

Abstract
3-hydroxypropionic acid (3-HP) is an important platform chemical used as a precursor for production of added-value compounds such as acrylic acid. Metabolically engineered yeast, Escherichia coli, cyanobacteria and other microorganisms have been developed for the biosynthesis of 3-HP. Attempts to overproduce this compound in recombinant Pseudomonas denitrificans revealed that 3-HP is consumed by this microorganism using the catabolic enzymes encoded by genes hpdH, hbdH and mmsA. 3-HP-inducible systems controlling the expression of these genes have been predicted in proteobacteria and actinobacteria. In this study, we identify and characterise 3-HP-inducible promoters and their corresponding LysR-type transcriptional regulators from Pseudomonas putida KT2440. A newly-developed modular reporter system proved possible to demonstrate that PpMmsR/PmmsA and PpHpdR/PhpdH are orthogonal and highly inducible by 3-HP in E. coli (12.3- and 23.3-fold, respectively) and Cupriavidus necator (51.5- and 516.6-fold, respectively). Bioinformatics and mutagenesis analyses revealed a conserved 40-nucleotide sequence in the hpdH promoter, which plays a key role in HpdR-mediated transcription activation. We investigate the kinetics and dynamics of the PpHpdR/PhpdH switchable system in response to 3-HP and show that it is also induced by both enantiomers of 3-hydroxybutyrate. These findings pave the way for use of the 3-HP-inducible system in synthetic biology and biotechnology applications.
Introduction
Chemicals and fuels can be produced from renewable or waste feedstocks using metabolically engineered microorganisms, which may aid to reducing greenhouse gas emissions1–3. Recently, significant research efforts have been directed towards developing microorganisms for the biosynthesis of value-added chemicals, including 3-hydroxypropionic acid (3-HP)4–7. 3-HP is a biotechnologically attractive platform chemical that can be used as a precursor for biosynthesis of acetaldehyde, acrylate, acrylamide, methylacrylate, and 1,3-propanediol6, as well as biodegradable polymer poly-3-HP8. A number of recombinant strains using Corynebacterium glutamicum, Escherichia coli, Klebsiella pneumoniae, Lactobacillus reuteri, Pseudomonas denitrificans, Synechocystis sp., Synechococcus elongatus and Saccharomyces cerevisiae as chassis9–16, and a few alternative metabolic pathways have been developed for 3-HP biosynthesis using intermediates such as β-alanine, malonyl-CoA, propionyl-CoA, glycerol and lactate17–21. Although, relatively high titres of 3-HP have been reported in E. coli and K. pneumoniae 18, 22, the challenge remains to develop a sustainable biotechnological production of this carboxylic acid23.
3-HP can be efficiently assimilated and utilised as a carbon and energy source by bacteria. P. denitrificans possesses 3-HP dehydrogenase and 3-hydroxyisobutyrate dehydrogenase activities which contribute to 3-HP degradation10, 24. The expression of some genes related to 3-HP metabolism in P. denitrificans have been shown to be strongly induced by 3-HP and putatively controlled by transcriptional regulators (TR)24, which belong to the family of LysR-type TRs (LTTRs)25.
LTTRs contain a conserved protein structure with a DNA binding helix-turn-helix motif at the N-terminus and an effector binding domain at the C-terminus. They are typically activated by small effector molecules and regulate the transcription of metabolism-related genes. Most often, LTTRs are encoded in opposite direction of the gene or operon that they regulate. They usually interact with at least two TR binding sites in the intergenic region25, 26. The regulatory binding site is located 60–80 nucleotides upstream with respect to the transcriptional start site of the ligand-metabolizing gene. This motif has been characterised as T-N11-A, which can vary in length and/or both nucleotide composition. Frequently, this site overlaps with the promoter of the LTTR gene mediating negative autoregulation. The activator binding site is usually adjacent or overlaps with the −35 box of the promoter25. Besides, examples with a single and multiple LTTR-binding sites have also been reported27, 28. Based on full length and domain analysis, in Pseudomonas LTTRs can be dissected into 9 evolutionary different phylogenetic groups29.
LTTR- and other TR-based switchable (inducible or repressible) systems regulate microbial gene expression in response to the change of intracellular levels of metabolites and play an important role in governing metabolic pathways and networks. Recently, a number of such switchable systems have been adapted as genetically-encoded biosensors and reporters that respond to a variety of native and non-native compounds30–34. Both the native and synthetic systems, in combination with a reporter gene, have been used to screen for metabolically engineered microbial strains enabling selection for microorganisms with improved production of target compounds35–38. They have also been applied as metabolite-responsive gene switches and dynamic regulators of metabolic pathways, in which levels of upstream or downstream gene expression are continuously adjusted to balance metabolic intermediate levels increasing the flux towards the product of interest37, 39.
Pseudomonas are an attractive source for exploration of novel gene targets such as TR-based switchable systems. Particularly, Pseudomonas putida KT2440 exhibits combinations of features characteristic to aquatic oligotrophs and terrestrial copiotrophs indicating that this bacterium has adapted functional capabilities permitting to thrive in various environments40. This genetically diverse microorganism contains a high affinity nutrient acquisition and metabolite efflux, as well as catabolic enzymes such as mono- and di-oxygenases, oxidoreductases and dehydrogenases. Additionally, P. putida also possesses a wide range of gene expression control systems involving various sigma factors and regulators which form a rich genomic basis for the exceptional metabolism versatility.
Here, we report the identification and characterisation of a 3-HP-inducible system from P. putida, which is composed of a LTTR and a corresponding 3-HP-responsive promoter. As demonstrated in this study, it can be used to control gene expression orthogonally in E. coli, a model gammaproteobacterium, and Cupriavidus necator, a chemolithoautotrophic betaproteobacterium with the potential to produce chemicals from carbon dioxide. A comprehensive analysis of the promoter region is performed to establish a consensus sequence required for potential TR binding. The characterised switchable system can be exploited as 3-HP biosensor, as it shows a high specificity and a wide induction range for this compound, as well as genetic element for the construction of autoregulated metabolic pathways aiming to improve the production of bio-based 3-HP.
Results
Identification of 3-hydroxypropionic acid-inducible systems in P. putida KT2440 and C. necator H16
Three enzymes have been identified in P. denitrificans to be involved in 3-HP degradation24. They are arranged in two operons. The 3-hydroxypropionate dehydrogenase (hpdH) in one operon and the methylmalonate-semialdehyde dehydrogenase (mmsA) and 3-hydroxyisobutyrate dehydrogenase (mmsB, also referred to as hbdH-4) in the second operon. In the opposite direction of each operon, a gene encoding a LTTR is located which was proposed to be required for 3-HP-inducible activation of gene transcription of its respective operon24. Homologues of the 3-HP catabolic genes have been found in various microbial genera including Cupriavidus 24. In this study we identified HpdH, MmsA and MmsB homologues in the genomes of P. putida KT2440 and C. necator H16 (Supplementary Table S1). In P. putida, the 3-HP catabolic genes and their respective LTTRs are as arranged as in P. denitrificans (Fig. 1a). In C. necator, however, the arrangement of genes is different. A P. denitrificans MmsA homolog with 49% protein sequence identity and 96% coverage is located between the C. necator hpdH and its putative LTTR (here termed HpdR). The short intergenic region of 27 bp between the mmsA homolog (here termed mmsA1) and hpdH suggests an operonic transcription of genes from a promoter located in the intergenic region between mmsA1 and hpdR. For mmsA (here termed mmsA2) and mmsB, both arranged in one operon, no LTTR was found upstream of the genes. An acyl-CoA dehydrogenase, acaD, is annotated upstream of mmsA2. Both genes are oriented in the same direction. They are separated only by a short intergenic region of 44 bp which suggests an operonic arrangement of genes. In the opposite orientation of acaD, a transcriptional regulator is located which is annotated to belong to the AraC family. The regulator does not share protein sequence similarity with the P. denitrificans LTTR, which is putatively required to activate transcription of the mmsAB operon. However, it was included in the analysis for 3-HP-inducible gene expression as potential regulator of the C. necator acaD-mmsA2-mmsB operon.
Figure 1.
