

Abstract
Microvascular dysfunction has been suggested to trigger adipose tissue dysfunction in obesity. This study investigates the hypothesis that glycation impairs microvascular architecture and expandability with an impact on insulin signalling. Animal models supplemented with methylglyoxal (MG), maintained with a high-fat diet (HFD) or both (HFDMG) were studied for periepididymal adipose (pEAT) tissue hypoxia and local and systemic insulin resistance. Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) was used to quantify blood flow in vivo, showing MG-induced reduction of pEAT blood flow. Increased adipocyte size and leptin secretion were observed only in rats feeding the high-fat diet, without the development of hypoxia. In turn, hypoxia was only observed when MG was combined (HFDMG group), being associated with impaired activation of the insulin receptor (Tyr1163), glucose intolerance and systemic and muscle insulin resistance. Accordingly, the adipose tissue angiogenic assay has shown decreased capillarization after dose-dependent MG exposure and glyoxalase-1 inhibition. Thus, glycation impairs adipose tissue capillarization and blood flow, hampering its expandability during a high-fat diet challenge and leading to hypoxia and insulin resistance. Such events have systemic repercussions in glucose metabolism and may lead to the onset of unhealthy obesity and progression to type 2 diabetes.
Introduction
Recent studies suggested different metabolic outcomes of metabolically healthy obesity (MHO) and metabolically unhealthy obesity (MUO), the latter being more likely associated with early development and progression of type 2 diabetes1–3. Adipose tissue dysfunction seems to be pivotal in this different outcome, given its contribution to insulin resistance and glucose and lipid dysmetabolism and the multilevel impact of adipokines4–9. Studies using genetic (ob/ob or db/db) and diet-induced models of obesity have shown that lipotoxicity-triggered inflammation and hypoxia govern adipose tissue dysfunction, causing lipolysis, ectopic FFA deposition and insulin resistance4, 6, 10–12. Models of high-fat (HF) diet-induced obesity are considered more physiological. However, hypercaloric diets are usually composed by a mixture of lipids and sugars, which may cause adipose tissue dysfunction without isolating the mechanisms involved.
Hypoxia was suggested to primarily derive from adipocyte hypertrophy, which would limit oxygen diffusion12, 13. However, recent evidences support the idea that hypoxia is not a primary event and may result from other factors than simple adipocyte growth14. Limited capillarization was recently shown in adipose tissue explants from obese donors, potentially causing hypoxia. However, reduced adipose tissue blood flow was observed to be independent of obesity per se, and shown to be more associated with insulin resistance15–18. Moreover, Goossens et al., recently demonstrated hyperoxia in the adipose tissue of obese patients, due to decreased metabolic rate and possibly mitochondrial dysfunction14. Thus, although these aspects remain to be addressed at the mechanistic level, such evidences suggest the modulation of angiogenesis and blood flow as potential targets in improving insulin sensitivity.
Methylglyoxal (MG) is a strong precursor of advanced glycation end products (AGE), which forms from glucose and thus its levels are elevated in prediabetic and diabetic patients and in sugar-rich foods19–23. Previous studies demonstrated MG-dependent inhibition of the insulin receptor pathway in 3T3-L1 adipocytes24, 25. The same was observed in animal models using fructose supplementation and different methods of intraperitoneal, intravenous and continuous subcutaneous MG administration26–28. In a study of Hofmann et al., the consumption of AGE- and MG-enriched diets caused glucose and insulin intolerance, but only in diabetic db/db mice, with much smaller effects on normal mice29. However, this was only observed with supraphysiological MG doses. Thus, the major limitation of the existing studies is that, to our knowledge, none of them assessed the effects of MG accumulation in comparison with similar endogenous levels observed in a diabetic model. Recently, we demonstrated that dietary MG supplementation increases AGE deposition in periepididymal adipose tissue (pEAT), causing structural and functional alterations30. Moreover, MG impairs metabolic adaptations after a surgery-induced decrease of blood supply and when reduced by pyridoxamine the microvascular lesions were improved31, 32. However, the effects of AGE deposition on the vascular adaptation in the course of adipose tissue expansion during obesity were not previously addressed. We hypothesized that progressive AGE accumulation in adipose tissue may hamper capillarization, blood flow and expandability, which could contribute to insulin resistance and thus further enhance AGE deposition in a positive feedback cycle. Understanding the mechanisms of microvascular dysfunction may definitively contribute to design strategies to improve adipose tissue function and thus prevent unhealthy obesity and type 2 diabetes development and progression.
Results
Glycation increases glycoconjugates and fibrosis in adipose tissue
Ne(carboxyethyl)lysine (CEL) is an AGE specifically derived from the MG reaction with lysine residues, which may accumulate in adipose tissue from local formation or dietary absorption of MG-lysine adducts. CEL levels were significantly superior in HFDMG and GK groups (Fig. 1A). No significant alterations were found for glyoxalase-1 (GLO-I) levels. Increased PAS (glycoconjugates), Masson Trichrome (fibrosis) and CEL staining were observed in MG-treated groups (MG and HFDMG) and in the non-obese type 2 diabetic GK rats (Fig. 1B,C,D).
Figure 1.

Increased pEAT levels of CEL in HFDMG and GK rats (A), calculated as percentage of Ct, while no differences were observed for GLO-1; representative WB are shown. Glycation increases glycoconjugates, fibrosis and CEL staining in pEAT. Histological analysis shows PAS (100X) (B), Masson Trichrome (100X) (C) and CEL staining (100X) (D). Ct - Wistar 12 m; MG - Wistar + MG supplementation; HFD - HF diet-fed Wistar; HFDMG - HF diet-fed Wistar + MG supplementation; GK - Goto-Kakizaki 12 m. Bars represent means ± SEM, n = 6–8. * vs Ct; $ vs HFD. 1 symbol p < 0.05; 2 symbols p < 0.01.
Glycation impairs adipose tissue blood flow and causes hypoxia in diet-induced obese rats
For the first time, we were able to develop a procedure to evaluate adipose tissue blood flow in vivo using DCE-MRI (Fig. 2). The accumulation curve of the contrast product was evaluated during 34 minutes. The fold increase in relation to the basal signal was calculated at each dynamic scan and the area under the curve (AUC) was determined. Enhancement curves of representative animals of each group (Fig. 2A) show rapid contrast product accumulation in pEAT, which is higher in control rats and reduced in groups submitted to MG supplementation (MG, HFDMG). Accordingly, the AUC was significantly reduced in these groups and in GK rats, showing decreased pEAT blood flow (Fig. 2B). Hypoxia was quantified through the accumulation of pimonidazole adducts and no significant differences were observed in HFD and MG groups (Fig. 2D). On the other hand, the HFDMG group showed increased pimonidazole accumulation (p < 0.001 vs Ct), which has also been demonstrated in histological analysis (Fig. 2D,E). Notably, hypoxia was also observed in non-obese diabetic GK rats.
