

Abstract
Whole-exome sequencing (WES), one of the next-generation sequencing (NGS), has become a powerful tool to identify exonic variants. Investigating causality of the sequence variants in human disease becomes an important part in NGS for the research and clinical applications. Recently, important guidelines on them have been published and will keep on updating. In our study, two Chinese families, with the clinical diagnosis of “Epilepsy”, which presented with seizures, psychomotor retardation, hypotonia and etc. features, were sequenced by Trio-WES (including the proband and the unaffected parents), and a standard interpretation of the identified variants was performed referring to the recently updated guidelines. Finally, we identified three novel mutations (c.71 C > T, p.P24L; c.1387-1389delGAG, p.E463-; c.134 G > A, p.W45*; NM_000026) in ADSL in the two Chinese families, and confirmed them as the causal variants to the disease-Adenylosuccinate Lyase Deficiency. Previous reported specific therapy was also introduced to the patients after our refined molecular diagnosis, however, the effect was very limited success. In summary, our study demonstrated the power and advantages of WES in exploring the etiology of human disease. Using the constantly updated guidelines to conduct the WES study and to interpret the sequence variants are a necessary strategy to make the molecular diagnosis and to guide the individualized treatment of human disease.
Introduction
As the rapid development and the huge decreased cost of next-generation sequencing (NGS) technologies, NGS has been used in an increasing number of studies for the research and clinical purpose of human disease1, 2. Exonic regions, account for about 1% of the genome, could cause approximate 85% of inherited disease3. Therefore, whole-exome sequencing (WES), which could well explore the exonic regions, has rapidly become a component of the clinical practice and is being applied to a wide range of clinical presentations that require a broad search for causal variants across the spectrum of genetically heterogeneous Mendelian disorders4, 5. However, when using the WES, it could offer about 25,000 variants in exome level per individual6. How to appropriately interpret the identified sequence variants and pinpoint the causal variants of the diseases are important parts in WES applications. Recently, The American College of Medical Genetics and Genomics (ACMG) has developed guidelines for the standard interpretation of sequence variants that cause Mendelian disorders7, 8. Nature 9 and the Clinical Sequencing Exploratory Research (CSER) consortium10 have also paid much attention to the standard interpretation of sequence variants as well.
Seizures and neurodevelopmental delay are common symptoms shared by a broad spectrum of diseases, and these genetically and clinically heterogeneous diseases are difficult to make a precise diagnosis with symptoms and signs alone in the clinical practice. In our study, we used Trio-WES (including the proband and the unaffected parents) and the related guidelines (Table 1) to explore them in Chinese population, and finally identified three novel mutations in ADSL gene in two Chinese families, and refined the diagnosis from “Epilepsy” to “Adenylosuccinate Lyase Deficiency”. This is the first report of ADSL mutations in Chinese population, which expanded the mutations spectrum of this gene. Our data also demonstrated that Trio-WES, combined with the objective interpretation of sequencing variants, is a preferable strategy to explore the genetics of human disease.
Table 1.
The important guidelines for WES applications in our clinical applications.
| No. | Title | Main content | Main applications | Main diseases | Ref. |
|---|---|---|---|---|---|
| 1 | Guidelines for investigating causality of sequence variants in human disease | Focus on investigating causality of sequence variants in human disease | Research and clinical applications | Rare and common diseases | 9 |
| 2 | Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology | Mainly focus on the interpretation of sequence variants in ever-known causative genes | Clinical applications | Mendelian disorders | 7 |
| 3 | Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium | Assess the performance of ACMG/AMP Variant-Interpretation Guidelines with detailed data | Clinical applications | Mendelian disorders | 10 |
| 4 | Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants | Improve on the detailed criteria on determining the “cosegregation” of ACMG | Clinical applications | Mendelian disorders | 11 |
| 5 | Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics | Focus on the secondary findings in clinical exome and genome sequencing | Clinical applications | Mendelian disorders | 8 |
| 6 | Points to Consider: Ethical, Legal, and Psychosocial Implications of Genetic Testing in Children and Adolescents | Focus on the statement on genetic testing in children and adolescents | Research and clinical applications | Diseases in children and adolescents | 22 |
Results
Clinical features
All of the four patients in the two families were partus matures with normal birth-weight. Then they began to present with neuropsychological clinical features characterized by severe psychomotor retardation, early onset of seizures, autism and etc.
