

Abstract
Identifying the genomic changes that control morphological variation and understanding how they generate diversity is a major goal of evolutionary biology. In Heliconius butterflies, a small number of genes control the development of diverse wing color patterns. Here, we used full genome sequencing of individuals across the Heliconius erato radiation and closely related species to characterize genomic variation associated with wing pattern diversity. We show that variation around color pattern genes is highly modular, with narrow genomic intervals associated with specific differences in color and pattern. This modular architecture explains the diversity of color patterns and provides a flexible mechanism for rapid morphological diversification.
Recent adaptive radiations, such as the Heliconius butterflies1, Galápagos finches2 and African cichlids3, offer insight into evolutionary and ecological forces that underlie diversification. Typically, ecological opportunities allow natural and sexual selection to drive adaptive change and speciation. At a genetic level, recruitment from ancient polymorphism, introgression of adaptive variants between populations and de novo mutation are important sources of variation. However, the genetic architecture of the traits under natural and sexual selection that facilitates rapid diversification remains largely unexplored.
In this study, we sequenced the genome of the Neotropical butterfly Heliconius erato and used re-sequence data from 116 additional individuals to dissect the architecture of genomic variation associated with their vividly colored wing patterns. With over 400 different wing color forms among 46 described species4, Heliconius represents one of the most visually diverse radiations in the animal kingdom and an excellent system for establishing a broad and integrative view of morphological diversification. The evolution of scale cells and the spatial coordinate system that controls wing pigmentation is a key innovation of the Lepidoptera. Wing patterns are often under strong natural and sexual selection and these forces probably shape much of the pattern diversity we see among the more than 160,000 butterfly and moth species5.
In Heliconius, conspicuous wing patterns are important for signaling toxicity to potential predators6 and play a role in mate selection7. Natural selection favors Müllerian mimicry among toxic butterflies, resulting in convergence between co-occurring species, as well as geographic divergence between populations of the same species8. Among Heliconius butterflies, the genetic basis of this wing diversity has been studied for nearly 60 years and more than 30 Mendelian loci have been described9. Over the past decade, however, genetic research has shown that most of the complexity of color variation across Heliconius is actually controlled by relatively few genes acting broadly across the fore- and hindwing10–16. These genes include the transcription factor optix14,17, the signaling ligand wntA15 and a cell cycle regulator cortex16. Hence, these studies have revealed that a limited set of “toolkit”18 genes has been repeatedly used for both highly divergent and convergent phenotypes in Heliconius, as well as other butterfly and moth species16,19,20. However, the key to wing pattern variation in Heliconius is not within the genes themselves, which are strongly conserved at the amino acid level, but at nearby non-coding regions that control expression during wing development14–16.
Here, we sequenced the genomes of 15 distinctly colored H. erato races and 8 closely related species to fully describe the regulatory architecture driving adaptive evolution of the major genes acting in Heliconius wing patterning (Figure 1). Our genomic survey included samples obtained near seven transition zones of hybridizing H. erato races with divergent wing patterns (Figure 2A). In these hybrid zones, the high rate of genetic admixture allows for detailed genotype by phenotype (G × P) association mapping to identify discrete genomic intervals associated with color and pattern variation on Heliconius wings21,22. We then further investigated these intervals with a novel phylogenetic method for identifying conserved non-coding regions in closely related non-hybridizing races and species. This combined strategy of association mapping and phylogenetic inference resulted in a distinct set of narrow genomic intervals that corresponded to loci described in early crossing experiments (Table S1 in SI section 1)9. All the intervals fell within non-coding regions adjacent to color pattern genes that affect forewing band shape (wntA; Figure 3), red pigmentation (optix; Figure 4) and a yellow hindwing bar (cortex; Figure 5). Our results underscore a highly modular regulatory architecture that provides a flexible mechanism for rapid morphological change (Figure 6).
Figure 1. Geographical distribution, phylogeny and color pattern diversity of the Heliconius erato adaptive radiation.
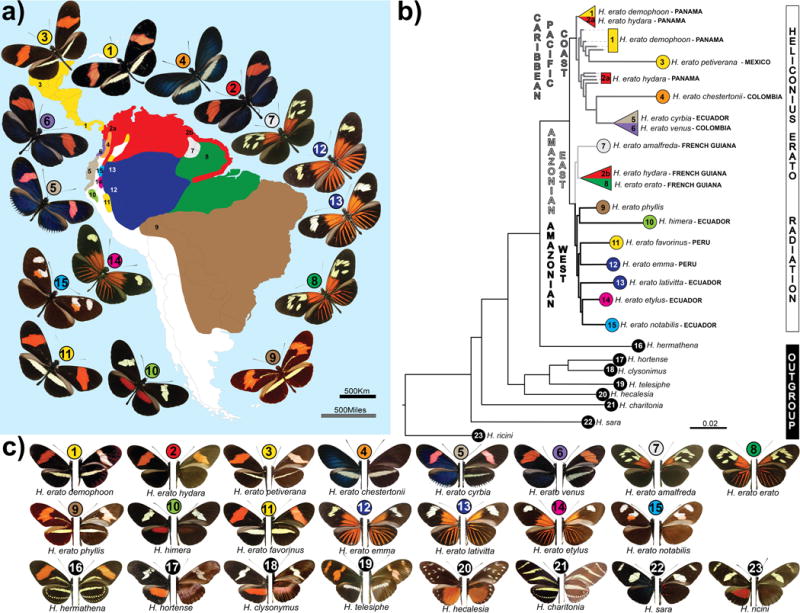
(a.) Geographical origin of samples; colors represent the distribution of the races; numbers are placed according to the sampling sites. (b.) Maximum likelihood tree based on autosomal sites located on chromosomes that do not show any marked FST peaks. All nodes shown had full local support based on the Shimodaira-Hasegawa test. Color and numbers represent, respectively, the geographical distribution and sampling site. On average five individuals were sequenced for each race and two for each outgroup species. All samples used in this study were included in the tree. There were three cases, (triangles) where individuals did not cluster together by racial designation (see Figure S5 for the full genome tree). (c.) Pictures of dorsal (left) and ventral (right) sides of the wings of races and species used in this study. Bottom row with black circles represent species that belong to the erato clade, but not to the H. erato adaptive radiation.
Figure 2. Genomic divergence across the Heliconius erato phenotypic transition zones.
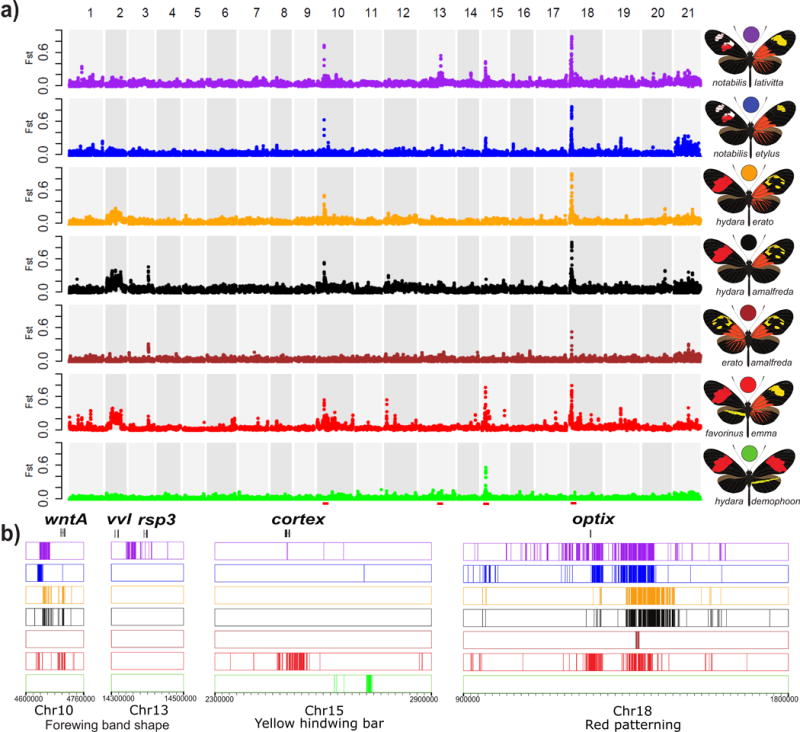
(a.) FST values were calculated between color morphs from each of seven hybrid zones (indicated at right) and averaged over 50 kb windows sliding in increments of 20 kb. Peaks represent regions of the genome with strongly divergent allele frequencies. Divergence at chromosome 10, 15 and 18 corresponds with, respectively, divergence near the color pattern genes wntA, cortex and optix (red dashes). These loci drive black forewing, yellow hindwing bar and red pigmentation patterns, respectively. Importantly, between hybridizing races that were divergently colored, the only regions of the genome in which we found fixed allelic differences were at the color pattern loci (see SI section 4.3 for a discussion of other regions of the genome with increased divergence). (b.) Distribution of genotypes fixed between hybridizing races located in the peaks of high divergence. This analysis revealed that, depending on the variable phenotype in the hybrid zone, clusters of fixed SNPs are found in different genomic intervals near color pattern genes.
