

Abstract
Neisseria meningitidis infections are a major issue for global health. The invasive MenC ST-103 clonal complex (CC103) has been the most prevalent in meningococcal outbreaks in Brazil, occurring also in several countries worldwide. Here we have analysed the population structure and accessory genome of MenC CC103 strains from a global perspective. An in-depth phylogenomic analysis revealed a lineage of N. meningitidis causing meningitis in Brazil and the United Kingdom. This lineage was also characterized as harbouring a particular accessory genome composed of CRISPR/Cas and restriction modification systems. This lineage was also characterized by a genomic island resembling an integrative and conjugative element. This island carried genes potentially associated with virulence and fitness. We propose this accessory gene repertoire could be contributing to the spatial-temporal persistence of the invasive MenC CC103 lineage.
Introduction
Neisseria meningitidis is part of the commensal microbiota in the upper human respiratory tract; however, it can occasionally invade the bloodstream causing meningitis and/or septicemia, which are associated with significant morbidity and mortality worldwide. N. meningitidis are classified into 12 serogroups based on antigenic properties of polysaccharide capsules. Most invasive meningococci belong to the A, B, C, W, and Y serogroups1. The control of meningococcal disease has been approached worldwide with vaccination against the A, C, W, and Y serogroups and, more recently, against serogroup B strains2.
In the last decade, invasive meningococci from serogroup C (MenC) has increased in Brazil, with the ST-103 clonal complex (CC103) corresponding to over 74% of the disease causing strains characterized in 20143–5. Due to this epidemiological scenario, the C serogroup conjugate vaccine was introduced in Brazil in 20106. Despite this, however, cases and outbreaks caused by MenC CC103 have continued to be reported in Brazil7.
The worldwide epidemiology of MenC has been dynamic considering the spatial-temporal distribution of distinct clonal complexes. In Europe, this serogroup is the second most widespread, and MenC CC11 has been responsible for outbreaks in several countries, even after the introduction of the MenC vaccination8.
Although horizontal gene transfers between Neisseria species and other bacteria have a pivotal role in meningococcal evolution9, the Neisseria population structure is mainly represented by lineages, which may be determined by the expansion of strains belonging to a clonal complex and/or presence of restriction-modification (RMS) and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) systems10, 11.
Comparisons of meningococcal genomes have allowed deep phylogenetic reconstructions and identification of accessory genomes that may be associated with invasive lineages; these accessory genomes represent a repertoire of genetic elements such as prophages, plasmids, and genomic islands12, 13. The Gonococcal genomic island (GGI) is a mobile element found in N. gonorrhoeae (80%) and in some N. meningitidis (17%) that is characterized by the presence of a particular Type 4 Secretion System (T4SS), which is supposed to play a role in Neisseria virulence and pathogenesis14–17.
The aim of this study was to determine the population structure and the accessory genome of invasive MenC CC103 genomes from Brazil under a global epidemiological perspective. We found that Brazilian MenC CC103 strains belong to a lineage characterized by CRISPR spacer sequences and a set of restriction modification systems. This lineage also harbour a genomic island resembling an integrative and conjugative-like element carrying genes predicted to code for virulence- and fitness-associated functions.
Results and Discussion
Genome statistics and phylogenomic analysis
The genome sequences of 24 invasive MenC strains from Brazil, belonging to the ST-103 clonal complex (CC103), were determined in this study. The genomes have been assembled into between 70 to 183 contigs, totaling, on average, ~2.17 Mb with a G + C content of ~51.5%; they are predicted to encode ~2,200 genes with 4 rRNA operons and 51 tRNA genes. A detailed statistics of these genomes is summarized in Table S1. The genomes were classified by multilocus sequence typing (MLST) as belonging to the CC103 (Table 1). Studies have shown that MenC of CC103 has caused outbreaks in Brazil in the last few years18.
Table 1.
MenC strains from Brazil sequenced in this study.
