

Abstract
Background
In addition to their classical role as detergents, bile acids function as signaling molecules to regulate gastrointestinal physiology, carbohydrate and lipid metabolism, and energy expenditure. The pharmacodynamic potential of bile acids is dependent in part on the tight pharmacokinetic control of their concentration and metabolism, properties governed by their hepatic synthesis, enterohepatic cycling, and biotransformation via host and gut microbiota-catalyzed pathways.
Key Messages
By altering the normal cycling and compartmentalization of bile acids, changes in hepatobiliary or intestinal transport can affect signaling and lead to retention of cytotoxic hydrophobic bile acids and cell injury. This review discusses advances in our understanding of the intestinal transporters that maintain the enterohepatic cycling of bile acids, signaling via bile acid-activated nuclear and G protein receptors, and mechanisms of bile acid-induced cell injury.
Conclusions
Dysregulated expression of the Asbt and Ostα-Ostβ alters bile acid signaling via the gut-liver FXR-FGF15/19 axis and may contribute to other bile acid-regulated metabolic and cell injury pathways.
Keywords: Enterohepatic circulation, intestine, liver, transporters, nuclear receptors
Introduction
Bile acids are synthesized from cholesterol and act as detergents to promote the digestion and intestinal absorption of fats and fat-soluble vitamins. Bile acids also function as ligands for nuclear and G protein-coupled receptors and have important roles as signaling molecules. The best-studied bile acid receptors are the farnesoid X receptor (FXR) and G protein-coupled receptor 1 (GPBAR1; also called TGR5 or membrane-type receptor for bile acids, M-BAR), but the list of bile acid receptors also includes other nuclear receptors, such as the pregnane X receptor (PXR) and vitamin D receptor (VDR), and other G protein-coupled receptors, such as the sphingosine-1-phosphate receptor 2 (S1PR2), and muscarinic receptors M2 and M3 [1,2]. It is important to note that bile acids also signal through α5,β1-integrin [3], and can induce membrane perturbations that activate pathways involving membrane-associated proteins such as NAD(P)H oxidases and Phospholipase A2 [4,5]. By direct and indirect mechanisms, bile acids activate Protein Kinase C, Epidermal Growth Factor Receptor, and the MAP kinase pathways, p38 MAPK, PI3K-kinase-Akt, extracellular signal-related kinase (ERK), and c-JNK N-terminal kinase (JNK) [6]. The ability of bile acids to signal through these myriad pathways depends in part on their local concentration and their chemical structure. This review discusses the contribution of an important component of the enterohepatic circulation, the intestinal bile acid transporters, to bile acid signaling.
Enterohepatic circulation of bile acids
After their synthesis in the hepatocyte, bile acids are secreted into bile and stored in the gallbladder or small intestinal lumen. In response to a meal, the gallbladder contracts and intestinal motility is stimulated. Bile acids then travel down the length of the small intestine to the ileum, where they are reabsorbed, and carried back to the liver in the portal circulation for uptake and resecretion into bile [7]. The enterohepatic circulation functions to safely store and then promptly deliver bile acids in high concentration to the intestinal lumen for digestion and absorption of lipids, while limiting the distribution and systemic signaling potential of these potentially cytotoxic detergent molecules. As a result of these tight pharmacokinetic controls, bile acid concentrations vary markedly between compartments in the body, ranging from high millimolar in the biliary tract and gallbladder, to low millimolar in the intestine and colon, to low micromolar in the systemic circulation [8]. The membrane carriers that maintain the enterohepatic circulation of bile acids are known and include specific transporters expressed on the sinusoidal and canalicular membranes of the hepatocyte, and on the apical brush border and basolateral membranes of the ileal enterocyte [9]. In ileum, the apical sodium-dependent bile acid transporter (Asbt or ibat; Slc10a2) and the heteromeric transporter Ostα-Ostβ (Ost = Organic solute transporter; Slc51a-Slc51b) mediate these steps [10]. These transporters help determine the turnover of bile acid ligands and control access to the intracellular bile acid receptors in important target cells.
