

Abstract
Kinesin motor proteins transport intracellular cargos throughout cells by hydrolyzing ATP and moving along microtubule tracks. Intramolecular autoinhibitory interactions have been shown for several kinesins in vitro, however, the physiological significance of autoinhibition remains poorly understood. Here, we identified four mutations in the stalk region and motor domain of synaptic vesicle kinesin, UNC-104/KIF1A, that specifically disrupt autoinhibition. These mutations augment both microtubule and cargo vesicle binding in vitro. In vivo, these mutations cause excessive activation of UNC-104, leading to decreased synaptic density, smaller synapses and ectopic localization of synaptic vesicles in the dendrite. We also show that the synaptic vesicle bound small GTPase ARL-8 activates UNC-104 by unlocking the autoinhibition. These results demonstrate that the autoinhibitory mechanism is utilized to regulate the distribution of transport cargoes and is important for synaptogenesis in vivo.
Graphical abstract
Introduction
Axonal transport is essential for neuronal function and morphogenesis (Hirokawa et al., 2010). Disruption of axonal transport underlies many neuronal diseases (Holzbaur and Scherer, 2011; Morfini et al., 2009; Niwa et al., 2013). Synaptic vesicle (SV) material is synthesized in the cell body and transported along the axon to synapses (Hirokawa et al., 2010). The axonal transport kinesin for SV precursors, UNC-104, was originally identified through genetic analysis in Caenorhabditis elegans (C. elegans) (Hall and Hedgecock, 1991; Otsuka et al., 1991). UNC-104 belongs to kinesin-3 family and is structurally and functionally well conserved among higher eukaryotes that have nervous systems. In the Drosophila Imac mutant, SVs and active zone proteins are largely missing from the axon (Barkus et al., 2008; Pack-Chung et al., 2007). In mammals, KIF1A and KIF1Bβ, the homologues of UNC-104, transport SV precursors as well (Niwa et al., 2008; Okada et al., 1995; Zhao et al., 2001).
Autoinhibition is a common mechanism for the regulation of molecular motors (Cheng et al., 2014; Coy et al., 1999; Friedman and Vale, 1999; Hackney, 1995; Liu et al., 2006; Torisawa et al., 2014). Several studies showed that KIF1A is inactivated by an autoinhibitory mechanism (Al-Bassam et al., 2003; Hammond et al., 2009; Lee et al., 2004). Coiled-coil 1 (CC1) and CC2 domains of KIF1A negatively affect the motor domain and inhibit the binding of KIF1A to MTs. Deletion of CC1 and CC2 domains constitutively activate the motor activity of KIF1A (Hammond et al., 2009). Structural studies have suggested that CC1 domain is required for the dimerization and activation of UNC-104/KIF1A (Huo et al., 2012; Yue et al., 2013). However, how the autoinhibition of UNC-104/KIF1A is unlocked remains completely elusive. Furthermore, the physiological roles of the autoinhibition of UNC-104/KIF1A remain totally unknown, while it has been suggested that the autoinhibition of molecular motors is required to avoid futile ATP hydrolysis when motors do not bind to cargos (Verhey and Hammond, 2009).
We have previously found that ARL-8, an arf-like small GTPase, is required for the axonal transport of SV precursors in C. elegans (Klassen et al., 2010). Axonal transport of SV precursors is perturbed and SVs ectopically accumulate at proximal sites along the axon in arl-8 mutants. In this paper, we report the identification of four dominant suppressors of arl-8 in C. elegans. These mutations are in the motor, CC1 and CC2 domains of UNC-104 and the nucleotide-binding domain of ARL-8 itself. By characterizing the synaptic phenotypes of these mutants, we showed that lack of this regulatory mechanism leads to defects of axon-specific sorting of synaptic materials, decreased synapse density and impaired cargo stopping in vivo.
Results
Identification of dominant suppressors of arl-8
We visualized the synaptic specializations in the DA9 motor neuron by expressing GFP∷RAB-3 under the itr-1 promoter (Klassen and Shen, 2007) (Figs 1A and B). Previously, we have shown that loss of ARL-8 activity in the arl-8(wy271) allele causes defects in axonal transport of SVs and abnormal accumulation of SVs to the proximal axon (Fig. 1C and D). arl-8(wy271) is a strong loss-of-function allele of arl-8 in which part of the promoter region and the first three amino acids are deleted (Klassen et al., 2010). To understand the molecular mechanisms of axonal transport, we performed an F1 suppressor screen to obtain dominant suppressors of arl-8(wy271). We isolated wy798, wy865 and wy873 as suppressors of arl-8(wy271) (Figs. 1C-E and Supplementary Figure S1A). In these suppressor mutants, GFP∷RAB-3 is more distally localized and the commissure mislocalization observed in arl-8(wy271) was diminished (Fig 1C - E). However, the expression level of a SV marker, SNB-1 (synaptobrevin), was not changed in these mutants (Fig. S1B). All three mutants were isolated by an F1 screen and behaved as dominant suppressors. Genetic mapping and sequencing analysis revealed that the causative mutations in wy798, wy865 and wy873 are all substitution mutations on conserved amino acids in unc-104. The substitution mutants were V6I (wy865), E412K (wy798) and E612K (wy873) (Fig. 1F). UNC-104 specifically transports SV precursors and active zone proteins but not other organelles in C. elegans (Hall and Hedgecock, 1991; Klassen et al., 2010; Klopfenstein et al., 2002; Maeder et al., 2014; Ou et al., 2010; Wu et al., 2013). The V6I mutation is in the motor domain, while the E412K and E612K mutations are in the CC1 and CC2 domain, respectively. Three lines of evidence suggest that all three mutations cause enhanced function of UNC-104/KIF1A. First, all three mutations are fully dominant: these mutants were isolated by the F1 screening and heterozygotes mutants suppress the arl-8 phenotypes. Second, overexpression of wild type version of UNC-104 can suppress arl-8 phenotypes in DA9 neurons cell autonomously (Fig. 1G). Third, we have also reported another gain-of-function (gf) allele of unc-104 (wy673), in which a point mutation in the region between FHA domain and CC2 domain (Wu et al., 2013). To quantify the suppressor mutant phenotypes, we measured the number of mis-accumulated puncta in the commissure and the length of the asynaptic region (Figs 1H-J). Misaccumulation of RAB-3 positive puncta in arl-8 was suppressed by unc-104 mutations (Fig. 1I). The length of the puncta-free dorsal axon is also correspondingly increased in the suppressor double mutants (Fig. 1J)
Figure 1. The arl-8 synapse phenotypes are suppressed by mutations in unc-104.