| Strain | State/Country1 | Year | porA | PorA VR1 | PorA VR22 | porB | fetA | nadA3 | nhba | fHbp | ST | Clonal Complex |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nm56 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F3-9 | 96 | 24 | 25 (25) | 3780 | ST-103 |
| Nm287 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F5-7 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| Nm288 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F5-7 | 161 | 24 | 25 (25) | 3780 | ST-103 |
| Nm292 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F5-7 | 161 | 24 | 25 (25) | 3780 | ST-103 |
| Nm612 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F5-7 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| Nm638 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F5-7 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| P3478 | BA/Brazil | 2009 | 10-1 | 5-1 | 10-1 | 2-23 | F3-9 | 85 | 24 | 25 (25) | 8436 | ST-103 |
| NmCRJ2 | RJ/Brazil | 2012 | 28 | 22 | 2-48 | 2-23 | F3-9 | 96 | 24 | 25 (25) | 3779 | ST-103 |
| Nm94 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F3-9 | 96 | 24 | 446 (377) | 3780 | ST-103 |
| Nm322 | MG/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F3-9 | 161 | 24 | 446 (377) | 3780 | ST-103 |
| P3966 | PE/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-253 | F3-9 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| P3978 | PE/Brazil | 2011 | 28 (22) | 22 | 2-48 | 2-23 | F3-9 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| P4005 | PE/Brazil | 2012 | 28 | 22 | 2-48 | 2-23* | F3-9 | 96 | 24 | 25 (25) | 3780 | ST-103 |
| P4144 | PE/Brazil | 2012 | 28 | 22 | 2-48 | 2-23 | F3-9 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| P3558 | PE/Brazil | 2010 | 28 | 22 | 2-48 | 2-23 | F3-9 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| P3965 | PE/Brazil | 2011 | 28 | 22 | 2-48 | 2-23 | F3-9 | 96 | 24 | 25 (25) | ND | - |
| P4077 | RJ/Brazil | 2012 | 28 | 22 | 2-48 | 2-23 | F3-9 | 85 | 24 | 25 (25) | 5122 | ST-103 |
| P4431 | NI/Brazil | 2014 | 28 | 22 | 2-48 | 2-23 | F3-9 | 161 | 24 | 25 (25) | 3780 | ST-103 |
| P4615 | NI/Brazil | 2015 | 714 | 21 | 16-36 | 3-106 | F5-8 | 85 | 24 | 490 (420) | 3327 | ST-865 |
| P4534 | NI/Brazil | 2015 | 28 | 22 | 2-48 | 2-23 | - | 96 | 24 | 25 (25) | 3779 | ST-103 |
| P4480 | NI/Brazil | 2015 | 28 | 22 | 2-48 | 2-23 | F3-9 | 85 | 24 | 25 (25) | 3780 | ST-103 |
| P4950 | NI/Brazil | 2015 | 28 | 22 | 2-48 | 2-23 | - | 85 | 24 | 25 (25) | 3779 | ST-103 |
| P4995 | NI/Brazil | 2015 | 28 | 22 | 2-48 | 2-23 | F3-9 | 85 | 24 | 25 (-) | 3780 | ST-103 |
| P4464 | NI/Brazil | 2014 | 28 | 22 | 2-48 | 2-23 | F3-9 | 85 | 24 | 789 (-) | 8730 | - |
The antigen-encoding gene profiles, sequence type and clonal complex designations were defined based on Neisseria pubMLST database (http://pubmlst.org/neisseria/). 1MG, Minas Gerais; BA, Bahia; RJ, Rio de Janeiro; PE, Pernambuco; NI, not informed. 2The 2-48 profile was defined by the closest match locus 3 nadA locus interrupted by insertion sequence (IS) element; the profile was defined by the closest match locus; -, not defined by PubMLST search.
An initial phylogenomic analysis was performed with 645 MenC genomes retrieved from the Bacterial Isolate Genome Sequence Database (BIGSdb) (see Fig. S1). To gain a better understanding of the evolution and population structure of CC103, 96 representative genomes were selected from this preliminary phylogeny for a more detailed analysis. Of these 96 MenC strain genomes, 24 were from Brazil (Fig. 1, Table S2). The great majority of CC103 genomes analysed belong to a cluster of invasive N. meningitidis strains from serogroups A (n = 1), Y (n = 1), B (n = 9), C (n = 36), Z (n = 6), and non-groupable (n = 5) strains identified in the last three decades in Brazil and European countries. The presence of different serogroups in this cluster indicated the occurrence of capsular switching events. All MenC CC103 Brazilian strains were placed in a subcluster, which allowed them to be classified to a defined lineage (Fig. 1).
Figure 1.
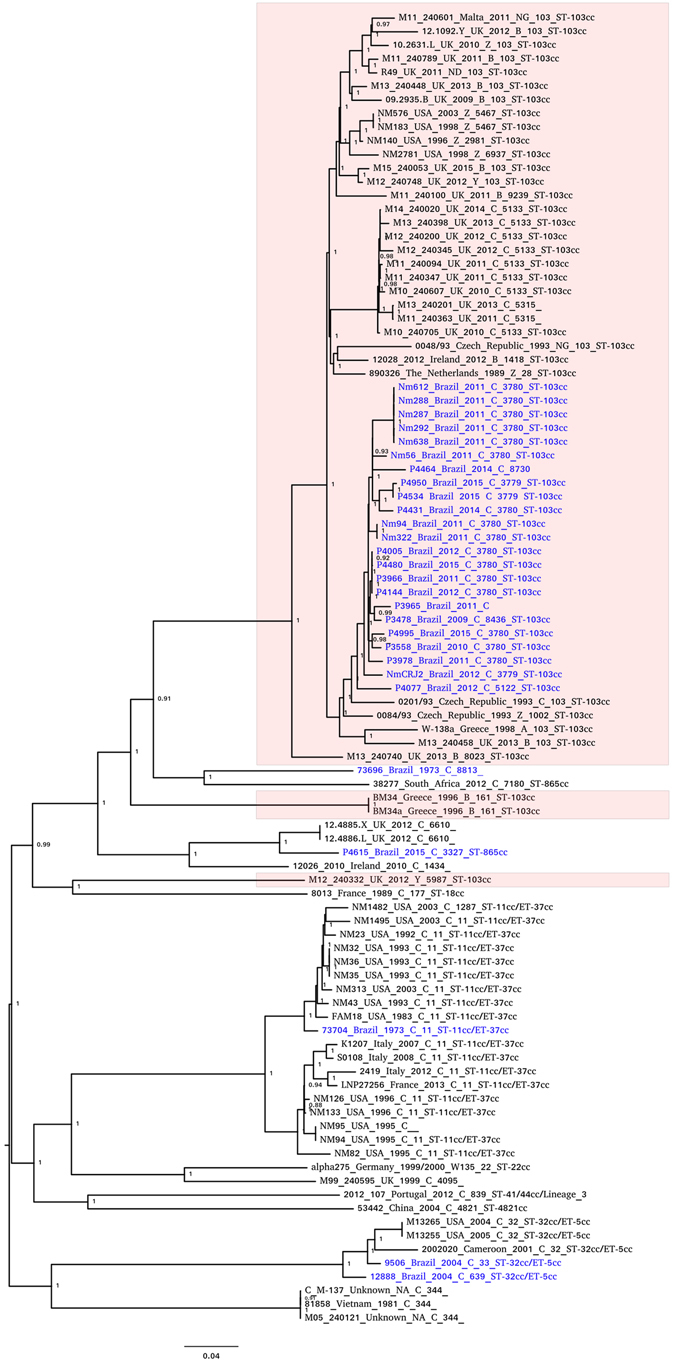
Genome phylogeny overview of N. meningitidis CC103 complex and close relatives. The phylogeny was determined considering the core genome alignment and maximum likelihood reconstruction phylogeny by using Parsnp program implemented in Harvest suite55. Highlighted boxes indicate the genomes assigned as CC103 complex. Brazilian MenC strains are depicted in blue.