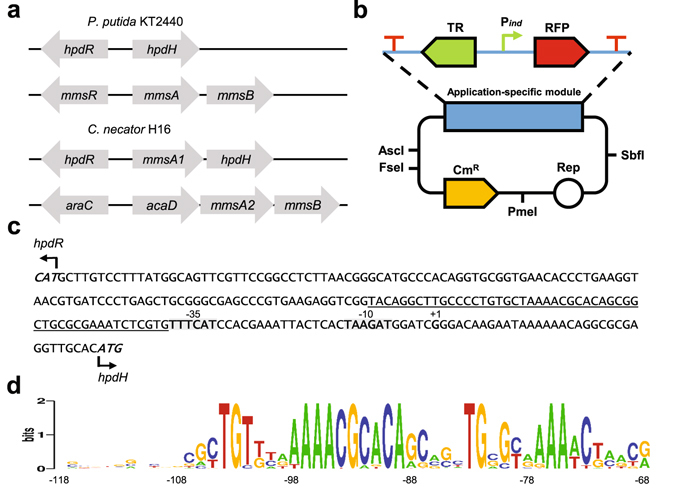
Identification of putative 3-HP-responsive genes in P. putida KT2440 and C. necator H16. (a) Operons putatively involved in 3-HP metabolism in P. putida KT2440 and C. necator H16. They are composed of the TR coding sequences hpdR, mmsR or araC, and divergently transcribed genes which are putatively required for 3-HP degradation: hpdH, 3-hydroxypropionate dehydrogenase; mmsA, methylmalonate-semialdehyde dehydrogenase; mmsB, 3-hydroxyisobutyrate dehydrogenase and acaD, acyl-CoA dehydrogenase. (b) Schematic illustration of the modular reporter system. It contains the four unique restriction sites AscI, FseI, PmeI and SbfI which are used for modular assembly. The application-specific module harbours the rfp reporter gene and the switchable system composed of transcriptional regulator and inducible promoter. (c) The P. putida KT2440 hpdR/hpdH intergenic region. Translational start sites are bold and italicised. The predicted hpdH −35 and −10 regions are bold and highlighted in grey. The nucleotide sequence between position −118 and −68 relative to the P. putida KT2440 hpdH translational start site is underlined. (d) A sequence similarity motif was generated which corresponds to the underlined sequence. It represents highly conserved nucleotides in the hpdR/hpdH intergenic regions of forty analysed Pseudomonas species.
Construction of a modular reporter system for evaluation of switchable systems
In order to determine the ability of different putative switchable systems to mediate gene expression in response to 3-HP, a modular reporter system was constructed (Fig. 1b). The reporter plasmid comprises the following features: (i) a pBBR1MCS derived broad-host-range vector replicon41, which allows for replication in Gram-negative bacteria, (ii) compatibility with the pMTL vector series enabling rapid exchange of replication origin and selection marker42, (iii) various restriction sites within the application-specific module to facilitate simple replacement of switchable system and rfp reporter gene43, and (iv) transcriptional terminators flanking the application-specific module to prevent background reporter gene expression. The reporter plasmid which has been employed as origin for all other constructs, pEH006, contains the arabinose-inducible system. It was used to validate the modular reporter construct and to make our results comparable to previously reported data. As a control, an identical plasmid lacking both the TR AraC and the arabinose-inducible promoter, pEH006E, was assembled. Single time point fluorescence measurements were performed in E. coli MG1655 and C. necator H16 harbouring the reporter plasmid. The arabinose-inducible system was tested and demonstrated more than a 1,200-fold increase of RFP expression in C. necator after six-hour induction with 0.1% (w/v) L-arabinose. This level of induction is significantly higher than what has been previously reported for a similar construct44. Fluorescence of cells containing pEH006E was similar to the background fluorescence of the medium, indicating no transcriptional read-though from the vector backbone. Therefore, its insulating backbone makes pEH006 a suitable original modular vector for construction of switchable systems.
P. putida KT2440 3-HP-inducible systems outperform systems derived from C. necator H16
Following the validation of the reporter construct, the putative 3-HP-inducible gene expression systems were cloned upstream of rfp into pEH006. They contain the TR and the intergenic region between the regulator coding sequence and the translational start site of the operons of the 3-HP catabolic genes. The two systems from P. putida KT2440 are referred to as PpMmsR/PmmsA (pEH007) and PpHpdR/PhpdH (pEH008) and those from C. necator H16 as CnAraC/PacaD (pEH009) and CnHpdR/PmmsA1 (pEH010). The constructs were tested in E. coli MG1655 and C. necator H16 for rfp reporter gene expression in response to 3-HP. Both 3-HP-inducible systems from P. putida exhibit statistically significant induction (p < 0.01) in E. coli and C. necator six hours after addition of 10 mM 3-HP to the culture (Table 1). PpHpdR/PhpdH demonstrates the highest level of normalized fluorescence and the strongest induction values; 23.3-fold in E. coli and more than 500-fold in C. necator. In contrast to C. necator in which CnHpdR/PmmsA1 shows a 88.4-fold induction, neither of the transcriptional regulators derived from Cupriavidus seem to be able to activate reporter gene expression from their proposed cognate promoters in E. coli. Besides, CnAraC/PacaD does not mediate statistically significant (p < 0.01) reporter gene expression after addition of 10 mM 3-HP in C. necator.
Table 1.
Comparison of normalized fluorescence of cells carrying the different versions of putative 3-HP-inducible gene expression systems from P. putida KT2440 (Pp) and C. necator H16 (Cn) in the presence or absence of 10 mM 3-HP.
| Switchable system | E. coli MG1655 | C. necator H16 | ||||
|---|---|---|---|---|---|---|
| Fluorescence intensity/OD600 | Induction ratio | Fluorescence intensity/OD600 | Induction ratio | |||
| Uninduced | Induced | Uninduced | Induced | |||
| PpMmsR/PmmsA | 59 ± 3 | 721 ± 24 | 12.3* | 444 ± 80 | 22,857 ± 1808 | 51.5* |
| PpHpdR/PhpdH | 1,259 ± 68 | 29,351 ± 756 | 23.3* | 304 ± 46 | 157,052 ± 8,409 | 516.6* |
| CnAraC/PacaD | 0 ± 0 | 0 ± 0 | 0 | 311 ± 16 | 464 ± 69 | 1.5 |
| CnHpdR/PmmsA1 | 22 ± 6 | 26 ± 5 | 1.2 | 847 ± 27 | 74,839 ± 1,763 | 88.4* |
| PpPmmsA | 193 ± 16 | 243 ± 23 | 1.3 | 44 ± 8 | 42 ± 4 | 1.0 |
| PpPhpdH | 908 ± 60 | 1,077 ± 47 | 1.2 | 42 ± 1 | 40 ± 3 | 1.0 |
Each system is composed of either a putative 3-HP-inducible promoter and its respective transcriptional regulator or a 3-HP-inducible promoter only. The mean values and standard deviations represent the normalized fluorescence of biological triplicates six hours after induction for E. coli MG1655 and C. necator H16. Asterisks indicate statistically significant induction values for p < 0.01 (unpaired t test).
The PpMmsR/PmmsA and PpHpdR/PhpdH switchable systems demonstrate a TR-dependent orthogonality in E. coli MG1655 and C. necator H16
Ideal TR-based switchable gene expression systems are specific to a certain metabolite or exogenously added inducer and are controlled by its corresponding TR only. In order to examine the cross-reactivity of host-originating transcription factors on reporter gene expression, the regulator coding sequences were removed from the plasmids that initially contained the PpMmsR/PmmsA and PpHpdR/PhpdH switchable systems. Constructs are solely composed of the 3-HP-inducible promoters PmmsA, or PhpdH upstream of the rfp reporter gene. CnHpdR/PmmsA1 and CnAraC/PacaD were not further investigated since they demonstrated no induction of reporter gene expression upon supplementation of 3-HP in E. coli and only in case of CnHpdR/PmmsA1 a statistically significant (p < 0.01) induction in C. necator.
In both E. coli and C. necator, neither of the solely P. putida derived promoters demonstrated statistically significant (p < 0.01) induction of RFP expression after addition of 3-HP (Table 1). This shows that their corresponding LTTRs are required for transcription activation and that E. coli and C. necator do not have cross-reacting regulator homologues. Since PhpdH demonstrated the highest induction level of the analysed 3-HP-inducible systems, and is controlled independently from host-originating TRs in E. coli and C. necator, it was chosen to be further characterised.