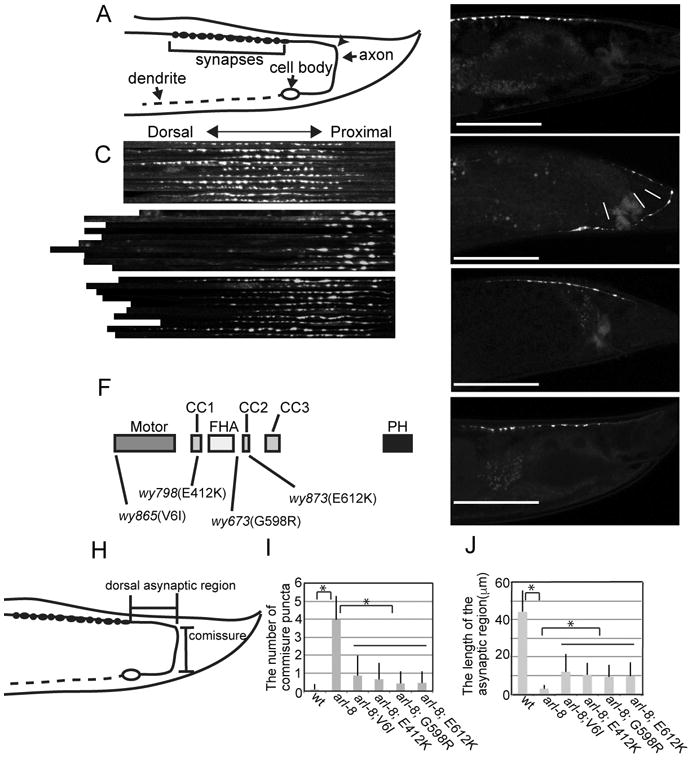
(A) A schematic drawing of the DA9 neuron.
(B) A representative image of the localization of GFP∷RAB-3 in wild type worms. GFP∷RAB-3 was expressed using an itr-1 pB promoter. The arrowhead shows where the DA9 commissure joins the dorsal nerve cord.
(C) Image montage of the dorsal synaptic regions in wild type, arl-8(wy271) and arl-8(wy271); wy873. 10 confocal images from each genotype were cropped, straightened and aligned using NIH image.
(D and E) Representative images of the localization of GFP∷RAB-3 in DA9 in (D) arl-8(wy271) and (E) arl-8(wy271); wy873, respectively.
(F) the domain structure of UNC-104 and the gain-of-function mutations.
(G) A representative image of arl-8(wy271) overexpressing UNC-104. Scale bars, 50 μm.
(H-J) Statistical analysis of suppressor mutants. (H) The definition of domains that were analyzed. (I) The number of puncta misaccumulated to the commisure region (J) The length of the asynaptic region. Mean ± Standard error of means (SEM) *, p < 0.01, compared to arl-8(wy271), Dunnett test, n = 50 animals. See also Figure S1.
Together, we have isolated multiple point mutations that can cause increase in UNC-104 activity.
Gain-of-function mutations disrupt the autoinhibition
It has been reported that the CC1 and CC2 domains can regulate the activity of KIF1A, a mammalian homologue of UNC-104, through autoinhibitory mechanisms (Al-Bassam et al., 2003; Hammond et al., 2009; Lee et al., 2004). To further understand the mechanisms of autoinhibition and how the point mutations result in increased UNC-104/KIF1A activity, we examined the localization of mutant kinesins in COS cells to evaluate their MT- and cargo-binding abilities. COS-7 cells are suitable to analyze the subcellular localization of motor proteins and assess motor states (Hammond et al., 2009). The mouse KIF1A is suitable for COS-7 experiments for the following reasons: The mutated residues are completely conserved between mouse KIF1A and C. elegans UNC-104 (Figure 2A and Figure S2A). Mammalian KIF1A and ARL8B efficiently rescued unc-104- and arl-8-mutant phenotypes in worms, including the defect of worm motility and the SV mislocalization (Figure S2B-G), suggesting that the functions of UNC-104 and ARL-8 in trafficking SVs are well conserved.
Figure 2. Gain-of-function KIF1A mutations and ARL8 disrupted the autoinhibition of KIF1A.