Figure 2.
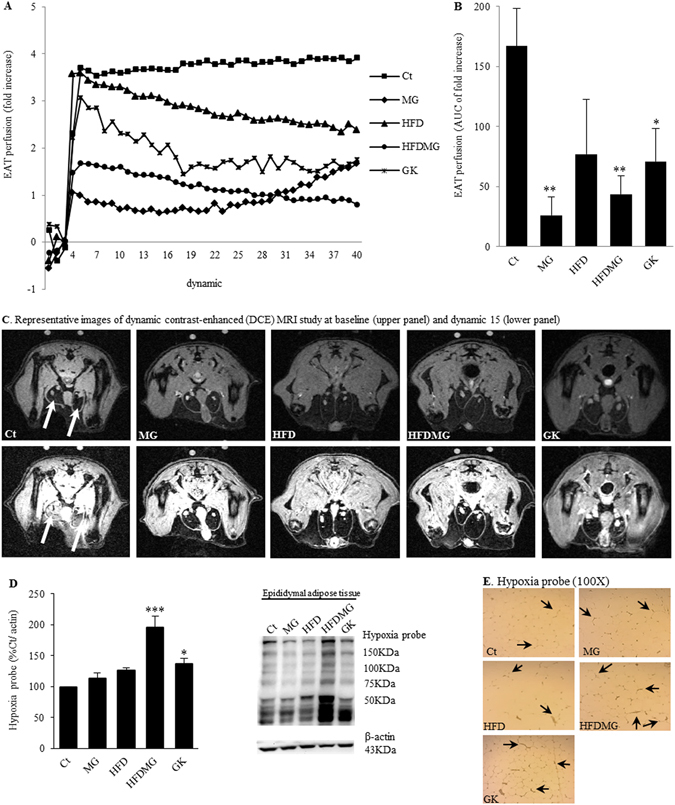
Glycation impairs pEAT blood flow and causes hypoxia in adipose tissue. Blood flow was evaluated through the area under the curve (B) of a dynamic contrast-enhanced (DCE) MRI study (A). Representative images at baseline and dynamic 15 (C). Hypoxia was assessed using an antibody against pimonidazole adducts by WB quantification (D) and IHC (E). Ct - Wistar 12 m; MG - Wistar + MG supplementation; HFD - HF diet-fed Wistar; HFDMG - HF diet-fed Wistar + MG supplementation; GK - Goto-Kakizaki 12 m. Bars represent means ± SEM, n = 6–8. * vs Ct. 1 symbol p < 0.05; 2 symbols p < 0.01; 3 symbols p < 0.001.
Glycation hampers adipose tissue pathways of adaptation to hypoxia, angiogenesis (capillarization) and expandability
pEAT expansion was assessed through the fat pad weight, adipocyte area and indirectly by circulating leptin. HFD rats had increased body and pEAT weight (Table 1). The HFDMG group showed a smaller increase of pEAT weight and no differences in body weight, although they had eaten similar amounts of food as compared to the HFD group (Table 1). GK rats had lower body weight than Wistar rats, without major differences in pEAT weight (Table 1). HF diet-induced adipose tissue expansion (HFD group) caused increased adipocyte area and circulating leptin levels (p < 0.001 vs Ct; p < 0.01 vs MG), which were not observed in the HFDMG group (leptin: p < 0.05 vs Ct) (Fig. 3A,B).
Table 1.
Food intake, body and periepididymal adipose tissue weight, fasting glycemia, HbA1c and serum triglycerides.
| Group | Ct | MG | HFD | HFDMG | GK |
|---|---|---|---|---|---|
| Food (g/rat/day) | 22.9 ± 0.7 | 24.3 ± 1.6 | 15.1 ± 1.1**,## | 14.4 ± 0.7**,## | 25.7 ± 0.6$$$,&&& |
| Body weight (g) | 508.8 ± 11.4 | 508.9 ± 18.4 | 652.7 ± 35.8**,## | 571.6 ± 27.3 | 398.7 ± 7.7*,#,$$$,&&& |
| pEAT weight (g) | 5.5 ± 0.4 | 5.9 ± 1.1 | 16.8 ± 2.5**,# | 14.2 ± 1.4* | 2.8 ± 0.3### , &&& |
| Fasting glycemia (mg/dl) | 68.5 ± 2.0 | 70.6 ± 1.4 | 70.9 ± 2.0 | 71.1 ± 1.6 | 91.7 ± 3.0***,#,$,& |
| HbA1c (%) (mmol/mol) | 3.2 ± 0.1 (11.8 ± 1.5) | 3.3 ± 0.1 (13 ± 2.1) | 3.3 ± 0.1 (12.8 ± 0.9) | 3.5 ± 0.1 (15.4 ± 1.9) | 5.6 ± 0.4 (36 ± 6.8)***,###,$$$,&&& |
| Triglycerides (mg/dl) | 77.2 ± 6.4 | 69.3 ± 10.7 | 77.8 ± 5.4 | 62.3 ± 3.2 | 160.1 ± 23.1***,###,$$$,&&& |
Ct - Wistar 12 m; MG - Wistar + MG supplementation; HFD - HF diet-fed Wistar; HFDMG - HF diet-fed Wistar + MG supplementation; GK - Goto-Kakizaki 12 m. Average ± SEM.* vs Ct; # vs MG; $ vs HFD; & vs HFDMG. 1 symbol p < 0.05; 2 symbols p < 0.01; 3 symbols p < 0.001.
Figure 3.