Feedback regulation of hepatic bile acid synthesis, intestinal bile acid transporters, and the FXR-FGF15/19 pathway
Bile acids are synthesized from cholesterol via 2 pathways, the major classical pathway (Cholesterol 7α-hydroxylase, Cyp7a1, pathway) that favors cholic acid biosynthesis, and a minor alternative pathway that favors biosynthesis of chenodeoxycholic acid (in humans) or muricholic acid (in mice) [11]. The expression of Cyp7a1, the rate-limiting step in bile acid biosynthesis, is tightly controlled by a complex network of regulatory mechanisms and has been the subject of intensive study. Investigation beginning in the 1960’s showed that hepatic bile acid synthesis and Cyp7a1 activity was suppressed after administration of exogenous bile acids and induced when bile acid return to the liver was blocked by administration of bile acid sequestrants or by biliary diversion [12]. This negative feedback regulation of Cyp7a1 was shown to be transcriptional and involve the bile acid-activated nuclear receptor FXR. Initially, it was thought that the mechanism for feedback regulation was restricted to FXR signaling in the liver. However, this model could not explain a series of puzzling experimental findings, including the observation that intravenous infusion of bile acids into bile-fistula rats was ineffective at down-regulating hepatic Cyp7a1 expression compared to intraduodenal infusion of bile acids [13].
More recent investigation determined that the major pathway for feedback regulation of bile acid synthesis under physiological conditions involves ileal FXR and gut-liver signaling via the polypeptide hormone fibroblast growth factor-19 (FGF19; mouse ortholog: FGF15) [14]. FGF15/19 is a ~25 kDa (216 amino acids in humans; 218 amino acids in mice) secreted protein, which acts not only locally but also systemically in an endocrine fashion. In the FXR-FGF15/19 pathway, the Asbt first transports bile acids from the intestinal lumen across the apical brush border membrane into the ileal enterocyte. Before their export from the cell via Ostα-Ostβ, bile acids bind and activate FXR, which induces transcription of FGF15/19. After its synthesis and release by the enterocyte, FGF15/19 is carried in the portal circulation to the hepatocyte where it binds a complex of the βKlotho accessory protein and fibroblast growth factor receptor-4 (FGFR4) [15]. This initiates signaling via the docking protein fibroblast growth factor substrate 2 (FRS2α) and a tyrosine phosphatase (tyrosine-protein phosphatase non-receptor type 11; Shp2) to stimulate extracellular-signal-regulated kinase (ERK1/2) activity and block activation of Cyp7a1 gene expression by HNF4α and LRH-1.
Changes in Asbt or Ostα-Ostβ expression has the potential to control the intracellular levels of bile acids and therefore influence FXR-mediated regulation of FGF15/19 production. This is nicely illustrated by comparing the effects of inhibiting ileal apical versus basolateral membrane bile acid transport on bile acid metabolism [16,17]. As illustrated in Figure 1, inactivation of the Asbt in mice produces a classical severe bile acid malabsorption phenotype whereby excretion of bile acids in the feces is increased more then 5-fold, whole body bile acid pool size is reduced, and whole body bile acid pool composition is altered. The ratio of taurocholate to tauromuricholate in the bile acid pool shifts from approximately 1:1, which is typically found in wild type mice, to greater than 5:1, suggesting a dramatic derepression of hepatic bile acid synthesis via the Cyp7a1 pathway [16]. In contrast to blocking apical bile acid uptake, inactivation of Ostα in mice failed to increase fecal bile acid excretion or significantly alter whole body bile acid pool composition. However, whole body bile acid pool size was reduced almost to the same extent as observed for inactivation of the Asbt [17]. Overall, the phenotype suggested impaired regulation of hepatic bile acid synthesis and as illustrated in Figure 2, can be explained by alterations in gut-liver FXR-FGF15 signaling. In Ostα−/− versus Asbt−/− mice, total ileal FGF15 mRNA expression is increased almost 10-fold, and this inversely correlates with approximately 8-fold reductions in liver Cyp7a1 mRNA expression, 9-fold reductions in liver microsomal Cyp7a1 enzyme activity, and 7-fold reductions in fecal bile acid excretion.