Figure 3. Association mapping in hybrid zones and phylogenetic comparisons identify the modular genetic architecture of black forewing variation.
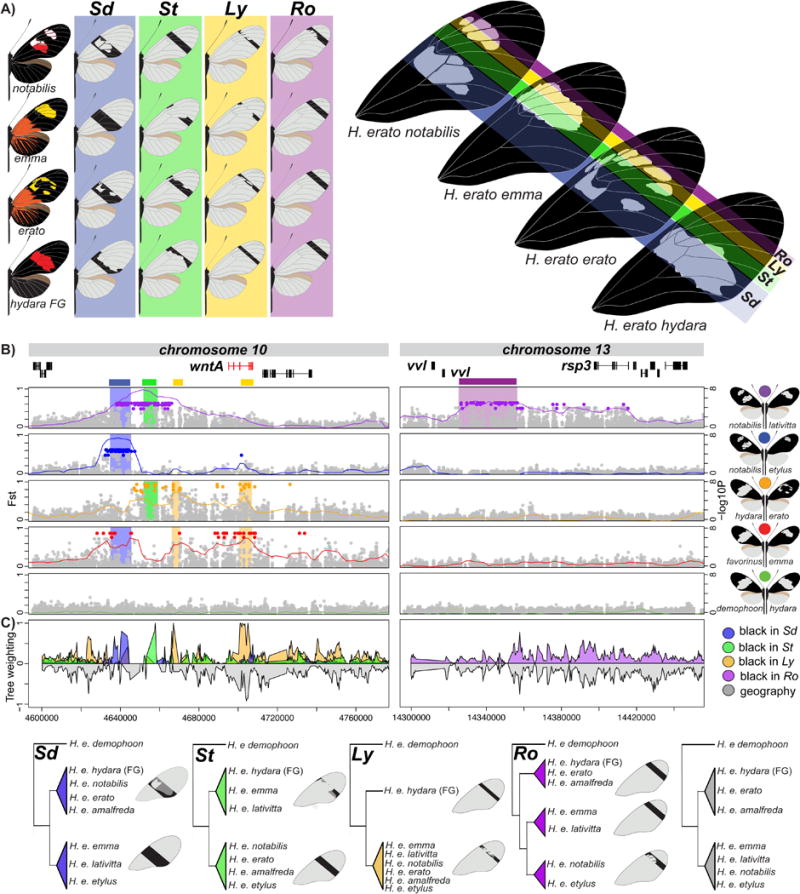
(a.) Variation in black forewing patterning in the H. erato races. Black shading in the forewings highlights variation in melanin production in different parts of the forewing. Color shading corresponds to shading in panel B and C. (b.) FST (lines; 20 kb window, 5 kb step size) and association (points) analysis at the peaks of divergence in chromosome 10 and 13. Colored points represent associations estimated from fixed SNPs. (c.) Phylogenetic weighting of phenotypic hypothesis consistent with the Sd, St, Ly and Ro elements. These weightings were obtained by summing weightings for topologies that were consistent with the hypothesized groupings presented in the phylogenies. Tree topologies consistent with a geographic grouping are represented negative in gray. Within the genomic regions with high phylogenetic weighting support for a particular phenotypic hypothesis, we defined the boundaries of the color pattern intervals as position 4,634,972-4,641,535 for Sd, 4,657,452-4,658,207 for St, 4,666,909-4,670,474 for Ly1 and 4,700,932-4,708,441 for Ly2 on chromosome 10 and Position 14,341,251- 14,412,364 for Ro on chromosome 13. It is possible to further subdivided the Sd interval into two narrow intervals based on the phylogenetic weighting support and patterns of shared genotypes (position 4,637,657-4,637,727 for Sd1, 4,639,853-4,641,535 for Sd2). See SI, section 4.1.2 and 4.2.2 for the full phylogenetic trees of the identified intervals including all H. erato samples and closely related outgroup species.
Figure 4. Modular architecture of red pattern variation.
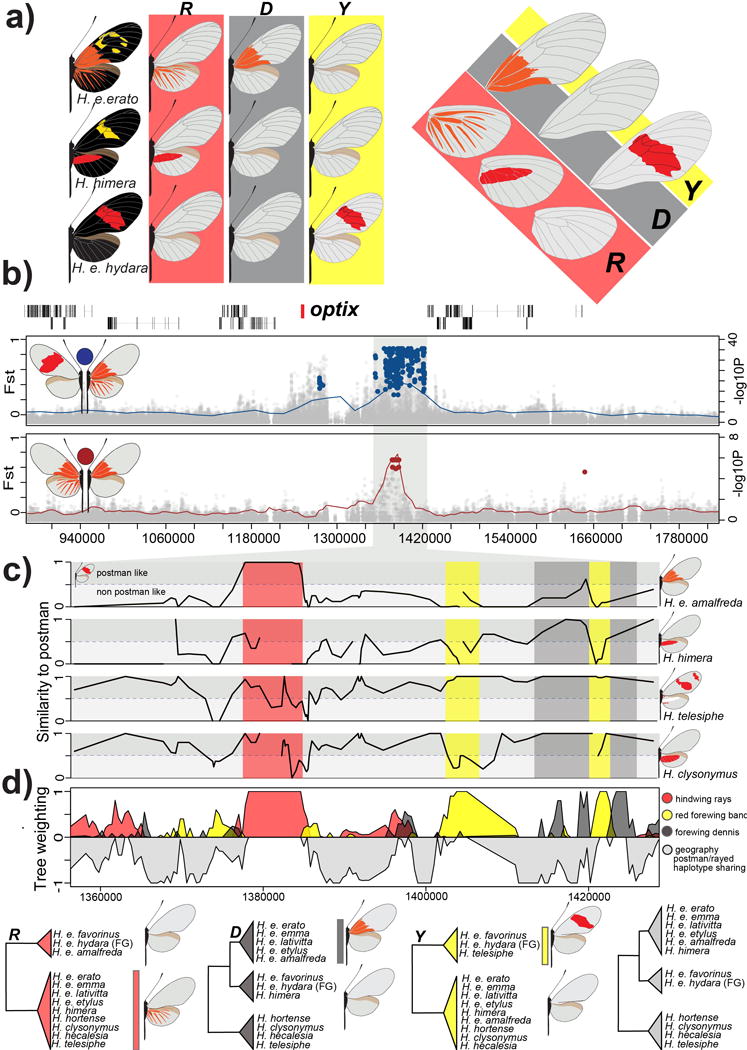
(A.) Variation in red color patterning in the H. erato races in the ray (R), band (Y) and dennis (D) region of the wings. (B.) FST (lines; 20 kb window, 5 kb step size) and association (points) analysis at the peaks of divergence in the optix genomic region on chromosome 18 between races with red rays and dennis patch (ray-dennis) versus races with a red forewing band (postman) (red; top panel) and H. e. amalfreda (no rays) versus H. e. erato (rays) (brown; bottom panel). Colored points represent associations estimated from fixed SNPs. (C.) Genotype weightings (10 SNP window, 5 SNP step size, 3 SNPs minimum genotyped in 50% of population) of the positions that were identified as fixed between ray-dennis versus postman. A weighting of 1, means races or species have the same genotypes as the postman races, whereas a weighting of 0 indicates completely different genotypes in the considered window of fixed SNPs. (D.) Phylogenetic weighting of phenotypic hypothesis consistent with the R, Y and D elements. These weightings were obtained by summing weightings for topologies that were consistent with the hypothesized groupings presented in the phylogenies. Due to haplotype sharing among Rayed/Dennis and Postman races, tree topologies consistent with geography are never supported in this genomic interval. Support for topologies consistent with a geographic tree that accounts for this haplotype sharing are represented upside-down in gray. We outlined the following positions: 1,377,801–1,384,841 for R, 1,403,328–1,412,865 for Y1, 1,420,912–1,422,355 for Y2, 1,412,888–1,419,375 for D1 and 1,422,585–1,428,307 for D2 on chromosome 18. See SI, section 3.3.2 for the full phylogenetic trees of the identified intervals including all H. erato samples and closely related outgroup species.
Figure 5. Independent modules generate convergent yellow hindwing bar phenotypes.