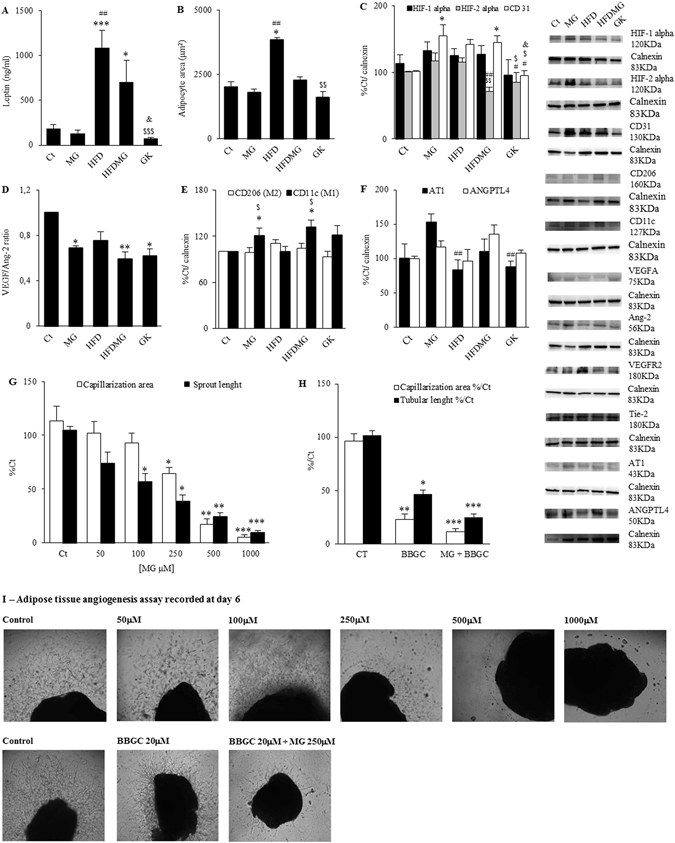
Glycation hampers adipose tissue pathways of adaptation to hypoxia, angiogenesis and expandability ratio. Serum leptin levels were assessed by ELISA (A). Adipocyte area was evaluated through the number of adipocytes counted in each field (10 fields/rat) (B). The levels of HIF-1alpha, HIF-2alpha, CD31 (C), VEGF, Ang-2 (D), CD11c, CD206 (E) ANGPTL4 and AT1 (F) were determined by WB. The VEGF/Ang-2 ratio was calculated. The adipose tissue angiogenesis assay was used to evaluate the effects of glycation in adipose tissue angiogenesis. MG concentrations higher than 100 μM decreased vascularization area, while lower concentrations decreased sprout length (G). Glyoxalase-1 inhibitor BBGC reduced vascularization area and sprout length, an effect which was further increased in combination with MG exposure (H). Representative images of all conditions are shown in (I). Ct - Wistar 12 m; MG - Wistar + MG supplementation; HFD - HF diet-fed Wistar; HFDMG - HF diet-fed Wistar + MG supplementation; GK - Goto-Kakizaki 12 m. Bars represent means ± SEM, n = 6–8. * vs Ct; # vs MG; $ vs HFD; & vs HFDMG; 1 symbol p < 0.05; 2 symbols p < 0.01; 3 symbols p < 0.001.
Adequate angiogenesis is determinant for adipose tissue expandability. When hypoxia regions are generated by physiological tissue expansion, hypoxia-inducible factors (HIFs) are activated as a mechanism to increase angiogenesis. Despite the fact that no changes were observed in HIF-1alpha, decreased HIF-2alpha expression in HFDMG and GK groups were observed when comparing with MG and HFD groups (Fig. 3C). Decreased HIF-2alpha expression in adipose tissue has been associated with activation of the macrophage M1 phenotype. Accordingly, HFDMG rats had increased M1 levels (Fig. 3E). No differences were observed for the M2 phenotype. Besides increased number of M1 macrophages, dysregulation of HIFs expression may hamper angiogenesis as well. Our group showed aberrant capillary formation in RPE cells and pEAT after MG-induced HIF-1alpha degradation and imbalance of VEGF/Ang-2 ratio30, 33. A similar imbalance of VEGF/Ang-2 ratio was here observed in the MG-supplemented groups and GK rats (Fig. 3D). This was coincident with increased levels of the endothelial cell marker CD31 in MG and HFDMG groups, what may denote a compensatory endothelial cell proliferation and formation of aberrant capillaries33 (Fig. 3C). Such results were corroborated by the adipose tissue angiogenesis assay. Incubation of adipose tissue explants with growing concentrations of MG showed progressive inhibition of endothelial cell migration in the collagen matrix only for concentrations higher than 100 μM (Fig. 3G). However, concentrations between 50 μM and 100 μM MG caused a significant decrease of sprout length, showing that glycation-induced vessel destabilization precedes inhibition of cell proliferation and migration in the adipose tissue (Fig. 3G). Selective inhibition of GLO-1 also caused a significant reduction of vascularization area and sprout length, an effect which was further increased when it was combined with 250 μM MG (Fig. 3H). Besides hampering angiogenesis, MG supplementation also increased angiotensin II receptor (AT1) expression, given that the HF diet decreased AT1 levels in adipose tissue but such decrease was not observed in HF diet-fat rats supplemented with MG (Fig. 3F). Such effects may increase angiotensin signalling decreasing blood flow.
Glycation impairs adipose tissue insulin signalling in high-fat diet-fed rats
Methylglyoxal supplementation and HF diet separately had no major effects in the insulin receptor, Akt, PPARgamma (regulator of lipid storage) and Perilipin-A (regulator of lipolysis) levels. However, HF diet with MG supplementation induced a significant decrease of activated insulin receptor form, similarly to non-obese type 2 diabetic rats (p < 0.05 vs Ct and p < 0.01 vs MG) (Fig. 4A). No major differences were however observed in phosphorylated Akt, PGC1alpha and the differentiation factors PPAR-gamma and C/EBPalpha (Fig. 4B,C). Perilipin-A degradation is strongly inhibited by insulin and, accordingly, its levels were significantly reduced in the HFDMG group suggesting insulin resistance (Fig. 4C). Thus, impairment of pEAT expandability induced by glycation results in impaired insulin signalling and lipid storage.
Figure 4.

Glycation impairs pEAT insulin signalling in high-fat diet-fed rats. Insulin signalling in pEAT was evaluated through WB quantification of activated and total forms of IR (A) and Akt (B). pEAT metabolism was assessed by WB quantification of PPARgamma and Perilipin-A levels (C). Ct - Wistar 12 m; MG - Wistar + MG supplementation; HFD - HF diet-fed Wistar; HFDMG - HF diet-fed Wistar + MG supplementation; GK - Goto-Kakizaki 12 m.Bars represent means ± SEM, n = 6–8.* vs Ct; # vs MG; 1 symbol p < 0.05; 2 symbols p < 0.01.
Glycation causes systemic dysmetabolism and impairs skeletal muscle insulin signalling in high-fat diet-fed rats
Methylglyoxal and HF diet effects on systemic metabolism were assessed through the evaluation of glycemia (fasting and IPGTT), HbA1c, FFA, triglycerides, insulin and adiponectin. MG supplementation alone had no effect on such systemic parameters (Fig. 5A–D; Table 1). Despite increased adiponectinemia, HFD rats developed glucose intolerance, with higher AUC during the IPGTT (p < 0.001 vs Ct and p < 0.01 vs MG), but no significant differences were observed for HbA1c, fasting glycemia, insulinemia and FFA levels (Fig. 5A–D; Table 1). In turn, HFDMG rats developed higher fasting FFA levels (p < 0.05 vs Ct), insulinemia (p < 0.05 vs Ct) and glucose intolerance (AUC) (p < 0.05 vs HFD; p < 0.001 vs Ct and MG) (Fig. 5B–D). Moreover, MG-induced glycation inhibited the increase of serum adiponectin levels observed in the HFD group (p < 0.05 vs HFD) (Fig. 5A). Such features were similar to the type 2 diabetic rats, which develop glucose intolerance, hypoadiponectinemia and increased FFA levels, as well as hypoinsulinemia, due to age-dependent impaired β-cell function (Fig. 5A–D). Altogether, such results show that glycation in HF diet fed-rats results in systemic insulin resistance and impaired glucose tolerance.