Identification of a conserved sequence motif within the hpdH promoter and in vivo analysis of the HpdR binding site
Control of gene expression in response to transcription-inducing metabolites (TIMs) is mainly mediated by TRs. They interact with conserved cis-acting regulatory elements (CRE) in the promoters of genes which are involved in TIM-converting pathways. A simple approach to identify CREs is to compare sequences from a variety of divergent species45. By multiple sequence alignment, conserved functional elements can easily be distinguished from sequences that are prone to evolve more quickly.
P. putida KT2440 HpdR and HpdH homologues were screened in other Pseudomonas species by searching the NCBI database for non-redundant protein sequences. Forty different Pseudomonas species were selected with HpdR and HpdH protein sequence identities of at least 70% (Supplementary Table S2). To aid in the identification of the putative HpdR binding site, the consensus sequence was determined in the intergenic region as described in Methods. The P. putida KT2440 hpdR/hpdH intergenic region is illustrated in Fig. 1c. Upstream of the hpdH −35 region, a range of nucleotides was found which are highly conserved across the analysed Pseudomonas species. This region is illustrated as a sequence similarity motif in Fig. 1d. It corresponds to the nucleotide sequence between position −118 and −68 relative to the P. putida KT2440 hpdH translational start site. Two partially similar and conserved sequence motifs can be identified. The first is an imperfect inverted repeat, sequence GCCCCTGTGC-n6-GCACAGCGGC in P. putida, located between positions −109 and −84 and is referred to as cis-acting regulatory element 1 (CRE1). The second motif is located between −83 and −68 and denoted as CRE2. In order to test if the highly conserved nucleotides in −109 to −68 are essential for promoter activation mediated by HpdR in response to 3-HP, the P. putida KT2440 hpdR/hpdH intergenic region was truncated and subsequently mutated.
Firstly, the −233 bp long intergenic region was analysed for secondary structures using the mfold web server46. A palindromic sequence was identified which can form a 97 bp spanning mRNA stem-loop structure (Supplementary Fig. S1). Interestingly, from the forty selected Pseudomonas species, this secondary structure is only present in P. putida KT2440 and the closely related Pseudomonas entomophila L48 and Pseudomonas plecoglossicida NBRC 103162. Likely, it has a species specific functional role, but is not evolutionary conserved. The reporter gene construct harbouring the native hpdR/hpdH intergenic region is referred to hpdH-233::rfp. The promoter truncation hpdH-118::rfp was generated by removing this palindromic sequence and incorporating an AvrII restriction site to allow further promoter modification (Fig. 2a). It contains the 118 bp long sequence upstream of the hpdH translational start site. Using hpdH-118::rfp, the promoter was truncated by additional twelve nucleotides, resulting in hpdH-106::rfp. The promoter-reporter gene constructs were analysed in E. coli MG1655 for their response to 3-HP (Fig. 2b). Compared to the native intergenic region (hpdH-233::rfp), the induction level slightly increased when the palindromic sequence was removed (hpdH-118::rfp). However, further truncation of the hpdH promoter by 12 nucleotides (hpdH-106::rfp) results in a 10-fold decrease of induction compared to that of hpdH-233::rfp.
Figure 2.
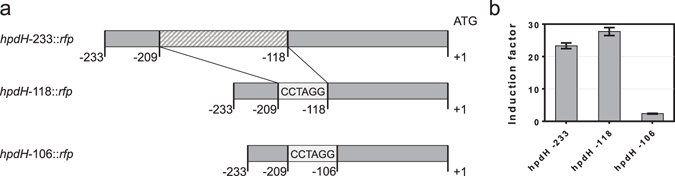
P. putida KT2440 hpdH promoter truncations. (a) Schematic illustration of the different hpdH promoter truncations that were evaluated for 3-HP-responsive activation of rfp reporter gene expression. The native promoter-reporter gene construct is denoted hpdH-233::rfp. The bioinformatically identified palindromic sequence is illustrated as a dashed box. This sequence was replaced by an AvrII restriction site, CCTAGG, resulting in promoter truncations hpdH-118::rfp and hpdH-106::rfp. (b) Induction levels mediated by the different promoter truncations. The various promoter-reporter gene constructs were analysed in E. coli MG1655 for RFP expression in the absence or presence of 10 mM 3-HP. Error bars represent standard deviations of three biological replicates.
Since hpdH-118::rfp demonstrates similar induction levels to the native, untruncated intergenic region, it was subsequently used to generate hpdH promoter mutants. The use of random mutagenesis was deemed to be a combinatorial challenge, seeking to achieve a full set of completely random mutants covering the 60 bp long conserved motif. Therefore, mutations were designed in such a way that they enabled a complete coverage by employing a mutagenesis strategy, which has been reported previously47. In total, twelve promoter variants were constructed, harbouring single or multiple nucleotide mutations between position −118 and −68 (Fig. 3a). They were analysed in E. coli MG1655 for reporter gene expression in the presence and absence of 3-HP (Fig. 3b). By comparing the sequence similarity motif (Fig. 1d) with the induction levels of the different promoter variants, it can be observed that the extent of nucleotide conservation correlates with their importance for inducible gene expression. Mutations 1, 2, and 3, which are upstream of the −109 - −68 region, show a minor impact on promoter activity (1.5- to 1.7-fold decrease in induction) compared to hpdH-118::rfp. Mutations 4, 8 and 11, which are located in less conserved stretches of the −109 - −68 region, decrease induction of reporter gene expression by 5.5-, 5.4- and 1.6-fold, respectively. The remaining five mutations, 5, 6, 7, 9 and 10 alter the most conserved nucleotides in either CRE1 or CRE2. These mutations abolish the ability of the promoter to mediate controllable gene expression by decreasing induction levels from 11.5- (mutation 6) to 25.2-fold (mutation 7). The in vivo analysis of various hpdH promoter truncations and mutations suggests that a 50 bp long sequence upstream of the −35 region is involved in 3-HP-inducible gene expression.
Figure 3.
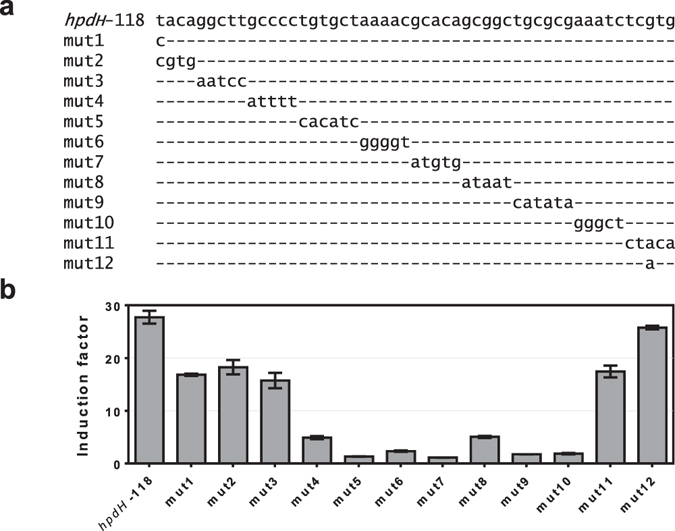
P. putida KT2440 hpdH-118::rfp promoter mutations. (a) Schematic illustration of the different hpdH-118::rfp promoter mutations that were evaluated for 3-HP-responsive activation of rfp reporter gene expression. Unchanged nucleotides are represented as dash. (b) Induction levels mediated by the different promoter mutations. The various promoter-reporter gene constructs were analysed in E. coli MG1655 for RFP expression in the absence or presence of 10 mM 3-HP. Error bars represent standard deviations of three biological replicates.
Finally, to confirm that HpdR binds directly to the hpdH promoter region, the TR was purified and electrophoretic mobility shift assay (EMSA) was performed as described in Supplementary Methods. A DNA shift can clearly be observed even in the absence of 3-HP (Supplementary Fig. S2), suggesting that regulator binding to the intergenic region occurs even under uninduced conditions. Addition of 3-HP to the binding reaction on the other hand results in formation of a slower migrating complex 2. A similar observation was made for catBC promoter, where a tighter and potentially higher-order TIM-protein-DNA complex was proposed to facilitate activation of gene expression48.
