

Abstract
Antibiotics are a relatively common disturbance to the normal microbiota of humans and agricultural animals, sometimes resulting in severe side effects such as antibiotic-associated enterocolitis. Gambusia affinis was used as a vertebrate model for effects of a broad-spectrum antibiotic, rifampicin, on the skin and gut mucosal microbiomes. The fish were exposed to the antibiotic in the water column for 1 week, and then monitored during recovery. As observed via culture, viable counts from the skin microbiome dropped strongly yet returned to pretreatment levels by 1.6 days and became >70% resistant. The gut microbiome counts dropped and took longer to recover (2.6 days), and became >90% drug resistant. The resistance persisted at ~20% of skin counts in the absence of antibiotic selection for 2 weeks. A community biochemical analysis measuring the presence/absence of 31 activities observed a 39% change in results after 3 days of antibiotic treatment. The antibiotic lowered the skin and gut microbiome community diversity and altered taxonomic composition, observed by 16S rRNA profiling. A 1-week recovery period did not return diversity or composition to pretreatment levels. The genus Myroides dominated both the microbiomes during the treatment, but was not stable and declined in abundance over time during recovery. Rifampicin selected for members of the family Comamonadaceae in the skin but not the gut microbiome. Consistent with other studies, this tractable animal model shows lasting effects on mucosal microbiomes following antibiotic exposure, including persistence of drug-resistant organisms in the community.
Keywords: microbiome, antibiotic, antibiotic resistance, Gambusia affinis, community disruption
Introduction
Antibiotics are potent tools in modern medicine, but select for resistance in pathogens and thus can become ineffective. An extensive study has been carried out and continues on drug-resistant mechanisms in pathogens. One area of research currently under intense investigation is the effects of antibiotic therapies on the normal microbiota of patients. Although the effects vary based on the particular antibiotic, the route of administration, and the composition of the preexisting microbial community, some general effects on human microbiota include lowering community diversity, change in community composition, loss of some rare members, alteration of enzymatic functions, selection of resistant members, and, in some but not all cases, lowering of total numbers.1 An antibiotic treatment in humans can have unintended consequences, such as diarrhea, antibiotic-associated enterocolitis, and opportunistic infections, most notably yeast and Clostridium difficile. In a study of three humans treated with ciprofloxacin,2 the gut microbiota was strongly and rapidly disrupted upon onset of treatment, and began to shift back to the initial state 1 week after cessation of the antibiotic. However, the composition did not return completely, instead obtained an alternative stable state. The drug treatment changed the abundance of approximately one-third of the gut taxa, and decreased the community richness, diversity, and evenness.3 In nine newborns, parenteral treatment with a combination of ampicillin and gentamicin at <2 days of age resulted in a decrease of the genera Bifidobacterium and Lactobacillus, and an increase in the class Proteobacteria, which persisted for at least 1 month.4
Studies in laboratory model organisms can be far more controlled and comprehensive, and complement clinical studies. Mice given two antibiotics and one biocide (ampicillin, metronidazole, and bismuth) in feed for 10 days demonstrated a dramatic shift in bacterial community composition in the cecea.5 The composition was stable in the absence of antibiotic pressure. Although the normal gut microbiota of mice was dominated by the phyla Firmicutes (74.4%) and Bacteroidetes (23.3%), Proteobacteria took over (initially 1.1%, changing to 75.5%) after multidrug treatment. After a 2-week recovery period, the gut returned almost completely to the initial composition. The treatment of mice with cefoperazone in the water for 10 days resulted in a less dramatic but still evident shift (loss of Bacteroidetes, increase in Firmicutes, and small change in Proteobacteria). Again, recovery was incomplete. Thus, the exact disruption of microbiome community composition caused by the antibiotics appears to vary according to each particular antibiotic and across model organisms.
This study examined the effect of a high dose of broad-spectrum antibiotic on community composition, biochemical activities, and drug resistance in the skin and gut microbiomes of a vertebrate model, Gambusia affinis (the Western mosquito fish). These small (sizes of 0.1–1.5 g) fish are a good model for mucosal microbiomes,6 easy to manipulate, and have vertebrate acquired immunity. Mucosal microbiomes have the highest densities of microbes and the most intimate interactions with the host. Rifampicin was chosen as a prototypical broad-spectrum antibiotic because the dominant resistance mechanism, substitutions in the beta subunit of the RNA polymerase (rpoB gene), is unlikely to be horizontally transferred between bacteria. It is also distributed well and evenly in tissues. Rifampicin, which is a primarily bactericidal member of the ansamycin family, has been used in human and veterinary medicine for many years, commonly against meningitis and mycobacterial infections. Because of rapid antibiotic accessibility and ability to sample without lethality to the host, this study focused on the skin mucosal microbiome. However, given that many published studies have examined the gut, this was included as well for comparison.
Materials and methods
Fish
For genetic homogeneity, the fish were caught from one location, a pristine pond in Walker County, Texas. Precautions were taken to avoid physically touching the fish by using a dip net, to not disturb the skin microbiome. The fish were maintained in 75 L aquaria with a 12-h light/12-h dark cycle until use (minimum 5-day acclimation to the laboratory after being caught) at 23°C–25°C. For experiments, groups of fish were added to a well-cleaned plastic bucket into 2 L of autoclaved artificial pond water (APW). APW is of 0.33 g/L CaCl2, 0.33 g/L MgSO4, and 0.12 g/L NaHCO3 in deionized water (Milli-Q system). During experiments, the fish were fed daily with 5 mg/fish of flake food (TetraFin) in the bucket. All animal experiments were performed under the approval of Institutional Animal Care and Use Committee’s protocols 13-04-29-1018-3-01 and 14-04-17-1018-3-01.
Antibiotic
Rifampicin stock solution (50 mg/mL in dimethyl sulfoxide) was stored at −20°C, and 500 µL stock added per liter of APW (25 µg/mL final concentration). Rifampicin activity was measured daily during 1 week in APW at 25°C and did not change, as measured by the size of a zone of inhibition (ZOI) on an overnight nutrient agar (NA) plate of Bacillus megaterium from a 50 µL sample of APW. Assuming the steady-state internal concentration in fish is close to the water concentration of the drug (25 µg/mL) and a mean fish size of 1 g (weights varied between 0.25 and 1.5 g), this would be equivalent to an approximate 25 mg/kg dose. By comparison, recommended human dosages are 10–20 mg/kg,7 so this is a high-dose model. Directly following a 3-day or 1-week exposure period, the remaining fish were transferred via dip net into a new tub habitat with fresh 1 L APW without rifampicin for recovery.