Figure 5.
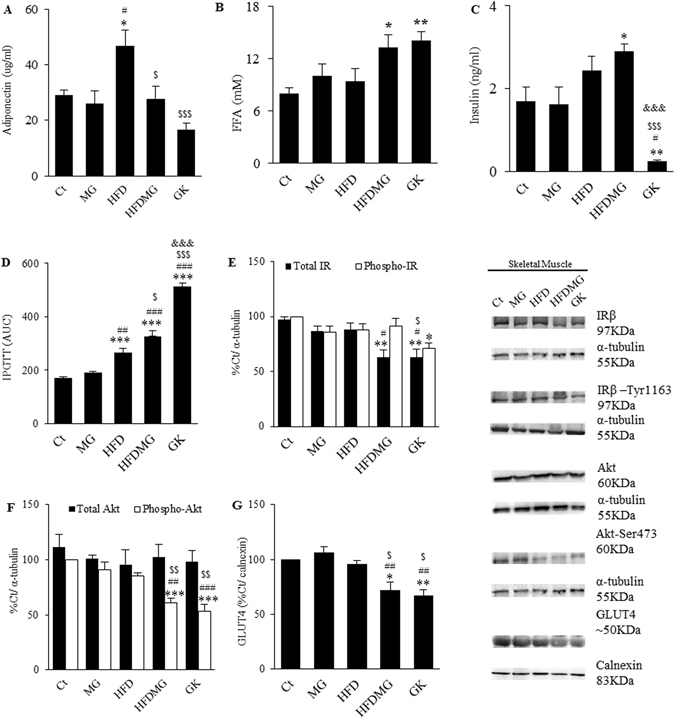
Glycation causes systemic dysmetabolism and impairs skeletal muscle insulin signaling in high-fat diet-fed rats. Circulating levels of adiponectin (A), FFAs (B) and insulin (C) were quantified by ELISA. Glucose intolerance was evaluated through the area under the curve of an IPGTT (D). Skeletal muscle insulin signalling was evaluated through WB quantification of activated and total forms of IR (E) and Akt (F) as well as GLUT4 transporter (G). Ct - Wistar 12 m; MG - Wistar + MG supplementation; HFD - HF diet-fed Wistar; HFDMG - HF diet-fed Wistar + MG supplementation; GK - Goto-Kakizaki 12 m. Bars represent means ± SEM, n = 6–8. * vs Ct; # vs MG; $ vs HFD; & vs HFDMG; 1 symbol p < 0.05; 2 symbols p < 0.01; 3 symbols p < 0.001.
As the adipose tissue, the skeletal muscle is a main target of insulin. MG supplementation or the HF diet separately had no effects in insulin signalling of skeletal muscle (Fig. 5E–G). However, HF diet fed-rats submitted to MG-supplementation demonstrated a significant decrease in the levels of the insulin receptor (p < 0.05 vs MG; p < 0.01 vs Ct), Akt active form (p < 0.01 vs MG and HFD; p < 0.001 vs Ct) and GLUT4 (p < 0.05 vs Ct and HFD; p < 0.01 vs MG) (Fig. 5E–G). Such effects were very similar to GK rats, which also showed decreased phopho-IR, total-IR, phospho-Akt and GLUT4 (Fig. 5E–G). Such results show that glycation also impairs skeletal muscle insulin signalling, contributing to systemic insulin resistance and glucose intolerance.
Discussion
In this study we investigated a new mechanism for adipose tissue dysfunction in obesity and type 2 diabetes. We demonstrate that glycation in pEAT has adverse vascular effects, impairing blood flow, hypoxia-response mechanisms and expandability which is tightly associated with local and systemic insulin resistance and glucose dysmetabolism. Adipose tissue dysfunction has been suggested to be caused by limited adipose tissue angiogenesis and expansion potential17, 34. This may be of particular relevance due to different metabolic outcomes of metabolically healthy (MHO) and unhealthy obesity (MUO). The latter is characterized by the earlier progression to insulin resistance and glucose dysmetabolism1, 2, 35.
Previously, we have shown that MG supplementation to Wistar rats impairs angiogenic markers and blood flow in adipose tissue, causing hypoxia, macrophage recruitment, hypoadiponectinemia and increased plasma FFA’s, but neither insulin resistance nor glucose dysmetabolism30. Such effects are reverted by pyridoxamine treatment, a dicarbonyl scavenger drug31. We have also demonstrated that glycation impairs adipocyte ability to adapt to hypoxia in a model of surgery-induced reduction of blood supply to the left pEAT, causing insulin resistance and adipocyte death32. Based on such observations, we hypothesized that adipose tissue glycation may induce microvascular lesions that hamper blood flow and expandability during a diet-induced expansion and lead to adipose tissue dysfunction and insulin resistance in obesity. This would therefore provide a strong mechanistic framework to the MUO phenotype. To test this conceptual model, we developed an animal model with HF diet-induced adipose tissue expansion and MG supplementation. We used a diet specifically enriched in triglycerides in order to induce physiological adipose tissue expansion and better isolate the variables of interest, while controlling for potential confounds. The effects of MG were compared with the endogenous glycation observed in diabetic GK rats.
MG supplementation in the diet increases the formation of stable MG adducts that are partially absorbed to the bloodstream, accumulate in different tissues and cause diabetes-like microvascular lesions36, 37. Our group demonstrated that our protocol results in MG levels (after derivatization) in plasma and adipose tissue that are similar to those of diabetic rats and here we demonstrate that MG supplementation results in CEL levels in adipose tissue similar to diabetic rats30.
In order to evaluate adipose tissue blood flow, we have developed, validated and applied a new in vivo quantitative DCE-MRI technique to assess pEAT blood flow. In the past, we evaluated pEAT blood flow through the accumulation of the fluorescent dye Evans Blue. However, this is a histological technique and is strongly influenced by adipocyte area, becoming inappropriate after HF diet-induced adipocyte hypertrophy30, 32. Using DCE-MRI, we demonstrate reduced pEAT blood flow in MG-supplemented groups and diabetic rats.
Several authors suggested that hypoxia could be caused by limited oxygen diffusion due to adipocyte hypertrophy, being a trigger to adipose tissue metabolic and endocrine dysfunction4, 11, 12, 38, 39. Nonetheless, the adipose tissue from obese patients was shown to be hyperoxic and to have only a very small proportion of adipocytes with a diameter superior to 100 µm, the oxygen diffusion distance, questioning such hypothesis14, 40. Our results demonstrate that HF diet-induced adipose tissue expansion does not cause significant alterations in adipose tissue blood flow and formation of hypoxic regions. Remarkably, hypoxia was found when the HF diet was combined with MG, showing that glycation-induced decreased blood flow leads to hypoxia when the adipose tissue is critically forced to expand.