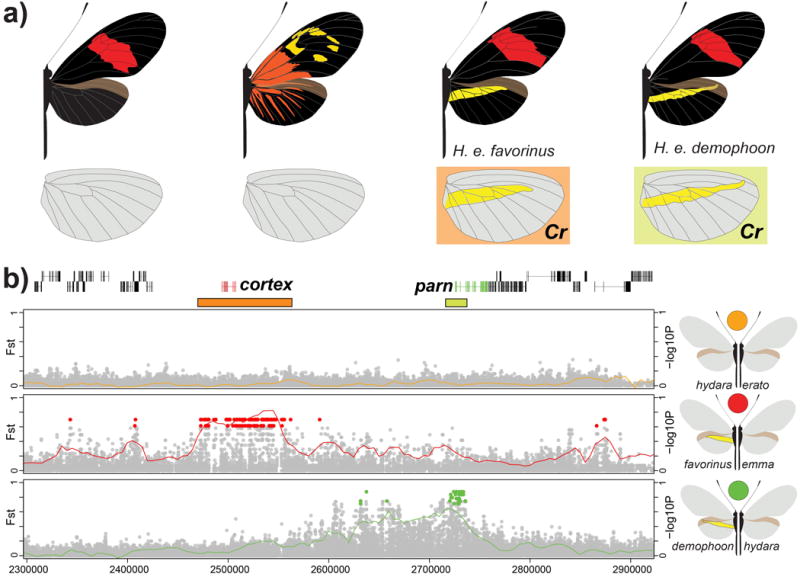
(a.) Variation in yellow hindwing bar in H. e. favorinus from Peru and H. e. demophoon from Panama. We note that the yellow hindwing bar morphology is not completely identical between these two races. While the yellow hindwing bar of H. e. demophoon is narrow, long and pointing up, H. e. favorinus exhibits a broader, shorter bar that points down. Shading corresponds to shading in panel b where two independent association peaks are identified. (b.) FST (lines; 20 kb window, 5 kb step size) and association (points) analysis near the cortex gene on chromosome 15. Comparison between H. e. favorinus and H. e. emma (red) shows a block of divergence different from the comparison between H. e. demophoon and H. e. hydara (green). The block of association between H. e. demophoon and H. e. hydara overlaps with the parn gene, but no functional link with color pattern variation has been identified for this gene16. Colored points represent associations estimated from fixed SNPs. Based on fixed SNP associations, we defined the positions of these two intervals as 2,053,037-2,171,230 for Cr1 (orange) and 2,211,881-2,315,926 for Cr2 (yellow). See SI, section 4.4.2 for the full phylogenetic trees of the identified intervals including all H. erato samples and closely related outgroup species.
Figure 6. Modular regulatory architecture characterizes color pattern diversity within the Heliconius erato radiation.
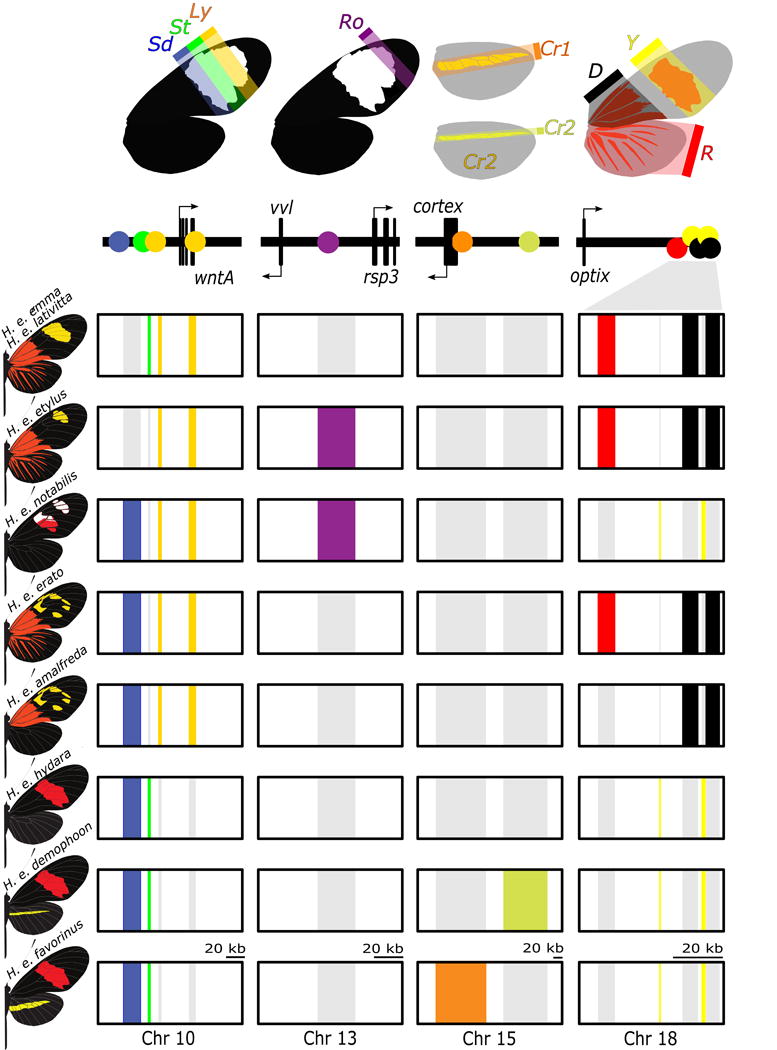
The upper panel provides a summary of color pattern variation found among H. erato butterflies that is related to spatial expression of the genes wntA (black forewing patterning; chromosome 10), cortex (yellow hindwing bar; chromosome 15), optix (red; chromosome 18) and a functionally uncharacterized genomic interval on chromosome 13 responsible for pattern variation in the most distal region of the forewing band (Ro; functional candidates vvl and rsp3). The boxes in the bottom panel represent chromosomal intervals that include regulatory modules. These regulatory modules are colored for butterflies in which the pattern is expressed. The regulatory modules have been rearranged among H. erato races to generate distinct wing phenotypes. Note that for Cr1 and Cr2 and rays (R), band (Y) and dennis (D) patterns are expressed when, respectively, cortex and optix are expressed, whereas for Sd, St and Ly pattern expression corresponds with absence of wntA expression.
Results and Discussion
Reference sequence and variants
With more than 25 different wing pattern races, H. erato provides exceptional opportunities to explore the links between genotype, phenotype, form and function. We first constructed a high-quality reference genome by a combination of hybrid assembly coupled with high-resolution linkage analysis. Our assembly and validation strategy generated one of the most contiguous and accurate Lepidopteran genomes assembled thus far (SI section 2), which is available on the LepBase genome browser. The final assembly consisted of 198 scaffolds with N50 length of over 10 Mb and a total assembly length of 383 Mb. A total of 13,678 genes were identified using RNA-seq and a thorough annotation process (SI section 3). To examine variation across our reference genome, we generated high (15–30x) coverage whole-genome resequence data from 116 individuals of H. erato and closely related species. For the 101 H. erato individuals sampled, we genotyped the majority of the non-repetitive portion of the genome (average of 62% per individual, SI section 4.1). For the 15 individuals from the 8 outgroup species, the number of positions that were genotyped for the outgroup species was lower, but above 40% for the most divergent comparison (SI section 4.1).
Genome-wide divergence across the H. erato color pattern radiation
Within H. erato, individuals clustered by geographic proximity rather than color pattern phenotype as has been previously reported23 (Figure 1B and C). For example, forewing red banded H. erato races were found in all three (Caribbean/Pacific Coast, East Amazonian, and West Amazonian) major geographic lineages (Figure 1). Even within these broad geographic regions, individuals used in this study grouped together by sampling location rather than wing morphology. Indeed, there was little genetic differentiation between H. erato individuals sampled across major phenotypic transition zones, except around the genomic regions already known to be involved in color pattern variation (Figure 2A). Genetic divergence as measured by FST was close to zero across most of the genome, supporting the hypothesis of unhindered gene flow except at the regions responsible for color pattern differences (FST < 0.1 in 97.07 ± 0.03% of 50 kb windows; SI section 3.3)22. This contrasted to three sharp peaks of genomic differentiation across known color pattern loci on chromosome 10 near the wntA gene, on chromosome 15 near cortex, and on chromosome 18 near optix (red in Figure 2B). As previously reported for the region around optix22, these regions showed the expected signatures of selection, including reduced nucleotide diversity and elevated dXY relative to genome-wide averages (SI section 4.3).
Associating genomic variation with color pattern diversity
Genetic differences at the regions controlling phenotypic variation in Heliconius are maintained by strong natural selection24–26. However, genotype by phenotype (G × P) associations were often complex between any pairwise comparison reflecting different histories of interactions between hybridizing taxa. Thus, at any specific comparison, associations often spanned hundreds of thousands of base pairs around each color pattern locus (Figure 2B). Nonetheless, by combining analysis of variation across multiple hybrid zones with phylogenetic analysis, we pinpointed specific genomic intervals associated with specific aspects of phenotypic variation. This combination of G × P association and phylogenetic analysis revealed a highly modular architecture to the variation around major color pattern loci.