Sampling
To extract the skin microbiome, the fish were vortexed in a sterile 15 mL conical tube in 2 mL of PBST (137 mM NaCl, 10 mM phosphate, and 0.1% Tween 20, pH 7.4) for 1 minute, pausing shortly every 10 seconds. To extract the gut microbiome, a fish was euthanized by snipping the nerve cord just behind the head, and then the entire gut canal was extracted by cutting just behind the mouth and just before the anus. The extracted gut was placed onto a sterile petri dish, and cut into short 1–2 mm sections. Gut contents from all the combined sections were extracted into 2 mL PBST by vortexing for 1 minute. For colony counts, tenfold serial dilutions of skin or gut extract were made in PBST, and then 100 µL aliquots spread onto NA (7.5 g/L of Bacto agar, 2.5 g/L of peptone, and 1.5 g/L of beef extract) plates in duplicate. NA containing 25 µg/mL rifampicin was used to quantify resistant organisms. The plates were checked after 48 h of incubation at 25°C.
All data except the community biochemical profiles and antibiograms came from one experiment. Fifty fish from the same aquarium were placed in 2 L of APW with 25 µg/mL of rifampicin for 1 week, and then moved to another bucket with 2 L of fresh APW for recovery. Two fish were sampled for metagenomics (one for skin and one for gut) on every day of the 7-day treatment, and 1, 2, 4, 6, and 14 days (skin only) afterward. Eight fish were sampled before treatment (four for skin and four for gut), and three aquarium water samples were taken at the same time. For a culture analysis, two fish (one for gut and one for skin) were sampled pretreatment, two fish on each day of the 7-day treatment, and two fish 7 and 14 days after treatment.
Metagenomics
For 16S rRNA gene profile analysis, bacteria were pelleted from each fish by a 2-minute spin in a centrifuge at room temperature at top speed (13,000 rpm). The bacterial pellet was frozen and stored at –80°C, and then shipped on dry ice to the Alkek Center for Metagenomics and Microbiome Research, Baylor College of Medicine. Genomic DNA was extracted using the PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA) following protocols benchmarked during the Human Microbiome Project and the Earth Microbiome Project. The V4 region of the 16S rRNA was amplified using the 515f/806r (barcoded) primer pair.8 Amplicons were pooled and sequenced in a single Illumina HiSeq run at the Human Genome Sequencing Center at Baylor College of Medicine.
Illumina reads 1 and 2 were quality filtered to remove any ambiguous bases and joined using a custom perl script with a minimum overlap of 15 base pairs (bps). Sequences were then quality trimmed, demultiplexed, and analyzed using the QIIME pipeline, version 1.8.0.9 Quality trimming used the following requirements: no ambiguous bases, maximum of one barcode error allowed, and a minimum phred score of 20. Median sequence length after trimming was 253 bp, and mean reads per sample were 204,325. The sequences were then binned into operational taxonomic units (OTUs) using the closed-reference OTU picking workflow, with a sequence identity of 97% against the Greengenes (August 2012 release) as the reference database and UCLUST10 as the underlying clustering algorithm. Singleton reads were then removed from the OTU table, which was rarefied to 31,856 sequences per sample before performing alpha- and beta-diversity analyses.
Community biochemical and resistance profiles
The same method of extracting the skin microbiome from the fish done in the previous experiments was used, but with 5 mL of 0.85% saline solution instead of PBST. For the control, three untreated fish from the aquarium were separately extracted and analyzed using API-20E and API-20NE strips (bioMerieux). The strips were placed into an incubator at 25°C, and read after 24 h. For the antibiotic exposure analysis, three fish from the same aquarium were exposed to the antibiotic rifampicin at 25 µg/mL in APW for 3 days, and then sampled.
The same protocol using six fish (three controls and three treated) with 3 days of rifampicin exposure was used for the community resistance profile. The antibiogram was determined using the disk diffusion method.
Results
Effects on number of culturable sensitive and resistant bacteria
As shown in Figure 1A, the proportion of the skin bacterial population that was naturally resistant to rifampicin was very low (was below the detection limit of ~20 colony-forming units [CFUs]/fish), and this was consistent across multiple experiments (data not shown). Initiation of antibiotic exposure resulted in a rapid loss of viability for the skin microbiome (73-fold drop in the CFU numbers at 17.5 h). However, culturable numbers were fully recovered and maintained after 38.5 h. During the 1-week exposure to antibiotic, the CFU numbers on rifampicin-containing plates were 71% (1.6 days), 73% (2.7 days), and 88% (3.9 days) those of nutrient agar. Most of the culturable bacteria were drug resistant during the exposure, with the proportion being relatively stable. Following the release of antibiotic selection pressure, the culturable resistance rate dropped to 18%, and was stable at that rate for at least 2 weeks (16.7 days was the last timepoint) in the absence of selection pressure.
Figure 1.

Culturable bacteria during antibiotic exposure. (A) Skin and (B) gut.
Notes: First samples are 5.5 h before rifampicin exposure began (control). Exposure started at time zero, and lasted 165 h (indicated by red bar), so the last three timepoints are 20.5, 165.5, and 400.5 h (skin only) after antibiotic removal by water change, respectively. No colonies were recovered on rifampicin-containing plates at the 5.5- and 17.5-h timepoints from skin. Scales of axes for panels (A) and (B) are identical, for ease of comparison. Viability is reported in CFUs/g of fish weight.
Abbreviations: CFU, colony-forming unit; NA, nutrient agar; rif-NA, rifampicin-containing NA.
Similarly, in the gut (Figure 1B), the resistance rate before exposure was very low (0.02%), and the exposure resulted in a dramatic 13,036-fold drop in the CFU numbers at 17.5 h. Recovery was slower, with only 0.01% of the pretreatment colony numbers appearing at 38.5 h, but numbers similar to pretreatment by 63.5 h and later. Assuming a slower exchange of bacteria between the fish gut and external environment than the fish skin and surrounding water, as well as a possible slower exchange of the antibiotic, this would be expected. Analogous with the skin, resistance rates were high during exposure (83% average of the three timepoints), and then dropped after the fish were moved to fresh drug-free water (29% at 185.5 h and 35% at 330.5 h). Resistance rates also appeared to be stable, although they were only sampled out to 6.9 days posttreatment, with no gut timepoint taken at 16.7 days. Interestingly, the resistance rates of the gut posttreatment were almost double that of the skin, suggesting either a slower exchange with the environment or greater competitive fitness of the resistant species/strains. The preliminary bioassay data suggest that fish clear rifampicin from their body in ~24 h (data not shown).
To confirm the resistance rates determined by comparison of CFU numbers on the NA plates to the NA plates containing rifampicin, one dozen colonies were selected at random from several NA plates from each timepoint. These colonies were then subcultured by streaking onto both NA and rifampicin-containing NA (rif-NA) plates, providing a separate determination of the proportion of resistant colonies that grew on the nonselective NA plates from the fish during exposure. The numbers determined were consistent. Although all 12 were resistant (regrew on NA and rif-NA plates) at both the skin and gut 38.5-h timepoints, only 40% of the gut and 17% of the skin colonies were resistant at the 330.5 h timepoint (6.9 days posttreatment).