Fig. 1. Schematic of differential effects of blocking ileal apical versus basolateral bile acid transport on bile acid metabolism.
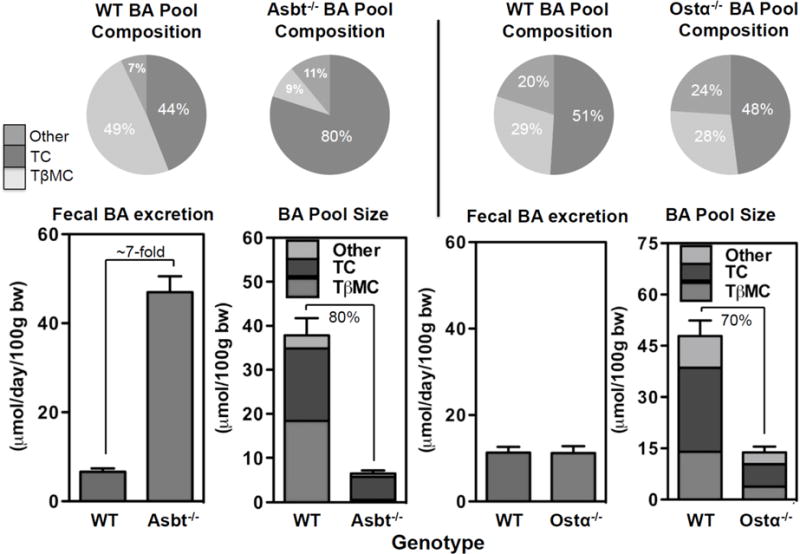
Relative to wild type mice, fecal bile acid excretion is increased almost 7-fold in Asbt knockout mice. The size of the whole body bile acid pool is reduced by almost 80% and its composition is altered. In contrast, fecal bile acid excretion and bile acid pool composition is unchanged in Ostα−/− versus wild type mice. However, whole body bile acid pool size is reduced. Values are calculated from references [16,17]. BA, bile acids; TC, taurocholate; TβMC, tauro-β-muricholate, WT, wild type.
Fig. 2. Schematic of differential effects of blocking ileal apical versus basolateral bile acid transport on the FXR-FGF15 signaling axis and bile acid metabolism.
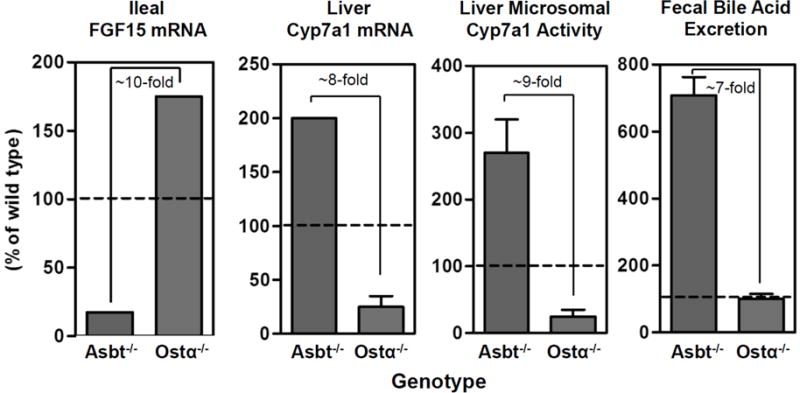
Relative to Asbt knockout mice, ileal total ileal FGF15 mRNA expression is elevated about 10-fold in Ostα knockout mice. This correlates with an approximately 8-fold reduction in liver Cyp7a1 mRNA expression, a 9-fold reduction in liver microsomal Cyp7a1 activity, and 7-fold reduction in fecal bile acid excretion. Values are calculated from references [16,17].