Regarding the mechanisms involved in glycation-induced vascular lesions, some groups have introduced the concept of targeting adipose tissue angiogenesis to improve insulin sensitivity, based on decreased angiogenic ability of explants from obese donors5, 15–18. This is in line with observations showing that adipose tissue blood flow was observed to be decreased in obese patients due to impaired arteriolar function41, 42. Given that hypoxia is the main trigger for angiogenesis, we evaluated the role of glycation in regulating the mechanisms involved in adaptations to hypoxia and angiogenesis, as well as vascular tone. MG was shown to hamper HIF-1alpha stabilization in hypoxia, impairing cell response to hypoxia43. Here we show no differences in HIF-1alpha levels. Nevertheless, recent studies demonstrated the involvement of HIF-2alpha in preventing hypoxia-induced insulin resistance. Choe et al.44, demonstrated that HIF-2alpha expression in macrophages prevents M1 phenotype and their proinflammatory activity in adipose tissue. Mice lacking HIF-2alpha had insulin resistance and glucose intolerance44. Thus, while HIF-1alpha is important for the proinflammatory activation of M1 macrophages through iNOS induction, HIF-2alpha contributes to metabolic homeostasis by inhibiting such mechanisms45. Here we show decreased HIF-2alpha expression in high-fat diet-fed rats with MG supplementation and diabetic rats. Moreover, we observed an increased number of M1 macrophages in pEAT, which is in accordance with decreased HIF-2alpha levels. Such events may create a pro-inflammatory environment in the adipose tissue.
Our group recently demonstrated that MG-induced imbalance of VEGF/Ang-2 ratio inhibits tube-like formation, conducting to dysregulated endothelial cell proliferation and formation of aberrant capillaries33. In the present study, we show similar VEGF/Ang-2 ratio in HF diet-fed and control rats, but a decreased VEGF/Ang-2 ratio in HF diet-fed rats submitted to MG supplementation. Moreover, increased levels of the endothelial cell marker CD31 were observed, suggesting a higher number of endothelial cells and vessel disarrangement. Our data are also consistent with the findings of Jörgens et al.46, and Wang et al.47, which observed VEGF downregulation and formation of aberrant vessels in methylglyoxal-treated zebrafish and inhibition of angiogenesis in MG-treated human umbilical vein endothelial cells (HUVEC). The adipose tissue angiogenesis assay, an adaptation of the aortic ring assay, was recently developed by Corvera’s laboratory, but only for human subcutaneous and mice periepididymal adipose tissues48, 49. Based on the original one, we developed an assay for rat periepididymal adipose tissue. Our results show that, higher MG concentrations or selective inhibition of its detoxification by GLO-1 block the angiogenic process. However, before inhibiting cell proliferation and migration, MG destabilizes sprouts structure and affects their growth. This is in accordance with the findings of Liu et al.50, and Jörgens et al.46, who have shown excessive endothelial cell proliferation in HUVECs and formation of aberrant vessels in zebrafish after MG exposure. However, the mechanisms involved are still controversial. While the authors of the first study have shown increased autophagy-dependent VEGFR2 degradation, the others have shown increased VEGFR2 autophosphorylation, which would lead to excessive endothelial cell proliferation46, 50. Thus, MG has been shown to impair capillary structure and growth and here we extend these observations to adipose tissue vessels. Such events are likely to decrease adipose tissue blood flow and contribute to insulin resistance, but the mechanisms should be further elucidated in the future.
Karpe et al.42, observed impaired postprandial blood flow in adipose tissue and demonstrated that such events were associated with lower insulin sensitivity. As well, Farb et al., described impaired arteriolar function in the adipose tissue of obese patients, which mechanisms remain to be uncovered. Vascular tone is strongly influenced by the renin-angiotensin system (RAS) and by factors influencing vascular integrity and permeability. RAS has been shown to be upregulated by AGEs, increasing vascular damage in different tissues (reviewed by Matafome9). As well, AGEs were recently shown to increase vascular permeability by upregulating angiopoietin-like 4 (ANGPTL4) expression. Our results are in accordance with such body of evidence, showing that adipose tissue glycation increases AT1 and ANGPTL4 expression, which may impair vascular function.
Insulin resistance in fat depots contributes to increased spill-over of FFA to the circulation, ectopic deposition and consequently the development of all body insulin resistance4, 51, 52. Our present study demonstrates for the first time that accumulation of glycated products during pEAT expansion is associated with impaired insulin signalling in adipose tissue, Perilipin-A loss and decreased adiponectin secretion. Moreover, these alterations observed in pEAT contributed to systemic alterations, namely decreased glucose tolerance, hyperinsulinemia, increased FFAs and skeletal muscle insulin resistance. Our observations are in accordance with previous data from our laboratory and the studies of Gaens et al.53, 54, and Uribarri et al.3, demonstrating that RAGE-mediated CML accumulation in adipose tissue is involved in adipokines dysregulation and suggesting the involvement of AGE in the progression from healthy to unhealthy obesity.
In sum, our results demonstrate that glycation impairs pEAT microcirculation and expandability, in particular when associated with high fat diets and ensuing obesity. Moreover, such observations were replicated in the subcutaneous adipose tissue, showing that this is not a localized effect in pEAT (Supplementary Figure). Thus, we propose the existence of an adipovascular coupling mechanisms, based on the fact that blood flow is critical for adipocyte function and when decreased causes insulin resistance. This coupling is disrupted by glycation, impairing blood flow, adaptation to hypoxia and expansion potential, thus causing hypoxia and local and systemic insulin resistance (Fig. 6). Although the mechanisms should be further addressed in the future, our results suggest promising therapeutic targets in preventing unhealthy obesity and metabolic disorders.
Figure 6.