Modular architecture of forewing black color variation
Recent genetic mapping coupled with studies of gene expression, suggest that a single gene, wntA, is driving much of the forewing pattern variation across Heliconius species27. Indeed, our G × P association highlighted a 100 kb non-coding region near wntA on chromosome 10 (Figure 3). Clusters of fixed SNPs defined discrete genomic intervals associated with the phenotypic effects of the Sd, St and Ly loci that were first described by Sheppard and colleagues more than 30 years ago9. Variation at Sd, St, and Ly was predicted to control patterning across the middle to the most distal sections of the forewing respectively (Figure 3A). Consistent with this hypothesis, we identified: 1) a 25 kb region of fixed differences between H. e. notabilis and H. e. lativitta that differed across the lower (Sd) and the middle (St) region of the forewing (purple in Figure 3B), 2) a narrow peak of association between H. e. notabilis and H. e. etylus that differed only in the lower forewing region (Sd) (blue in Figure 3B), and 3) a broad region of association that spans roughly 60 kb and appears to be composed of several distinct peaks between H. e. erato and H. e. hydara from French Guiana that differed in St and Ly (orange in Figure 3B). Comparisons between races with identical forewings showed no G × P association across any of these regions (green in Figure 3B).
To further refine the regions associated with forewing band pattern, we used a novel tree weighting approach called Twisst (Topology weighting by iterative sampling of subtrees; see methods)28, to explore how phylogenetic relationships varied around wntA. We hypothesize that the genomic variation underlying wing pattern differences should cluster individuals by wing pattern rather than geographic proximity. Sliding window phylogenetic comparisons identified four narrow genomic intervals near wntA that were strongly associated with changes in the spatial distribution of black scales on the forewing (Figure 3C). The first region was a 10 kb interval roughly 50 kb upstream of wntA (blue in Figure 3C) that supported the monophyletic grouping of races that are partially black in the lower midsection of the forewing extending just distal of the discal cell region. Similarly, a separate 8 kb interval roughly 35 kb upstream of wntA grouped geographically distant individuals with similar distribution of black scales across most of the distal mid-section of the forewing (St interval) (green in Figure 3C). Finally, two additional regions, one 25 kb upstream of wntA and another centered on wntA, grouped all individuals that were partially black in the upper section of the forewing (Ly intervals) (orange in Figure 3C). Although, the region centered on wntA showed some support for tree topologies based on geographic proximity, we still considered it a possible color pattern interval because the phenotypic grouping is more strongly supported than geographic grouping. Other areas across this region supporting the phenotypic tree also showed similar support for tree topologies based on geographic proximity and were not considered as candidate color pattern intervals.
Our genomic analysis also confirmed a new locus (Ro) responsible for pattern variation in the most distal region of the forewing band29. Comparisons of H. e. notabilis and H. e. lativitta showed an approximately 71 kb region associated with pattern differences in the upper forewing (purple in Figure 3B). Similar to the wntA region, G × P associations were localized to non-genic regions near two genes, the Heliconius homolog of ventral veins lacking gene (vvl) and the homolog of radial spoke head protein 3 (rsp3). The transcription factor vvl is involved in the formation of specific wing veins, neuronal differentiation and steroid production in Drosophila30–32. The rsp3 gene encodes a kinase A-anchoring protein that scaffolds the cAMP-dependent protein kinase holoenzyme (PKA) and is involved in numerous regulatory events in the cell33. The absence of geographically independent hybrid zones for this phenotype limited our ability to further resolve this region with phylogenetic weighting. Although spatial expression patterns of wntA in Heliconius have been shown to prefigure variation in this upper region of the forewing15, it is likely that one or both of these genes interact with wntA to shape this variation. Such epistatic interactions are commonly observed in color pattern variation in Heliconius34–36.
Modular architecture of red pattern variation
Regulation of red patterns across the fore- and hindwing of H. erato, known to be under control of the gene optix14,17, was also highly modular. We identified discrete genomic intervals near optix that were associated with the presence of red hindwing rays, a red patch (“dennis”) in the proximal part of the forewing and a red forewing band. We use the original nomenclature in H. erato for these different pattern elements: R for red hindwing “rays”, D for a red “dennis” forewing patch and Y for forewing “band” color (Figure 4A)9.
Associations between individuals that differed across all three pattern elements, the so-called “dennis-rayed” and “postman” phenotypes, were strongly clustered in a 69 kb region downstream of optix (Figure 4B)26. Within this 69 kb region, G × P associations between hybridizing H. e. amalfreda and H. e. erato, which differ only in absence/presence of hindwing rays, were clustered in a 7 kb interval (Figure 4B). In this interval, H. e. amalfreda possessed the postman haplotype, which contrasts with the rest of the 69 kb region where H. e. amalfreda shared a haplotype with H. e. erato. Phylogenetic trees constructed from this region, grouped H. e. amalfreda with postman phenotypes that lack rays (red shading in Figure 4C). Unexpectedly, the tree across this interval clustered the outgroup species, H. telesiphe, H. hortense, H. hecalesia, H. clysonymus, and H. sara on a derived node with all rayed H. erato races (SI section 5.3.2). Heliconius hecalesia, H. hortense, and H. clysonymus all have large red hindwing patches, whereas, H. sara and H. telesiphe possess much smaller red spots on the underside of their hindwing. This pattern contrasts with the phylogenetic placement of these species in the tree constructed with data from the rest of the genome (Figure 1A), possibly reflecting historical introgression of modular elements among species closely related to H. erato. Such patterns of introgression have also been observed in other closely related Heliconius species1,37.
Genomic intervals strongly associated with forewing band color (Y) and the red dennis patch (D) could be similarly localized using the combination of G × P association and phylogenetic weighting. For forewing band color, we identified two distinct and narrow intervals separated by approximately 20 kb (yellow in Figure 4B and C). In these regions, there were 15 fixed SNPs that distinguished butterflies with a red forewing band from those that lacked red. Phylogenetic trees from this region strongly supported clustering of the red banded phenotypes H. telesiphe, H. hermathena, H. e. favorinus and H. e. hydara, whereas H. himera, H. hortense, H. clysonymus and H. hecalesia, all of which lack red on the forewing, grouped with the yellow banded H. erato races (Figure 4C and SI section 5.3.2). Finally, we identified several intervals associated with the red dennis patch. For this analysis, we focused primarily on genetic variation within H. himera. Heliconius himera has red on the hindwing similar to rays, but lacks the dennis patch. Therefore, comparing H. himera and H. erato races with a dennis/rays phenotype allowed us to separate the dennis from the rays elements. Across the 69 kb region, there was a 12 kb area where H. himera genotypes were similar to the postman haplotype (grey in Figure 4B). Phylogenetic weighting analysis in this area strongly supported the grouping of H. himera individuals by color pattern phenotype with postman races from both sides of the Amazon basin (grey in Figure 4C).
Independent modules generate convergent yellow hindwing bar phenotypes
Recent association and expression data implicated the gene cortex as an important gene controlling a variety of pattern elements across the Heliconius wing, including presence or absence of yellow hindwing bar in H. erato, known as the Cr locus9,16. In H. erato, we identified two discrete regions containing clusters of fixed sites associated with a yellow hindwing bar in two geographically isolated, yet phenotypically similar, H. erato races (Figure 5). The Peruvian races H. e. favorinus and H. e. emma differed across an interval consisting of 269 fixed SNPs over 100 kb roughly centered on cortex (red in Figure 5). Eight of these SNPs fell within the coding region of cortex, but only one resulted in amino acid substitution (an arginine to lysine at scaffold Herato1505 position 2,087,610). Curiously, a different region distinguished the Panamanian races, H. e. demophoon and H. e. hydara (green in Figure 5), which show a similar difference in the presence/absence of a yellow hindwing bar. In this hybrid zone, there was a cluster of fixed differences located roughly 100 kb away and centered on the Heliconius homolog of parn, a poly(A)-specific ribonuclease. These association differences are consistent with the independent evolution of the yellow hindwing bar on either side of the Andes34,38.
In H. erato, there are other color pattern elements controlled by variation at this locus, including the presence/absence of white hindwing fringes and yellow forewing line39, but our sampling of H. erato races did not allow us to distinguish these elements (SI section 5.4). The hybrid zone comparisons H. e. notabilis/H. e. lativitta and H. e. notabilis/H. e. etylus also showed increased FST estimates near the cortex gene, but no pattern of perfect association was observed for these comparisons. Crossing experiments have suggested possible epistatic interactions between cortex and wntA38,40, which provides a possible explanation for this increased divergence without any phenotypic effect known to be directly controlled by the cortex locus. Furthermore, the phenotypic effects of alleles at this locus can be dramatic in other Heliconius species16, suggesting that this locus interacts broadly with the other Heliconius patterning loci10,41.
Modular regulatory architecture and pattern diversity within H. erato
Less than 0.2% of the genome was associated with wing pattern diversity across the H. erato radiation. This variation was highly modular and fell in non-coding regions near color patterning genes, including optix, wntA and cortex14–16 and a less well-documented color pattern locus (Ro) that controls spatial variation of melanin in the upper forewing. Based on the proximity of these mostly non-coding intervals to known patterning genes, it is likely they represent cis-regulatory regions modulating the spatial expression of key patterning genes in discrete areas of the developing wing. In Heliconius, this modularity of cis-regulatory architecture provides a readily adopted mechanism for rapid evolution of novel morphologies.