Bile acid-regulation of metabolism, bile acid transporters, and the FXR-FGF15/19 pathway
Bile acids have been shown to modulate a variety of metabolic pathways involved in controlling glucose, lipid, and energy homeostasis. The regulation is complex and involves a variety of bile acid receptors and signaling pathways. This includes FXR-mediated control of glucose metabolism through regulation of hepatic gluconeogenesis and glycogenolysis, control of intestinal glucose absorption, and actions in adipose and pancreas [18–20]. There is also an expanding role for TGR5. The bile acid-activated G-protein coupled receptor TGR5 is expressed by nonparenchymal cells of the liver, macrophages, brown adipose tissue, enteroendocrine cells, spinal and enteric neurons, and a variety of cell types in brain. At these different sites, TGR5 has actions that are anti-inflammatory, increase energy expenditure [21], modulate glucose metabolism [22], control gallbladder filling and intestinal motility, and mediate itch and analgesia [23].
The bile acid receptors and mediators that contribute to these physiological and metabolic effects will vary depending in part on the enterohepatic circulation of bile acids. This is nicely illustrated by the finding of beneficial metabolic effects with the seemingly contradictory approaches of administering bile acids (or FXR agonists) or removing bile acids using a bile acid sequestrant. The beneficial metabolic actions of bile acids or FXR agonists may be mediated in part by induction of ileal FGF15/19 production [24]. Several lines of evidence suggest that FGF15/19 functions as a postprandial hormone to regulate glucose metabolism [25,26]. In addition, delivery of pharmacological doses of FGF19, either by transgenic overexpression or administration of recombinant FGF19 protein, induces resistance to diet-induced weight gain, increases metabolic rate, and improves glucose tolerance [27,28].
The opposite strategy of sequestering bile acids also decreases plasma glucose and increases insulin sensitivity in patients with type 2 diabetes [29]. The mechanisms responsible for the beneficial metabolic effects of bile acid sequestrants have been explored in mouse models. One line of evidence suggests that bile acid sequestrants induce hepatic bile acid synthesis and alter the bile acid species present in the systemic circulation, thereby increasing activation of TGR5 in brown adipose tissue and increasing energy expenditure [30]. Interestingly, changes in systemic levels of different bile acid species have also been implicated in the beneficial metabolic effects of bariatric surgery [31–33]. A second line of evidence suggests that bile acid sequestrants act by increasing delivery to the colon of bile acids, which signal through TGR5 on enteroendocrine L-cells to increase production of GLP-1 [34,35], an incretin hormone with potent anti-diabetic actions. Finally, sequestering bile acids or blocking Asbt-mediated uptake may produce beneficial metabolic effects by inhibiting FXR signaling in the intestine to reduce biosynthesis of intestinally derived ceramides. Whereas there is evidence that intestinal-selective FXR agonist has metabolic benefit [36], other studies in high fat diet-fed mouse models suggest that activation of FXR in the intestine increases production of ceramides, which act systemically to stimulate Srebp1c-driven hepatic lipogenesis and reduce beige fat thermogenic function [37,38].