In family 1 (Fig. 1A), the two patients have the similar features. The proband (II 2) is 0.5-year old now, while her sister (II 1) 2.5-year old. They firstly presented generalized seizures within 1-week old, which manifested with abnormal multifocal spike and ware wave in EEG (Fig. 1D). The average frequency of the seizures (including myoclonus) were 1–3 times per day before the treatment, however, it could reach to 8 times in some days. Valproate sodium and levetiracetam were adopted to them to control the seizures. Both II 1 and II 2 have marked psychomotor retardation, hypotonia and lack of eye-to-eye contact. Brain MRI examination showed obvious myelination in the white matter of the proband (II 2, 1-month old). No abnormal signals were observed in the brain MRI of the patient II 1 when she was 1-month old (Fig. 1C).
Figure 1.
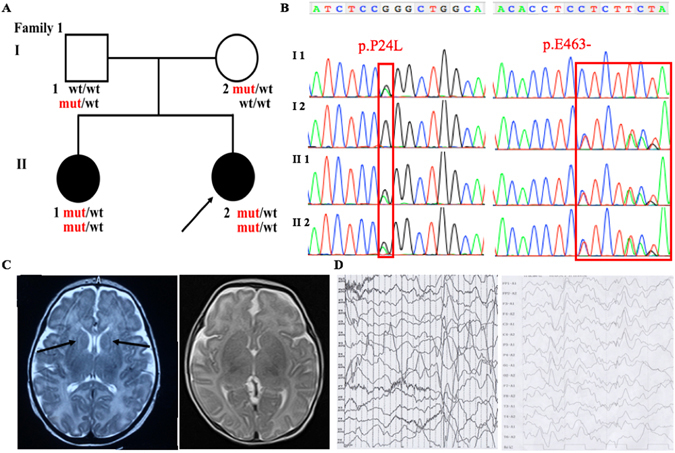
Clinical features of the patients and family 1 with the c.71 C > T, p.P24L; c.1387-1389delGAG, p.E463- in ADSL. (A) Pedigree structure of the studied family 1. In the family 1, WES was performed in I:1, I:2, II:2. Also, the compound heterozygous mutations were presented in the pedigree. (B) The PCR products were sequenced with the reverse primers (ADSL c.71 C > T, p.P24L; c.1387-1389delGAG, p.E463-). (C) The brain MRI shows the abnormal myelination of white matter in the proband (1-month old) (Left), and normal signal of the II 1 (Right) with T2-weighted. (D) EEG of the patients show the abnormal multifocal spike and ware wave in the proband (Left) and II 2 (Right).
The two patients in family 2 (Fig. 2A) are monozygotic twins who are 3-year old now. Similar to the family 1, they firstly presented generalized seizures within 1-week old, manifesting with abnormal multifocal spike and ware wave in EEG (Fig. 2D). The frequency of the seizures (including myoclonus) were 2–5 times per day before the treatment. Interestingly, there were some difference in phenotypes between the twins. For example, the clinical manifestation of II 3 was more severe than his elder brother and the two patients in family 1, such as showing recurrent temper tantrums, crying and poor sleep. Both II 2 and II 3 had marked psychomotor retardation, hypotonia and autistic features, etc. (Fig. 2C), which is similar to family 1. Although with different symptoms, they had same abnormal changes on brain MRI (2-month old), which presented with enlarged extracerebral space (MRI images were lost, with the scan reports left), indicating the atrophy of the cerebral cortex.
Figure 2.