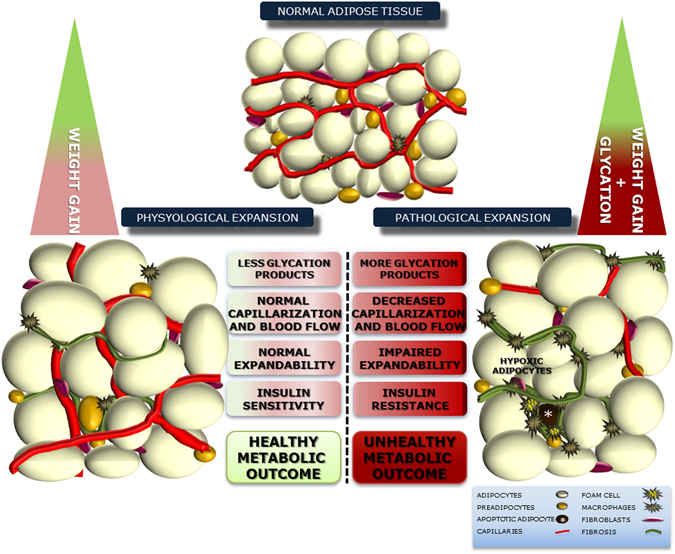
Impact of glycation in adipose tissue expandability, causing pathological expansion with impaired capillarization and blood flow, formation of hypoxc regions and insulin resistance. Such mechanisms may contribut to the poorer metabolic outcome of unhealthy obesity due to glucose dysmetabolism and alterations of the secretome.
Research Design and Methods
Antibodies
Calnexin, VEGFA, CD31, HIF-1 alpha, (AB0037, AB0063, AB0092, AB0112, Sicgen, Portugal), β-actin, α-tubulin (A5316, T6199, Sigma, USA), PPARγ, AKT, p-AKT(Ser473), VEGFR2, PI3K (#2443, #9272, #4058, #2479, #4249, Cell Signaling, USA), ANG-2, F4/80, Perilipin-A, GLUT4, p-IR(Y1361), GLO-I, C/EBPalpha, PGC1alpha, ANGPTL4, AT1, HIF-2 alpha (Ab8452, Ab74383, Ab3526, Ab65267, Ab60946, Ab96032, Ab40764, Ab191838, Ab2920, Ab9391, Ab8365, Abcam, UK), CD11c, CD206 (bs-2058R, bs-2664R Bioss, USA) TIE-2, IRβ (sc-324, sc-57342, SantaCruz Biotechnology, USA), CEL (KH025, TransGenic Inc, Japan).
Animal maintenance
Wistar and Goto-Kakizaki (GK) rats from our breeding colonies (Faculty of Medicine, University of Coimbra) were kept under standard conditions30, 32. The experimental protocol was approved by the local Institutional Animal Care and Use Committee (ORBEA IBILI-FMUC 03-2015). All the procedures were performed in accordance with the European Union Directive for Protection of Vertebrates Used for Experimental and Other Scientific Ends (2010/63/EU) and by users licensed by the Federation of Laboratory Animal Science Associations (FELASA).
Experimental groups
Male Wistar rats were randomly divided in four groups (n = 12/group): (1) Control (Ct) with standard diet AO3 (5% triglycerides, 21% proteins, 45% carbohydrates, SAFE, France); (2) Methylglyoxal group (MG) with standard diet and MG administration; (3) High-fat diet-fed group (HFD); (4) High-fat diet group with MG supplementation (HFDMG). A group (GK) of age-matched non-obese type 2 diabetic GK rats feeding the standard diet AO3 was used as a model of endogenous glycation.
Diet and MG administration
High-fat (HF) diet (40% triglycerides, 10% carbohydrates and 26% proteins, 231 HF, SAFE, France) was administered during 18 weeks (8 to 12 months old). MG (75 mg Kg−1day−1) was administered orally as before30, 32, 37.
Body weight and glycemic profile
In overnight (18 h) fasted rats, body weight was recorded and HbA1c, glycemia (fasting and 1 and 2 hours after i.p. glucose administration; 1.8 g Kg−1; IPGTT) were measured in the tail vein.
Magnetic resonance imaging
A dynamic contrast-enhanced (DCE) MRI study was performed using a BioSpec 9.4 T MRI scanner (Bruke, Biospin, Ettlingen, Germany). Rats (n = 6/group) were kept anesthetized by isoflurane (2–3%) with 100% O2 with body temperature and respiration monitoring (Intruments SA, Stony Brook, USA). A body quadrature transmit/receive volume coil with 71/112 mm of inner/outer diameter was used. Images were acquired with a fat-saturated T1-weighted gradient-echo sequence with parameters: TR/TE = 301.5/2.5 ms, FA = 50°, FOV = 65 × 65 mm2, matrix size = 169 × 169, 40 slices (axial orientation), slice thickness = 1.0 mm, 40 dynamics, scan time per dynamic = 51 s, total scan time = 34 mins. The contrast agent (Gadovist®, LUSAL, Portugal) was administered intraperitoneally, after the acquisition of 3 baseline dynamics. Tissue enhancement curves were obtained offline using homemade software implemented in Matlab (v2013a, Mathworks, Natick, Mass). Intensity variation as a function of time was quantified in regions of interest (ROIs) in skeletal muscle, pEAT and subcutaneous adipose tissue. The area under the curve (AUC) was calculated to indirectly quantify blood flow.
Blood and adipose tissue collection
Animals were anesthetized and serum and plasma were collected as described before30, 32. After sacrifice by cervical displacement, adipose and muscle tissue samples were frozen (−80 °C) or stored in 10% formalin.
Analysis of adipose tissue hypoxic regions
Hypoxic regions in the adipose tissue were determined through intra-peritoneal injection of pimonidazole (60 mg−1 kg, 40 minutes, n = 3/group). The Hypoxia Probe Kit (Millipore, USA) was used to assess hypoxic regions by Western blotting (WB) and immunohistochemistry (IHC).
Blood analyses
Serum triglyceride levels were determined using commercial kits (Olympus-Diagnóstica, Portugal). Plasma levels of FFA and insulin were assessed using the FFA Assay Kit (ZenBio, NC, USA) and the Rat Insulin ELISA Kit (Mercodia, Sweden). Serum adiponectin and leptin were determined using the Rat Adiponectin Immunoassay Kit and the Rat Leptin Immunoassay Kit (Invitrogen, USA).
Western Blotting
Adipose tissue (300 mg) and skeletal muscle (100 mg) (n = 6) were homogenized and assayed as before30, 32. The secondary antibodies were anti-mouse (GE Healthcare, UK), anti-rabbit and anti-goat (Bio-Rad, USA). Membranes were revealed using ECL substrate in a Versadoc system (Bio-Rad, USA) and analyzed with Image Quant® (Molecular Dynamics, USA).
Histological colorimetric assays
Tissue sections (4 µm) from paraffin-embedded pEAT (n = 3/group) were stained with Periodic Acid-Schiff (PAS) or Masson Trichrome staining. Images were captured in a Zeiss microscope with incorporated camera (Germany). The number of adipocytes was determined in at least 10 fields/slice and the mean adipocyte area was determined.
Immunohistochemistry
Immune staining of pimonidazole adducts and CEL was performed after paraffin removal, hydration and blocking. Sections were incubated overnight (4 °C) with primary antibody and with secondary antibody-peroxidase (2 hours, RT) (IHC peroxidase Kit, Chemicon, USA). DAB (diaminobenzidine) was used as substrate. Sections were stained with hematoxylin before mounting.