Role of intestinal bile acid transporters in cytoprotection against bile acid-induced injury
The cellular and molecular mechanisms of hepatocyte injury caused by retention of hydrophobic bile acids in cholestatic disease have been the subject of intensive study [39]. Bile acids can activate hepatocyte death receptors, induce generation of reactive oxygen species and oxidative damage, induce mitochondrial dysfunction, and endoplasmic reticulum stress. With regard to the transporters that maintain the enterohepatic circulation, bile acid-associated injury is perhaps best described for Progressive Familial Intrahepatic Cholestasis Type 2 (PFIC2). In PFIC2, inherited mutations in the bile salt export pump (BSEP; ABCB11) lead to loss of canalicular bile acid export, hepatocellular accumulation of bile acids, and subsequent injury [40]. By contrast, less is known regarding the mechanisms and signaling pathways underlying the potential cytotoxic effects of bile acids in other cell types and in extra-hepatic tissues [4,5]. In theory, dysregulated expression of the Asbt and Ostα-Ostβ may lead to bile acid accumulation and injury in ileal enterocytes or in the bile acid transporting epithelium of the biliary tract and renal proximal tubules, which express the same complement of transporters. Important clues to those potential mechanisms of epithelial injury may come from study of Asbt and Ostα-Ostβ deficiency. Loss-of-function ASBT mutations in humans or mice yield a classical primary bile acid malabsorption phenotype, with impaired intestinal bile absorption in the absence of ileal histological changes [16,41,42]. Although loss of Ostα-Ostβ expression also blocks intestinal bile acid absorption, the phenotype of Ostα−/− mice includes a dramatic remodeling of the ileal morphology, with features typically associated with epithelial damage and subsequent healing [17,43]. The molecular mechanisms linking the imbalance of Ostα-Ostβ and Asbt expression and the altered ileal morphology have not yet been identified. However, a similar imbalance has also been described in some animal models of necrotizing enterocolitis, a disease characterized by necrosis of the distal ileum and proximal colon in premature infants [44]. Outside of Inflammatory Bowel Disease, Ileal atrophy is not often seen in the absence of duodenojejunal atrophy [45]. However, cases of apparent Primary Ileal Villous Atrophy (PIVA) and bile acid malabsorption of unknown etiology have been reported [46–48]. The relationship of the intestinal bile acid transporters to the pathogenesis of PIVA and similar disorders remains to be determined.
Acknowledgments
This work was supported by National Institutes of Health grant DK047987.
References
- 1.Copple BL, Li T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res. 2016;104:9–21. doi: 10.1016/j.phrs.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiang JY. Bile acid metabolism and signaling. Comprehensive Physiology. 2013;3:1191–1212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gohlke H, Schmitz B, Sommerfeld A, Reinehr R, Haussinger D. Alpha5 beta1-integrins are sensors for tauroursodeoxycholic acid in hepatocytes. Hepatology. 2013;57:1117–1129. doi: 10.1002/hep.25992. [DOI] [PubMed] [Google Scholar]
- 4.Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–1689. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrasa JI, Olmo N, Lizarbe MA, Turnay J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol In Vitro. 2013;27:964–977. doi: 10.1016/j.tiv.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 6.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyer JL. Bile formation and secretion. Comprehensive Physiology. 2013;3:1035–1078. doi: 10.1002/cphy.c120027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann AF. The enterohepatic circulation of bile acids in mammals: Form and functions. Front Biosci (Landmark Ed) 2009;14:2584–2598. doi: 10.2741/3399. [DOI] [PubMed] [Google Scholar]