Determination of inducer-dependent orthogonality of the PpHpdR/PhpdH switchable system in E. coli MG1655 and C. necator H16
In addition to host-originating TRs, which may interfere with the heterologous switchable system, other metabolites than the primary inducer may be able to activate gene expression. These can be either compounds that are involved in the cells metabolism, constituents of the growth medium or additional TIMs that were supplemented to control independent gene expression from other switchable promoters. Compounds that were investigated for cross-reactivity with the PpHpdR/PhpdH switchable system (Fig. 4a) include: (i) the commonly used inducers L-arabinose and IPTG, (ii) fructose as alternative carbon source, (iii) the 3-HP precursors glycerol (2), pyruvate (3) and β-alanine (4) (shown in blue)49, and (iv) a broad range of compounds that are structurally similar to 3-HP such as mono- and dihydric alcohols (shown in red), mono- and dicarboxylic acids (shown in green), as well as α- and β-hydroxycarboxylic acids (shown in purple). Evaluation of these compounds may aid in the determination of the structural features of the ligand which are required to interact with the TR. Additionally, it may assist to identify an analogue of 3-HP as inducer if 3-HP is metabolized as shown in P. denitrificans 10. In order to achieve a sustained gene expression from the hpdH promoter, either 3-HP metabolizing genes need to be deleted or a metabolically inert analogue inducer should be employed.
Figure 4.
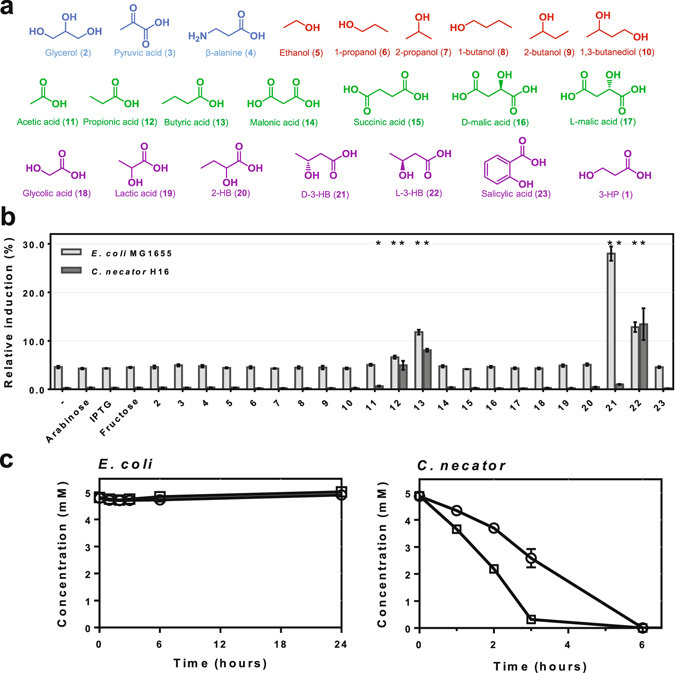
Determination of TIM-dependent orthogonality. (a) Compounds that were investigated for cross-reactivity with the PpHpdR/PhpdH switchable system: 3-hydroxypropionic acid (1), glycerol (2), pyruvic acid (3), β-alanine (4), ethanol (5), 1-propanol (6), 2-propanol (7), 1-butanol (8), 2-butanol (9), 1,3-butanediol (10), acetic acid (11), propionic acid (12), butyric acid (13), malonic acid (14), succinic acid (15), D-malic acid (16), L-malic acid (17), glycolic acid (18), lactic acid (19), 2-hydroxybutyric acid (20), D-3-hydroxybutyric acid (21), L-3-hydroxybutyric acid (22) and salicylic acid (23). (b) Relative induction levels of the PpHpdR/PhpdH switchable system which was subjected to a variety of putative TIMs. The various compounds were tested in E. coli MG1655 (light grey) and C. necator H16 (dark grey) harbouring pEH008. Error bars represent standard deviations of three biological replicates. Asterisks indicate statistically significant induction values for p < 0.01 (unpaired t test). (c) Consumption of D- (21, square) and L-3-HB (22, circle) in E. coli MG1655 and C. necator H16. Error bars represent standard deviations of three biological replicates.
Single time point fluorescence measurements were performed in E. coli MG1655 and C. necator H16 carrying the reporter plasmid with the native PpHpdR/PhpdH switchable system, pEH008. The compounds were supplemented at a concentration of 5 mM, except for DL-2-hydroxybutyrate (20) which, due to toxic effects, was used at 2.5 mM in C. necator. The response of the reporter system to the different molecules is illustrated as induction level relative to the one which was achieved by adding 5 mM 3-HP to the culture (Fig. 4b).
In addition to 3-HP, the monocarboxylic acids propionate (12) and butyrate (13), along with the structurally very similar D-3-hydroxybutyrate (D-3-HB, 21) and L-3-hydroxybutyrate (L-3-HB, 22), demonstrate statistically significant (p < 0.01) induction of reporter gene expression in both E. coli and C. necator. The highest level of induction relative to 3-HP was achieved with the D-enantiomer of 3-HB (21) in E. coli. It was able to induce RFP expression almost to one third of the level that had been obtained with 3-HP. The fact that in E. coli the L-enantiomer of 3-HB (22) only induces to half of the level of D-3-HB (21) indicates a stereospecific preference of HpdR for the D-enantiomer.
Whereas the relative induction levels for propionate (12) and butyrate (13) are roughly the same in both microorganisms when RFP expression under uninduced conditions is subtracted, a similar correlation cannot be observed for the 3-HB enantiomers. The relative induction mediated by D-3-HB (21) is 1% in C. necator as opposed to 28% in E. coli, whereas the L-enantiomer demonstrates a higher relative induction in Cupriavidus. This antagonistic behaviour in both investigated microorganisms was hypothesized to be caused by rapid metabolism of the natural D-enantiomer in C. necator. It encodes two D-3-hydroxybutyrate dehydrogenases (E.C. 1.1.1.30), which can convert D-3-HB (21) into acetoacetate. E. coli MG1655 completely lacks these enzymes. To test this hypothesis, C. necator and E. coli were cultivated in the presence of D- or L-3-HB (21, 22, respectively). Consumption of these compounds was monitored by HPLC analysis of cell-free supernatant samples.
As hypothesized, the concentration of D-3-HB (21) in the supernatant of the C. necator culture decreased rapidly (Fig. 4c). From the initial concentration of 5 mM D-3-HB (21), only 0.3 ± 0.1 mM remained in the culture supernatant three hours after it had been added. Surprisingly, the non-natural L-enantiomer was consumed as well. Its concentration does not decrease as quickly as D-3-HB (21), however, even L-3-HB (22) is fully depleted six hours after its supplementation. E. coli on the other hand consumes neither of the 3-HB enantiomers. Varying consumption rates of D- and L-3-HB (21, 22, respectively) in C. necator may explain the discrepancy between the relative induction levels of both microorganisms. In C. necator, the rate of D-3-HB (21) consumption appears to be higher than the rate of TIM-regulator-DNA complex formation. This subsequently results in decreased gene transcription. Even though it is metabolized, the concentration of L-3-HB (22) remains still high enough over the period of cultivation to mediate transient reporter gene expression.
3-HP-inducible system kinetics and dynamics
The PpHpdR/PhpdH switchable system was analysed for fluorescence output over time at different concentrations of 3-HP. This time course experiment was performed in both microorganisms and provides information about the time that is required to activate reporter expression, the influence of different inducer levels on growth and the dynamic range32, 50. In both microorganisms, RFP expression above the level of the uninduced culture started 30 minutes after addition of 3-HP (Fig. 5a, Supplementary Table S3). However, the profile of induction kinetics differs strongly in E. coli and C. necator. The variability can be explained by their growth kinetics and their dose response. In E. coli, growth is not affected by the 3-HP concentrations that were tested. In C. necator, however, the growth profile changes as a result of increase in 3-HP concentration. The addition of 10 mM 3-HP has a growth-retarding effect at the beginning of cultivation. The stationary growth phase is reached earlier in the presence of higher than with lower inducer concentrations, most likely due to an increased 3-HP metabolism as it has been reported for P. denitrificans 10 and other species that possess mmsR-mmsA-hbdH and hpdR-hphH regulons24. The metabolism of inducer in C. necator in turn results in transient gene expression and only higher levels of 3-HP are able to maintain a fluorescence output over the time course of experiment. This behaviour is reflected in the dose response curve for C. necator, which illustrates the correlation between inducer concentration and fluorescence output 4, 6 and 8 hours after 3-HP addition (Fig. 5b). Since 3-HP is not metabolized in E. coli, a sustained RFP expression can be observed throughout the time course of experiment. As it can be seen in the dose response curve, HpdR demonstrates a high induction cooperativity and gene expression can be tuned precisely in the range of 0.1–3 mM for a linear fluorescence output.