Both shuffling of existing modules and de novo evolution of new modules is associated with phenotypic diversity in H. erato. Indeed, we can recreate the color pattern diversity across the H. erato radiation using a combination of ten non-genic regions, near four color pattern genes (Figure 6). This conclusion is perhaps best exemplified in the distribution of genetic variation around wntA, where different color pattern races have different combinations of four distinct genomic intervals. These different intervals likely regulate the expression of wntA in different areas of the forewing to adjust the position, size, and shape of the forewing to closely match patterns in other co-occurring warningly colored butterfly species. Within this modular framework, recombination can reshuffle existing regulatory variation to generate new combinations of regulatory elements and new wing pattern phenotypes. Recombination of color pattern modules and introgression into other populations is likely driven by high rates of gene flow between adjacent populations. For example, H. e. amalfreda appears to have evolved via recombination of regulatory variation between rayed (H. e. erato) and red-banded (H. e. hydara) haplotypes that instantaneously generated a novel wing pattern, a process which closely mirrors the one recently described in the co-mimetic forms of H. melpomene37.
New regulatory modules associated with wing pattern variation can also evolve de novo, further increasing the flexibility of these regions to generate pattern diversity. This was evident in the independent evolution on the yellow hindwing bar in the H. erato clade (Figure 5), but also in the comparison of regulatory variation around the red patterning locus between H. erato and its co-mimic H. melpomene. Red pattern variation in the two species is similarly generated by regulatory differences at the optix locus14, and the genomic position and order of its cis-regulatory elements is broadly similar26. Furthermore, in both species distinct intervals were associated with different red pattern elements and ‘enhancer shuffling’ through recombination has similarly generated novel red pattern phenotypes37. This implies considerable conservation of function of optix cis-regulatory regions that were re-used to generate the convergent patterns that underlie mimicry. Nonetheless, the precise elements associated with placement of red in discrete areas of the fore- and hindwing are not homologous in the two species (SI, section 5.3.3). Thus, convergent patterns are clearly independently derived in the two radiations by the parallel evolution of new enhancer variation.
Conclusions
Our results reconcile decades of genetic and genomic studies of Heliconius color pattern variation9,42. For the first time, we were able to place an entire radiation within a single genomic framework. We reinforce the role of a simple toolkit of a few color pattern genes and demonstrate that pattern diversity is likely generated by the regulatory complexity around these genes. We characterized a discrete number of 1–7 kb intervals that modulate phenotypic variation, and show that divergent and convergent morphologies, are the product of enhancer shuffling and de novo independent evolution of these modules. Overall, our work provides a genomic framework to further explore this regulatory complexity. The regions we identified may contain a number of distinct regulatory elements that may be further resolved with chromatin accessibility data43 and studied in detail with targeted genome editing. Such an integrated genomic view promises to accelerate our understanding of the links between genotype and phenotype and how they play out on a developing butterfly wing. This research has broader ramifications because the small number of genes shown to generate wing pattern variation across Heliconius have been implicated in pattern variation in other butterflies and moths16,19,44. Thus, the Heliconius wing pattern loci appear to be ‘genomic hotspots’ that underlie the evolution of phenotypic diversity in Lepidoptera. The radiation of warning colors in H. erato provides an example of regulatory complexity generated by a small toolkit of genes. This may well be a common hallmark of rapid morphological diversification in adaptive radiations.
Methods
Scaffold assembly and validation
The H. erato (race demophoon) genome was assembled using Illumina paired-end reads with different insert sizes and partially gap filled with PacBio data (Table S2 in SI section 2.1). Illumina data was produced according to the ALLPATHS-LG assembly protocol45 with the paired-end library originating from a single individual and the mate pair libraries from a second, sibling, individual. An initial assembly was performed with ALLPATHS-LG using default parameters and the reads were mapped back to the assembly to acquire accurate distributions of fragment size for each library. Next, contaminant small fragment sequences were purged from the paired-end and mate-pair libraries. Reads were error-corrected using the software Blue46. A kmer database was built from the raw paired-end data and used to remove unsupported reads from mate-paired libraries. This step reduced polymorphism that may cause erroneous assembly. The PacBio data were error-corrected using the Illumina data and the LoRDEC software47.
Five assemblies were obtained using different combinations of raw or error-corrected Illumina data. Each assembly was quality checked against approximately 4 Mb of BAC sequences using nucmer48. All assemblies gave similar amounts of gapped sequence (about 10% of the base pairs), which reflects long simple repeats scattered across the genome. The assembly with the best statistics (i.e. highest N50’s and best alignment to BAC) was then post-processed to replace putative tandem repeats with Ns. Small repetitive scaffolds and putative redundant haplotype sequences were removed and based on a combination of “all-versus-all” alignments and depth of coverage estimates prior to performing ALLPATHS-LG scaffolding. Gaps were then filled using the filled fragment pairs, the corrected PacBio data and the small scaffolds that had been previously removed using PBJelly49. PBJelly was run three times iteratively to balance sensitivity and specificity and the final assembly, called Hera_Stage1, had a length of 402.8 Mb and scaffold N50 of 612 kb, respectively. The assembly process with associated statistics are provided in Table S2 and Figure S1 in SI section 2.2.
Linkage mapping
We generate a high-resolution linkage map by sequencing a backcross family generated from our focal genomic line (Figure S2 in SI section 2.3). Our strategy was to identify markers by coupling high-coverage, whole-genome sequencing (30–40x) of each parent with low coverage (5x–10x) sequencing of their offspring. The low sequencing coverage of the offspring makes it difficult to determine individual genotypes with high accuracy. We therefore developed an in-house pipeline utilizing the mpileup command in SAMtools50 to produce genotype posteriors over a candidate set of 6.7 million SNPs. These genotype posteriors were used to construct a linkage map with Lep-Map3 (sourceforge.net/projects/lep-map3/) a new linkage mapping software developed from the Lep-Map1/2 software51,52.
The linkage map was constructed with Lep-Map3 as follows (see Figure S3 in SI section 2.3): First, to obtain the most accurate parent genotypes, we calculated the parental genotype posteriors using the combined information from parents and offspring using the ParentCall module (Lep-Map2). Next, we calculated pair-wise LOD scores between markers with zero recombination rate (theta=0) using the module SeparateIdenticals (Lep-Map3) with lodLimit=26.5, informativeMask=12 and numParts=20. This step identified markers that segregated identically. The 20 most abundant identical maternal markers were used as the chromosome prints (each maternal marker in a chromosome segregates identically as there is no recombination in the female in Heliconius butterflies). In this step, we could identify 20 of the 21 chromosomes, because we found that chromosome 2 was completely homozygous in the mother. To identify chromosomes, especially chromosome 2, in the paternal linkage map, identical paternal markers were joined using module JoinLGs (Lep-Map3) with recombination rate theta=0.01 and LOD score limit lodLimit=20. More precisely, the linkage groups could be linked together for chromosome 2 by inspecting the markers at nearby positions in the assembly. These paternal markers clustered to 21 linkage groups identifying chromosome 2 and the same 20 chromosomes that were found in the maternal map. Next, the module ShortPath (Lep-Map3) was run on the identical paternal markers. This module finds the longest shortest path in a marker graph (i.e. the longest path in graph for which the shortest path is chosen between pairs of markers), where markers are nodes and each marker pair have been connected with an edge of length 4n -3, if there are n detected recombinations (different genotypes considering both phases in this case) between the markers. The best paths were manually checked to determine the final order of the markers. After the maternal and paternal markers were placed within a linkage framework (Table S3 in SI section 2.3), we added the remaining markers into this framework using JoinIdenticals (Lep-Map3), with LOD score limits of 25 and 20, for paternal and maternal markers, respectively. The 1.2 million markers that were heterozygous in both parents were discarded (informativeMask=12). Finally, the identified linkage groups (chromosomes) were named to reflect the nomenclature of the H. melpomene genome. We were able to easily identify homologous chromosomes by mapping the flanking regions of each marker to the H. melpomene genome1. Our final linkage map covered all 21 chromosomes, including the Z chromosome.
Assembly correction and chromosomal scaffolding
We used our high-resolution linkage map to error correct and improve our genome assembly. To do this, we first manually identified scaffolds that were inconsistent with our linkage map. About 10% of the scaffolds, representing 62 Mb, had such errors. Due to the high-density of markers on our linkage map, most errors were localized within a few kb. These errors generally fell at a gap sequence meaning that the scaffolding step of the assembly process, rather than the creation of contigs, caused most misassembles. The scaffolds in the assembly with errors were cut to produce an error-free assembly. The assembly was also separated into chromosomes at this point. There was about 16 Mb of gapped sequence in the Herato_stage1 assembly. The 34 scaffolds that failed to map to chromosomes totaled 3.7 Mb, 3.5 Mb of which were bacterial genome sequence and the rest was mainly very highly repetitive haplotypes that failed to create substantially long (> 3 kb) contigs.