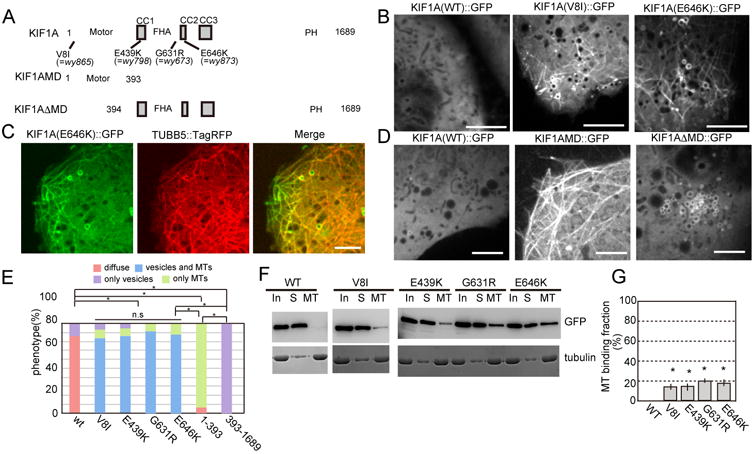
(A) Domain structures of the full-length and mutant KIF1A used in Fig. 2 and Fig. S2.
(B) Confocal images of wt and mutant KIF1A expressed in COS-7 cells. Note that KIF1A(WT)∷GFP is mostly diffuse in the cytoplasm. Introduction of a motor domain mutation (V8I) or a tail domain mutation (E646K) causes KIF1A∷GFP to show MT-like and vesicular staining pattern. Bars, 5 μm.
(C) Fluorescence images of KIF1A(E646K)∷GFP and TUBB5∷TagRFP co-expressed in COS-7 cells. Note the colocalization of KIF1A and tubulin. Pearson's R value is 0.68 ± 0.13 (Mean ± Standard deviation). Bar, 5 μm.
(D) Subcellular localization of truncation mutants of KIF1A fused with GFP and expressed in COS-7 cells. Note that the full length KIF1A is diffuse in the cytoplasm, the motor domain alone mutant (KIF1AMD) is localized on MTs. Conversely, the motor domain was deleted (KIF1ADMD) showed vesicular structure. Scale bar, 5 μm.
(E) Quantification of subcellular localization of WT and mutant forms of KIF1A in COS-7 cells.. n = 103 cells for each genotype. *, p < 0.05, n.s, p > 0.05, Chi-square test with Bonferroni corrections.
(F and G) Western blot analyses of tubulin's ability to precipitate WT and mutant forms of KIF1A. (F) Representative results. KIF1A∷GFP and tubulin were detected by anti-GFP antibody and CBB staining. “In” is total input. “S” is supernatant. “MT” is the microtubule bound fraction. (G) Quantification of western blot analysis. Mean ± SEM, *, p < 0.01, Dunnett test. N = 5. See also Figure S2
In COS-7 cells, wild type (wt) KIF1A was mostly diffuse in the cytoplasm, which can attributed to the autoinhibitory mechanisms shown previously (Fig. 2B, left panel) (Hammond et al., 2009). All four amino acid changes (V8I, E439K, G631R and E646K corresponding to wy865, wy798, wy673 and wy873 mutations respectively) induced KIF1A to exhibit both tubular and intracellular membrane localization patterns (Fig 2B, center and right panels and Fig S2H). This tubular structure was characteristic of a MT localization pattern. Subsequent visualization of MTs using a MT marker TUBB5∷TagRFP (Niwa et al., 2013) confirmed that KIF1A indeed localized to MTs (Fig 2C, Pearson's R value is 0.68 ± 0.13, Mean ± Standard deviation). The donut-shaped internal membrane fluorescence pattern is reminiscent of the localization of endosomal proteins such as RAB5 (Sönnichsen et al., 2000). These results suggest that the mutated KIF1A can bind to both MTs and internal membranes more readily compared with the wild type controls. To further understand the domain requirement of MT and membrane binding, we expressed truncated versions of KIF1A in COS-7 cells (Figs 2A and 2D). Consistent with previous studies (Hammond et al., 2009; Lee et al., 2004; Niwa et al., 2013), the motor domain alone (KIF1AMD) exhibited a strong MT localization pattern, suggesting that the motor domain is responsible and sufficient for the MT binding and that this binding activity is masked by the tail domains in the full length construct (Fig 2D, central panel) (Hammond et al., 2009). Conversely, we found that the tail domains alone (KIF1ADMD) showed exclusive internal membrane localization pattern (Figs 2D right panel), indicating that the tail domains are sufficient to bind to the vesicular cargoes. Statistical analysis supports these observations (Fig. 2E).
Moreover, we performed MT-binding assays (Niwa et al., 2013). Consistent with its cellular localization, wt KIF1A did not show strong binding to MTs (Fig 2F and G); most wt KIF1A was recovered from the supernatant fraction. However, when each of the four mutations was introduced, mutant KIF1A could be recovered from the MT pellet fractions, further confirming the fact that the point mutations enhance the MT binding of KIF1A (Figs 2F and G).
Taken together, these data suggest that the motor domain and the tail domain mutually inhibit each other's respective abilities to bind to the MTs and cargo membranes. Introduction of mutations into the motor domain (V6I) or the tail domain (E412K, G598R and E612K) disrupts the autoinhibition and enables the mutant kinesins to bind to both MTs and vesicles. Since V6I is in the motor domain, we examined whether this mutation changes the ability of the motor domain to move on MTs. We expressed truncated versions of UNC-104 with or without the V6I mutation (1-371 a.a) in E. coli and performed in vitro MT gliding assays (Pierce et al., 1999). We found that the V6I mutation did not significantly change the speed of MT gliding, suggesting that neither the ATPase nor the affinity to MTs is affected by this mutation (Figure S2I, wt = 1.68 ± 0.38 mμ/sec, V6I = 1.35 ± 0.44 μm/sec, n = 25).