The accessory genome of MenC CC103 - Prophage and CRISPR/Cas identification
Comparative genomic analyses of the MenC CC103 from Brazil and the FAM18 reference genomes revealed the presence of four genomic islands (NmGI-1 to 4) and ten prophages (Nf-C1, Nf-C2, IHT-E, Nf-C3, Nf-C4, Nmpp-1, phast-1, phast-2, Nmpp-2, and Nmpp3) (Fig. 2). All four genomic islands and 4/10 prophage sequences (Nmpp-1, phast-1, Nmpp-2, and Nmpp3) were found in the MenC CC103 genomes from Brazil while all these genomic regions were present in FAM18, except NmGI-4. Although the prophages Nf-C1, Nf-C2, IHT-E, Nf-C3, Nf-C4 and phast-2 identified in FAM18 reference genome had already been associated with different invasive serogroups19–21, none of them were present in the Brazilian invasive MenC CC103 lineage (Fig. 2, Fig. S3), corroborating the polygenic character of meningococcal virulence13.
Figure 2.

Circular representation of the genomes from N. meningitidis serogroup C (MenC) strains isolated from Brazil. The draft genomes were aligned against FAM18 reference genome. The inner circle (histogram) shows G + C content variations throughout the genome. The gray circle represents the compared chromosomal regions and the black region placed in the terminus region (,200 bp) represents regions not allocated as part of the chromosomal genome. Each genome is represented by a colored circle. The red circles represent Brazilian MenC genomes from CC103 clonal complex harbouring the ICENmCC103. The variable regions such as prophages and genomic islands, including the NmGI-4 (ICENmCC103 element), are highlighted.
Bacteria possess CRISPR/Cas systems that can restrict horizontal gene transfer, such as phage insertion by transduction22. We identified a CRISPR/Cas system in the MenC CC103 from Brazil (Table S1) that was absent in the FAM18 genome. This identified system consisted of Cas proteins and arrays of a 36 nucleotide identical direct repeat (ATTGTAGCACTGCGAAATGAGAATGGGAGCTACAAC), separated by variable spacer regions. The number of direct repeats was seen to be variable among these genomes (vary from 12 to 28) as was the sequence composition of the spacer regions, which ranged from 28 to 42 bp in length. Interestingly, some of these spacer sequences corresponded to 5/6 prophages identified in FAM18 that were missing in the Brazilian MenC CC103 lineage (Nf-C1, Nf-C2, Nf-C3, phast-2 and Nf-C4). This finding suggests that this CRISPR/Cas system could limit the acquisition of these phages by the Brazilian MenC CC103 lineage (Fig. 2), and could be contributing to its population structure delineation.
General features of the NmGI-4 genomic island
The NmGI-1-3 was shared by all genomes included in this comparative analysis. Conversely, the NmGI-4 was specific for MenC CC103 lineage from Brazil (Fig. 2). This genomic island is ~52.3 kb in length, has a G+C content of 44% and it was flanked by identical 23 bp direct repeat sequences. It was observed that the NmGI-4 had several features in common with the Gonococcal Genomic Islands (GGIs), which are mobile elements characterized by the presence of a particular T4SS23. For example, the NmGI-4 shared synteny and gene content with GGIs and they were inserted in the same chromosomal context (dif site) (Fig. 3, Fig. S2).
Figure 3.

Genetic organization of the GGI from N. gonorrhoeae (Ng) compared with the N. meningitidis GGI-like elements characterized in this study. Schematic comparison of the GGI versions of Ng, Nm56 (ICENmCC103), Nm576 and Nm322 genomes. Genes are presented as arrows, with the arrowhead indicating the direction of the transcription. Black arrows indicate the chromosomal genes flanking the GGI-like elements, which are represented by colored arrows. Areas between the GGIs shaded as horizontal blocks indicate similar regions based on sequence identity.
Different GGI configurations have already been identified in N. meningitidis 12, 23, 24 and, in fact, the comparative genomic analysis performed here revealed the presence of different GGI-like versions among the MenC CC103 strains. The NmGI-4 (complete version recovered from Nm56 strain) shared 97% coverage and 99% nucleotide identity with the GGI-like T4SS characterized in N. meningitidis serogroup W alpha-275 strain25. Two GGI variants were also identified (recovered from Nm576 and Nm322), and they differ from the NmGI-4 and from the types previously described in N. meningitidis 25 by the presence of large deletions (Fig. 3).