Ex vivo adipose tissue angiogenic assay
The rat adipose tissue angiogenic assay was developed based on the method described by Gealekman et al.48, and Rojas-Rodríguez et al.49. Periepididymal adipose tissue from 4 week old Wistar rats was collected and cut in ~1 mm3 pieces. Explants were then immediately embedded in 60 μl of collagen in 96-well plates and cultured with EGM-2 MV (BulletKit CC-3202; Lonza, Allendale, NJ, USA). Explants were incubated with control medium, MG-supplemented (50 μM, 100 μM, 250 μM, 50 μM and 1 mM), treated with the inhibitor of GLO-1 S-p-bromobenzylglutathione cyclopentyl diester (BBGC, 20 μM) or with BBGC 20 μM in combination with MG 250 μM (n = 15 explants/condition) (n = 4 experiments). After 6 days, images were captured in a Zeiss Axio Observer Z1 with an incorporated camera (Zeiss, Germany). The area of capillarization was calculated and normalized for the area of the explant. Tubular (sprout) length was also determined as a measure of cell organization and capillary integrity, as described before55.
Statistical analysis
Results are presented as mean ± SEM per group. Given the relatively small sample size (n = 6–12), the non-parametric Kruskal-Wallis test (all pairwise multiple comparisons) was applied to determine all statistical differences between the groups, using the SPSS software (IBM, NY, USA). The alpha level of significance for all experiments was 0.05 and p < 0.05 was considered as the criterion for significance.
Electronic supplementary material
Acknowledgements
This study was supported by Portuguese Foundation for Science and Technology (project UID/NEU/04539/2013), QREN- COMPETE (project DoIT – Diamarker: a consortium for the discovery of novel biomarker in diabetes), POCI-01-0145-FEDER-007440 and by the Faculty of Medicine, University of Coimbra. T. R. and P. M. are supported by a PhD (SFRH/BD/101172/2014) and a Post-Doc Grant (SFRH/BPD/104881/2014). This study was granted by the Portuguese Society of Diabetology (Portuguese National Prize of Diabetes).
Author Contributions
P.M., as a FELASA user, was responsible for in vivo experiments, supervised the project and is the guarantor of this work, having full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. T.R., J.A., L.G., C.C., A.A., E.W., C.N. and R.F. were responsible for animal treatments and performed most of the biochemical analyses of blood, muscle and adipose tissue. J.S., J.C. and S.G. were responsible for magnetic resonance data acquisition and analysis, under the supervision of M.C.B. I.S. developed all the experiments involving histological analyses. P.M., S.V.C. and R.S. are the guarantors and were responsible for data discussion and the final revision of the manuscript. T.R. wrote this manuscript, which was also reviewed by all the other authors.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01730-3
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Boonchaya-anant P, Apovian CM. Metabolically Healthy Obesity—Does it Exist? Curr. Atheroscler. Rep. 2014;16:1–9. doi: 10.1007/s11883-014-0441-1. [DOI] [PubMed] [Google Scholar]
- 2.Hinnouho G-M, et al. Metabolically healthy obesity and the risk of cardiovascular disease and type 2 diabetes: the Whitehall II cohort study. Eur. Hear. J. 2014;36:551–559. doi: 10.1093/eurheartj/ehu123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uribarri J, et al. Dietary Advanced Glycation End Products and Their Role in Health and Disease. Adv. Nutr. An Int. Rev. J. 2015;6:461–473. doi: 10.3945/an.115.008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008;114:715–28. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corvera S, Gealekman O. Adipose tissue angiogenesis: Impact on obesity and type-2 diabetes. Biochim. Biophys. Acta. 2014;1842:463–472. doi: 10.1016/j.bbadis.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006;6:772–83. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 7.Rodrigues T, Matafome P, Seiça R. A vascular piece in the puzzle of adipose tissue dysfunction: mechanisms and consequences. Arch. Physiol. Biochem. 2013;120:1–11. doi: 10.3109/13813455.2013.838971. [DOI] [PubMed] [Google Scholar]
- 8.Hocking S, Wu L, Guilhaus M. Intrinsic Depot-Specific Differences in the Secretome of Adipose Tissue, Preadipocytes, and Adipose Tissue–Derived Microvascular Endothelial Cells. Diabetes. 2010;59:3008–3016. doi: 10.2337/db10-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matafome P, Rodrigues T, Seiça R. Glycation and hypoxia: two key factors for adipose tissue dysfunction. Curr. Med. Chem. 2015;22:1–21. doi: 10.2174/0929867322666150209155633. [DOI] [PubMed] [Google Scholar]
- 10.Wellen K, Hotamisligil G. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Invest. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am. J. Physiol. Endocrinol. Metab. 2007;293:E1118–28. doi: 10.1152/ajpendo.00435.2007. [DOI] [PubMed] [Google Scholar]
- 12.Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br. J. Nutr. 2008;100:227–35. doi: 10.1017/S0007114508971282. [DOI] [PubMed] [Google Scholar]
- 13.Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013;93:1–21. doi: 10.1152/physrev.00017.2012. [DOI] [PubMed] [Google Scholar]
- 14.Goossens GH, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation. 2011;124:67–76. doi: 10.1161/CIRCULATIONAHA.111.027813. [DOI] [PubMed] [Google Scholar]
- 15.Gealekman O, et al. Effect of rosiglitazone on capillary density and angiogenesis in adipose tissue of normoglycaemic humans in a randomised controlled trial. Diabetologia. 2012;55:2794–2799. doi: 10.1007/s00125-012-2658-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mannerås-Holm L, Krook A. Targeting adipose tissue angiogenesis to enhance insulin sensitivity. Diabetologia. 2012;55:2562–4. doi: 10.1007/s00125-012-2651-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rutkowski JM, Davis KE, Scherer PE. Mechanisms of obesity and related pathologies: the macro- and microcirculation of adipose tissue. FEBS J. 2009;276:5738–46. doi: 10.1111/j.1742-4658.2009.07303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 2013;18:478–89. doi: 10.1016/j.cmet.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 20.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 21.Goldin A, Beckman J, Schmidt AM, Creager M. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 22.Negre-Salvayre A, et al. Hyperglycemia and glycation in diabetic complications. Antioxid. Redox Signal. 2009;11:3071–109. doi: 10.1089/ars.2009.2484. [DOI] [PubMed] [Google Scholar]
- 23.Matafome P, Sena C, Seiça R. Methylglyoxal, obesity, and diabetes. Endocrine. 2013;43:472–84. doi: 10.1007/s12020-012-9795-8. [DOI] [PubMed] [Google Scholar]