Figure 5.
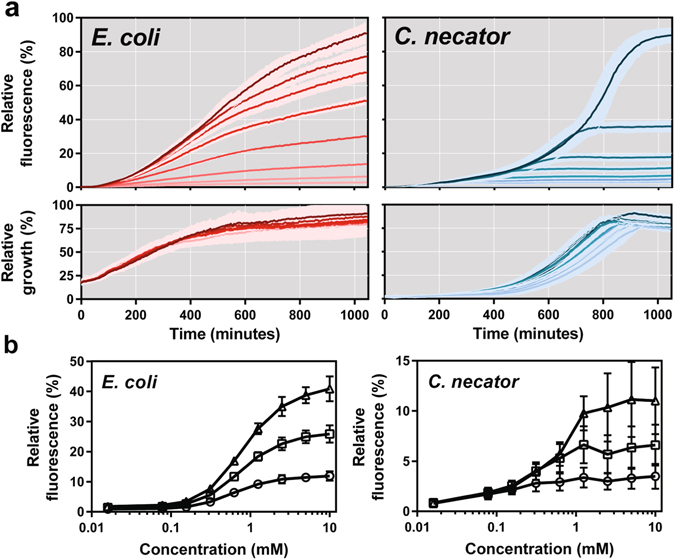
Induction dynamics and kinetics of the PpHpdR/PhpdH switchable system. (a) Relative fluorescence and absorbance curves of E. coli MG1655 and C. necator H16 cultures harbouring the native PpHpdR/PhpdH switchable system, pEH008. 3-HP was supplemented at time zero to the final concentrations of 10, 5, 2.5, 1.25, 0.625, 0.156, 0.016 mM and no inducer. The darker the shade of colour, the higher the level of 3-HP. The standard error of three biological replicates is illustrated as band. (b) Dose response curve of the PpHpdR/PhpdH switchable system in E. coli MG1655 and C. necator H16. It illustrates the relation between inducer concentration and fluorescence output 4 (circle), 6 (square) and 8 (triangle) hours after 3-HP addition. Inducer levels range from 0, 0.016, 0.078, 0.156, 0.313, 0.625, 1.25, 2.5, 5 and 10 mM. Error bars represent standard deviations of three biological replicates.
Discussion
Biosustainable production of chemicals and fuels is becoming increasingly important in the global need to reduce air pollution and greenhouse gas emissions. 3-HP is an important platform chemical used as a precursor for the synthesis of acrylic acid and production of other value-added compounds. Recently, substantial metabolic engineering efforts have been made to develop microbial chassis and biosynthetic pathways for the biological production of 3-HP.
TR-based switchable systems are suitable for controlling metabolic pathways and screening microorganisms with improved production of target compounds. They have been shown to be indispensable in the iterative design–build–test cycles of synthetic biology and biotechnology, and can lead eventually to the development of improved and sustainable biosynthesis processes31, 35. In this study, we identified potential 3-HP-inducible systems in P. putida KT2440 and C. necator H16, each encoding two regulons putatively involved in 3-HP metabolism. They were analysed for functionality in the well-characterised microorganism E. coli and the chemolithoautotrophic bacterium C. necator which is of industrial interest due to its ability to utilise CO2 gas as sole carbon source.
The mmsR-mmsA-hbdH and hpdR-hphH regulons are widespread amongst proteobacteria and actinobacteria as shown previously24. In this study, we established that neither the PpmmsA nor PphpdH promoter can be induced by the chromosomally encoded CnHpdR in the absence of P. putida MmsR or HpdR homologues, respectively. This suggests that MmsR and HpdR homologues and/or their respective mmsA and hpdH promoter regions in C. necator (betaproteobacteria) and P. putida (gammaproteobacteria) have diverged to the extent that their corresponding species-specific LTTRs are required for transcription activation. These findings indicate that the PpMmsR/PmmsA and PpHpdR/PhpdH switchable systems can be utilised for TR-dependent orthogonal gene expression control in biotechnologically relevant microorganisms from two different bacterial classes, gammaproteobacterium E. coli and betaproteobacterium C. necator. It was observed that absolute reporter gene expression from both P. putida switchable systems is much higher in C. necator than in E. coli, which results in overall higher induction levels in Cupriavidus. This behaviour may be due to a better expression of the P. putida transcriptional regulators in C. necator. The GC content of the TR coding sequences (62% and 64% for PpMmsR and PpHpdR, respectively) is more similar to the median whole genome GC content of C. necator (66.3%) when compared to E. coli (50.6%). Codon-optimisation of the regulator coding sequences for E. coli may result in higher absolute reporter gene expression in this microorganism. Since PpHpdR/PhpdH showed the highest induction level of the analysed 3-HP-inducible systems (516.6-fold induction in C. necator) and is controlled independently from host-originating TRs in both analysed organisms, it was subjected to further characterisation.
Although in this study our aim was to uncover an orthogonal 3-HP-inducible system that can be utilised for synthetic biology and biotechnology applications, a significant attempt was made to characterise the molecular mechanisms involved in PpPhpdH activation and PpHpdR-PhpdH interaction. A thorough understanding of these mechanisms is crucial for genetic part design in order to build synthetic metabolic pathways for the production of added-value compounds such as 3-HP. The bioinformatically identified conserved 40-nucleotide sequence within PhpdH and the in vivo analysis of various promoter mutations suggests, that two proposed cis-acting regulatory elements are potentially required for promoter region interaction with HpdR to mediate transcriptional activation. The length, location and the motif complexity of the TR binding site is common for almost all LysR-type inducible promoters reported so far26. All Pseudomonas LTTRs have been clustered into 9 phylogenetic groups on the basis of full length and domain sequence analysis29. Interestingly, HpdR belongs to group VI, which includes only a few LysR-type proteins. None of the regulators or their potential target DNA-binding sites have been characterised to date. The novelty of the motif identified in this study can be further highlighted by the observation that neither CRE1 nor CRE2 contains the typical LysR T-N11-A binding motif. Furthermore, the two motifs T-N11-A and TTA-N7/8-GAA which were proposed by Zhou et al.24 to be the HpdR regulatory binding site in P. denitrificans ATCC 13867 could neither be detected in the P. putida hpdH promoter upstream of the predicted −35 box, nor do these motifs seem to be conserved across the analysed Pseudomonas species. It should be noted that we confirmed in vitro binding of HpdR to the native hpdH promoter region by EMSA. Protein-DNA complex formation occurs even under uninduced conditions likely indicating that HpdR is involved in negative autoregulation of hpdR transcription. The mechanism of negative autoregulation is typical for LTTRs25. Interestingly, a stronger retardation effect of protein-DNA complex was observed in the presence of 3-HP. Ligand binding may facilitate formation of tighter and higher order protein-DNA complexes required for transcription activation as reported for catBC promoter48.
In addition to characterisation of molecular mechanisms involved in promoter activation, we determined the ligand-specificity of the system as well as induction kinetics and dynamics. The analysis of structurally similar compounds revealed a set of ligand features that are necessary to interact with HpdR. The minimum requirement for the ligand to be functional is one carboxyl group, as in propionate (12) and butyrate (13). The presence of a hydroxyl group at the β-position enhances regulator activity. However, replacing the hydroxyl group by an amine- or a carboxyl group, changing the position of the hydroxyl group from β to α, or adding more functional groups, renders the ligand inactive. Cross-induction by these compounds appears to be caused by a lack of specificity for HpdR, rather than by their conversion into the primary inducer 3-HP. In this context, we uncovered the structurally similar 3-HB to be a metabolically inert analogue inducer in E. coli. However, due to their metabolism in C. necator, none of the compounds that were able to activate the PpHpdR/PhpdH switchable system can be applied as 3-HP analogue inducer for control of sustained gene expression. Metabolism and growth-retarding effects of the primary inducer were demonstrated to impact induction and growth kinetics in C. necator. Finally, we propose that the PpHpdR/PhpdH switchable system can be a useful genetic tool to: (i) build a 3-HP or 3-HB biosensor which reports intracellular metabolite concentrations by fluorescence output, (ii) implement directed-evolution strategies for high-throughput screening of strains with improved production titres, and (iii) to balance enzyme levels in metabolic pathways that accumulate these compounds as final product or utilise them as intermediate compound.