The unique genetic repertoire of NmGI-4 genomic island
NmGI-4 harboured a set of genes including some that could have an adaptive role in strains carrying it such as umuC, umuD, rdgC, and pinR (Fig. 3).
Meningococci residing in the nasopharynx are exposed to oxidative stress that can cause DNA damage, reflecting the need of DNA repair mechanisms. However, this species lacks the SOS response system such as the UmuDC-dependent mutagenic repair26, 27. Interestingly, NmGI-4 carried the umuDC system, which represents an additional DNA repair pathway/mechanism and could be associated with fitness innovation28. The rdgC gene found in NmGI-4 is a homolog of the DNA recombination-dependent growth factor C (RdgC), whose deduced protein shared 70–90% amino acid identity with other RdgC alleles present in the chromosome of several Neisseria species. RdgC modulates RecA activity, being involved in several cell functions such as pilin antigenic variation, natural DNA transformation, and SOS DNA repair induction29–31. The presence of rdgC together with the umuDC system in the context of NmGI-4 suggests that the former may control the SOS response evoked by UmuDC, contributing to the adaptive fitness of MenC CC103. Finally, the pinR gene encodes a site-specific resolvase from the Serine Recombinase Superfamily, which is involved in excision/insertion of entire genomic islands. Therefore, contrasting with the GGIs so far described in N. gonorrhoeae, which lack any site-specific recombinase gene and depend on an in trans activity of the XerCD recombinase to be excised/integrated32, the presence of pinR in the NmGI-4 potentially provide the ability of self-excision/insertion through in cis recombination events.
Integrative and conjugative elements (ICEs) are defined by the presence of genes involved in integration/excision, conjugation and replication functions33. Therefore, in spite of the similarities between NmGI-4 and GGIs (Fig. 3), the former contained a serine recombinase pinR gene (integration/excision module), the entire set of T4SS genes (conjugation/secretion module), the parA gene and the transfer origin oriT (replication module) that, altogether, would characterize the NmGI-4 as an ICE (Fig. 3)33. Based on these features, we named this NmGI-4 version (complete version found in Nm56 strain) as ICENmCC103 (relative to the ST-103 clonal complex).
Together with the umuDC genes, potentially involved in adaptive traits, several genes that could contribute to the success of a pathogen in colonizing and infecting its host were found in ICENmCC103, such as the eppA gene and toxin-antitoxin (TA) systems. The eppA encodes a zinc metallopeptidase/endopeptidase from the M23 family. Proteins from this enzyme class may act in a variety of processes, including pilus DNA secretion process, cleavage of the extracellular matrix fibronectin from the host’s tissue and pilus biogenesis34–36.
TA modules comprise toxin proteins, which disrupt the cell’s own molecular processes, and cognate antitoxins that block this poisonous activity. These systems can be found in chromosomes and within mobile genetic elements including plasmids, prophages, transposons or superintegrons37. TA systems are activated in response to environmental stress, and confer advantageous bacterial behaviours such as biofilm formation, phase variation, virulence regulation, bacterial persistence, genetic competence and plasmid maintenance during cell division, which, in turn, may contribute to the dissemination of antibiotic resistance and virulence determinants harboured by such mobile genetic elements37, 38. TA systems are part of N. meningitidis genetic background and vary among lineages39. Using TA-Finder software (http://202.120.12.133/TAfinder/), we verified that MenC CC103 and FAM18 share TA systems from the RelE, Xre, MazF, and AbrB families while the PIN-PHD system was restricted to the FAM18 genome. Moreover, two additional TA systems, prlF-yhaV 40 and yecA-yeb, were identified embedded in ICENmCC103 (Table S3). Therefore, the presence of two other TA systems in ICENmCC103 suggests that, besides their contribution to ICE maintenance, they can provide an adaptive increment to the CC103 lineage.
Genomic epidemiology of ICENmCC103
We performed an analysis focusing on the prevalence and worldwide distribution of ICENmCC103 using this element as a query to search the Neisseria sequence repository at the PubMLST database. A total of 247 meningococci genomes harbouring the ICENmCC103 were identified (Fig. 4). The highest ICE prevalence was in serogroup W (n = 98), followed by B (n = 55), C (n = 19), and Y (n = 16) with a bias to genomes from European countries, particularly the United Kingdom (Fig. 4). A phylogenomic analysis using 96 genomes from CC103 and closely related clonal complexes revealed that most of them belong to a monophyletic cluster and carried ICENmCC103 (Fig. 4), showing a global association of ICENmCC103 with CC103 strains. This cluster was mainly composed of genomes from the United Kingdom and Brazil recovered from meningitis cases between 1973 and 2015. Interestingly, the presence of other serogroups (A, B, W and Z), besides MenC, in this CC103 cluster indicates the occurrence of capsular switching events involving strains from this clonal complex.
Figure 4.

ICENmCC103 geographical distribution and its abundance among N. meningitidis genomes from distinct serogroups. This figure has been created in R51 using the Rworldmap package52. The legend on the top left side shows the number of genomes positive for ICENmCC103 element relative to the total number of genomes of each serogroup. On the top right the prevalence of this element in genomes is shown for different serogroups (different colored bar sections) and year of isolation.
Two other islands related to ICENmCC103 were also identified: one characterized by a 37 kb deletion, was found in the Brazilian Nm94 and Nm322 genomes (Fig. 3), and the other, characterized by a 20 kb deletion, was found in 26 genomes mainly derived from the United Kingdom strains (Fig. 5).