Suppressor mutations activate UNC-104 in vivo
We examined the localization of GFP-fused C. elegans UNC-104 (UNC-104∷GFP) in the DA9 neuron in vivo. First, we asked if these mutations affect the protein expression or stability of UNC-104 by measuring the level of UNC-104. A western blot analysis showed that the mutant UNC-104 proteins are expressed at similar levels compared with that of the wild type control (Fig. 3A). Next, we examined the subcellular distribution UNC-104 variants. Wild type UNC-104∷GFP was distributed throughout the DA9 cell body and axon (Figs 3B and C), but was under the detectable level in the dendrite (data not shown). Within the axon, diffuse GFP signal can be detected in the synaptic region, and the distal axon, with a mild enrichment at the axonal tip (Figs 3B and C, WT). In contrast, UNC-104(E612K)∷GFP was strongly accumulated at the axonal tip and under the detectable level in the cell body and the rest of the axonal regions (Figs. 3B and C, E612K). Expression of wild type and mutant UNC-104∷GFP did not affect the DA9 axonal and dendritic morphology. The other three mutants, UNC-104(V6I)∷GFP, UNC-104(E412K)∷GFP, UNC-104(G598R)∷GFP, also showed similar tip accumulation (Figure S3 and Figure 3D). This tip accumulation observed in DA9 neuron is similar to those observed in mammalian neurons in which autoinhibition-disrupted kinesins were expressed (Hammond et al., 2009; Lee et al., 2004; Nakata and Hirokawa, 2003; Niwa et al., 2013). Taken together, these biochemical experiments in COS cells and cell biological experiments in vivo suggest that full length UNC-104 is regulated by an autoinhibitory interaction between the motor domain and the tail domains. Specific mutations in the coiled coil domain and motor domain strongly activate UNC-104 by alleviating the autoinhibition.
Figure 3. UNC-104 is activated by autoinhibition mutations in DA9.
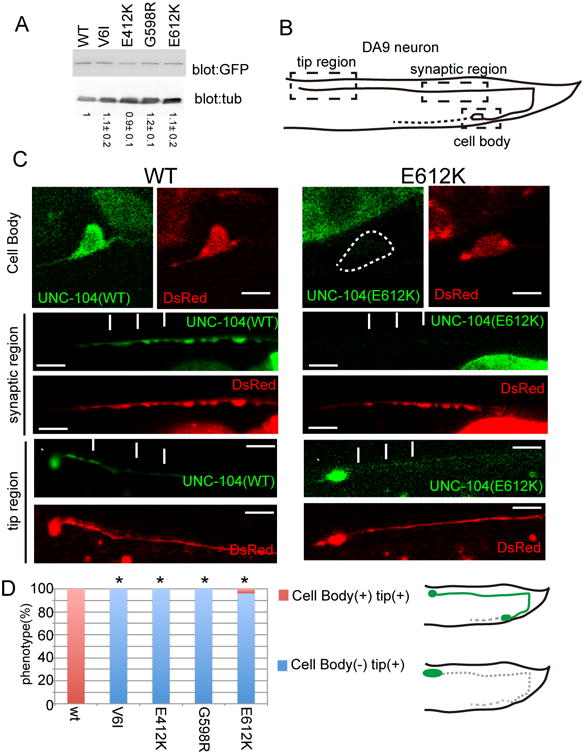
GFP∷UNC-104(WT), GFP∷UNC-104(V6I), GFP∷UNC-104(E412K),GFP∷UNC-104(G598R) and GFP∷UNC-104((E612K) were co-expressed with DsRed in DA9 using the itr-1 pB promoter.
(A) Western blot analysis for expression level of GFP∷UNC-104 transgenes. The number indicate the relative expression level compared to wt. n = 3.
(B and C) Subcellular localization of UNC-104∷GFP and UNC-104(E612K)∷GFP. (B) Schematic drawing of the DA9 cell body and axon. Three boxed regions were shown in (C). (C) Green, the localization of UNC-104 (WT)∷GFP and UNC-104 (E612K)∷GFP. Red, images of the DsRed channel. Note that UNC-104(WT)∷GFP is diffusely localized in the DA9 cell, but UNC-104(E612K)∷GFP is strongly enriched at the distal tip of DA9. Arrows, axonal shafts. Arrow heads, axonal distal tip. Bars, 5 μm.
(D) Quantification of the subcellular distribution of wildtype and mutant UNC-104. n = 103 for each genotype. p < 0.05, Chi-square test with Bonferroni corrections. Schema shows the distribution of UNC-104∷GFP. See also Figure S3.