One significant finding (in Figure 2) of the first major report11 from the Human Microbiome Project was that the samples from the same body sites of different humans varied greatly with respect to phylogenetic diversity, yet were highly consistent with respect to presence of genes for certain metabolic pathways. This suggests that physiological functionality drives the microbiome community composition at each body site. Disruption of community biochemical activities may therefore better reflect microbiome dysfunction than taxonomic changes.12 Accordingly, biochemical tests were used to examine the pathways that were phenotypically active in the whole microbiome consortium before and after antibiotic exposure.
Figure 2.

Wild-type skin microbiome composition.
Notes: Average percent normalized abundance of the top 11 taxa from skin of four control fish, shown at the genus level. “Candidatus” are candidate names not yet officially approved. Captivus is a proposed genus in the Holosporaceae family. Captivus1 is identified as an “uncultured alpha proteobacterium,” whereas the best match to Captivus2 sequences is identified as “uncultured bacterium.” FukuN18 is an uncultured bacterium within the class Spartobacteria, isolated from a lake in Germany.26 B is an unnamed genus within the family Comamonadaceae. LD28 is a genus within the family Methlophilaceae.
As expected for a complex community, the skin micro-biome extracted before treatment was biochemically active, with 23 of 31 tests being positive (Table 1). Eight (35%) of these enzyme activities were lost after 3 days of antibiotic exposure, whereas four new activities were gained. The 3-day timepoint was selected because it was the earliest timepoint at which the total number of culturable bacteria was very similar to pretreatment. When results from four of the tests that were duplicated between the API-20E and API-20NE systems conflicted, the positive result was assumed to be more reflective of the capabilities of the microbiome. Although host-derived enzymes in the skin mucus could potentially cause false positive results in these systems, it is unlikely since these enzymes would be at very low concentrations compared to the bacterial enzymes in this culture-based system.
Table 1.
Biochemical culture results from skin microbiome samples
| Test | Activity | Controla | Treatb |
|---|---|---|---|
| ONPG/PNPG | β-Galactosidase | + | + |
| ADH/ADH | Arginine dihydrolase | + | +/− |
| LDC | Lysine decarboxylase | + | − |
| ODC | Ornithine decarboxylase | − | − |
| CIT/CIT | Citrate use | − | +/− |
| H2H | H2S from thiosulfate | + | − |
| URE/URE | Urease | − | − |
| TDA | Tryptophane deaminase | + | + |
| IND/TRPc | Indole production | + | − |
| VP | Acetoin production | − | + |
| GEL/GEL | Gelatinase | −/+ | − |
| GLU/GLUd | Glucose ox/ferm | + | + |
| GLU | Glucose fermentation | + | − |
| MAN/MAN | D-mannitol ox/ferm | + | + |
| IND Inositol ox/ferm | − | − | |
| SOR | D-sorbitol ox/ferm | − | + |
| RHA | L-rhamnose ox/ferm | + | + |
| SAC | D-saccharose ox/ferm | + | + |
| MEL | D-melibiose ox/ferm | − | + |
| AMY | Amygdaline ox/ferm | + | + |
| ARA/ARA | L-arabinose ox/ferm | + | +/− |
| NO3 | Nitrate reduction | + | + |
| ESC | Esculin β-glucosidase | + | + |
| MNE | D-mannose ox/ferm | + | − |
| NAG | N-acetyl-glucosamine ox/ferm | + | + |
| MAL | D-maltose ox/ferm | + | + |
| GNT | Gluconate ox/ferm | + | + |
| CAP | Capric acid ox/ferm | + | − |
| ADI | Adipic acid ox/ferm | + | − |
| MLT | Malic acid ox/ferm | + | + |
| PAC | Phenylacetic acid ox/ferm | − | − |
Notes: Results between all three fish in each group matched 100% of the time. Bold indicates the test result from the API-20E strip, and italics indicates the test result from the API-20NE strip.
Control is three untreated fish.
Treat is three fish after 3 days of rifampicin exposure.
IND and TRP are two names for the same test.
Glucose ox/ferm was positive in both API-20E and API 20NE before and after, but glucose fermentation was lost after exposure to air. Four tests that were shared between the two strips had conflicting results: ADH, CIT, GEL, and ARA.
Abbreviation: Ox/ferm, oxidation or fermentation of the respective carbon source.
To further examine the phenotypical properties of the skin microbiome consortium following antibiotic exposure, the susceptibility profile of the consortium was determined before and after 3 days of exposure (Table 2). Eighteen different antibiotics that target six different cellular pathways/processes were used. As expected, the microbiota was sensitive to rifampicin before exposure but fully resistant afterward. No change was seen with five drugs (AMP, OXA, BAC, VAN, and G; antibiotic codes are given in Table 2), but the microbiome consortium was completely resistant to four of those both before and after rifampicin exposure. All of the four target cell wall synthesis, so it may be reflective of the almost exclusive Gram-negative composition of the skin community (Figures 2 and 3). Interestingly, the one other drug examined that targets cell wall synthesis was ceftriaxone, and RIF selection resulted in a switch from sensitive to fully resistant. Given the polymicrobial nature of the microbiome, the reason for this is unclear. For antibiotics (GEN, KAN, and PMB) in which the difference between the ZOI before and after exposure was less than the standard deviations of the two measurements, the results were not significant. Community susceptibility increased more than the measured ZOI standard deviations for two drugs, AMK and TET, which both target protein synthesis. The rifampicin-selected microbiome community had a trend towards more sensitivity (larger ZOIs) to the aminoglycosides (AMK, GEN, KAN, and STR), which target the A-site on the 30S subunit of the ribosome.13,14 In contrast, erythromycin is a macrolide that targets the 50S subunit, which showed decreased susceptibility. Chloramphenicol, also targeting the 50S, showed decreased susceptibility. Although the microbiome was sensitive at the same level both before and after rifampicin selection against sulfisoxazole, at least some resistance to trimethoprim/sulfamethoxazole was selected.
Table 2.
Antibiogram of skin microbiome
| Untreated fish | Treated fish | Code | Antibiotic | Antibiotic target |
|---|---|---|---|---|
| 2.57±0.12 | 3.07±0.2 | AMK-30 | Amikacin | Protein synthesis |
| 2.7±0.35 | 3.17±0.15 | GEN-10 | Gentamicin | |
| 2.6±0.17 | 3.3±0.54 | KAN-30 | Kanamycin | |
| 2.0±0.56 | 2.37±0.25 | STR-10 | Streptomycin | |
| 4.07±0.12 | 2.33±0.42 | CHL-30 | Chloramphenicol | |
| 2.23±0.46 | 3.33±0.06 | TET-30 | Tetracycline | |
| 1.8±0.1 | 1.27±0.32 | ERY-15 | Erythromycin | |
| 0 | 0 | AMP-10 | Ampicillin | Cell wall synthesis |
| 4.07±0.38 | 0 | CRO-30 | Ceftriaxone | |
| 0 | 0 | OXA-1 | Oxacillin | |
| 0 | 0 | BAC-10 | Bacitracin | |
| 0 | 0 | VAN-30 | Vancomycin | |
| 1.97±0.15 | 2.33±0.58 | PMB-300 | Polymyxin B | Cell membrane |
| 4.23±0.21 | 2.9±0.36 | CIP-5 | Ciprofloxacin | DNA gyrase |
| 4.4±0.17 | 3.77±0.06 | NAL-30 | Nalidixic acid | |
| 1.9±0.1 | 0 | RIF-5 | Rifampicin | RNA polymerase |
| 2.97±0.25 | 3±0.01 | G-.25 | Sulfisoxazole | Folic acid synthesis |
| 3.5±0.3 | 0 | SXT | Trimethoprim/sulfamethoxazole |
Figure 3.