Figure 5.

Phylogeny of N. meningitidis from CC103 and close relatives associated with a heatmap of the ICENmCC103 gene content. The genes listed in the top correspond to ICENmCC103 genes. The gene content is represented as presence (red) or absence (white). Most of CC103 genomes, including genomes from Brazil, belong to a monophyletic cluster and carried the ICENmCC103 element. All genomes from Brazil are shaded in blue.
Restriction modification systems (RMS) in MenC CC103
In order to determine if the CC103 lineage is characterized by a specific combination of RMS, we applied a hierarchical clustering algorithm using the presence/absence of genes from the REBASE database. In general, the clusters defined by RMS patterns corresponded to those revealed by the phylogenomic analysis (Fig. 6). The CC103 lineage was characterized by a specific RMS pattern: M.NmeAI, M.Nme18I, NgoAX, M.NgoAX, NgoAV, M.Zmoll, M.NgoAV, M.Zmo29192I, NflHI, NmeAIII, NmeBL869I, NgoAVI, and NmeAII. A previous study demonstrated that the population structure of the CC41/44, PC32/69, and PC8/11 phyloclades is shaped by specific RMS patterns, and the authors hypothesized that this could be a global phenomenon in populations of N. meningitidis 10. Our results showed that a lineage from another clonal complex (CC103) is also delineated by these systems. Therefore, our findings are adding evidences to the role of RMS in maintaining the population structure of N. meninigitidis.
Figure 6.
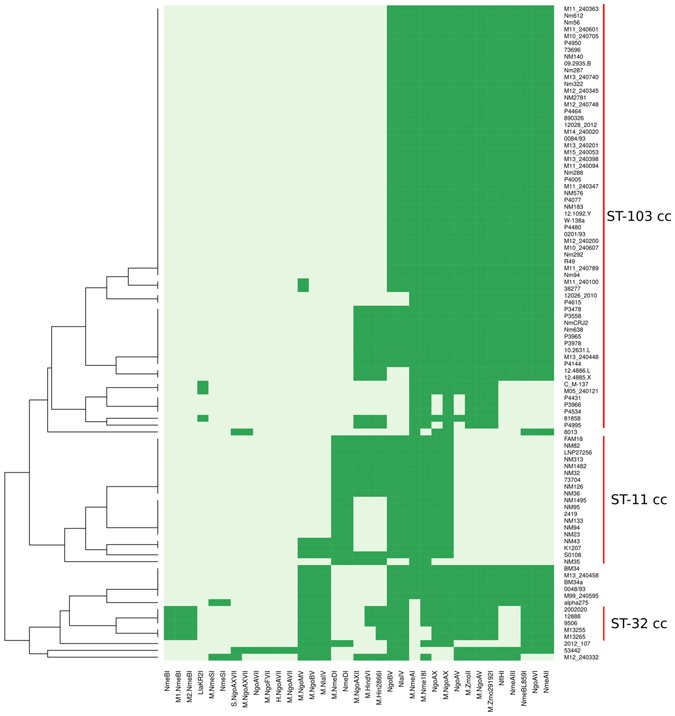
Heatmap showing the presence/absence of RMSs in N. meningitidis genomes. Each row in the matrix represents one genome and each column represents one of 35 RMSs identified. The names of RMSs correspond to the reference names used in REBASE database. A dark green square indicates the presence of an RMS in the genome, while light green indicates absence. The dendrogram on the left was generated by applying a hierarchical clustering algorithm where the genomes are grouped according to their RMS content.
In the present study, we performed an in-depth phylogenetic analysis using the core genome information of N. meningitidis. This analysis revealed that N. meningitidis strains from CC103 that were characterized by an accessory genome belong to several different serogroups (Fig. S4). This study contributes to the understanding of N. meningitidis clonal complexes evolution based on their pangenomes. Therefore, population studies on meningococci should include analyses of the core as well as the accessory genomes, which drive the fast evolution of lineages and also contributes to the emergence of new phenotypes.
Methods
Strain collection, bacterial culture, and DNA isolation
The 24 invasive N. meningitidis strains used in this study (Table 1) were recovered from several meningitis cases treated in hospitals in four Brazilian regions: Bahia (2009), Pernambuco (2010–2012), Minas Gerais (2011), and Rio de Janeiro (2012). The isolates were grown on 5% sheep blood agar. Each subcultivation was started from a single colony, and several colonies were used for genomic DNA extraction with the ChargeSwitch gDNA Mini Bacteria Kit (Invitrogen), according manufacturer’s instructions. Approvals from the institutional review board of the hospitals and the Research Ethics committee of the Meningitis Advisory Committee of the State Department of Health from Rio de Janeiro, Brazilian Ministry of Health in Bahia, and Ezequiel Dias Foundation in Minas Gerais were obtained to conducting the study. All methods were performed in accordance with the required guidelines and regulations.
Genome sequencing, assembly annotation, and analysis
Genomic DNA was sequenced on an Illumina HiSeq 2500 sequencer using Nextera XT paired-end run with a 500-bp insert library at the High-Throughput Sequencing Platform of the Oswaldo Cruz Foundation (Fiocruz, Rio de Janeiro, Brazil). Following quality assessment of the reads with FASTQC, assemblies were generated using the SPAdes 3.5 assembler41. Improved assemblies were produced with the PAGIT pipeline using default settings42.