Methods
Bacterial strains, medium and growth conditions
E. coli TOP10 (Invitrogen, Carlsbad, CA, USA) was used for cloning and plasmid propagation. RFP fluorescence assays were performed in wild type E. coli MG165551 and C. necator H16 (ATCC 17669). Bacterial strains were propagated in lysogeny broth (LB). For reporter gene assays, E. coli MG1655 was grown in M9 minimal growth medium supplemented with 1 µg/ml thiamine, 20 µg/ml uracil52 and 0.4% (w/v) glucose. C. necator reporter gene assays were performed in minimal medium containing 0.4% (w/v) sodium gluconate as carbon source53. If appropriate, antibiotics were added to the growth medium at the following concentrations: 25 µg/ml chloramphenicol or 50 µg/ml kanamycin for E. coli and 50 µg/ml chloramphenicol for C. necator. E. coli TOP10 was grown at 37 °C. C. necator H16 was cultivated at 30 °C. For comparison, both E. coli MG1655 and C. necator H16 RFP fluorescence assays were performed at 30 °C.
Cloning and transformation
Plasmid minipreps were carried out using the New England BioLabs miniprep kit (NEB, Ipswich, MA, USA). Microbial genomic DNA was extracted employing the SigmaEluteTM bacterial genomic DNA kit (Sigma, St. Louis, MO, USA). For cloning, DNA was amplified by PCR using Phusion High-Fidelity DNA polymerase from New England BioLabs in 50 µl reactions under recommended conditions. Restriction enzymes and NEBuilder Hifi DNA assembly master mix were purchased from NEB and reactions were set up according to the manufacturers’ protocol. The ZymocleanTM gel DNA recovery kit (Zymo, Irvine, CA, USA) was used to extract gel purified linearized DNA which was subsequently used for cloning.
Chemical competent E. coli were prepared and transformed by heat shock as previously described54. Electrocompetent C. necator were prepared and transformed as reported by Ausubel et al.55.
Plasmid construction
Oligonucleotide primers were synthesized by Eurofins Genomics (Ebersberg, Germany). Primer sequences are listed in Supplementary Table S4. Plasmids were constructed using either the NEBuilder Hifi DNA assembly method according to the manufacturers’ protocol or by conventional restriction-based cloning procedures54. Based on the modular vector system which has been developed for the genus Clostridium 42, a modular C. necator-E. coli shuttle vector was designed. All plasmids contain a chloramphenicol resistance gene flanked by PmeI and FseI restriction sites, a pBBR1 origin of replication flanked by SbfI and PmeI41 and an application-specific module flanked by SbfI and AscI restriction recognition sites.
The application-specific module contains the switchable system and a rfp reporter gene. The coding sequences for the transcriptional regulator and RFP are arranged in opposite direction. To prevent transcriptional read-through, transcription of the regulator and the reporter gene is terminated by a rrnB1 and a double terminator, respectively44, 56. Promoter and regulator sequences were verified by DNA sequencing (Source BioScience, Nottingham, UK). Plasmids used and generated in this study are listed in Supplementary Table S5. The nucleotide sequence of plasmids pEH006, pEH007, pEH008, pEH009, and pEH010 have been deposited in the public version of the JBEI registry (https://public-registry.jbei.org) under the accession numbers JPUB_008750, JPUB_008751, JPUB_008752, JPUB_008753, and JPUB_008754, respectively.
In order to compare gene expression in response to 3-HP to frequently applied switchable systems, a modular broad-host range vector containing the arabinose-inducible gene expression system was assembled. This vector was subsequently employed to generate all other constructs. pEH006 was assembled by using the NEBuilder Hifi DNA assembly method. Oligonucleotide primers EH001_f and EH002_r, EH003_f and EH004_r, EH005_f and EH006_r were used to amplify the replication origin and the chloramphenicol resistance gene, the rfp reporter gene and the arabinose-inducible system from pBBR1MCS-2-PphaC-eyfp-c1 and pKTrfp, respectively44, 57. The primer overhangs were designed to contain PmeI, FseI, AscI and SbfI restriction sites to allow for modular assembly and AatII and NdeI sites to be able to replace the switchable system. Since the transcriptional start site (+1) from the arabinose inducible promoter was known, the sequence downstream of +1 was replaced with a T7 mRNA stem-loop and a ribosome binding site from E. coli to improve mRNA stability and ribosome binding44.
pEH006E contains the promoter-less rfp reporter gene. This plasmid was employed to evaluate the impact of transcriptional read-through from the vector backbone on reporter gene expression. The rfp reporter gene was amplified with oligonucleotide primers EH013_f and EH148_r from pKTrfp44 and cloned into pEH006 by AatII and SbfI restriction sites.
The putative 3-HP-inducible systems PpMmsR/PmmsA, PpHpdR/PhpdH, CnAraC/PacaD and CnHpdR/PmmsA1 were amplified with oligonucleotide primers EH017_f and EH018_r, EH019_f and EH020_r, EH021_f and EH022_r, EH023_f and EH024_r, respectively, from P. putida KT2440 (Pp) and C. necator H16 (Cn) genomic DNA and cloned into pEH006 by AatII and NdeI restriction sites. The 3-HP-inducible promoters PpPmmsA, PpPhpdH and CnPmmsA1 were amplified with oligonucleotide primers EH096_f and EH095_r, EH059_f and EH060_r, EH098_f and EH097_r, respectively, from P. putida KT2440 and C. necator H16 genomic DNA and cloned into pEH006 by AatII and NdeI restriction sites.
PpPhpdH promoter truncations and mutations are based on vector pEH036 (hpdh-118::rfp) which was assembled by using the NEBuilder Hifi DNA assembly method. Oligonucleotide primers EH061_f and EH062_r, EH099_f and EH100_r were used to generate a plasmid identical to pEH008; however, the bioinformatically identified mRNA stem loop structure within the hpdH promoter was replaced by an AvrII restriction site to allow further promoter modifications. The truncated hpdH promoter version hpdh-106::rfp was amplified with oligonucleotide primers EH138_f and EH139_r from pEH008 and cloned into pEH036 by AvrII and NdeI. Promoter mutations were incorporated by amplifying the promoter region from pEH008 with oligonucleotide primers that contain the desired mutation in their overhang. Generally, A’s were replaced by G’s, T’s were replaced by C’s and vice versa. Promoter versions hpdh-118::rfp_mut1 – mut12 were constructed with reverse primers EH178, EH165 – EH174, EH179, respectively, and forward primer EH175. The PCR product was cloned into pEH036 by AvrII and NdeI.
RFP fluorescence assay
For evaluation of RFP fluorescence at a single time point, single colonies of freshly transformed E. coli MG1655 or C. necator H16 were used to inoculate 2 ml of minimal medium in 30 ml culture tubes. Cells were grown for 18 or 24 hours, respectively, before being diluted 1:20 into 2 ml fresh minimal medium. After the stock inducer was added at an OD600 of 0.5, cultures were cultivated for six hours at 30 °C and 200 rpm. Subsequently, 100 µl of cells were transferred to a 96-well clear-bottom plate (Greiner Bio One International, Germany; Cat. no 655090) and fluorescence was measured using an Infinite® M1000 PRO (Tecan, Switzerland) micro plate reader with 585 nm as excitation wavelength and an emission wavelength of 620 nm. The gain factor for fluorescence was set manually to 100%. Additionally, the OD600 was determined to normalize fluorescence by optical density.
For the growth and fluorescence time course experiments, E. coli MG1655 and C. necator H16 precultures were prepared as for the single time point measurements. They were diluted 1:40 into fresh minimal medium and incubated at 200 rpm and 30 °C until an OD600 of 0.2 was reached. 142.5 µl of the log-phase cells were transferred to a well of a 96-well plate. To the cultures, 7.5 µl fresh minimal medium were added containing the stock inducer at the desired concentration. Fluorescence and absorbance were measured as described earlier using Tecan micro plate reader every 5 min for 16 hours.