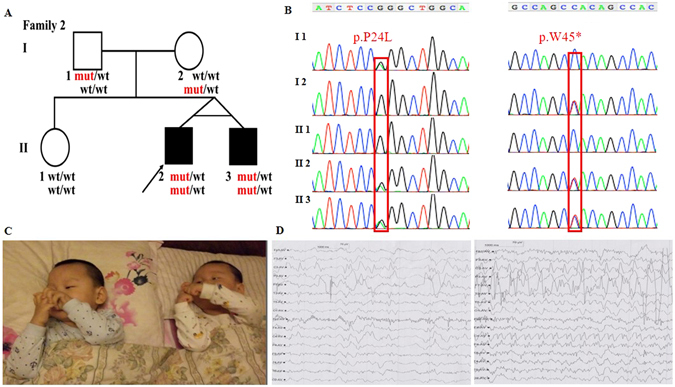
Clinical features of the patients and family 2 with the c.71 C > T, p.P24L; c.134 G > A, p.W45* in ADSL. (A) Pedigree structure of the studied family 2. In the family 2, WES was performed in I:1, I:2, II:2. Also, the compound heterozygous mutations were presented in the pedigree. (B) The PCR products were sequenced with the reverse primers (ADSL c.71 C > T, p.P24L; c.134 G > A, p.W45*). (C) Both of the patients have obvious hypotonia (could not sit by himself), autistic features (lack of eye-to-eye contact, repetitive movements, impaired verbal communication, etc.), etc. (3-year old). (D) EEG of the patients show the abnormal multifocal spike and ware wave in the proband (Left) and II 3 (Right).
Genetic findings
All of the four patients have a normal G-banding and karyotype. Using Trio-WES, more than 97% of the target regions were covered by at least 10 times for all the six individuals. After sequencing data analysis and variants filtering, we got 12 candidate genes with homozygous variants (C1orf86, RNF19B, CYP2C18, BEST1, SLAIN1, IGHJ6, KCNC3, LILRA3, P2RY1, CANX, LCA10), 5 candidate genes with compound heterozygous variants (ADSL, RHOBTB1,TTN, HELZ2, SYNE1), and 6 candidate genes with de novo variants (FBLIM1, VPS33A, SMURF2, HIF3A, SCD5, HDAC9) in family 1 (Supplementary Table S1); 7 candidate genes with homozygous variants (AL359195.1, AGAP3, EGFL6, ZNF182, PFKFB1, ARHGAP36, MAMLD1), 11 candidate genes with compound heterozygous variants (ADSL, LPHN2, ABLIM1, DCHS1, A2ML1, PKD1L2, DCC, SIGLEC12, SLC3A1, ZFHX4, DMRT1), and 8 candidate genes with de novo variants (FOXO6, SKA3, ZFPM1, HES6, PIGU, CLCN2, TRBV5-4, CACNA1B) in family 2 (Supplementary Table S2). Sanger sequencing was conducted with priority to the compound heterozygous variants (c.71 C > T, p.P24L and c.1387-1389delGAG, p.E463- in family 1; c.71 C > T, p.P24L and c.134 G > A, p.W45* in family 2;NM_000026) in ADSL (the only gene associated with seizures and neurodevelopmental delay among the candidate genes list above) for excluding the false positive of WES. The results of the Sanger sequencing are showed in Figs 1B and 2B. The novel variants in ADSL co-segregated well in the families (Figs 1A and 2A) and were not found in more than 60,000 people (Table 2). They are predicted as “damaging” by PolyPhen, SIFT, CADD or Mutation Taster (Table 2). According to ACMG guideline7, 11, our identified novel compound heterozygous variants are categorized to be “pathogenic variant” due to belonging to PVS1 (pathogenic very strong 1: null variant in a gene where loss of function is a known mechanism of disease), PS4 (pathogenic strong 4: the prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls), PM2-4 (pathogenic moderate 2–4: absent from controls, or at extremely low frequency if recessive, in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium; for recessive disorders, detected in trans with a pathogenic variant; protein length changes as a result of in-frame deletions/insertions in a nonrepeat region or stop-loss variants), and PP1-3 (pathogenic supporting 1-3: cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease; missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease; multiple lines of computational evidence support a deleterious effect on the gene or gene product), which indicates that they are the etiology of the disease.
Table 2.