Mobile elements
We identified putative genomic islands and prophage regions by using the IslandViewer (http://www.pathogenomics.sfu.ca/islandviewer/)43 and PHAST (http://phast.wishartlab.com/)44 software, respectively. Genome comparisons were performed in the BRIG program (http://brig.sourceforge.net/)45.
Putative ICEs were identified by the presence of a modular organization of a typical ICE33, such as the presence of oriT, DNA recombinase, relaxase, and T4SS. Contigs from Nm56 genome that harboured an ICE-like region were delimited, concatenated, and compared with other assembled genomes to reconstruct a putative ICE. A putative ordered ICE-region was used as a template for mapping the reads using Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2)46. Paired-end reads, corresponding to the ICE target, were recovered with SAMtools (http://samtools.sourceforge.net)47 and finally assembled with SPAdes 3.5 using default settings. The final contig was confirmed to be complete by PCR using custom primers; PCR amplification products were sequenced by Sanger sequencing. The ICE-like region of Nm56 was annotated using Prokka 1.848 with subsequent manual curation in Artemis (www.sanger.ac.uk/). The final representation and comparison with other ICE-like elements were performed using genoplotR49. We performed Blast searches using the ICE-like element of Nm56 as a query against all BIGSdb genomes. The element was considered present when hits with sequence identities above 70% and more than 10,000 identical residues were obtained. The LS-BSR pipeline50 and the R Package51 were used to detect the presence of ICE-like elements in MenC genomes (Fig. S1), as well for genomic comparison with the GGI from N. gonorrhoeae (Fig. S2). The geographical visualization of N. meningitidis genomes carrying ICE-like region was done with rworldmap52.
Phylogenomic analysis
All 5,531 genomes of N. meningitidis and the metadata used in this study were retrivied from BIGSdb database53 (www.pubmlst.org/neisseria) in September 2015. The csc gene, which encodes a particular poly sialyltransferase that defines the serogroup C, was used as a query in the Blastn search against BIGSdb to select all MenC genomes. A preliminary genome phylogeny with 645 MenC genomes was performed by feature frequency profiles - FFPs (http://ffp-phylogeny.sourceforge.net/)54 (Fig. S1). From the resulting phylogeny, 96 representative MenC genomes and other N. meningitidis CC103 close relatives (considering the presence in GenBank and their position at the phylogenetic tree) were selected for a more datailed analysis (Table S2). Core genome SNPs were identified in these genomes and used for the construction of a phylogenetic tree with the Parsnp program from the Harvest suite55 with the “-p 16 -x -c” parameters. The Gegenees software56 was used for the comparative analysis of gene contents among genomes. The software performs a pairwise comparison of genome fragments creating a distance matrix based on shared fragments, which was exported as a Nexus file; the split tree gene content was produced using SplitsTree4 through the GeneContentDistance method57.
Identification of DNA methyltransferase genes/RMS analysis
All nucleotide sequences of RMSs (DNA methylase and DNA endonuclease) identified in N. meningitidis were obtained from REBASE (rebase.neb.com). Total sequence redundancy (100% identity) were removed using the CD-HIT-EST program58. An RMS presence/absence matrix, considering the 35 non-redundant RMSs sequences, was constructed with the LS-BSR pipeline using default settings50. The resulting presence/absence binary table was converted into a distance matrix and clustered by means of a complete linkage hierarchical cluster analysis using heatmap.2 function from the gplots R package51.
CRISPR identification and analyses
Putative CRISPR arrays were identified in the Brazilian MenC CC103 using PILER-CR v1.06 with default parameters59. Spacer target sequences were clustered with CD-HIT v4.6 using a 90% sequence identity cutoff (parameter: cd − hit − est − i[spacer sequences] − r0 − c0.9 − d100 − [result]); the non-redundant spacer sequences were subsequently compared with prophage regions found in the FAM18 genome by similarity comparisons.
Electronic supplementary material
Acknowledgements
This work was supported by the Programa Nacional de Pós Doutorado/Capes (PNPD/CAPES); Brasil Sem Miséria (BSM/CAPES); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq); FNS/SVS (25030001256201331) and the Instituto Oswaldo Cruz, IOC/FIOCRUZ. This publication made use of the Neisseria Multi Locus Sequence Typing website (http://pubmlst.org/neisseria/) developed by Keith Jolley and sited at the University of Oxford53. The development of this site has been funded by the Wellcome Trust and European Union. We thank Dr. Lee Crainey for the English revision.