Autoinhibition of UNC-104 determines synaptic density
Having established that unc-104 gain-of-function (gf) mutations disrupt the autoinhibition, we next analyzed the synaptic phenotypes of these mutants to clarify the consequence of overactive UNC-104 and the function of autoinhibition in axonal transport. It was found that weak but significant amount of SVs visualized with GFP-RAB3 were observed in the dendritic shaft in unc-104(gf) mutants while SVs were largely restricted to the dorsal synapse domain in the axon (Fig 4A, A′, B and B′). This fully penetrant phenotype suggests that overactivation of UNC-104 can lead to excessive trafficking of SVs to the dendrite. Within the synaptic domain, we found that the density of presynaptic boutons visualized with both RAB-3 and SYD-2 (an active zone protein) was reduced compared to the wild type controls (Figs 4C-I). In both genotypes, RAB-3 and SYD-2 were similarly and strongly colocalized at the synaptic region (Fig. 4E, Pearson's R values are 0.72 ± 0.13 and 0.72 ± 0.14 in wt and wy873, respectively. Not statistically significantly changed in t-test). The average intersynaptic distance was 3.1 ± 1.5 μm, 4.4 ± 2.4 μm and 5.2 ± 2.7 μm in WT, unc-104(wy865[V6I]) and unc-104(wy873 [E612K]), respectively (n = 69, p < 0.01, Mann–Whitney U test). While only about 10 % of the intersynaptic distance was more than 5 μm in wt, about 40 % of intersynaptic distance was more than 5 mμ in unc-104(gf) mutants (Fig. 4F). The number of synaptic boutons was not significantly changed in unc-104(gf) alleles (Fig 4G, 23.2 ± 1.9, 22.4 ± 1.8 and 22.7 ± 1.8 synaptic boutons in wt, unc-104(wy865[V6I]) and unc-104(wy873[E612K]) respectively, n = 13 animals, Mean ± SD). These data suggest that the autoinhibition of UNC-104 regulates the synaptic density, but not the number of synaptic boutons in DA9 neuron. We also measured the fluorescent intensity and size of RAB-3∷GFP and SYD-2∷RFP puncta. Both RAB-3 and SYD-2 puncta were weaker and smaller in the unc-104(wy873[E612K]) compared with the wild type controls (Figs 4H and I). In order to further characterize the synaptic phenotypes, we used a microfluidic device coupled with an automated image analysis system to quantify various parameters of the DA9 synapses (Crane et al., 2012). In addition to the synapse density phenotype, we found that the mean synaptic area and the mean fluorescence intensity visualized with SNB-1∷YFP was subtly but significantly reduced in all four unc-104(gf), compared to wt, suggesting that the mutant synapses are smaller (Fig. 4J and K). As a control, we observed jkk-1(km2), a loss-of-function mutant of a MAP kinase kinase MKK7 that we have shown to regulate synaptic marker intensity (Wu et al., 2013). Consistent with the previous observation, jkk-1(km2) also showed weaker puncta.
Figure 4. Autoinhibition mutants show synapse organization and trafficking phenotypes.
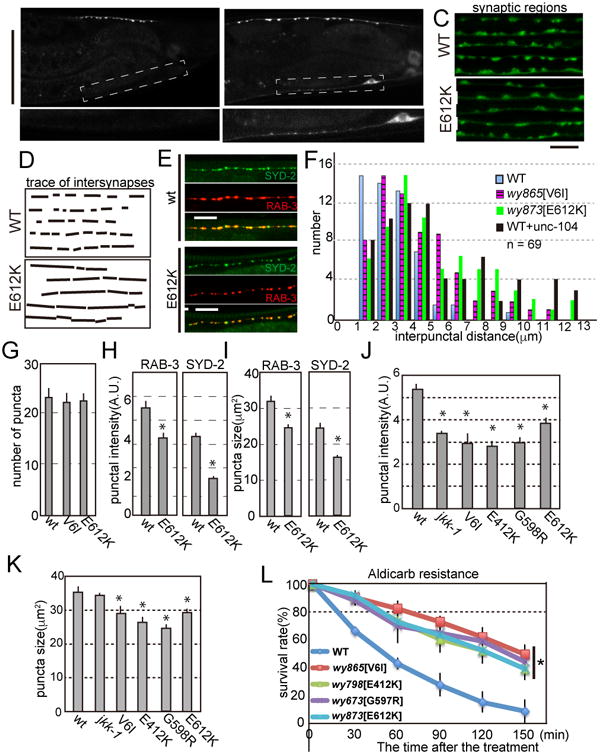
(A and B) Confocal images of (A) DA9 synapses in wild type (WT) and (B) unc-104(wy873[E612K]), labeled with GFP∷RAB-3. (A′ and B′) Representative images of DA9 dendritic region in wild type (A′) and unc-104(wy873[E612K]) (B′). Boxed area in panel (A) and (B) are zoomed. Scale Bars, 50 μm.
(C and D) (C) Image montages of part of the synaptic region in WT and the mutant. Note that the interpunctal distance is larger in unc-104(wy873[E612K]) than wild type. Bars, 5 μm. (D) Tracing of interpuncta in panel (C).
(E) Confocal images of DA9 synaptic region labeled by mCherry∷RAB-3 (SV marker, red) and GFP∷SYD-2 (active zone marker, green). While the inter-synapse distance varies between different genotypes, SVs and the active zone are well colocalized in both wt and unc-104(wy873[E612K]). Pearson's R values are 0.72 ± 0.13 and 0.72 ± 0.14 in wt and unc-104(wy873[E612K]), respectively (n= 8 worms). Not significant in t-test.
(F) The distribution of interpunctal distance in wild type, unc-104(wy865[V6I]) and unc-104(wy873[E612K]). In unc-104(wy865[V6I]) and unc-104(wy873[E612K]), the mean intersynapse distance is significantly longer compared to wild type. (p < 0.01, Mann–Whitney U test, n = 69 intersynapses from 8 animals)
(G) The number of synaptic boutons visualized by GFP∷Rab-3. Mean ± S.D. n = 13 animals. Not significant, compared to wt.
(H and I) Fluorescent intensity (H) and size (I) of mcherry∷RAB-3 and SYD-2∷GFP signals at synaptic boutons in wild type and unc-104(wy873[E612K]). Mean ± SEM, p < 0.01, Dunnett test, n = 30 synaptic boutons.