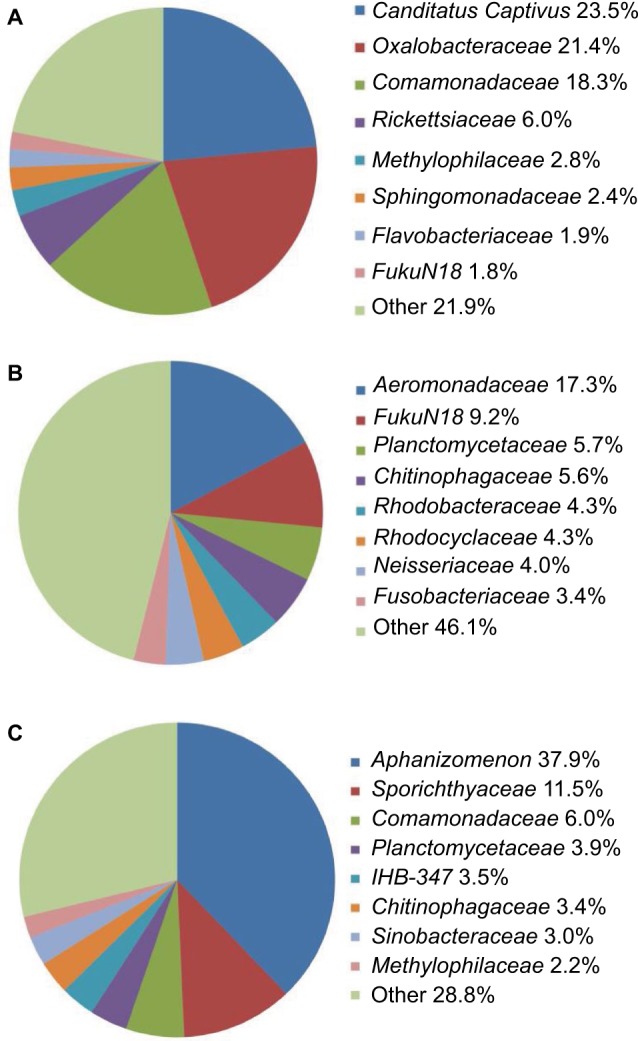
Comparison of fish skin, fish gut, and water microbiomes. (A) Eight most abundant families from fish skin (average normalized abundance from four fish). (B) Eight most abundant families from fish gut (average normalized abundance from four fish). (C) Eight most abundant families from water (average normalized abundance from two separate water samples).
Notes: Average percent abundances shown next to family name. IHB-347 is within phylum Cyanobacteria.
The many susceptibility changes strongly suggest that a major change in the microbiome composition is caused by rifampicin selection. The DNA sequencing, via 16S profiling, was used to confirm this. Given the inherent limitations of PCR amplification and next-generation sequencing with accurate identification and quantification of rare sequences,15 this analysis focused on the most abundant taxa (≥1% of population).
The skin microbiota from untreated or fish exposed to rifampicin for 3 days was inoculated onto Mueller–Hinton plates with six antibiotic disks/plates and incubated at 25°C for 48 h. The mean and standard deviation ZOI (in centimeters) for three fish from each condition are shown. The number in the code is the amount of antibiotic in the disk in micrograms, except for G-.25 (250 mg) and SXT (1.25 mg of trimethoprim and 23.75 mg of sulfamethoxazole).
Consistent with previous studies of animal microbiomes, the fish skin microbiome is diverse, but not equitable, being dominated by a few taxa.11,16,17 The dominant five taxa comprise 57.8% of all recovered sequences, and the top eleven are 70.4% of all sequences (Figure 2). In total, 648 Eubacterial taxa were recovered from the four control fish, with 419 of those taxa shared by at least two of the four fish. The consistency between fish was high, as only one taxa (18th-most abundant, Xanthomonas, lacking from one fish) within the 58 most abundant (representing 91.5% of all sequences) not being shared among all four fish, whereas all of the 229 taxa identified from just one out of the four fish were rare (below the abundance level of 0.04%). Fish from the same aquarium have very similar skin microbiome compositions.
Fish skin, fish gut, and lake water microbiome communities contain mostly the same families, but at strongly different abundances (Figure 3). Of the 308 total bacterial families recovered on the skin, 237 are shared between skin and water, representing 99.8% of the sequence abundance on skin. Of the 398 families in the gut, 242 are shared between the gut and skin, which is 98.5% of the gut sequence abundance. In contrast, only one family in the top eight most abundant is present in both the gut (9.2%) and skin (1.8%) microbiomes, which is FukuN18. These two microbiomes also only share one abundant order, the one containing FukuN18, which is Chthoniobacterales. For both skin and gut, all of the eight families are within the Gram-negative lineage. Two of the top eight skin-derived families are shared with the surrounding water, Comamonadaceae (6.0% of water and 18.3% of skin) and Methylophilaceae (2.2% of water and 2.8% of skin). Only one water family, Sporichthyaceae, is a Gram-positive clade.
Rifampicin exposure causes dramatic changes in the skin microbiome composition. The top five genera that dominate (57.8%) on fish skin before rifampicin introduction at the six exposure timepoints only comprise 17.9%, 1.2%, 12.2%, 8.9%, 5.1%, and 5.1% of the skin composition (Figure 4). The genus Myroides (in the family Flavobacteriaceae), a consistently minor component (0.007%±0.001%) before exposure, is selected during the first three timepoints, comprising 91.1%, 34.8%, and 15.4% of the population. Myroides dominates the microbiome during the first 3 days of antibiotic selection, but is not stable. The top five genera that dominate after 7 days of antibiotic exposure (28.7% Variovorax, 21.6% Hydrogenophaga, 7.5% Mitsuaria, 7.2% Pelomonas, and 3.9% Ottowia), only total 0.038% of the pretreated microbiome. Interestingly, four of those five genera are in the family Comamonadaceae (Mitsuaria is uncertain at family level, but in same order of Burkholderiales). Seven taxa increase in abundance in the majority of the treatment timepoints, especially at the end. These genera (Variovorax, Vibrio, Pelomonas, Hydrogenophaga, Pseudomonas, Mitsuaria, and family Spirochaeta) are apparently selected by the antibiotic.