- 24.Dhar A, Dhar I, Jiang B, Desai KM, Wu L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes. 2011;60:899–908. doi: 10.2337/db10-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Unoki H, et al. Advanced glycation end products attenuate cellular insulin sensitivity by increasing the generation of intracellular reactive oxygen species in adipocytes. Diabetes Res. Clin. Pract. 2007;76:236–44. doi: 10.1016/j.diabres.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Jia X, Wu L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 2007;306:133–139. doi: 10.1007/s11010-007-9563-x. [DOI] [PubMed] [Google Scholar]
- 27.Cook LJ, et al. Effects of methylglyoxal on rat pancreatic beta-cells. Biochem. Pharmacol. 1998;55:1361–7. doi: 10.1016/S0006-2952(97)00619-9. [DOI] [PubMed] [Google Scholar]
- 28.Fiory F, et al. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia. 2011;54:2941–52. doi: 10.1007/s00125-011-2280-8. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann SMS, et al. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes. 2002;51:2082–2089. doi: 10.2337/diabetes.51.7.2082. [DOI] [PubMed] [Google Scholar]
- 30.Matafome P, et al. Methylglyoxal causes structural and functional alterations in adipose tissue independently of obesity. Arch. Physiol. Biochem. 2012;118:58–68. doi: 10.3109/13813455.2012.658065. [DOI] [PubMed] [Google Scholar]
- 31.Rodrigues T, Paulo M, Santos-Silva D, Sena C, Seiça R. Reduction of methylglyoxal-induced glycation by pyridoxamine improves adipose tissue microvascular lesions. J. Diabetes Res. 2013;2013:690650. doi: 10.1155/2013/690650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodrigues T, Matafome P, Seiça R. Methylglyoxal further impairs adipose tissue metabolism after partial decrease of blood supply. Arch. Physiol. Biochem. 2013;119:209–218. doi: 10.3109/13813455.2013.812121. [DOI] [PubMed] [Google Scholar]
- 33.Bento CF, et al. Methylglyoxal-induced imbalance in the ratio of vascular endothelial growth factor to angiopoietin 2 secreted by retinal pigment epithelial cells leads to endothelial dysfunction. Exp. Physiol. 2010;95:955–70. doi: 10.1113/expphysiol.2010.053561. [DOI] [PubMed] [Google Scholar]
- 34.Moreno-Indias I, Tinahones FJ. Impaired Adipose Tissue Expandability and Lipogenic Capacities as Ones of the Main Causes of Metabolic Disorders. J. Diabetes Res. 2015;2015:970375–12. doi: 10.1155/2015/970375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uribarri J, et al. Elevated serum advanced glycation endproducts in obese indicate risk for the metabolic syndrome: a link between healthy and unhealthy obesity? J. Clin. Endocrinol. Metab. 2015;100:1957–66. doi: 10.1210/jc.2014-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golej J, Hoeger H, Radner W, Unfried G, Lubec G. Oral administration of methylglyoxal leads to kidney collagen accumulation in the mouse. Life Sci. 1998;63:801–7. doi: 10.1016/S0024-3205(98)00336-1. [DOI] [PubMed] [Google Scholar]
- 37.Berlanga J, et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. 2005;109:83–95. doi: 10.1042/CS20050026. [DOI] [PubMed] [Google Scholar]
- 38.Hosogai N, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–11. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 39.Halberg N, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 2009;29:4467–83. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goossens GH, Blaak EE. Adipose Tissue Dysfunction and Impaired Metabolic Health in Human Obesity: A Matter of Oxygen? Front. Endocrinol. 2015;6:1–5. doi: 10.3389/fendo.2015.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farb MGM, et al. Arteriolar function in visceral adipose tissue is impaired in human obesity. Arterioscler. Thromb. Vasc. Biol. 2012;32:467–73. doi: 10.1161/ATVBAHA.111.235846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karpe F, et al. Impaired Postprandial Adipose Tissue Blood Flow Response Is Related to Aspects of Insulin Sensitivity. Diabetes. 2002;51:2467–2473. doi: 10.2337/diabetes.51.8.2467. [DOI] [PubMed] [Google Scholar]
- 43.Bento CF, et al. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1α for degradation in the presence of methylglyoxal. PLoS One. 2010;5:1–13. doi: 10.1371/journal.pone.0015062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choe SS, et al. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes. 2014;63:3359–3371. doi: 10.2337/db13-1965. [DOI] [PubMed] [Google Scholar]
- 45.Aouadi M. HIF-2 Blows Out the Flames of Adipose Tissue Macrophages to Keep Obesity in a Safe Zone. Diabetes. 2014;63:3169–3171. doi: 10.2337/db14-0878. [DOI] [PubMed] [Google Scholar]
- 46.Jörgens K, et al. High Tissue Glucose Alters Intersomitic Blood Vessels in Zebrafish via Methylglyoxal Targeting the VEGF Receptor Signaling Cascade. Diabetes. 2015;64:213–225. doi: 10.2337/db14-0352. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, et al. Glycation of vitronectin inhibits VEGF-induced angiogenesis by uncoupling VEGF receptor-2 − α v β 3 integrin cross-talk. Cell death Dis. 2015;6:1–8. doi: 10.1038/cddis.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gealekman O, et al. Enhanced angiogenesis in obesity and in response to PPARgamma activators through adipocyte VEGF and ANGPTL4 production. Am. J. Physiol. Endocrinol. Metab. 2008;295:E1056–64. doi: 10.1152/ajpendo.90345.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rojas-Rodriguez R, et al. Adipose Tissue Angiogenesis Assay Raziel. Methods Enzym. 2014;537:75–91. doi: 10.1016/B978-0-12-411619-1.00005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu H, Yu S, Zhang H, Xu J. Angiogenesis Impairment in Diabetes: Role of Methylglyoxal-Induced Receptor for Advanced Glycation Endproducts, Autophagy and Vascular Endothelial Growth Factor Receptor 2. PLoS One. 2012;7:e46720. doi: 10.1371/journal.pone.0046720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith U. Abdominal obesity: a marker of ectopic fat accumulation. J clin invest. 2015;125:1790–1792. doi: 10.1172/JCI81507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goossens GH. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol. Behav. 2008;94:206–18. doi: 10.1016/j.physbeh.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 53.Gaens KHJ, Stehouwer CDA, Schalkwijk CG. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Curr. Opin. Lipidol. 2013;24:4–11. doi: 10.1097/MOL.0b013e32835aea13. [DOI] [PubMed] [Google Scholar]
- 54.Gaens KHJ, et al. Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2014;34:1199–1208. doi: 10.1161/ATVBAHA.113.302281. [DOI] [PubMed] [Google Scholar]
- 55.Santos-Oliveira P, et al. The Force at the Tip–Modelling Tension and Proliferation in Sprouting Angiogenesis. PLoS Comput. Biol. 2015;11:e1004436. doi: 10.1371/journal.pcbi.1004436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.