(J and K) Characterization of synaptic boutons visulalized with SNB-1∷YFP in wild type and unc-104(gf) alleles revealed by the computer-based automatic analysis. (J) The size of synaptic area and (K) The mean fluorescent intensity of synaptic boutons visualized by SNB-1(synaptobrevin)∷GFP. Note that both the size and the mean fluorescent intensity were reduced in unc-104(gf) alleles compared to wild type animals. Mean ± SEM, p < 0.01, Dunnett test, n = 49 synaptic boutons.
(L) The aldicarb resistance assay. Worms were treated by aldicarb and the ratio of worms that were not paralyzed at indicated time points were plotted. *, p < 0.01 in all the mutants compared to wt. Dunnet test. Three independent experiments. 25 worms were tested in each experiment. Note that unc-104(gf) alleles were more resistant to aldicarb compared to wt.
To test whether the changes of synaptic localization and size affect synaptic transmission, we performed an aldicarb resistance assay. Aldicarb inhibits synaptic choline esterase and paralyzes worms. Aldicarb resistance is suggestive of defects in the cholinergic synaptic transmission (Mahoney et al., 2006). We found that all four autoinhibition mutants of unc-104 were aldicarb resistant, suggestive of reduced synaptic transmission in the mutants (Fig. 4L). This synaptic transmission phenotype is consistent with the reduced synaptic size observed in these mutants.
ARL-8 and UNC-104 regulate the pause duration of transport vesicles
To better understand the effect of autoinhibition mutants on SV transport in vivo, we performed time-lapse imaging experiments after photobleaching a segment of the axon (Fig. 5A). We found that mobile transport RAB-3 puncta pause frequently along the axonal shaft in wt (Fig. 5B). The photobleaching procedure allows us to unambiguously determine the number of movement events, the duration of pauses and the directionality of re-initiated movements after pause. This procedure revealed that most transported vesicles stopped at vesicle pools do not fuse with other vesicles and restart movements (Fig 5B). Then, we measured the pause duration in wt, arl-8(wy271) and unc-104(gf). Compared to the wild type controls, the arl-8 (wy271) mutants have longer pauses (Fig 5C, 8.3 ± 0.4 sec and 15.5 ± 1.1 sec in wt and arl-8, respectively. Mean ± SEM). unc-104(gf) mutations tended to reduce the pause duration in both wt and arl-8(wy271) background (Fig 5C, 5.5 ± 0.47 sec, 6.9 ± 0.54 sec, 7.9 ± 0.47 sec, 11.6 ± 0.64 sec, 14.3 ± 0.85 sec in unc-104(wy673[G598R]), unc-104(wy873[E612K]), unc-104(wy865[V6I]), arl-8;unc-104(wy673[G598R]), arl-8; unc-104(wy873[E612K]), respectively). The frequency of anterograde movement was reduced in arl-8(wy271) (Fig 5D, 0.27 ± 0.013 /sec and 0.17 ± 0.009 / sec respectively in wt and arl-8). Similarly, the frequency of retrograde transport was reduced in arl-8(wy271) compared to wt (Fig. 5E, 0.13 ± 0.008 /sec and 0.06 ± 0.007 /sec respectively in wt and arl-8). The G598R and E612K mutations completely suppressed the movement number defect of arl-8(wy271) in the anterograde transport and partially suppressed in the retrograde transport (Fig. 5D and E). While G598R and E612K mutations did not change the transport frequency in the wt background, V6I mutation significantly increased the frequency of the vesicular movement (Figs 5D and E, 69 % and 60 % increase in the anterograde and retrograde transport, compared to wt). In addition, we analyzed the direction of the vesicle movement after a pause (Fig 5F). It was found that anterograde transport events were much less likely to reverse direction after a pause in unc-104(wy673[G598R]), unc-104(wy873[E612K] (Fig 5F). This phenomena was observed in both wild type and arl-8(wy271) background, suggesting that more vesicles are transported to distal when these autoinhibition mutations are introduced to unc-104. Taken together, these data suggest that these tail-domain autoinhibition mutants of UNC-104 (G598R and E612K) are more likely to reactivate motor transport after pausing, and bias re-initiated transport toward movement in the anterograde direction. V6I showed different properties compared to other two mutants but the transport is increased.
Figure 5. Kymograph analysis.
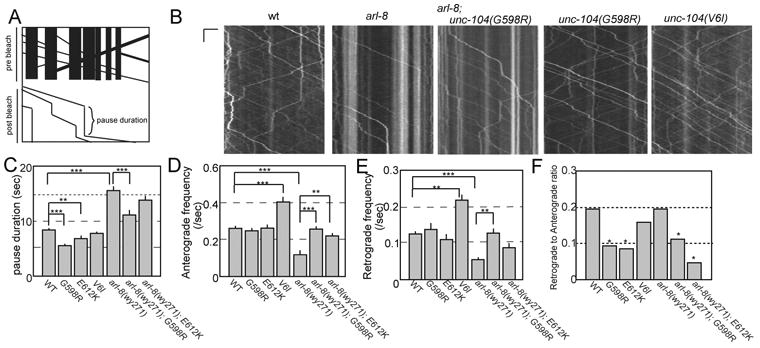
(A) Schematic showing kymograph with transport events obscured by existing puncta inupper panel (pre-bleach). In lower panel, transport events and their pause durations arerevealed by bleaching existing puncta (post-bleach).
(B) Representative kymographs of transport events. Time and length is on the y-axisand x-axis. Scale bars, 6 sec and 5 μm, respectively.