We produced the final assembly by integrating information from two independent de novo assemblies to gap fill our oriented stage2 assembly. The first was an ALLPATHS-LG assembly generated from the same Illumina dataset paired-end and mate-paired dataset, and assembled as follows. Illumina paired-end and mate-pair data were subsampled to prescribed coverage depth according to Gnerre et al. 201145 and assembled using ALLPATHS-LG with “HAPLOIDIFY = TRUE” and “CLOSE_UNIPATH_GAPS = False”. The resulting assembly was improved by performing 3 iterations of PBJelly49, incorporating prior PBJelly assemblies into subsequent iterations. The second was an assembly of an additional sibling female individual using approximately 100× coverage of 2 × 250 Illumina data generated from PCR free libraries. The genome of this individual was assembled using DISCOVAR de novo53,54. The scaffolds that spanned gaps in our assembly were extracted from the BWA-MEM55 produced bam files using in-house software. This software used a variant of Smith-Waterman local alignment56 to compute the best alignment to fix gaps. Both positive and negative gaps were considered. The alignment parameters used were +1 for nucleotide match, −4 for mismatch, −8 for gap open and −1 for gap extension. Gaps were filled iteratively, using the independent ALLPATHS assembly first. Here we required an alignment score of 100 across a 4 kb region on each side of a gap for the gap to be filled. Regions with multiple gaps were joined as if they contained a single large gap. Finally, we filled remaining gaps using the DISCOVAR assembly. In this case, we used alignment to 2 kb regions around each gap. Using this strategy, we reduced the number of gaps in our assembly to 5.2 Mb. Assembly completeness, as assessed against a benchmarked set of 2,675 single-copy orthologues using BUSCO57 was 82% (2,179) in the H. erato genome and a further 11% were present, but marked as ‘fragmented’. These BUSCO results were similar to those for other high quality lepidopteran genomes (Table S8 in SI section 2.4). We assembled 5 of 20 autosomes and the Z chromosome into single scaffolds. We failed to identify a W chromosome, likely because of its highly repetitive nature. See Figure S4 in the SI section 2.3 for the completeness of the scaffolding in the final H. erato genome assembly.
Genome Annotation
Annotation of the genome was performed using Just_Annotate_My_Genome (JAMg; https://github.com/genomecuration/JAMg). To facilitate annotation, we used RNASeq data generated from different life stages and tissue types (Table S9 in SI section 3). These data include recent Illumina 2×250 data, 454 data, and archival Illumina 2x50 data. All data were preprocessed using “justpreprocessmyreads” (http://justpreprocessmyreads.sourceforge.net) and were error corrected using Blue46 with a ‘reference’ kmer dataset derived the most recently collected 2×250 Illuminia RNA-seq data and a coverage cut-off of 2. The Illumina RNA-Seq data was assembled using Trinity RNA-Seq version 2.1.158 with both the ‘de-novo’ and ‘genome-guided’ options. The 454 data alongside all mRNA data acquired from GenBank and public Illumina data acquired from NCBI SRA were assembled and clustered using MIRA 4.9.559. The Trinity de-novo, Trinity genome-guided and the MIRA assemblies were aligned and assembled against the genome using a new version of PASA (Haas et al. 2003; Haas, Papanicolaou et al. in preparation), thus, creating a non-redundant, intron-aware transcript set referred here as PASA cDNA contigs. The new Illumina RNA-Seq were aligned against the reference H. erato genome using GSNAP v.2015-09-2961 providing high-quality information of intron coordinates. Repetitive content was identified (simple, complex/transposable, de-novo, tRNA and rRNA elements) using trf62, RepeatModeler63, RepeatScout64, RepeatMasker63, RepBase data65, tRNAScan66 and Aragorn67. This masked dataset was provided at the last stage of the pipeline only.
We used two de novo gene modelers, GeneMark-ET68 and Augustus 3.2.169 for gene prediction. Both used the intron co-ordinates as external evidence. In addition, Augustus used further external evidence as hints including the RNA-seq coverage derived from the Illumina reads, protein domains acquired from searching the genome against Swissprot using the HHBlits program70, a high-quality subset of the PASA cDNA contigs as determined by JAMg, alignments of Uniref50 and the Heliconius melpomene predicted protein set71. The Augustus HMM models were trained and evaluated using a ‘training’ and ‘test’ subsets of the high-quality PASA cDNA contigs. Following this, the external evidence was weighted using the JAMg optimization method and the same training and test cDNA contig datasets. At this point, we determined that the repeat masking data provided inferior prediction results and were not used in the final prediction. Finally, Augustus was run with UTR prediction enabled to reduce false positive exons. Resulting UTRs were removed from the final prediction.
The Repeat masking information, GenMark-ET, Augustus, PASA cDNA contigs, the Uniref50 and H. melpomene protein alignments were provided to EvidenceModeler72 to derive a consensus gene dataset. This consensus dataset was then twice edited with PASA2 in order to add alternative splicing information and the UTRs as supported by cDNA evidence. This formed our Official Gene Set (OGS1). The OGS1 proteins were then functionally annotated using Just_Annotate_My_Proteins (JAMp; https://github.com/genomecuration/JAMp) searched against Hidden Markov Profiles of known proteins with manually curated metadata (Swissprot; clustered at 70% identity and aligned). For each significant hit (using the default settings of JAMp such as an e-value of 1e-10 and p-value of 1e-12), any Gene Ontology, ENZYME and KEGG ontology terms of the known Swissprot proteins were linked to the H. erato predicted proteins but only if the annotation evidence was experimentally derived and not inferred (i.e. terms with the evidence codes of ‘IEA’, ‘ISS’, ‘IEP’, ‘NAS’, ‘ND’, ‘NR’ were ignored). The RNA-Seq data was finally aligned against the OGS1 CDS data and processed with DEW (https://github.com/alpapan/DEW) to infer the expression profiles for each gene. The functional and expression annotations are available from http://annotation.insectacentral.org/heliconius_erato.
Sequence alignment and variant calling
We collected and sequenced 101 individual H. erato butterflies from Peru (n = 15), French Guiana (n = 14), Suriname (n = 5), Ecuador (n = 29), Colombia (n= 12), Bolivia (n = 4), Mexico (n = 6) and Panama (n = 16). We collected phenotypically pure (i.e. phenotypes resembling the geographical H. erato races) individuals of each color pattern race from admixed populations where the ranges of two color pattern races overlap. Additionally, we collected individuals from 8 different closely related species including H. ricini, H. sara, H. charithonia, H. hecalesia, H. telesiphe, H. hortense, H. clysonimus, and H. hermathena (Figure 1; Table S10,11 in SI section 4.1).
Whole genome 100 bp paired-end Illumina resequencing data of these individuals was aligned to the H. erato v1 reference genome using BWA v0.7.1373 with default parameters. PCR duplicated reads were removed using Picard v1.138 (http://picard.sourceforge.net) and sorted using SAMtools74. Genotypes were called using the Genome Analysis Tool Kit (GATK) Haplotypecaller75 with default parameters. Individual genomic VCF records (gVCF) were jointly genotyped using GATK’s genotypeGVCFs with default parameters, except for setting expected heterozygosity to 0.025 to match the populations high heterozygosity and grouping individuals according to race and sampling location. Genotype calls were only considered in downstream analysis if they met the following criteria: Quality (QUAL) ≥ 30, minimum depth ≥ 10, maximum depth ≤ 100 (to avoid false SNPs due to mapping in repetitive regions), overall depth ≤ 100*number of samples, strand bias (FS) < 200, Quality by depth ≥ 5, and for variant calls, genotype quality (GQ) ≥ 30.
Divergence and association analysis
We estimated levels of relative (FST)76 and absolute genetic divergence (dXY)77, and nucleotide diversity (π)77 between populations in sliding windows using python scripts and egglib78. In all our analyses, we only considered windows for which at least 10% of the positions were genotyped for at least 75% of the individuals within each population. For the whole genome analysis of the seven hybrid zones, on average 96.4% (SD = 1.1%) of windows met these criteria. Genotype by phenotype (G × P) associations were tested for each variant position using a two-tailed Fisher’s exact test. Positions were excluded if less than 75% of individuals were genotyped for each phenotype. The sliding window approach and the identification of distinct blocks of associated SNPs provides a robust approach for identifying genomic regions of interests in our study system79.
Phylogenetic analysis
We used FastTree v2.180 to infer an approximately maximum-likelihood phylogeny from the entire genome using the default parameters. In this analysis, we only used concatenated SNP data from chromosome 4–9, 11–14, 16, 17 and 20, because these chromosomes did not show any genetic divergence peaks in our population analysis. FastTree computes support values on nodes using the Shimodaira–Hasegawa test. Phylogenetic relationships of individuals across defined color pattern intervals were constructed using Maximum Likelihood (ML) trees with RAxML v8.0.2681. The best likelihood tree was chosen from 100 trees generated from a distinct starting tree using a GTR model with CAT approximation of rate heterogeneity and the support values of this tree was inferred with 100 bootstrap replicates.