Author Contributions
M.A.A. and A.C.V. conceived and designed the study; D.A.C., R.S.C. and I.F. selected and collected the strains, F.F. and I.F. performed the wet lab experiments, M.A.M. and F.E. conducted the bioinformatic analyses, M.A.M., E.F., F.E. and A.C.V. analyzed the results. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01671-x
Accession codes: The whole genome sequences from 24 Brazilian strains sequenced in this study have been submitted to NCBI genome database with the following accession numbers: P4615 (MBLW00000000), P3966 (MBLX00000000), P3978 (MBLY00000000), P4077 (MBLZ00000000), P4431 (MBMA00000000), P4464 (MBMB00000000), P4480 (MBMC00000000), P4534 (MBMD00000000), P4950 (MBME00000000), P4995 (MBMF00000000), P4005 (MBMG00000000), P3558 (MBKM00000000), P3965 (MBKN00000000), P4144 (MBKO00000000), P3478 (JYIS00000000), Nm56 (MCAC00000000), Nm287 (MCAD00000000), Nm288 (MCAE00000000), Nm292 (MCAF00000000), Nm612 (MCAG00000000), Nm638 (MCAH00000000), Nm94 (MCAI00000000), Nm322 (MCAJ00000000) and NmCRJ2 (MCAK00000000).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Harrison OB, et al. Description and nomenclature of Neisseria meningitidis capsule locus. Emerging infectious diseases. 2013;19:566–73. doi: 10.3201/eid1904.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crum-Cianflone N, Sullivan E. Meningococcal Vaccinations. Infectious diseases and therapy. 2016;5:89–112. doi: 10.1007/s40121-016-0107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibarz-Pavón AB, et al. Laboratory-based surveillance of Neisseria meningitidis isolates from disease cases in Latin American and Caribbean countries, SIREVA II 2006–2010. PloS one. 2012;7:e44102. doi: 10.1371/journal.pone.0044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Filippis I, et al. Molecular epidemiology of Neisseria meningitidis serogroup B in Brazil. PloS one. 2012;7:e33016. doi: 10.1371/journal.pone.0033016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorla MCO, et al. Phenotypic and molecular characterization of serogroup C Neisseria meningitidis associated with an outbreak in Bahia, Brazil. Enfermedades infecciosas y microbiologia clinica. 2012;30:56–9. doi: 10.1016/j.eimc.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sáfadi MAP, et al. Carriage rate and effects of vaccination after outbreaks of serogroup C meningococcal disease, Brazil, 2010. Emerging infectious diseases. 2014;20:806–11. doi: 10.3201/eid2005.130948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brasil. Guia de Vigilância Epidemiológica. Ministerio da Saude (2015).
- 8.Stefanelli P, et al. Genome-based study of a spatio-temporal cluster of invasive meningococcal disease due to Neisseria meningitidis serogroup C, clonal complex 11. The Journal of infection. 2016;73:136–44. doi: 10.1016/j.jinf.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Rotman E, Seifert HS. The genetics of Neisseria species. Annual review of genetics. 2014;48:405–31. doi: 10.1146/annurev-genet-120213-092007. [DOI] [PubMed] [Google Scholar]
- 10.Budroni S, et al. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4494–9. doi: 10.1073/pnas.1019751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Molecular cell. 2013;50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison OB, Bray JE, Maiden MCJ, Caugant DA. Genomic Analysis of the Evolution and Global Spread of Hyper-invasive Meningococcal Lineage 5. EBioMedicine. 2015;2:234–243. doi: 10.1016/j.ebiom.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joseph B, et al. Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome. PloS one. 2011;6:e18441. doi: 10.1371/journal.pone.0018441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dillard JP, Seifert HS. A variable genetic island specific for Neisseria gonorrhoeae is involved in providing DNA for natural transformation and is found more often in disseminated infection isolates. Molecular microbiology. 2001;41:263–77. doi: 10.1046/j.1365-2958.2001.02520.x. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton HL, Domínguez NM, Schwartz KJ, Hackett KT, Dillard JP. Neisseria gonorrhoeae secretes chromosomal DNA via a novel type IV secretion system. Molecular microbiology. 2005;55:1704–21. doi: 10.1111/j.1365-2958.2005.04521.x. [DOI] [PubMed] [Google Scholar]
- 16.Zweig M, et al. Secreted single-stranded DNA is involved in the initial phase of biofilm formation by Neisseria gonorrhoeae. Environmental microbiology. 2014;16:1040–1052. doi: 10.1111/1462-2920.12291. [DOI] [PubMed] [Google Scholar]
- 17.Zola TA, Strange HR, Dominguez NM, Dillard JP, Cornelissen CN. Type IV secretion machinery promotes ton-independent intracellular survival of Neisseria gonorrhoeae within cervical epithelial cells. Infection and immunity. 2010;78:2429–2437. doi: 10.1128/IAI.00228-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sardinha G, et al. Replacement of Neisseria meningitidis C cc11/ET-15 variant by a cc103 hypervirulent clone, Brazil 2005–2011. Diagnostic microbiology and infectious disease. 2013;76:524–5. doi: 10.1016/j.diagmicrobio.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Kawai M, Uchiyama I, Kobayashi I. Genome comparison in silico in Neisseria suggests integration of filamentous bacteriophages by their own transposase. DNA research: an international journal for rapid publication of reports on genes and genomes. 2005;12:389–401. doi: 10.1093/dnares/dsi021. [DOI] [PubMed] [Google Scholar]
- 20.Bille E, et al. A chromosomally integrated bacteriophage in invasive meningococci. The Journal of experimental medicine. 2005;201:1905–13. doi: 10.1084/jem.20050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunning Hotopp JC, et al. Comparative genomics of Neisseria meningitidis: core genome, islands of horizontal transfer and pathogen-specific genes. Microbiology (Reading, England) 2006;152:3733–49. doi: 10.1099/mic.0.29261-0. [DOI] [PubMed] [Google Scholar]