The prediction of the identified variants in ADSL.
| Gene name | Position | Transcript | Substitution | ExAC | 1000 G | dbSNP ID | ESP | SIFT score | Polyphen score | Grantham score | GERP score | CADD score | Mutation Taster |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADSL | Chr 22: 40742633 | NM_000026.2 | c.71 C > T/p.P24L | novel | novel | novel | novel | 0.067 | 0.001 | 98 | 0.153 | 14.87 | Disease causing |
| ADSL | Chr 22: 40762457 | NM_000026.2 | c.1387-1389delGAG/p.E463- | novel | novel | novel | novel | NA | NA | NA | 5.470 | NA | Disease causing |
| ADSL | Chr 22: 40742696 | NM_000026.2 | c.134 G > A/p.W45* | novel | novel | novel | novel | NA | NA | NA | 3.240 | 29.20 | Disease causing |
Abbreviations: ExAC, Exome Aggregation Consortium; 1000 G, 1000 genomes; ESP, Exome Sequencing Project; NA, not available.
Secondary findings
As to the potential interest of the patients and medical actionability, informed consent of analyses of secondary findings (SFs) were got from the parents. The new updated SFs minimum list include 59 medically actionable genes recommended by the ACMG8. None of the known or expected pathogenic variants in theses genes were found in our patients.
Specific therapy
After the molecular diagnosis of the patients, we refined the diagnosis from “Epilepsy” to “Adenylosuccinate Lyase Deficiency”. To date, there has no enzyme replacement therapy for Adenylosuccinate Lyase Deficiency, however, ketogenic diet12 and D-ribose13 were reported to be effective to some patients. In family 2, both II 2 and II 3 were given a ketogenic diet (a high-fat, adequate-protein, low-carbohydrate diet) for 14 months (beginning from 3-month old to 1.5-year old). During the ketogenic diet therapy, the seizures were eliminated without taking other antiepileptic drugs. However, the ketogenic diet therapy was finally given up and and replaced by sodium valproate due to the reoccurrence of the seizures. Patients in Family 1 did not adopt ketogenic diet therapy, but took levetiracetam and sodium valproate to control seizures. While taking the antiepileptic drugs, D-ribose administration was also introduced to both family 1 and 2, recently. D-ribose was administrated orally (four times daily) to the patients with an initial dose of 1 mmol/kg/day at beginning, then increased progressively up to 10 mmol/kg/day13. Interestingly, II 2 in family 2, who has the slightest features among the four patients, has an improvement in motor nimbleness and wish of playing and expressing himself after 1-month therapy. Now he has been treated with D-ribose for 3 months. However, II 3 in family 2 has no improvements after 1-month D-ribose administration, and gave it up. Both two patients in Family 2 gave up the D-ribose administration after 1-week treatment because of the increased frequency of seizures.
Discussion
Adenylosuccinate lyase deficiency (OMIM #103050) is an autosomal recessive defect of purine metabolism. Since the first mutation in ADSL reported in 199214, several mutations have been reported in ADSL in European15, 16, however, there is few reports in Asian. In 2010, Chen et al. firstly reported two novel ADSL mutations in a Malaysian patient17. Here, we firstly reported three novel ADSL mutations in Chinese, which will expand the spectrum of mutations in ADSL in Asian. So far, there has been more than 50 mutations in ADSL been reported associating with Adenylosuccinate Lyase Deficiency (Fig. 3). Among these mutations, R426H was the most frequent one, indicating it is a hot spot in European18. The ever-known mutations locate in different parts/domains of the protein, and there is no minor allele frequency of missense/loss of function variants in ADSL larger than 0.005 in the Exome Aggregation Consortium (http://exac.broadinstitute.org), which may indicate the high conservation of ADSL sequence and its mutations predisposition to human disease.
Figure 3.