Phylogenetic weighting
We applied a phylogenetic strategy for identifying shared or conserved genomic intervals akin to ‘phylogenetic shadowing’82. We evaluated the support for alternative phylogenetic hypotheses in the regions of peaks of divergence around color pattern loci using a novel method called Topology Weighting by Iterative Sampling of Subtrees (Twisst: https://github.com/simonhmartin/twisst)28. This method solves the problem of describing the relationships between groups that are not necessarily monophyletic. Given a tree and a set of pre-defined groups (in this case races) Twisst determines a weighting for each possible topology describing the relationship of the groups (e.g. 6 groups yield 105 possible unrooted topologies and therefore 105 weightings). Topology weightings are determined by sampling a single member of each group and then identifying the topology matched by the resulting subtree. This sampling is iterated over a large number of subtrees and weightings are calculated as the frequency of occurrence of each topology. This method therefore reduces tree complexity caused by imperfect clustering of samples within groups. The ability to consider all possible topologies at each window provides an advantage over more commonly used likelihood ratio tests that only compare two topologies, which is especially relevant for taxa that have potentially many distinct evolutionary histories across their genomes. Weightings were estimated from 500 sampling iterations and averaged over ten bootstrap trees produced by RAxML v8.0.2681 for each 2 kb window. Averaging weightings over bootstrap trees is expected to reduce false support for certain phylogenetic groupings from trees with low bootstrap support.
For phylogenetic weighting along the wntA (chromosome 10) and Ro (chromosome 13) interval, we compared weightings of topologies defined by samples from the following six groups: H. e. demophoon, H. e. etylus, H. e. notabilis, H. e. lativitta/emma, H. e. erato/amalfreda and H. e. hydara (FG). To partly control for the strong phylogeographic signal within H. erato, we focused these analyses on eastern Andean and Amazonian races, which also show the most variation in forewing band shape, size and position. For the optix (chromosome 18) interval, we compared weightings of topologies defined by samples from the following six groups: H. e. amalfreda, H. e. favorinus/hydara (FG), H. e. etylus/lativitta/emma/erato, H. himera, H. telesiphe and H. clysonymus/hortense/hecalesia. To obtain weightings for hypothesized phylogenetic groupings of specific color pattern forms, we summed the counts of all topologies that were consistent with the hypothesized grouping.
Genotype weighting optix
We evaluated genotypic similarity of species/races to the reference “postman” haplotype using a sliding window analysis. The “postman” haplotype was defined based on the consensus of fixed SNPs between all ‘postman’ (H. e. demophoon, H. e. hydara (Panama), H. hydara (French Guiana), H. e. notabilis and H. e. favorinus) and all ‘rayed’ (H. e. erato, H. e. etylus, H. e. emma and H. e. lativitta) H. erato races. In total there were 264 fixed SNPs across a 69 kb window on chromosome 18 near optix. For each species/race evaluated, the proportion of SNPs that were identical to the postman haplotype was calculated over windows of ten fixed SNPs, with a minimum coverage of 3 SNPs called in all individuals. The window size and minimum coverage was chosen to best capture the turn-over of the genotypic similarity along the genomic interval.
Defining boundaries of color pattern intervals
Our argument for identifying regulatory modules was hierarchical. The association peaks, or regions of the genome containing clusters of sites perfectly associated with wing pattern phenotype, marked the genomic intervals that likely contained the functional variation responsible for phenotypic differences. We further resolved these intervals combining data across independent transition zones. The rationale is that independent recombination events in the distinct locations break down the pattern of associations, except at those very narrow intervals responsible for pattern differences. Thus, in these areas individuals should group by color pattern phenotype rather than geographic proximity, which is the pattern evident across the bulk of the genome. This is the basis of the Twisst analyses described above. Specific boundaries are defined by a combination of Twisst and G × P association. For example, near wntA and optix, we defined the boundary positions of the regulatory modules by overlaying the phylogenetic weighting with genotype tables of the fixed allelic differences in the hybrid zone comparisons. More precisely, at the regions where phylogenetic weighting support for phenotypic grouping shifted and increased rapidly, we conservatively identified the boundaries of the intervals by looking for patterns of shared genotypes between samples with similar phenotypes. It should be noted that this approach assumes a single origin for functional alleles that are shared across similar phenotypes and will miss regions where patterning alleles evolved independently. The boundaries of the regulatory modules near Ro and cortex were defined only using the fixed SNP associations because the geographic distribution of the phenotypes does not allow phylogenetic weighting to distinguish between geography and phenotypic grouping for these loci.
Supplementary Material
Acknowledgments
We thank Adriana Tapia for maintaining the H. erato genome line and for generating our mapping family and Marta Vargas and Claudia Rosales for Illumina library preparation. We acknowledge the University of Puerto Rico, the Puerto Rico INBRE grant P20 GM103475 from the National Institute for General Medical Sciences (NIGMS), a component of the National Institutes of Health (NIH); CNRS Nouraugues and CEBA awards (BAC); National Science Foundation awards DEB-1257839 (BAC), DEB-1257689 (WOM), DEB-1027019 (WOM); awards 1010094 and 1002410 from the Experimental Program to Stimulate Competitive Research (EPSCoR) program of the National Science Foundation (NSF) for computational resources; and the Smithsonian Institution. This research was supported in part by Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute, and in part by the Indiana METACyt Initiative. The Indiana METACyt Initiative at IU is also supported in part by Lilly Endowment, Inc.
Footnotes
Data accessibility
Sequencing data was submitted to the Sequence Read Archive (SRA) with BioProject accession PRJNA324415; Genome assembly data: SAMN05578372-SAMN05578377, RNAseq data: SRR616674-SRR616691, SAMN05578182-SAMN05578206, Linkage map data: SAMN05572290-SAMN05572390 and re-sequencing data: SAMN05224096- SAMN05224211.
Author contributions
S.M.V.B., B.A.C., W.O.M. and R.P. designed the study and wrote the paper. P.R., A.P. and J.J.M conducted genome assembly. P.R. conducted linkage map and genome quality assessment. A.P. conducted genome annotation. S.M.V.B. conducted population genomic, phylogenetic and comparative genomic analyses. M.R, M.A.S, H.H. and J.J.H. conducted comparative genomic analyses. S.H.M. contributed scripts for Twisst analyses. B.A.C., W.O.M., R.P., H.H., C.D.J., J.M., M.L., C.S., C.F.A. and G.M. collected samples for sequencing.