- 9.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56:1085–1099. doi: 10.1194/jlr.R054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, Ballatori N. The heteromeric organic solute transporter alpha-beta, ostalphaostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 12.Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res. 2009;50(Suppl):S120–125. doi: 10.1194/jlr.R800026-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandak WM, Heuman DM, Hylemon PB, Chiang JY, Vlahcevic ZR. Failure of intravenous infusion of taurocholate to down-regulate cholesterol 7 alpha-hydroxylase in rats with biliary fistulas. Gastroenterology. 1995;108:533–544. doi: 10.1016/0016-5085(95)90083-7. [DOI] [PubMed] [Google Scholar]
- 14.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Katafuchi T, Esterhazy D, Lemoff A, Ding X, Sondhi V, Kliewer SA, Mirzaei H, Mangelsdorf DJ. Detection of fgf15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry. Cell Metab. 2015;21:898–904. doi: 10.1016/j.cmet.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, Maeda N, Parks JS. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem. 2003;278:33920–33927. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- 17.Rao A, Haywood J, Craddock AL, Belinsky MG, Kruh GD, Dawson PA. The organic solute transporter alpha-beta, ostalpha-ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A. 2008;105:3891–3896. doi: 10.1073/pnas.0712328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prawitt J, Abdelkarim M, Stroeve JH, Popescu I, Duez H, Velagapudi VR, Dumont J, Bouchaert E, van Dijk TH, Lucas A, Dorchies E, Daoudi M, Lestavel S, Gonzalez FJ, Oresic M, Cariou B, Kuipers F, Caron S, Staels B. Farnesoid x receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. 2011;60:1861–1871. doi: 10.2337/db11-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dufer M, Horth K, Wagner R, Schittenhelm B, Prowald S, Wagner TF, Oberwinkler J, Lukowski R, Gonzalez FJ, Krippeit-Drews P, Drews G. Bile acids acutely stimulate insulin secretion of mouse beta-cells via farnesoid x receptor activation and k(atp) channel inhibition. Diabetes. 2012;61:1479–1489. doi: 10.2337/db11-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 22.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. Tgr5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, Cevikbas F, Steinhoff M, Nassini R, Materazzi S, Guerrero-Alba R, Valdez-Morales E, Cottrell GS, Schoonjans K, Geppetti P, Vanner SJ, Bunnett NW, Corvera CU. The tgr5 receptor mediates bile acid-induced itch and analgesia. The Journal of clinical investigation. 2013;123:1513–1530. doi: 10.1172/JCI64551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D. Efficacy and safety of the farnesoid x receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013 doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 25.Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, Suino-Powell K, Xu HE, Gerard RD, Finck BN, Burgess SC, Mangelsdorf DJ, Kliewer SA. Fgf15/19 regulates hepatic glucose metabolism by inhibiting the creb-pgc-1alpha pathway. Cell metabolism. 2011;13:729–738. doi: 10.1016/j.cmet.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, Xu HE, Shulman GI, Kliewer SA, Mangelsdorf DJ. Fgf19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331:1621–1624. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, Tsai SP, Powell-Braxton L, French D, Stewart TA. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143:1741–1747. doi: 10.1210/endo.143.5.8850. [DOI] [PubMed] [Google Scholar]
- 28.Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, Soriano R, Corpuz R, Moffat B, Vandlen R, Simmons L, Foster J, Stephan JP, Tsai SP, Stewart TA. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145:2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 29.Out C, Groen AK, Brufau G. Bile acid sequestrants: More than simple resins. Curr Opin Lipidol. 2012;23:43–55. doi: 10.1097/MOL.0b013e32834f0ef3. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe M, Morimoto K, Houten SM, Kaneko-Iwasaki N, Sugizaki T, Horai Y, Mataki C, Sato H, Murahashi K, Arita E, Schoonjans K, Suzuki T, Itoh H, Auwerx J. Bile acid binding resin improves metabolic control through the induction of energy expenditure. PLoS One. 2012;7:e38286. doi: 10.1371/journal.pone.0038286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohli R, Setchell KD, Kirby M, Myronovych A, Ryan KK, Ibrahim SH, Berger J, Smith K, Toure M, Woods SC, Seeley RJ. A surgical model in male obese rats uncovers protective effects of bile acids post-bariatric surgery. Endocrinology. 