser J, Marchetti S, Bruno B, Luini A. Glucocorticoid stabilization of actin filaments: a possible mechanism for inhibition of corticotropin release. Proc Natl Acad Sci USA. 1992;89:3775–3779. doi: 10.1073/pnas.89.9.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Easom RA. CaM kinase II: a protein kinase with extraordinary talents germane to insulin exocytosis. Diabetes. 1999;48:675–684. doi: 10.2337/diabetes.48.4.675. [DOI] [PubMed] [Google Scholar]
- 52.Ferreira A, Rapoport M. The synapsins: beyond the regulation of neurotransmitter release. Cellular and molecular life sciences: CMLS. 2002;59:589–595. doi: 10.1007/s00018-002-8451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Revest JM, et al. The enhancement of stress-related memory by glucocorticoids depends on synapsin-Ia/Ib. Molecular psychiatry. 2010;15(1125):1140–1151. doi: 10.1038/mp.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turgeon JL, Kimura Y, Waring DW, Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Molecular endocrinology. 1996;10:439–450. doi: 10.1210/mend.10.4.8721988. [DOI] [PubMed] [Google Scholar]
- 55.Mellon PL, et al. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-E. [DOI] [PubMed] [Google Scholar]
- 56.Viengchareun S, et al. Osmotic stress regulates mineralocorticoid receptor expression in a novel aldosterone-sensitive cortical collecting duct cell line. Molecular endocrinology. 2009;23:1948–1962. doi: 10.1210/me.2009-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Romieu R, et al. Critical stages of tumor growth regulation in transgenic mice harboring a hepatocellular carcinoma revealed by distinct patterns of tumor necrosis factor-alpha and transforming growth factor-beta mRNA production. International immunology. 1997;9:1405–1413. doi: 10.1093/intimm/9.10.1405. [DOI] [PubMed] [Google Scholar]
- 58.Danielsen M, Northrop JP, Ringold GM. The mouse glucocorticoid receptor: mapping of functional domains by cloning, sequencing and expression of wild-type and mutant receptor proteins. EMBO J. 1986;5:2513–2522. doi: 10.1002/j.1460-2075.1986.tb04529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levy AD, et al. Subcellular Golgi localization of stathmin family proteins is promoted by a specific set of DHHC palmitoyl transferases. Mol Biol Cell. 2011;22:1930–1942. doi: 10.1091/mbc.E10-10-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garrel G, et al. Unsaturated fatty acids stimulate LH secretion via novel PKCepsilon and -theta in gonadotrope cells and inhibit GnRH-induced LH release. Endocrinology. 2011;152:3905–3916. doi: 10.1210/en.2011-1167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.