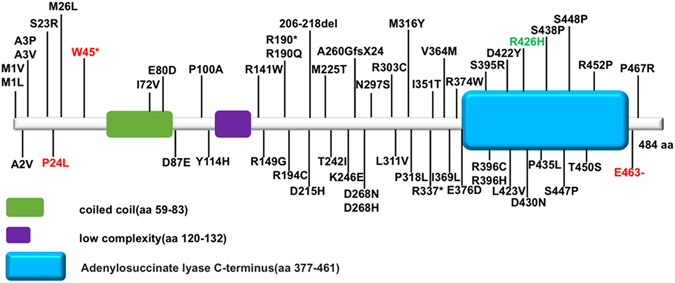
Mutations in ADSL protein. Note: aa, amino acid. Mutations in red are identified in our patients, mutation in green is the hot spot in European population. c.-49T > C, IVS5 -37 C > T and IVS12 + 1 G > C are not included in the figure. Schematic diagram was referred to SMART database (http://smart.embl-heidelberg.de). Mutations are referred to ADSL database (http://www1.lf1.cuni.cz/udmp/adsl/) and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php).
Patients with Adenylosuccinate lyase deficiency were reported with significant clinical heterogeneity. In our patients, the clinical heterogeneity was also presented in clinical manifestations and brain MRI. For clinical manifestation, the symptoms of II 3 in family 2 was more severe (such as having recurrent temper tantrums, crying and poor sleep) than other patients, even than his twin-brother. Also, the patients’ brain MRI images showed different changes, without presenting corpus callosum missing, cerebellar vermis missing19, 20 or lissencephaly21, indicating the heterogeneity in brain MRI.
In the genetic test, we referred to the updated guidelines to ensure the accuracy of our study (Table 1). Our purpose of the test was according to American Society of Human Genetics recent suggestions for genetic testing in children and adolescents22, 23. Also, the attention should be paid when using WES, as nonstandard applications and interpretation of WES will cause serious effects on the doctor’s decision and patient’s therapy, etc.24. The process of our analysis and the interpretation of the sequencing variants are consisted to the guidelines published by MacArthur et al.9 and ACMG7, 8. Quality controls were conducted in the whole process, including DNA library creation, raw sequence data processing and base calling as described in previous paper25. After the standard quality controls of the WES, candidate genes associated with seizures and neurodevelopmental delay were preferentially considered to be the causative genes. As it is suggested by MacArthur et al.9 that “Investigators should begin by examining sequence variation in genes known to be associated with that phenotype, and assessing sequence coverage of the coding sequences and splice junctions for these genes before exploring the possibility of new candidate genes in the affected individuals”, Sanger sequencing was conducted with priority to ADSL. In the light of ACMG guidelines, we clinicians, laboratory scientists, and bioinformaticians, with collaborative interactions, objectively determined the identified novel variants in ADSL belong to “Pathogenic”, as they meet the criteria of PVS1, PS4, PM2-4 or PP1-3 in ACMG7, 10, 11, and then determined them to the causal mutations of the two families. The whole process of WES and the sequence variants interpretation in our study were standard.
Recently, secondary findings or incidental findings, which include genetic variants found incidentally or accidentally, have been paid much attention by the doctors, geneticist, and researchers8, 26. After obtaining the written informed consent from all the families, we also explored the secondary findings for the patients. Luckily, both the two probands have no identified mutations in 59 medically actionable genes suggested by ACMG8, indicating their low predisposition to the related disorders.
We also tried to conduct individual treatment after we refined the diagnosis from “Epilepsy” to “Adenylosuccinate Lyase Deficiency”. However, due to the limited research on Adenylosuccinate Lyase Deficiency therapy, no exciting effects were observed on all of our patients and the effect of D-ribose administration to Adenylosuccinate Lyase Deficiency was still ambiguous. This also indicates the eagerness to accelerate the evidence-based practice of genomic medicine and precision medicine27.
In conclusion, using Trio-WES, we identified three novel mutations in ADSL gene and using molecular diagnosis, we refined the diagnosis from “Epilepsy” to “Adenylosuccinate Lyase Deficiency”. This is a paradigm to do molecular diagnosis of rare Mendelian disease. Trio-WES, combining with the guidelines for investigating causality of sequence variants in human disease, has become a preferable option to explore the pathogeny and to make clinical practice of rare Mendelian disorders.