To determine the background autofluorescence, a culture was included in a separate well containing a control strain transformed with the empty plasmid pEH006E. Prior calculation, fluorescence and absorbance values were corrected by medium fluorescence and absorbance. Normalized fluorescence values for each measurement were obtained by dividing fluorescence by absorbance and subtracting the normalized background autofluorescence. Induction factors represent the ratio of normalized fluorescence of induced cells to normalized fluorescence of uninduced cells. Relative induction levels (%) are calculated by dividing the normalized fluorescence of cells treated with an inducer by the normalized fluorescence of cells that were induced with 3-HP.
Metabolite consumption assay
To analyse the consumption of 3-HB by E. coli MG1655 and C. necator H16, single colonies of the wild type strains were used to inoculate 2 ml of minimal medium. From the saturated overnight culture, 6 ml of fresh medium were inoculated 1:100 in 50 ml Falcon tubes and incubated at 30 °C and 200 rpm. At an OD600 of 0.4–0.5, stock solutions of D- (21) or L-3-HB (22) were added to the cultures to a final concentration of 5 mM. 0.5 ml samples were taken immediately, 1, 2, 3, 6 and 24 hours after supplementation of 3-HB. The OD600 was determined for every time point. Subsequently, samples were centrifuged for 5 min and 13,000 rpm. The supernatant was removed and subjected to HPLC analysis as described previously58 with slight modifications. Briefly, to the cell-free supernatant samples, an equal volume of mobile phase was added which was spiked with 50 mM valerate as internal standard. The mobile phase was composed of 0.005 M H2SO4. Subsequently, the mixture was passed through a 0.22 µm pore size membrane filter. Samples were analysed using a Thermo Scientific Ultimate 3000 HPLC system equipped with a Phenomenex Rezex ROA-organic acid H+ (8%) 150 mm × 7.8 mm × 8 µm column and a diode array detector with the wavelength set at 210 nm. The column was operated at 35 °C with an isocratic flow rate of 0.5 ml/min. Samples were run for 30 min and the injection volume was 20 µl.
Determination of consensus sequence
The nucleotide sequences of all retrieved hpdR/hpdH intergenic regions were aligned by using Clustal Omega59, 60. Subsequently, a sequence similarity motif was generated with WebLogo61. Putative RNA polymerase binding sites and the hpdH transcriptional start site were predicted by using the programs BPROM and NNPP, respectively62, 63.
Electronic supplementary material
Acknowledgements
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) [grant number BB/L013940/1]; and Engineering and Physical Sciences Research Council (EPSRC). We thank University of Nottingham for providing SBRC-DTProg PhD studentship to E.H., Stephan Heeb for gifting P. putida genomic DNA and all members of SBRC who helped in carrying out this research.
Author Contributions
E.H. and N.M. designed the study. E.H. performed the experiments. E.H., N.M. and N.P.M. analysed the data and wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01850-w
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Grousseau E, Lu J, Gorret N, Guillouet SE, Sinskey AJ. Isopropanol production with engineered Cupriavidus necator as bioproduction platform. Appl. Microbiol. Biotechnol. 2014;98:4277–4290. doi: 10.1007/s00253-014-5591-0. [DOI] [PubMed] [Google Scholar]
- 2.Latif H, Zeidan AA, Nielsen AT, Zengler K. Trash to treasure: Production of biofuels and commodity chemicals via syngas fermenting microorganisms. Curr. Opin. Biotechnol. 2014;27:79–87. doi: 10.1016/j.copbio.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Clomburg, J. M., Crumbley, A. M. & Gonzalez, R. Industrial biomanufacturing: The future of chemical production. Science355 (2017). [DOI] [PubMed]
- 4.Erickson B, Nelson, Winters P. Perspective on opportunities in industrial biotechnology in renewable chemicals. Biotechnol. J. 2012;7:176–185. doi: 10.1002/biot.201100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi S, Song CW, Shin JH, Lee SY. Biorefineries for the production of top building block chemicals and their derivatives. Metab. Eng. 2015;28:223–239. doi: 10.1016/j.ymben.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Becker J, Lange A, Fabarius J, Wittmann C. Top value platform chemicals: Bio-based production of organic acids. Curr. Opin. Biotechnol. 2015;36:168–175. doi: 10.1016/j.copbio.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 7.Valdehuesa KNG, et al. Recent advances in the metabolic engineering of microorganisms for the production of 3-hydroxypropionic acid as C3 platform chemical. Appl. Microbiol. Biotechnol. 2013;97:3309–3321. doi: 10.1007/s00253-013-4802-4. [DOI] [PubMed] [Google Scholar]
- 8.Andreeßen B, Taylor N, Steinbüchel A. Poly(3-hydroxypropionate): A promising alternative to fossil fuel-based materials. Appl. Environ. Microbiol. 2014;80:6574–6582. doi: 10.1128/AEM.02361-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathnasingh C, Raj SM, Jo JE, Park S. Development and evaluation of efficient recombinant Escherichia coli strains for the production of 3-hydroxypropionic acid from glycerol. Biotechnol. Bioeng. 2009;104:729–739. doi: 10.1002/bit.22429. [DOI] [PubMed] [Google Scholar]
- 10.Zhou S, Catherine C, Rathnasingh C, Somasundar A, Park S. Production of 3-hydroxypropionic acid from glycerol by recombinant Pseudomonas denitrificans. Biotechnol. Bioeng. 2013;110:3177–3187. doi: 10.1002/bit.24980. [DOI] [PubMed] [Google Scholar]
- 11.Burgé G, et al. Diversity of Lactobacillus reuteri strains in converting glycerol into 3-hydroxypropionic acid. Appl. Biochem. Biotechnol. 2015;177:923–939. doi: 10.1007/s12010-015-1787-8. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Bao J, Kim IK, Siewers V, Nielsen J. Coupled incremental precursor and co-factor supply improves 3-hydroxypropionic acid production in Saccharomyces cerevisiae. Metab. Eng. 2014;22:104–109. doi: 10.1016/j.ymben.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Lan EI, et al. Metabolic engineering of cyanobacteria for photosynthetic 3-hydroxypropionic acid production from CO2 using Synechococcus elongatus PCC 7942. Metab. Eng. 2015;31:163–170. doi: 10.1016/j.ymben.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L, Lin J, Wang H, Xie J, Wei D. Development of a two-step process for production of 3-hydroxypropionic acid from glycerol using Klebsiella pneumoniae and Gluconobacter oxydans. Bioprocess Biosystems Eng. 2015;38:2487–2495. doi: 10.1007/s00449-015-1486-4. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, et al. Biosynthesis of platform chemical 3-hydroxypropionic acid (3-HP) directly from CO2 in cyanobacterium Synechocystis sp. PCC 6803. Metab. Eng. 2016;34:60–70. doi: 10.1016/j.ymben.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, et al. Metabolic engineering of Corynebacterium glutamicum for the production of 3-hydroxypropionic acid from glucose and xylose. Metab. Eng. 2017;39:151–158. doi: 10.1016/j.ymben.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Borodina I, et al. Establishing a synthetic pathway for high-level production of 3-hydroxypropionic acid in Saccharomyces cerevisiae via β-alanine. Metab. Eng. 2015;27:57–64. doi: 10.1016/j.ymben.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Kim K, Kim SK, Park YC, Seo JH. Enhanced production of 3-hydroxypropionic acid from glycerol by modulation of glycerol metabolism in recombinant Escherichia coli. Bioresour. Technol. 2014;156:170–175. doi: 10.1016/j.biortech.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Rathnasingh C, et al. Production of 3-hydroxypropionic acid via malonyl-CoA pathway using recombinant Escherichia coli strains. J. Biotechnol. 2012;157:633–640. doi: 10.1016/j.jbiotec.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Gokarn, R.R. et al. In US8999700 (2015).