Figure 4.

Antibiotic effect on skin microbiome.
Notes: Abundance of dominant genera across time during the week of antibiotic exposure. At 5.5 h is represented the average of four fish pre-exposure. Exposure timepoints are at 50.5, 63.5, 88.5, 115.5, 136.6, and 161.5 h.
Rifampicin selection also alters the gut microbiome composition. Of the 16 dominant OTUs shown in Figure 5, the first eight have a declining pattern (highest abundance in the untreated gut, lower at the 17.5-h timepoint, and least at the ending 115.5-h timepoint). These eight are abundant in the untreated gut (together 46.6% of all sequences), dropping to 19.6% at 17.5 h, and at 115.5 h only compose 0.76% of all sequences, an approximately 60-fold final decline. Rifampicin exposure thus strongly selects against this originally dominating group. Only two of the six gut samples collected during antibiotic exposure had an acceptable sequence quality. As was observed on the skin, the genus Myroides comes to dominate (0.0054% pretreatment, 63.3% at 115.5 h), although this effect is significantly delayed. Five of the OTUs (family Cetobacterium along with genera Variovorax, Myroides, Achromobacter, and Vibrio) have an increasing pattern (lowest abundance in the pretreatment condition, more at 17.5 h, and then maximum at 115.5 h). These five account for a total of 4.0% (with Cetobacterium being 3.4% by itself) of the control gut composition, yet together compose almost all (94.2%) of the gut community at 115.5 h. Overall, following the rifampicin treatment, the gut exhibits a clear selection away from the initial abundant community members to an almost completely different composition. Although the skin is dominated by the family Comamonadaceae (top five genera totaling 68.9%) at the last 161.5-h timepoint (and already having 32.4% at 115.5 h), the gut is not at 115.5 h (contains only the abundant genus Variovorax at 15.5%).
Figure 5.

Antibiotic effect on gut microbiome.
Notes: Abundance of dominant genera across time during the week of antibiotic exposure. At 5.5 h is represented the average of four fish pre-exposure. Exposure timepoints are at 17.5 and 115.5 h. *For operational taxonomic units, where the genus is uncertain, the family name is given, except for Cyanobacteria, which is a phylum.
Selection by rifampicin has a similar effect on the gut and skin microbiomes. Comparing the two 115.5-h timepoints (Tables 3 and 4), the most abundant genus is the same, Myroides (63.3% of gut and 15.4% of skin). Six of the eight most abundant genera in the gut are also present within the 30 most abundant genera of the skin. Although the skin only has 87 genera present, 68 of those are shared with the gut (contains 275 genera) at the same timepoint.
Table 3.
Skin microbiome diversity before, during, and after treatment
| Timepoint | αa | Top 20 | 1% Ab | 0.1% Ab | Dominate | β | Relate |
|---|---|---|---|---|---|---|---|
| C1 | 294 | 90.6% | 12 | 46 | 32.3% Captivus1, 24.7% Massilia, 7.0% Captivus2 | – | – |
| C2 | 345 | 80.9% | 18 | 79 | 23.6% Massilia, 12.1% Ottowia, 9.0% Captivus1 | – | – |
| C3 | 414 | 78.7% | 19 | 82 | 19.0% Massilia, 17.0% Captivus1, 6.8% Ottowia | – | – |
| C4 | 471 | 80.5% | 18 | 71 | 20.5% Captivus1, 17.7% Massilia, 7.2% Rickettsia | – | – |
| AvgC | 381±78 | 80.1% | 17±3 | 70±16 | 21.2% Massilia, 19.7% Captivus1, 6.0% Rickettsia | 0 | 80.1% |
| T50.5 | 155 | 97.0% | 11 | 22 | 60.8% Myroides, 13.1% Massilia, 3.5% Captivus2 | 9 | 30.0% |
| T63.5 | 64 | 99.4% | 2 | 16 | 91.1% Myroides, 3.1% Vibrio, 0.7% Captivus2 | 12 | 2.5% |
| T88.5 | 75 | 94.9% | 14 | 33 | 34.8% Myroides, 13.4% Variovorax, 11.2% Hydrogenophaga | 10 | 39.0% |
| T115.5 | 87 | 93.1% | 15 | 44 | 15.4% Myroides, 13.8% Hydrogenophaga, 8.7% Ottowia | 14 | 21.7% |
| T136.5 | 136 | 91.0% | 17 | 39 | 17.5% Variovorax, 13.8% Hydrogenophaga, 13.0% Myroides | 13 | 26.6% |
| T161.5 | 128 | 91.0% | 15 | 43 | 28.7% Variovorax, 21.6% Hydrogenohaga, 7.5% Mitsuaria | 14 | 37.0% |
| AvgT | 108±37 | 90.6% | 12±5 | 33±12 | 36.5% Myroides, 11.3% Variovorax, 10.2% Hydrogenophaga | 12±2 | 26.1% |
| R20.5 | 166 | 98.1% | 12 | 23 | 46.7% Comamonas, 22.1% Vibrio, 8.5% Pseudomonas | 12 | 7.7% |
| R44.5 | 180 | 97.1% | 13 | 26 | 37.5% Variovorax, 13.9% Pseudomonas, 12.6% Comamonas | 14 | 54.6% |
| R68.5 | 184 | 98.2% | 13 | 24 | 34.9% Variovorax, 10.6% Flavobacterium, 9.7% Hydrogenophaga | 16 | 46.5% |
| R120.5 | 148 | 97.4% | 14 | 29 | 24.0% Mitsuaria, 18.8% Flavobacterium, 14.0% Hydrogenophaga | 15 | 27.1% |
| R165.5b | 153 | 94.7% | 13 | 33 | 38.0% Flavobacterium, 9.9% Pseudomonas, 9.7% Mitsuaria | 17 | 44.2% |
| AvgR | 166±16 | 95.2% | 13±0.7 | 27±4 | 17.7% Variovorax, 15.0% Comamonas, 14.1% Flavobacterium | 15±2 | 33.5% |
Notes: All numbers are based on genus-level identification of sequences. Top 20 is the cumulative percent abundance of the 20 most abundant taxa. 1% Ab is the number of taxa with an abundance of ≥1%, whereas 0.1% Ab is the number of taxa with abundance of ≥0.1%. Dominate is the percent abundance and identity of the three most abundant genera. β is the absence/presence difference in the top 20 taxa between the sample and the average of the control samples (AvgC). Relate is the cumulative percent abundance of the same 20 taxa that are most abundant in the average of the control samples, thus how related the dominant taxa are to the pretreatment samples.
α is richness, the total number of OTUs in the sample.