Methods and Materials
Subjects
More than 200 Chinese families with neurodevelopmental delay were enrolled in our study (data unpublished). In this manuscript, we reported two nonconsanguineous Chinese families (Figs 1A and 2A), and other than the four patients, there are no history of psychiatric or neurologic disorders reported in the families. Genomic DNA was extracted from peripheral blood leukocytes by standard phenol-chloroform extraction methods from the probands, and their unaffected parents and siblings. This study was approved by the ethics committee of Xiangya Hospital affiliated to Central South University and written informed consent was obtained from all the parents. Informed consent to publish identifying information/images was obtained from all the parents. The methods in this study were performed in accordance with the approved guidelines.
Whole exome sequencing and Sanger sequencing
All of the four patients were conducted chromosome G-banding and karyotype analysis. After that, we took a Trio-WES strategy to identify the causal variants of the disease. TruSeq Exome Enrichment kit (Illumina) was used for exome capture and Hiseq 3000 platform (Illumina), with 2 × 150 bp pair-end reads, was used for sequencing the genomic DNA of the proband and the parents in WuXi AppTec (Shanghai, China).
Illumina’s Pipeline (version 1.3.4) with default parameters were used for raw image analysis and base calling. Sequence data were aligned to the reference human genome (hg19 version) using Burrows-Wheeler Aligner (BWA)28, duplicate reads were removed using Picard tool. We also used a comprehensive bioinformatics pipeline to analyze the exome sequencing data, including preprocessing, variant calling, annotation, and filtering according to our previous study25, and standard quality controls of the sequencing data were conducted in the process as well.
Considering the characteristics of the pedigree, the rare recessive, compound heterozygous and de novo variations in the probands were considered to be candidate causal variations with priority to the disease. Three rare novel mutations (c.71 C > T, p.P24L; c.1387-1389delGAG, p.E463-; c.134 G > A, p.W45*; NM_000026) in ADSL gene identified by WES were confirmed by Sanger sequencing. Two pairs of the primers for them are “Forward Primer TGGTCAAAGAAGCGAACCAA, Reverse Primer CGCAGCTCCTCAGGACAG” (c.71 C > T, p.P24L; c.134 G > A, p.W45*) and “Forward Primer TGTATTTGCTTTCCTCTGGCA, Reverse Primer AGGAAAGCACCATGGGAAGA” (c.1387-1389delGAG, p.E463-).
Electronic supplementary material
Acknowledgements
The authors are grateful to all subjects for their participation in our study. We also thank Prof. Costantino Salerno (University of Roma La Sapienza) for help us in the patient’s therapy, and thank all the clinicians (including pediatricians), laboratory scientists, and bioinformaticians for discussion on the interpretation of sequencing variants. This work was supported by grants 81361120404, 81430023 and 81130021 from the National Natural Science Foundation of China (to Dr. Bei-sha Tang), grants 81371405, 81571248 from the National Natural Science Foundation of China (to Dr. Ji-feng Guo), grant 2016CX025 from innovation-driven plan of Central South University (to Dr. Ji-feng Guo).
Author Contributions
Conceived and designed the experiments: K.L., X.M., J.F.G. Performed the experiments: K.L., X.M. Analyzed the data: K.L., X.M., B.S.T., J.F.G. Contributed reagents/materials/analysis tools: K.L., X.M., B.S.T., Y.L., D.X.D., Y.W.Z., C.R.W., X.T.Z., Z.H.L., Y.Z., P.Z.W., Q.X., Q.Y.S., K.X., X.X.Y., H.J., L.S., J.F.G. Contributed to the writing of the manuscript: K.L., X.M. Reviewed and critiqued the first manuscript: K.L., X.M., B.S.T., J.F.G.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01637-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014;370:2418–2425. doi: 10.1056/NEJMra1312543. [DOI] [PubMed] [Google Scholar]
- 2.Lewis R. Exome sequencing comes to the clinic. JAMA. 2015;313:1301–1303. doi: 10.1001/jama.2015.1389. [DOI] [PubMed] [Google Scholar]