- 21.Luo, H. et al. Production of 3-hydroxypropionic acid via the propionyl-CoA pathway using recombinant Escherichia coli strains. PLoS ONE11 (2016). [DOI] [PMC free article] [PubMed]
- 22.Li, Y., Wang, X., Ge, X. & Tian, P. High production of 3-hydroxypropionic acid in Klebsiella pneumoniae by systematic optimization of glycerol metabolism. Sci. Rep. 6 (2016). [DOI] [PMC free article] [PubMed]
- 23.Chen Y, Nielsen J. Biobased organic acids production by metabolically engineered microorganisms. Curr. Opin. Biotechnol. 2016;37:165–172. doi: 10.1016/j.copbio.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Zhou, S., Ainala, S.K., Seol, E., Nguyen, T.T. & Park, S. Inducible gene expression system by 3-hydroxypropionic acid. Biotechnol. Biofuels8 (2015). [DOI] [PMC free article] [PubMed]
- 25.Maddocks SE, Oyston PCF. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 26.Oliver, P., Peralta-Gil, M., Tabche, M.L. & Merino, E. Molecular and structural considerations of TF-DNA binding for the generation of biologically meaningful and accurate phylogenetic footprinting analysis: The LysR-type transcriptional regulator family as a study model. BMC Genomics17 (2016). [DOI] [PMC free article] [PubMed]
- 27.MacLean AM, Anstey MI, Finan TM. Binding site determinants for the LysR-type transcriptional regulator PcaQ in the legume endosymbiont Sinorhizobium meliloti. J. Bacteriol. 2008;190:1237–1246. doi: 10.1128/JB.01456-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porrúa O, et al. An A-tract at the AtzR binding site assists DNA binding, inducer-dependent repositioning and transcriptional activation of the PatzDEF promoter. Mol. Microbiol. 2013;90:72–87. doi: 10.1111/mmi.12346. [DOI] [PubMed] [Google Scholar]
- 29.Reen FJ, Barret M, Fargier E, O’Muinneacháin M, O’Gara F. Molecular evolution of LysR-type transcriptional regulation in Pseudomonas aeruginosa. Mol. Phylogen. Evol. 2013;66:1041–1049. doi: 10.1016/j.ympev.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 30.Michener JK, Thodey K, Liang JC, Smolke CD. Applications of genetically-encoded biosensors for the construction and control of biosynthetic pathways. Metab. Eng. 2012;14:212–222. doi: 10.1016/j.ymben.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogers JK, Church GM. Genetically encoded sensors enable real-time observation of metabolite production. Proc. Natl. Acad. Sci. USA. 2016;113:2388–2393. doi: 10.1073/pnas.1600375113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogers JK, et al. Synthetic biosensors for precise gene control and real-time monitoring of metabolites. Nucleic Acids Res. 2015;43:7648–7660. doi: 10.1093/nar/gkv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Jensen MK, Keasling JD. Development of biosensors and their application in metabolic engineering. Curr. Opin. Chem. Biol. 2015;28:1–8. doi: 10.1016/j.cbpa.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 34.Zhang F, Carothers JM, Keasling JD. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 2012;30:354–359. doi: 10.1038/nbt.2149. [DOI] [PubMed] [Google Scholar]
- 35.Dietrich JA, Shis DL, Alikhani A, Keasling JD. Transcription factor-based screens and synthetic selections for microbial small-molecule biosynthesis. ACS Synth. Biol. 2013;2:47–58. doi: 10.1021/sb300091d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eggeling L, Bott M, Marienhagen J. Novel screening methods-biosensors. Curr. Opin. Biotechnol. 2015;35:30–36. doi: 10.1016/j.copbio.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 37.Qian S, Cirino PC. Using metabolite-responsive gene regulators to improve microbial biosynthesis. Curr. Opin. Chem. Eng. 2016;14:93–102. doi: 10.1016/j.coche.2016.08.020. [DOI] [Google Scholar]
- 38.Mahr R, et al. Biosensor-driven adaptive laboratory evolution of L-valine production in Corynebacterium glutamicum. Metab. Eng. 2015;32:184–194. doi: 10.1016/j.ymben.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 39.Fang M, et al. Intermediate-sensor assisted push-pull strategy and its application in heterologous deoxyviolacein production in Escherichia coli. Metab. Eng. 2016;33:41–51. doi: 10.1016/j.ymben.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Dos Santos VAPM, Heim S, Moore ERB, Strätz M, Timmis KN. Insights into the genomic basis of niche specificity of Pseudomonas putida KT2440. Environ. Microbiol. 2004;6:1264–1286. doi: 10.1111/j.1462-2920.2004.00734.x. [DOI] [PubMed] [Google Scholar]
- 41.Kovach, M. E., Phillips, R. W., Elzer, P. H., Roop I, R. M. & Peterson, K. M. pBBR1MCS: A broad-host-range cloning vector. BioTechniques16 (1994). [PubMed]
- 42.Heap JT, Pennington OJ, Cartman ST, Minton NP. A modular system for Clostridium shuttle plasmids. J. Microbiol. Methods. 2009;78:79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Campbell RE, et al. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bi, C. et al. Development of a broad-host synthetic biology toolbox for Ralstonia eutropha and its application to engineering hydrocarbon biofuel production. Microb. Cell Fact. 12 (2013). [DOI] [PMC free article] [PubMed]
- 45.Hughes JR, et al. Annotation of cis-regulatory elements by identification, subclassification, and functional assessment of multispecies conserved sequences. Proc. Natl. Acad. Sci. USA. 2005;102:9830–9835. doi: 10.1073/pnas.0503401102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffmann, J. & Altenbuchner, J. Functional characterization of the mannitol promoter of Pseudomonas fluorescens DSM 50106 and its application for a mannitol-inducible expression system for Pseudomonas putida KT2440. PLoS ONE10 (2015). [DOI] [PMC free article] [PubMed]
- 48.Parsek MR, Shinabarger DL, Rothmel RK, Chakrabarty AM. Roles of CatR and cis,cis-muconate in activation of the catBC operon, which is involved in benzoate degradation in Pseudomonas putida. J. Bacteriol. 1992;174:7798–7806. doi: 10.1128/jb.174.23.7798-7806.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang X, Meng X, Xian M. Biosynthetic pathways for 3-hydroxypropionic acid production. Appl. Microbiol. Biotechnol. 2009;82:995–1003. doi: 10.1007/s00253-009-1898-7. [DOI] [PubMed] [Google Scholar]
- 50.Van Der Meer JR, Belkin S. Where microbiology meets microengineering: Design and applications of reporter bacteria. Nature Rev. Microbiol. 2010;8:511–522. doi: 10.1038/nrmicro2392. [DOI] [PubMed] [Google Scholar]
- 51.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 52.Jensen KF. The Escherichia coli K-12 ‘wild types’ W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J. Bacteriol. 1993;175:3401–3407. doi: 10.1128/jb.175.11.3401-3407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlegel HG, Kaltwasser H, Gottschalk G. Ein Submersverfahren zur Kultur wasserstoffoxydierender Bakterien: Wachstumsphysiologische Untersuchungen. Archiv für Mikrobiologie. 1961;38:209–222. doi: 10.1007/BF00422356. [DOI] [PubMed] [Google Scholar]
- 54.Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular cloning: a laboratory manual (Cold Spring Harbor Laboratory Press, New York, 1989).
- 55.Ausubel, F. M. et al. Current Protocols in Molecular Biology (John Wiley & Sons, 2003).
- 56.Orosz A, Boros I, Venetianer P. Analysis of the complex transcription termination region of the Escherichia coli rrnB gene. Eur. J. Biochem. 1991;201:653–659. doi: 10.1111/j.1432-1033.1991.tb16326.x. [DOI] [PubMed] [Google Scholar]
- 57.Pfeiffer D, Jendrossek D. Interaction between poly(3-hydroxybutyrate) granule-associated proteins as revealed by two-hybrid analysis and identification of a new phasin in Ralstonia eutropha H16. Microbiology. 2011;157:2795–2807. doi: 10.1099/mic.0.051508-0. [DOI] [PubMed] [Google Scholar]
- 58.Sheng, L., Kovács, K., Winzer, K., Zhang, Y. & Minton, N. P. Development and implementation of rapid metabolic engineering tools for chemical and fuel production in Geobacillus thermoglucosidasius NCIMB 11955. Biotechnol. Biofuels10 (2017). [DOI] [PMC free article] [PubMed]
- 59.Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7 (2011). [DOI] [PMC free article] [PubMed]
- 60.Goujon M, et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Solovyev, V. & Salamov, A. In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies 62–78 (2011).
- 63.Reese MG. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 2001;26:51–56. doi: 10.1016/S0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.