(C) Pause duration. A pause was defined as a clear lack of movement at motor driven transport speeds (> 1um/sec), which lasted longer than 1 second. Mean ± S.E.M., *, p <0.05, Kruskal-Wallis test. wt, 902 pauses from 76 worms. unc-104(wy673[G598R]), 271 pauses from 28 worms. unc-104(wy873[E612K]), 259 pauses from 23 worms. unc-104(wy865[V6I]), 398 pauses from 29 worms. arl-8(wy271), 197 pauses from 30 worms. arl-8(wy271); unc-104(wy673[G598R]), 476 pauses from 33 worms. arl-8(wy271); unc-104(wy873[E612K]), 311 pauses from 24 worms.
(D and E) Anterograde (D) and retrograde (E) event frequency was measured by isolating a specific one micron section of the axon and calculating the number of events moving anterogradely or retrogradely that passed through this one micron zone during the one minute transport movies. Kruskal-Wallis test *, p <0.05. Significance was computed using the Kruskal-Wallis test. Mean ± S.E.M. N = wt, 76 worms. unc-104(wy673[G598R]), 28 worms. unc-104(wy873[E612K]), 23 worms. unc-104(wy865[V6I]), 29 worms. arl-8(wy271), 30 worms. arl-8(wy271); unc-104(wy673[G598R]), 33 worms. arl-8(wy271); unc-104(wy873[E612K]), 24 worms.
(F) The retrograde to anterograde ratio was calculated for transport re-initiation events that emerged from an anterogradely moving event pausing. After the anterograde event paused, and then re-initiated transport the ratio of reversed direction (retrograde) to continued direction (anterograde) was computed. Pearson's chi-squared test with Bonferroni correction for multiple-comparisons. n = wt, 75 retrograde and 314 anterograde. unc-104(wy673[G598R]), 13 retrograde and 131 anterograde. unc-104(wy873[E612K]), 10 retrograde and 109 anterograde. unc-104(wy865[V6I]), 20 retrograde and 108 anterograde. arl-8(wy271), 16 retrograde and 66 anterograde. arl-8(wy271); unc-104(wy673[G598R]), 25 retrograde and 205 anterograde. arl-8(wy271); unc-104(wy873[E612K]), 6 retrograde and 131 anterograde.
Coexpression of ARL8B mimics gain of function mutations
We postulated that ARL-8 might bind to UNC-104 and activate it by relieving the autoinhibition. To test this hypothesis, we cotransfected KIF1A-GFP with ARL8B in COS-7 cells (Figure 6A-D). Mammalian ARL8B is suitable for this assay because KIF1A is a mammalian protein and ARL8B can rescue arl-8 phenotypes when expressed in C.elegans (Supplemental Figure S2D-G). Unlike the diffuse localization pattern of KIF1A∷GFP alone (Fig 6A, left panel), when cotransfected with ARL8B, KIF1A∷GFP displayed internal-membrane localization (Fig 6A, right panel). While about 80 % cells showed totally diffuse localization when only wt KIF1A was transfected, most cells showed MT- and/or internal membrane localization when co-transfected with ARL8B (Fig. 6B). Consistent with these results, using a MT-binding assay, the amount of KIF1A in the MT pellet was significantly increased in the presence of ARL8B compared to the KIF1A alone (Fig. 6C and D). Together, this evidence indicates that ARL-8 binding to UNC-104 unlocks the intramolecular autoinhibitory interaction.
Figure 6. Identification of an intragenic suppressor of arl-8.
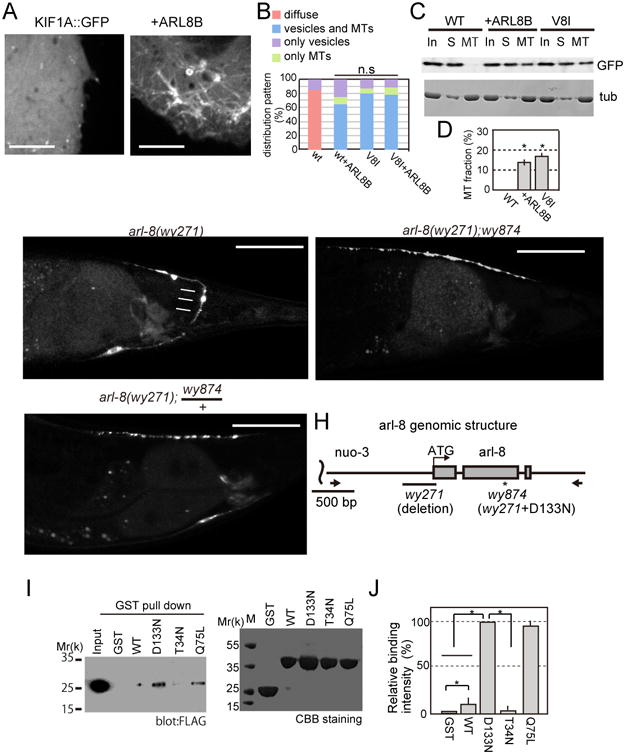
(A and B) KIF1A or KIF1A∷GFP and HaloTag∷ARL8B were transfected to COS-7 cells and the GFP signals were observed by spinning disc confocal microscopy.
(C and D) MT binding assay was performed as described in Figure 2E and F. (C) A representative image of western blot and (D) Quantification of western blot analysis. Mean ± SEM, *, p < 0.01, compared to the WT expression. Dunnett test. N = 5.
(E-G) GFP∷RAB-3 localization in DA9 neurons in (E) arl-8(wy271), (F) arl-8; wy874 and (G) arl-8(wy271); wy874/+. Bar, 50 μm.
(H) A schematic graph showing the nature of arl-8(wy271) and arl-8(wy874). Arrows indicate the position of PCR primers used to amplify the arl-8 genomic DNA fragment. Bar, 500 bp.