2013 doi: 10.1210/en.2012-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohli R, Bradley D, Setchell KD, Eagon JC, Abumrad N, Klein S. Weight loss induced by roux-en-y gastric bypass but not laparoscopic adjustable gastric banding increases circulating bile acids. J Clin Endocrinol Metab. 2013;98:E708–712. doi: 10.1210/jc.2012-3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmad NN, Pfalzer A, Kaplan LM. Roux-en-y gastric bypass normalizes the blunted postprandial bile acid excursion associated with obesity. Int J Obes (Lond) 2013 doi: 10.1038/ijo.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Potthoff MJ, Potts A, He T, Duarte JA, Taussig R, Mangelsdorf DJ, Kliewer SA, Burgess SC. Colesevelam suppresses hepatic glycogenolysis by tgr5-mediated induction of glp-1 action in dio mice. Am J Physiol Gastrointest Liver Physiol. 2013;304:G371–380. doi: 10.1152/ajpgi.00400.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harach T, Pols TW, Nomura M, Maida A, Watanabe M, Auwerx J, Schoonjans K. Tgr5 potentiates glp-1 secretion in response to anionic exchange resins. Sci Rep. 2012;2:430. doi: 10.1038/srep00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, Atkins AR, Khvat A, Schnabl B, Yu RT, Brenner DA, Coulter S, Liddle C, Schoonjans K, Olefsky JM, Saltiel AR, Downes M, Evans RM. Intestinal fxr agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21:159–165. doi: 10.1038/nm.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, Tanaka N, Desai D, Amin SG, Albert I, Patterson AD, Gonzalez FJ. Intestinal farnesoid x receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125:386–402. doi: 10.1172/JCI76738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, Liu Y, Gavrilova O, Patterson AD, Gonzalez FJ. Intestine-selective farnesoid x receptor inhibition improves obesity-related metabolic dysfunction. Nature communications. 2015;6:10166. doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: A clear and present danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oude Elferink RP, Paulusma CC, Groen AK. Hepatocanalicular transport defects: Pathophysiologic mechanisms of rare diseases. Gastroenterology. 2006;130:908–925. doi: 10.1053/j.gastro.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 41.Heubi JE, Balistreri WF, Fondacaro JD, Partin JC, Schubert WK. Primary bile acid malabsorption: Defective in vitro ileal active bile acid transport. Gastroenterology. 1982;83:804–811. [PubMed] [Google Scholar]
- 42.Oelkers P, Kirby LC, Heubi JE, Dawson PA. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (slc10a2) J Clin Invest. 1997;99:1880–1887. doi: 10.1172/JCI119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lan T, Rao A, Haywood J, Kock ND, Dawson PA. Mouse organic solute transporter alpha deficiency alters fgf15 expression and bile acid metabolism. J Hepatol. 2012;57:359–365. doi: 10.1016/j.jhep.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halpern MD, Holubec H, Saunders TA, Dvorak K, Clark JA, Doelle SM, Ballatori N, Dvorak B. Bile acids induce ileal damage during experimental necrotizing enterocolitis. Gastroenterology. 2006;130:359–372. doi: 10.1053/j.gastro.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halphen M, Galian A, Certin M, Ink F, Filali A, Rambaud JC. Clinicopathological study of a patient with idiopathic villous atrophy and small vessel alterations of the ileum. Dig Dis Sci. 1989;34:111–117. doi: 10.1007/BF01536164. [DOI] [PubMed] [Google Scholar]
- 46.Popovic OS, Kostic KM, Milovic VB, Milutinovic-Djuric S, Miletic VD, Sesic L, Djordjevic M, Bulajic M, Bojic P, Rubinic M, et al. Primary bile acid malabsorption. Histologic and immunologic study in three patients. Gastroenterology. 1987;92:1851–1858. [PubMed] [Google Scholar]
- 47.Marteau P, Lavergne-Slove A, Lemann M, Bouhnik Y, Bertheau P, Becheur H, Galian A, Rambaud JC. Primary ileal villous atrophy is often associated with microscopic colitis. Gut. 1997;41:561–564. doi: 10.1136/gut.41.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milutinovic-Djuric S, Popovic OS, Milovic V, Necic D, Kostic KM. Postcholecystectomy diarrhea from villous atrophy of the terminal ileum. J Clin Gastroenterol. 1993;16:227–230. doi: 10.1097/00004836-199304000-00013. [DOI] [PubMed] [Google Scholar]