For the R165.5 timepoint, if the abundance of Flavobacterium is subtracted, the relate value drops to 6.2%. C and a number, the four control fish before treatment; R followed by a number, the hours of recovery after removal of rifampicin; T and a number, the hours of rifampicin treatment.
Abbreviations: Ab, abundance; AvgC, mean and standard deviation of the controls; AvgR, mean and standard deviation of the recovery samples; AvgT, mean and standard deviation of the six treatment samples; OTU, operational taxonomic unit.
Table 4.
Gut microbiome diversity before, during, and after treatment
| Timepoint | αa | Top 20 | 1% Ab | 0.1% Ab | Dominate | β | Relate |
|---|---|---|---|---|---|---|---|
| G1 | 538 | 64.7% | 23 | 99 | 12.9% Aeromonas, 7.4% FukuN18, 5.3% Rhodobacter | – | – |
| G2 | 508 | 75.4% | 14 | 80 | 26.3% Aeromonas, 10.7% FukuN18, 10.2% Cetobacterium | – | – |
| G3 | 436 | 61.2% | 21 | 111 | 15.6% Aeromonas, 7.2% X1b, 3.6% Rhodobacter | – | – |
| G4 | 402 | 74.4% | 22 | 80 | 16.5% FukuN18, 14.5% Aeromonas, 5.1% IHB-347c | – | – |
| AvgG | 471±63 | 65.5% | 20±4 | 93±15 | 17.3% Aeromonas, 9.2% FukuN18, 4.2% X1b | 0 | 65.5% |
| gT17.5 | 168 | 89.5% | 10 | 58 | 39.3% Ottowia, 14.0% Aeromonas, 9.0% Cetobacterium | 8 | 68.1% |
| gT115.5 | 282 | 97.6% | 4 | 21 | 63.3% Myroides, 15.5% Variovorax, 13.2% Cetobacterium | 7 | 14.2% |
| AvggT | 225 | 7 | 40 | 31.7% Myroides, 19.7% Ottowia, 11.1% Cetobacterium | 7 | 41.7% | |
| gR20.5 | 218 | 97.3% | 8 | 23 | 47.5% Variovorax, 11.3% Hydrogenophaga, 11.3% Cetobacterium | 3 | 12.0% |
| gR68.5d | 142 | 99.2% | 5 | 17 | 65.2% Cetobacterium, 19.9% Variovorax, 5.65% Shewanella | 1 | 65.4% |
| gR120.5d | 96 | 99.8% | 5 | 14 | 69.8% Cetobacterium, 19.9% Variovorax, 5.65% Shewanella | 3 | 75.5% |
| gR165.5 | 234 | 92.4% | 17 | 42 | 34.7% Oryza, 17.8% Flavobacterium, 10.5% Mitsuaria | 3 | 2.9% |
| AvggR | 173±65 | 96.1% | 9±6 | 24±13 | 36.7% Cetobacterium, 17.4% Variovorax, 5.7% Shewanella | 2 | 65.5% |
Notes: All numbers are based on genus-level identification of sequences. Top 20 is the cumulative percent abundance of the 20 most abundant taxa. 1% Ab is the number of taxa with an abundance of ≥1%, whereas 0.1% Ab is the number of taxa with abundance of ≥0.1%. Dominate is the percent abundance and identity of the three most abundant genera. β is the absence/presence difference in the top 20 taxa between the sample and the average of the control samples (AvgC). Relate is the cumulative percent abundance of the same 20 taxa that are most abundant in the average of the control samples, thus how related the dominant taxa are to the pretreatment samples.
α is richness, the total number of OTUs in the sample.
X1 is an uncultured genus from the family Chitinophagaceae.
IHB-347 is an organism of indeterminate placement in the phylum Cyanobacteria.
If the one genus Cetobacterium is removed from the analysis, then the relate value for sample gR68.5 is 0.2% and for gR120.5 is 5.7%. G and a number, the four control fish before treatment; gR followed by a number, the hours of recovery after removal of rifampicin; gT and a number, the hours of rifampicin treatment.
Abbreviations: Ab, abundance; AvgG, mean and standard deviation of the controls; AvgT, mean (standard deviation cannot be obtained from two numbers) of the two treatment samples; AvggT, mean of two treatment samples (SD cannot be determined from less than 3 samples); OTU, operational taxonomic unit.
Rifampicin exposure causes a loss of over half of the OTUs from the skin after 50.5 h and thus a major decrease in diversity (Table 3). There is only a small return of diversity during the week of recovery. The proportions of the most abundant organisms likewise change during exposure with little return to the pretreatment state during recovery. The effects are also clear at the level of order (two taxonomic levels above genus). Before exposure, the four fish contain 172 different orders among them (gamma diversity), with 63 of those (37%) being present on the skin of all four fish. Forty-nine orders were recovered from one of the fish only, yet they are all rare (highest abundance of 0.05%). In contrast, from the six treated fish, only 116 orders were recovered, with only 30 present (26%) in all six fish, and 37 found on one fish only (top abundance of 0.1%). During the 1-week recovery after the antibiotic, the diversity loss continued, with only 85 orders being recovered from the five sampled fish. Thirty-one of those orders were present (36%) on all five fish, and 22 on one fish only (highest abundance of 0.01%).
The microbiome of the gut is affected more severely than that of the skin (Table 4). Before exposure, the gut microbiome of individual fish is more diverse (higher number of OTUs) and is more even (more OTUs with ≥0.1% abundance and the 20 most abundant OTUs making a smaller proportion of the total). The gut microbiome loses even more diversity (48% of OTUs lost) during rifampicin exposure, and this does not return but continues to decline during the 1-week recovery period. The composition of abundant microbes also changes even more than the skin. Neither the skin microbiomes nor the gut microbiomes return to the pretreatment composition during the recovery period, and recovery communities are more similar to the treated than the untreated (Figure 6).
Figure 6.