References
- 1.Dasmahapatra KK, et al. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamichhaney S, et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature. 2015;518:371–375. doi: 10.1038/nature14181. [DOI] [PubMed] [Google Scholar]
- 3.Brawand D, et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature. 2014;513:375–381. doi: 10.1038/nature13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamas G. In: Hesperioidea – Papilionoidea. Lamas G, editor. Gainesville, Florida: Association for Tropical Lepidoptera; Scientific Publisher; 2004. pp. 261–274. [Google Scholar]
- 5.Nijhout HF. The development and evolution of butterfly wing patterns. Smithsonian Institution Press; 1991. [Google Scholar]
- 6.Chouteau M, Arias M, Joron M. Warning signals are under positive frequency-dependent selection in nature. Proc Natl Acad Sci. 2016;113:2164–2169. doi: 10.1073/pnas.1519216113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naisbit RE, Jiggins CD, Mallet J. Disruptive sexual selection against hybrids contributes to speciation between Heliconius cydno and Heliconius melpomene. Proc Biol Sci. 2001;268:1849–1854. doi: 10.1098/rspb.2001.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner JRG. A tale of two butterflies. Nat Hist. 1975;84:28–37. [Google Scholar]
- 9.Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC. Genetics and the evolution of Muellerian mimicry in Heliconius Butterflies. Philos Trans R Soc B Biol Sci. 1985;308:433–610. [Google Scholar]
- 10.Joron M, et al. A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol. 2006;4:e303. doi: 10.1371/journal.pbio.0040303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papa R, et al. Multi-allelic major effect genes interact with minor effect QTLs to control adaptive color pattern variation in Heliconius erato. PLoS One. 2013;8:e57033. doi: 10.1371/journal.pone.0057033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kronforst MR, Kapan DD, Gilbert LE. Parallel genetic architecture of parallel adaptive radiations in mimetic Heliconius butterflies. Genetics. 2006;174:535–539. doi: 10.1534/genetics.106.059527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapan DD, et al. Localization of müllerian mimicry genes on a dense linkage map of Heliconius erato. Genetics. 2006;173:735–757. doi: 10.1534/genetics.106.057166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reed RD, et al. optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science. 2011;333:1137–1141. doi: 10.1126/science.1208227. [DOI] [PubMed] [Google Scholar]
- 15.Martin A, et al. Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proceedings of the National Academy of Sciences. 2012;109:12632–12637. doi: 10.1073/pnas.1204800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nadeau N, et al. The gene cortex controls mimicry and crypsis in butterflies and moths. Nature. 2016;534:106–110. doi: 10.1038/nature17961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin A, et al. Multiple recent co-options of Optix associated with novel traits in adaptive butterfly wing radiations. Evodevo. 2014;5:7. doi: 10.1186/2041-9139-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll SB. Evo-Devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008;134:25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 19.Gallant JR, et al. Ancient homology underlies adaptive mimetic diversity across butterflies. Nat Commun. 2014;5:1–10. doi: 10.1038/ncomms5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van’t Hof AE. The industrial melanism mutation in British peppered moths is a transposable element. Nature. 2016;534:102–105. doi: 10.1038/nature17951. [DOI] [PubMed] [Google Scholar]
- 21.Rosser N, Dasmahapatra KK, Mallet J. Stable Heliconius butterfly hybrid zones are correlated with a local rainfall peak at the edge of the Amazon basin. Evolution. 2014;68:3470–3484. doi: 10.1111/evo.12539. [DOI] [PubMed] [Google Scholar]
- 22.Supple M, Papa R, Hines HM, McMillan WO, Counterman BA. Divergence with gene flow across a speciation continuum of Heliconius butterflies. BMC Evol Biol. 2015;15:204. doi: 10.1186/s12862-015-0486-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hines HM, et al. Wing patterning gene redefines the mimetic history of Heliconius butterflies. Proc Natl Acad Sci U S A. 2011;108:19666–19671. doi: 10.1073/pnas.1110096108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mallet J, Barton NH. Strong natural selection in a warning-color hybrid zone. Evolution. 1989;43:421–431. doi: 10.1111/j.1558-5646.1989.tb04237.x. [DOI] [PubMed] [Google Scholar]
- 25.Kapan DD. Three-butterfly system provides a field test of müllerian mimicry. Nature. 2001;409:18–20. doi: 10.1038/35053066. [DOI] [PubMed] [Google Scholar]
- 26.Supple Ma, et al. Genomic architecture of adaptive color pattern divergence and convergence in Heliconius butterflies. Genome Res. 2013;23:1248–57. doi: 10.1101/gr.150615.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin A, et al. Diversification of complex butter flywing patterns by repeated regulatory evolution of a Wnt ligand. Proc Natl Acad Sci U S A. 2012;109:12632–12637. doi: 10.1073/pnas.1204800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin SH, Van Belleghem SM. Exploring evolutionary relationships across the genome using topology weighting. BioRxiv. 2016 doi: 10.1534/genetics.116.194720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nadeau NJ, et al. Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Res. 2014;24:1316–1333. doi: 10.1101/gr.169292.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danielsen ET, et al. Transcriptional control of steroid biosynthesis genes in the Drosophila prothoracic gland by Ventral veins lacking and Knirps. PLoS Genet. 2014;10:e1004343. doi: 10.1371/journal.pgen.1004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Celis JF, Llimargas M, Casanova J. ventral veinless, the gene encoding the Cf1a transcription factor, links positional information and cell differentiation during embryonic and imaginal development in Drosophila melanogaster. Development. 1995;121:3405–3416. doi: 10.1242/dev.121.10.3405. [DOI] [PubMed] [Google Scholar]
- 32.Meier S, Sprecher SG, Reichert H, Hirth F. ventral veins lacking is required for specification of the tritocerebrum in embryonic brain development of Drosophila. Mech Dev. 2006;123:76–83. doi: 10.1016/j.mod.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Jivan A, Earnest S, Juang Y-C, Cobb MH. Radial spoke protein 3 is a mammalian protein kinase A-anchoring protein that binds ERK1/2. J Biol Chem. 2009;284:29437–29445. doi: 10.1074/jbc.M109.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiggins CD, Mcmillan WO. The genetic basis of an adaptive radiation: warning colour in two Heliconius species. Proc R Soc B. 1997;264:1167–1175. [Google Scholar]
- 35.Baxter SW, Johnston SE, Jiggins CD. Butterfly speciation and the distribution of gene effect sizes fixed during adaptation. Heredity (Edinb) 2009;102:57–65. doi: 10.1038/hdy.2008.109. [DOI] [PubMed] [Google Scholar]
- 36.Huber B, et al. Conservatism and novelty in the genetic architecture of adaptation in Heliconius butterflies. Heredity (Edinb) 2015;114:515–524. doi: 10.1038/hdy.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallbank RWR, et al. Evolutionary novelty in a butterfly wing pattern through enhancer shuffling. PLoS Biol. 2016;14:e1002353. doi: 10.1371/journal.pbio.1002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maroja LS, Alschuler R, Mcmillan WO, Jiggins CD. Partial complementarity of the mimetic yellow bar phenotype in Heliconius butterflies. PLoS One. 2012;7:e48627. doi: 10.1371/journal.pone.0048627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC. Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Philos Trans R Soc B Biol Sci. 1985;308:433–610. [Google Scholar]
- 40.Mallet J. The genetics of warning colour in Peruvian hybrid zones of Heliconius erato and H. melpomene. Proc R Soc B. 1989;236:163–185. [Google Scholar]
- 41.Joron M, et al. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature. 2011;477:203–206. doi: 10.1038/nature10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kronforst MR, Papa R. The functional basis of wing patterning in Heliconius butterflies: The molecules behind mimicry. Genetics. 2015;200:1–19. doi: 10.1534/genetics.114.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis JJ, et al. ChIP-Seq-annotated Heliconius erato genome highlights patterns of cis-regulatory evolution in Lepidoptera. CellReports. 2016;16:2855–2863. doi: 10.1016/j.celrep.2016.08.042. [DOI] [PubMed] [Google Scholar]
- 44.Martin A, Reed RD. Wnt signaling underlies evolution and development of the butterfly wing pattern symmetry systems. Dev Biol. 2014 doi: 10.1016/j.ydbio.2014.08.031. [DOI] [PubMed] [Google Scholar]
- 45.Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–8. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenfield P, Duesing K, Papanicolaou A, Bauer DC. Sequence analysis Blue: correcting sequencing errors using consensus and context. Bioinformatics. 2014;30:2723–2732. doi: 10.1093/bioinformatics/btu368. [DOI] [PubMed] [Google Scholar]
- 47.Salmela L, Rivals E. Sequence analysis LoRDEC: accurate and efficient long read error correction. Bioinformatics. 2014;30:3506–3514. doi: 10.1093/bioinformatics/btu538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.English AC, et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One. 2012;7:e47768. doi: 10.1371/journal.pone.0047768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rastas P, Paulin L, Hanski I, Lehtonen R. Lep-MAP: fast and accurate linkage map construction for large SNP datasets. Bioinformatics. 2013;29:3128–3134. doi: 10.1093/bioinformatics/btt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rastas P, Calboli FCF, Guo B, Shikano T, Merilä J. Construction of ultradense linkage maps with Lep-MAP2: Stickleback F2 recombinant crosses as an example. Genome Biol Evol. 2015;8:78–93. doi: 10.1093/gbe/evv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weisenfeld NI, et al. Comprehensive variation discovery in single human genomes. Nat Genet. 2014;46:1350–1355. doi: 10.1038/ng.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love RR, Weisenfeld NI, Jaffe DB, Besansky NJ, Neafsey DE. Evaluation of DISCOVAR de novo using a mosquito sample for cost-effective short-read genome assembly. BMC Genomics. 2016;17:187. doi: 10.1186/s12864-016-2531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 1303.3997v1. [Google Scholar]
- 56.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 57.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 58.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chevreux B, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haas BJ, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smit AFA, Hubley R, Green P. RepeatMasker. 2014 http://www.repeatmasker.org/
- 64.Price AL, Jones NC, Pevzner Pa. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 65.Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 66.Lowe TM, Eddy S. R tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lomsadze A, Burns PD, Borodovsky M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014;42:e119. doi: 10.1093/nar/gku557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;11:1–11. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Remmert M, Biegert A, Hauser A, Johannes S. HHblits : Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 71.Davey JW, et al. Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3. 2016;6:695–708. doi: 10.1534/g3.115.023655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H, et al. The Sequence Alignment/Map (SAM) Format and SAMtools 1000 Genome Project Data Processing Subgroup. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van der Auwera Ga, et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;UNIT 11.10:1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992;132:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nei M, Jin L. Variances of the average numbers of nucleotide substitutions within and between populations. Mol Biol Evol. 1989;6:290–300. doi: 10.1093/oxfordjournals.molbev.a040547. [DOI] [PubMed] [Google Scholar]
- 78.De Mita S, Siol M. EggLib: processing, analysis and simulation tools for population genetics and genomics. BMC Genet. 2012;13:27. doi: 10.1186/1471-2156-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nadeau NJ, et al. Genomic islands of divergence in hybridizing Heliconius butterflies identified by large-scale targeted sequencing. Philos Trans R Soc Lond B Biol Sci. 2012;367:343–353. doi: 10.1098/rstb.2011.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Price MN, Dehal PS, Arkin AP. FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boftelli D, et al. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.