- 22.Westra ER, et al. The CRISPRs, they are a-changin’: how prokaryotes generate adaptive immunity. Annual review of genetics. 2012;46:311–39. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 23.Ramsey ME, Woodhams KL, Dillard JP. The Gonococcal Genetic Island and Type IV Secretion in the Pathogenic Neisseria. Frontiers in microbiology. 2011;2:61. doi: 10.3389/fmicb.2011.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pachulec E, et al. Functional analysis of the Gonococcal Genetic Island of Neisseria gonorrhoeae. PloS one. 2014;9:e109613. doi: 10.1371/journal.pone.0109613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woodhams KL, Benet ZL, Blonsky SE, Hackett KT, Dillard JP. Prevalence and detailed mapping of the gonococcal genetic island in Neisseria meningitidis. Journal of bacteriology. 2012;194:2275–85. doi: 10.1128/JB.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutton MD, Smith BT, Godoy VG, Walker GC. The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annual review of genetics. 2000;34:479–497. doi: 10.1146/annurev.genet.34.1.479. [DOI] [PubMed] [Google Scholar]
- 27.Davidsen T, Tønjum T. Meningococcal genome dynamics. Nature reviews. Microbiology. 2006;4:11–22. doi: 10.1038/nrmicro1324. [DOI] [PubMed] [Google Scholar]
- 28.Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. The EMBO journal. 2000;19:6259–65. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehr IJ, Long CD, Serkin CD, Seifert HS. A homologue of the recombination-dependent growth gene, rdgC, is involved in gonococcal pilin antigenic variation. Genetics. 2000;154:523–32. doi: 10.1093/genetics/154.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drees JC, Chitteni-Pattu S, McCaslin DR, Inman RB, Cox MM. Inhibition of RecA protein function by the RdgC protein from Escherichia coli. The Journal of biological chemistry. 2006;281:4708–17. doi: 10.1074/jbc.M513592200. [DOI] [PubMed] [Google Scholar]
- 31.Obergfell, K. P. & Seifert, H. S. Mobile DNA in the Pathogenic Neisseria. Microbiology spectrum3, MDNA3–0015–2014 (2015). [DOI] [PubMed]
- 32.Domínguez NM, Hackett KT, Dillard JP. XerCD-mediated site-specific recombination leads to loss of the 57-kilobase gonococcal genetic island. Journal of bacteriology. 2011;193:377–88. doi: 10.1128/JB.00948-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carraro, N. & Burrus, V. Biology of Three ICE Families: SXT/R391, ICEBs1, and ICESt1/ICESt3. Microbiology spectrum2 (2014). [DOI] [PubMed]
- 34.Kohler PL, et al. Mating pair formation homologue TraG is a variable membrane protein essential for contact-independent type IV secretion of chromosomal DNA by Neisseria gonorrhoeae. Journal of bacteriology. 2013;195:1666–79. doi: 10.1128/JB.02098-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nair DB, et al. Identification of an additional minor pilin essential for piliation in the archaeon Methanococcus maripaludis. PloS one. 2013;8:e83961. doi: 10.1371/journal.pone.0083961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szabó Z, et al. Identification of diverse archaeal proteins with class III signal peptides cleaved by distinct archaeal prepilin peptidases. Journal of bacteriology. 2007;189:772–8. doi: 10.1128/JB.01547-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kedzierska, B. & Hayes, F. Emerging Roles of Toxin-Antitoxin Modules in Bacterial Pathogenesis. Molecules (Basel, Switzerland) 21 (2016). [DOI] [PMC free article] [PubMed]
- 38.Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science (New York, N.Y.) 2003;301:1496–1499. doi: 10.1126/science.1088157. [DOI] [PubMed] [Google Scholar]
- 39.Jamet A, et al. A new family of secreted toxins in pathogenic Neisseria species. PLoS pathogens. 2015;11:e1004592. doi: 10.1371/journal.ppat.1004592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt O, et al. prlF and yhaV encode a new toxin-antitoxin system in Escherichia coli. Journal of molecular biology. 2007;372:894–905. doi: 10.1016/j.jmb.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology: a journal of computational molecular cell biology. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swain MT, et al. A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nature protocols. 2012;7:1260–84. doi: 10.1038/nprot.2012.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dhillon BK, et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic acids research. 2015;43:W104–8. doi: 10.1093/nar/gkv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic acids research. 2011;39:W347–52. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alikhan N-F, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics (Oxford, England) 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 49.Guy L, Kultima JR, Andersson SG. E. genoPlotR: comparative gene and genome visualization in R. Bioinformatics (Oxford, England) 2010;26:2334–5. doi: 10.1093/bioinformatics/btq413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sahl JW, Caporaso JG, Rasko DA, Keim P. The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. PeerJ. 2014;2:e332. doi: 10.7717/peerj.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria URL https://www.R-project.org (2016).
- 52.South A. rworldmap: A New R package for Mapping Global Data. The R Journal. 2011;3:35–43. [Google Scholar]
- 53.Jolley KA, Maiden MCJ. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sims GE, Kim S-H. Whole-genome phylogeny of Escherichia coli/Shigella group by feature frequency profiles (FFPs) Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8329–34. doi: 10.1073/pnas.1105168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome biology. 2014;15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agren J, Sundström A, Håfström T, Segerman B. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PloS one. 2012;7:e39107. doi: 10.1371/journal.pone.0039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huson DH, Steel M. Phylogenetic trees based on gene content. Bioinformatics (Oxford, England) 2004;20:2044–9. doi: 10.1093/bioinformatics/bth198. [DOI] [PubMed] [Google Scholar]
- 58.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics (Oxford, England) 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 59.Edgar RC. PILER-CR: fast and accurate identification of CRISPR repeats. BMC bioinformatics. 2007;8:18. doi: 10.1186/1471-2105-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.