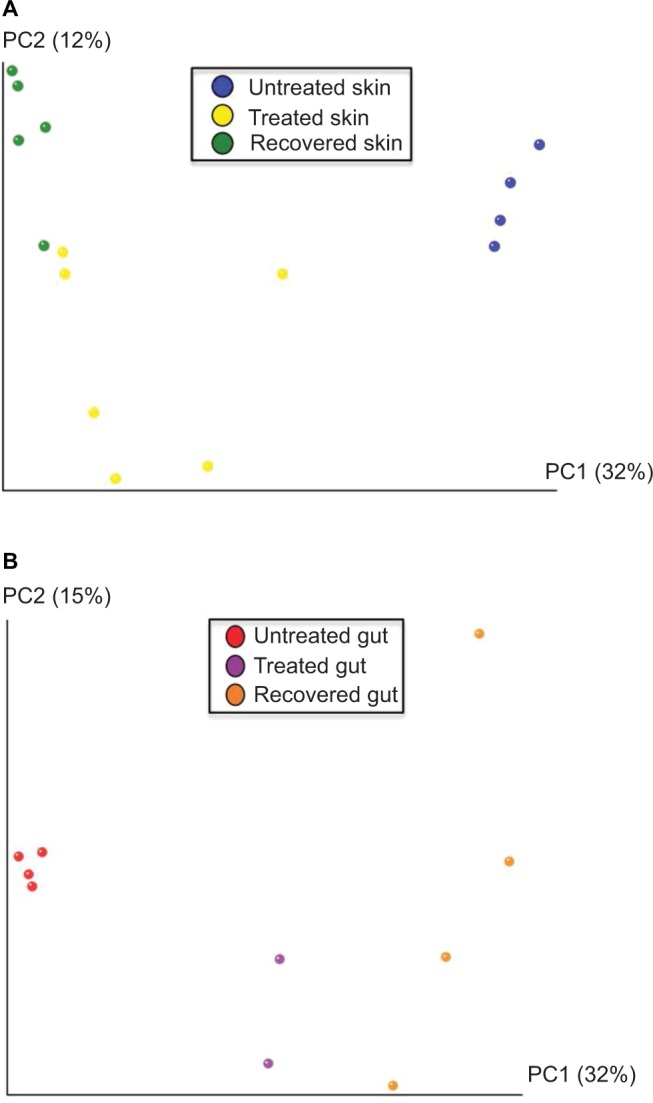
Recovered skin and gut microbiomes do not return to pretreated composition. Unweighted UniFrac-based principal components analysis. (A) Skin and (B) gut.
Abbreviation: PC, principal components analysis.
Discussion
To date, this is the most extensive study of antibiotic effects on the microbiome using a fish model. This is also the first study specifically of rifampicin. The gut microbiome of captive and wild-caught zebra fish (Danio rerio) contains similar genera16 as the mosquito fish gut, but at different abundances. The dominant genera from three zebra fish were Aeromonas (average abundance of 29.2%), Streptophyta (13.5%), Cetobacterium (13.3%), Pseudomonas (10.4%), and Vibrio (4.7%), with the four control fish exhibiting 17.3%, 0%, 3.4%, 0.6%, and 0.08% of those genera, respectively. It should be noted that Streptophyta is in the class Cyanobacteria, and Gambusia fish gut contained 3.3% of an uncultured Cyanobacteria, unclassified at the genus and family level. Zebra fish (family Cyprinidae) and mosquito fish (family Poeciliidae) do not join taxonomically until the level of order, Cyprinodontiformes, and thus are somewhat genetically distant, with host factors potentially selecting for different gut microbiota compositions. A significant physiological and anatomical difference also exists; zebrafish lay eggs, whereas mosquitofish have internal fertilization and give live birth.
Although most of the same genera are shared between the Gambusia skin and the gut, the dominant organisms in high abundance are very different. And although in fish both environments are mucosal surfaces, one likely major difference is oxygen level, with the skin being more aerobic and the gut more anaerobic. Of the abundant families in the gut, Aeromonadaceae and Rhodobacteraceae are facultative anaerobes, Rhodocyclaceae contains both facultative and obligate anaerobes, Planctomycetaceae, Chitinophagaceae, and Neisseriaceae are obligate aerobes, and Fusobacteriaceae contains obligate anaerobes. FukuN18 is uncultured and the metabolic classification is unknown. Of the abundant skin families, Oxalobacteraceae includes both obligate aerobes and anaerobes, Comamonadaceae, Sphingomonadaceae, and Methylophilaceae are obligate aerobes, and Flavobacteriaceae includes obligate aerobes and microaerobic species. Rickettiaceae are obligate intracellular organisms. Captivus is uncultured and has unknown oxygen requirements.
A previous study on the Gambusia skin microbiome suggested that this mucosal surface may be selective, not simply a passive recipient of bacteria from the water column.6 The differences in community composition between the skin and water observed in this study (Figure 3) support this conclusion.
Disruption of the fish skin microbiome by rifampicin results in only a transient (<38.5 h) loss of culturable bacteria; however, drug-resistant bacteria are rapidly selected for and persist for at least 2 weeks following the removal of antibiotic. Not only is the taxonomic distribution of the skin bacterial community strongly altered, the biochemical functions (of the culturable fraction) are also altered (43% change in results, 12 of 28 tests) during exposure. The biochemical function is not measured in most studies of antibiotic effects on microbiomes, so this interesting potential functional difference needs further investigation. The extent of gut microbiome loss of culturability is greater than the skin, and takes longer to recover (63.5 h). Antibiotic exposure not only changes the skin and gut community composition, but also reduces the overall diversity. Neither the microbiome (gut or skin) diversity nor the composition returns to pretreatment values during the 1-week recovery period without antibiotic. Grouping of communities on principal components analysis (Figure 6) suggests the communities stabilize to a different composition during recovery. It remains to be seen if, given more time, the communities would return to the pretreatment composition. In addition to the loss of diversity, the evenness is also reduced during antibiotic treatment (Table 3; rise in percent abundance of the Top 20), and drop in both 1% abundance (Ab) and 0.1% Ab). Rifampicin also causes a major loss of diversity and evenness in the gut microbiome (Table 4). Treatment of salmon with oxytetracycline in food for up to 25 days did not alter overall plate counts from intestine, but did select for culturable resistant organisms and reduced community diversity as measured by RFLP.18
At 115.5 h of treatment, the genera present on the gut and skin overlap more than the pretreatment state. This may be due to an initial rapid depletion of skin organisms, and then recolonization of the skin by organisms from the gut via feces through the water column. It should be noted that in a recent study on rainbow trout, about 50% of the microbiome community diversity was present within the skin epithelium, so this may be serving as a major reservoir to reseed the mucosa, and was not examined in this study.19
Rifampicin-resistant organisms are likely present at low levels in the Gambusia skin microbiome (the detection limit iŝ0.1%–0.01% of the culturable population) and selected by the antibiotic exposure. In this case, the genus Myroides was selected earliest and to the greatest degree. Rifampicin resistance was expected, but resistance to ceftriaxone and trimethoprim/sulfamethoxazole also appeared, for as yet unclear reasons. There is very little literature on the susceptibilities of Myroides spp., and none on fish-derived strains. Three patients with urinary tract infections from Myroides odoratimimus were successfully treated with a combination of ciprofloxacin and rifampicin.20 Myroides spp. are noted to be commonly found in aquatic environments, and can be opportunistic pathogens.21 Myroides organisms are reported to often be intrinsically resistant to β-lactam antibiotics. It is unclear at this time if the Myroides spp. present in fish used in this study are acting as a commensal or are pathogenic.