- 3.Choi M, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee H, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singleton AB. Exome sequencing: a transformative technology. Lancet Neurol. 2011;10:942–946. doi: 10.1016/S1474-4422(11)70196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalia, S. S. et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet. Med., doi:10.1038/gim.2016.190 (2016). [DOI] [PubMed]
- 9.MacArthur DG, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amendola LM, et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 2016;98:1067–1076. doi: 10.1016/j.ajhg.2016.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarvik GP, Browning BL. Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. Am. J. Hum. Genet. 2016;98:1077–1081. doi: 10.1016/j.ajhg.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefevre F, Aronson N. Ketogenic diet for the treatment of refractory epilepsy in children: A systematic review of efficacy. Pediatrics. 2000;105:E46. doi: 10.1542/peds.105.4.e46. [DOI] [PubMed] [Google Scholar]
- 13.Salerno C, et al. Effect of D-ribose administration to a patient with inherited deficit of adenylosuccinase. Adv. Exp. Med. Biol. 1998;431:177–180. doi: 10.1007/978-1-4615-5381-6_34. [DOI] [PubMed] [Google Scholar]
- 14.Stone RL, et al. A mutation in adenylosuccinate lyase associated with mental retardation and autistic features. Nat. Genet. 1992;1:59–63. doi: 10.1038/ng0492-59. [DOI] [PubMed] [Google Scholar]
- 15.Donti TR, et al. Diagnosis of adenylosuccinate lyase deficiency by metabolomic profiling in plasma reveals a phenotypic spectrum. Mol Genet Metab Rep. 2016;8:61–66. doi: 10.1016/j.ymgmr.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jurecka A, Zikanova M, Kmoch S, Tylki-Szymańska A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015;38:231–242. doi: 10.1007/s10545-014-9755-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen BC, et al. Adenylosuccinate lyase deficiency in a Malaysian patient, with novel adenylosuccinate lyase gene mutations. J. Inherit. Metab. Dis. 2010;33(Suppl 3):S159–162. doi: 10.1007/s10545-010-9056-z. [DOI] [PubMed] [Google Scholar]
- 18.Marie S, et al. Mutation analysis in adenylosuccinate lyase deficiency: eight novel mutations in the re-evaluated full ADSL coding sequence. Hum. Mutat. 1999;13:197–202. doi: 10.1002/(SICI)1098-1004(1999)13:3<197::AID-HUMU3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 19.Van den Berghe G, Vincent MF, Jaeken J. Inborn errors of the purine nucleotide cycle: adenylosuccinase deficiency. J. Inherit. Metab. Dis. 1997;20:193–202. doi: 10.1023/A:1005304722259. [DOI] [PubMed] [Google Scholar]
- 20.Edery P, et al. Intrafamilial variability in the phenotypic expression of adenylosuccinate lyase deficiency: a report on three patients. Am. J. Med. Genet. A. 2003;120A:185–190. doi: 10.1002/ajmg.a.20176. [DOI] [PubMed] [Google Scholar]
- 21.Stathis SL, Cowley DM, Broe D. Autism and adenylosuccinase deficiency. J Am Acad Child Adolesc Psychiatry. 2000;39:274–275. doi: 10.1097/00004583-200003000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Botkin JR, et al. Points to Consider: Ethical, Legal, and Psychosocial Implications of Genetic Testing in Children and Adolescents. Am. J. Hum. Genet. 2015;97:6–21. doi: 10.1016/j.ajhg.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Botkin JR. Ethical issues in pediatric genetic testing and screening. Curr. Opin. Pediatr. 2016;28:700–704. doi: 10.1097/MOP.0000000000000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berg JS. Genome-scale sequencing in clinical care: establishing molecular diagnoses and measuring value. JAMA. 2014;312:1865–1867. doi: 10.1001/jama.2014.14665. [DOI] [PubMed] [Google Scholar]
- 25.Ding D, et al. Identification of a de novo DYNC1H1 mutation via WES according to published guidelines. Sci Rep. 2016;6:20423. doi: 10.1038/srep20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green RC, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roundtable on Translating Genomic-Based Research for Health, Board on Health Sciences Policy, Health and Medicine Division & National Academies of Sciences, Engineering, and Medicine. Applying an Implementation Science Approach to Genomic Medicine: Workshop Summary. (National Academies Press (US), 2016). [PubMed]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.