An interesting result in this study was the loss of Rickettsia from the fish. In the skin microbiome before treatment, the genus Rickettsia represented 6.04% of all sequences. This was the only genus recovered in the Rickettsiaceae family, whereas 14 OTUs were present in the order Rickettsiales, comprising 32.9% of all sequences (25.6% coming from two OTUs of Captivus). Thus, these organisms are a dominant component in the normal fish skin microbiome, with Rickettsia being the third-most abundant genus, and Rickettsiales counting for almost one-third of all sequences. At the first treatment timepoint of 50.5 h, Rickettsia had dropped to 0.25% and the Rickettsiales were down to 6.11%. At the last timepoint of 161.5 h, Rickettsia was at only 0.013% and Rickettsiales was at only 0.093%, and contained only five OTUs, a drastic loss of abundance and diversity. Across the five fish during the week of post-antibiotic recovery, no Rickettsia sequences were found and Rickettsiales only comprised an average of 0.013% of all sequences. Likewise, the Rickettsia genus was present in the four control fish gut samples at an average of 0.073%, declined in the treatment samples to 0.003%, and was at 0.0004% in the first recovery timepoint of 20.5 h, and lacking in the latter two recovery points. Rifampicin cleared Rickettsia from the fish skin and gut. Given that Rickettsia is an obligate intracellular organism, this highlights the ability of rifampicin to reach this niche. Whether Rickettsia plays a pathogenic, commensal, or other role with the fish is unclear at this point in the literature.
The API-based approach to examine community biochemical activities is limited to organisms that can live in those culture conditions, so it may not capture all of the biodiversity present. Thus, it determines the biochemical capabilities of community members, but not certainty that those pathways are active in the mucosal microbiome environment. The alignment of API-derived biochemical results with community 16S-derived identifications is unfeasible, because prediction of biochemical profiles from genus-level identification is too variable. Although methods are developing for predicting the biochemical pathways in a community from only 16S profile data, such as PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states),22 this bioinformatic approach is based on genomic data of human-derived strains, and lacks power with the fish-derived organisms of this study. However, the net loss of four of the community API biochemical activities at 3 days of treatment is consistent with the diversity loss observed by the 16S sequencing data.
In mice treated with one of three different antibiotics (clindamycin, ampicillin, or enrofloxacin) for 2 days, susceptibility to a following dose of C. difficile varied from high to moderate to low, respectively.23 This suggests that individual antibiotics affect a key function of the normal microbiome, namely colonization resistance, very differently. The controllable vertebrate model used in this study is now being used to investigate the effects on the host organism resulting from significant antibiotic-induced microbiome community changes,24,25 and driving factors during the recovery of the microbiome after antibiotic stress. A 3-day exposure of fish to rifampicin at the same concentration and same method used in this study resulted in reduced resistance to the pathogen Edwardsiella ictaluri, reduced weight gain over a month, and increased susceptibility to osmotic stress, compared to controls. The mechanisms behind these negative effects are being investigated.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Ferrer M, Méndez-Garcia C, Rojo D, Barbas C, Moya A. Antibiotic use and microbiome function. Biochem Pharmacol. 2016 doi: 10.1016/j.bcp.2016.09.007. In press. [DOI] [PubMed] [Google Scholar]
- 2.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011;108:4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dethlefsen L, Huse S, Sogin ML, Relman DA. The correct pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fouhy F, Guinane CM, Hussey S, et al. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob Agents Chemother. 2012;56:5811–5820. doi: 10.1128/AAC.00789-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonopoulos DA, Huse SM, Morrison HG, Schmidt TM, Sogin ML, Young VB. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun. 2009;77:2367–2375. doi: 10.1128/IAI.01520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leonard AB, Carlson JM, Bishoff DE, et al. The skin microbiome of Gambusia affinis is defined and selective. Adv Microbiol. 2014;4:335–343. [Google Scholar]
- 7. Drugs.com [webpage on the Internet] Rifampin Dosage. Available from http://www.drugs.com/dosage/rifampin.html.
- 8.Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 11.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Integrative HMP (iHMP) Research Network Consortium The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;16:276–289. doi: 10.1016/j.chom.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong CH, Hendrix M, Priestley ES, Greenberg WA. Specificity of aminoglycoside antibiotics for the A-site of the decoding region of ribosomal RNA. Chem Biol. 1998;5:397–406. doi: 10.1016/s1074-5521(98)90073-4. [DOI] [PubMed] [Google Scholar]
- 14.Mehta R, Champney WS. 30S ribosomal subunit assembly is a target for inhibition by aminoglycosides in Escherichia coli. Antimicrob Agents Chemother. 2002;46:1546–1549. doi: 10.1128/AAC.46.5.1546-1549.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CK, Herbold CW, Polson SW, et al. Groundtruthing next-gen sequencing for microbial ecology-biases and errors in community structure estimates from PCR amplicon pyrosequencing. PLoS One. 2012;7:e44224. doi: 10.1371/journal.pone.0044224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roeselers G, Mittge EK, Stephens WZ, et al. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011;5:1595–1608. doi: 10.1038/ismej.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR. Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 2016;14:e2000225. doi: 10.1371/journal.pbio.2000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarrete P, Mardones P, Opazo R, Espejo R, Romero J. Oxytetracycline treatment reduces bacterial diversity of intestinal microbiota of Atlantic salmon. J Aquat Anim Health. 2008;20:177–183. doi: 10.1577/H07-043.1. [DOI] [PubMed] [Google Scholar]
- 19.Lowrey L, Woodhams DC, Tacchi L, Salinas I. Topographical mapping of the Rainbow Trout (Oncorhynchus mykiss) microbiome reveals a diverse bacterial community with antifungal properties in the skin. Appl Environ Microbiol. 2015;81:6915–6925. doi: 10.1128/AEM.01826-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ktari S, Mnif B, Koubaa M, et al. Nosocomial outbreak of Myroides odoratimimus urinary tract infection in a Tunisian hospital. J Hosp Infect. 2012;80:77–81. doi: 10.1016/j.jhin.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Mammeri H, Bellais S, Nordmann P. Chromosome-encoded β-lactamases TUS-1 and MUS-1 from Myroides odoratus and Myroides odoratimimus (formerly Flavobacterium odoratum), new members of the lineage of molecular subclass B1 metalloenzymes. Antimicrob Agents Chemother. 2002;46:3561–3567. doi: 10.1128/AAC.46.11.3561-3567.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buffie CG, Bucci V, Stein RR, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlson JM, Hyde ER, Petrosino JF, Manage AB, Primm TP. The host effects of Gambusia affinis with an antibiotic-disrupted microbiome. Comp Biochem Physiol C Toxicol Pharmacol. 2015;178:163–168. doi: 10.1016/j.cbpc.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Carlson JM, Chavez O, Aggarwal S, Primm TP. Examination of host phenotypes in Gambusia affinis following antibiotic treatment. J Vis Exp. 2017;120:e55170. doi: 10.3791/55170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glöckner FO, Zaichikov E, Belkova N, et al. Comparative 16S rRNA analysis of lake bacterioplankton reveals globally distributed phylogenetic clusters including an abundant group of actinobacteria. Appl Environ Microbiol. 2000;66:5053–5065. doi: 10.1128/aem.66.11.5053-5065.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]