(I and J) Binding of various forms of ARL-8 to KIF1A. GST-ARL8B(Δ1-17) and ARL8B(Δ1-17) mutants were expressed in E.coli and attached to Glutathione Sepharose beads. Proteins were separated by SDS-PAGE and detected by western blot or CBB staining. (I) Representative results of GST pull down and (J) Relative protein binding compared to D133N. n = 3 independent experiments, *, p < 0.01, t-test. See also Figure S4.
D133N mutant is an intragenic suppressor of arl-8(wy271)
To further test the hypothesis that ARL-8 binding releases the autoinhibition of UNC-104, we set out to identify mutant versions of ARL-8 that have increased binding affinity with UNC-104. arl-8(wy271) is a strong loss-of-function allele, caused by the deletion of its the promoter region and first three amino acid of the coding region which dramatically reduced ARL-8 expression (Klassen et al., 2010). Using an F1 suppressor screen in the arl-8(wy271) background, we identified wy874. wy874 was not an unc-104 allele. In arl-8; wy874 allele, SV mislocalization, the presence of ectopic SV clusters in proximal axon and absence of synapses in the distal axon, were suppressed (Figs 6E-G and supplementary Figure S4A-C). While arl-8(wy271) has reduced body length, grows slower and gives smaller number of progenies than wild type, these phenotypes are largely suppressed in arl-8; wy874. Axonal transport parameters were also recovered in arl-8; wy874 (Figs S4D-F).
It was found that wy874 is an intragenic suppressor of arl-8 for the following reasons (Fig 6H). First, this mutation causes an amino acid substitution D133N on a conserved GTP binding residue of ARL-8. Second, F2 animals obtained by backcrossing of this strain with wt N2 worms did not give any arl-8 animals (from n = 132 F2 animals), suggesting that the suppressor mutation is closely linked with arl-8 gene. Third, injection of an arl-8 genomic fragment amplified from the arl-8(wy874) allele could rescue arl-8(wy271) mutants (supplemental Fig S4G-J), while a genomic fragment amplified from arl-8(wy271) mutant could not. In addition, whole genome sequencing shows that there are no other mutations in the rescuing fragments. Taken together, these pieces of genetic evidence argue that the D133N mutation causes a strong gain-of-function in arl-8 and compensates for the severe reduction of arl-8 expression in the wy271 allele.
ARL-8/ARL8B(D133N) strongly binds to and activates UNC-104/KIF1A
Previously, we have shown that ARL-8 directly binds to the CC3 region of UNC-104/KIF1A (Wu et al., 2013). To test whether the binding between ARL-8 and UNC-104 is augmented by the D133N mutation, we tested the strength of binding with GST-pull down experiments. ARL8B and KIF1A, mammalian orthologs of arl-8 and unc-104, were used because mammalian cells are more suitable for biochemical assays compared to C. elegans. Compared to wild type ARL8B controls, ARL8B(D133N) showed stronger binding to KIF1A (Figs 6I and J). We also tested predicted GTP and GDP forms of ARL-8. ARL8B(Q75L), a mutant that mimics the GTP-bound form of ARL8B, strongly binds to KIF1A as the D133N form. In contrast, ARL8B(T34N), a GDP form mutant, show minimal binding. These results suggest that the binding between ARL8B and KIF1A depends on the nucleotide states of ARL8B and that D133N likely represents an activating mutation for ARL-8/ARL8B. The tip accumulation assay in DA9 neurons and the MT binding assay in COS cells also indicated that D133N mutation activates UNC-104/KIF1A in cells (Supplementary Figure S4K-N). These data strongly suggest that the binding between ARL-8/ARL8B and UNC-104/KIF1A correlates with the activation of UNC-104/KIF1A by ARL-8/ARL8B in vivo.
ARL-8(D133N) single mutant phenocopies unc-104(gf) mutants
As arl-8(wy874) contains both the promoter deletion and the D133N mutation, both protein level and ARL-8 activity are likely affected in this mutant (Fig 6H). To tease out the contribution of ARL-8 GTPase activity to synaptic phenotypes in the clean genetic background, we used CRISPR/CAS9 and homologous recombination to introduce the D133N mutation in the wild type background without the promoter mutation (Fig. 7A) (Arribere et al., 2014). The resultant arl-8(jpn1[D133N]) allele showed increased interpunctal distance compared to the wt control, suggesting that activated ARL-8 leads to a reduced synapse density and weak dendritic mislocalization of GFP∷RAB-3 signal (Figs. 7B-G), a phenotype that is also found in the unc-104(gf) (Fig. 4A-D). The SV punctal intensity was much weaker and synaptic area was much smaller in arl-8(jpn1[D133N]) compared to those of wt controls (Figs 7D-G). A double mutant containing both the D133N mutation and the unc-104(wy798[E412K]) mutation showed no further enhancement of the synapse size phenotype, suggesting that arl-8(gf) and unc-104(gf) are in the same pathway (Fig. 7F and G). We have previously shown that the arl-8(wy271) can be partially suppressed by loss of jkk-1 (Wu et al., 2013). As a single mutant, jkk-1 showed reduced punctal intensity of synaptic markers (Fig. 7F). To understand the relationship between the jkk-1 and unc-104 pathways, we analyzed synaptic intensities and area in unc-104(wy798[E412K]); jkk-1 double mutant. The synaptic punctual intensity and area of the double mutant are enhanced suggesting that UNC-104 and JKK-1 function in parallel pathways to regulate synapse size (Fig. 7F and G).
Figure 7. The phenotype of the gain of function mutant of arl-8.
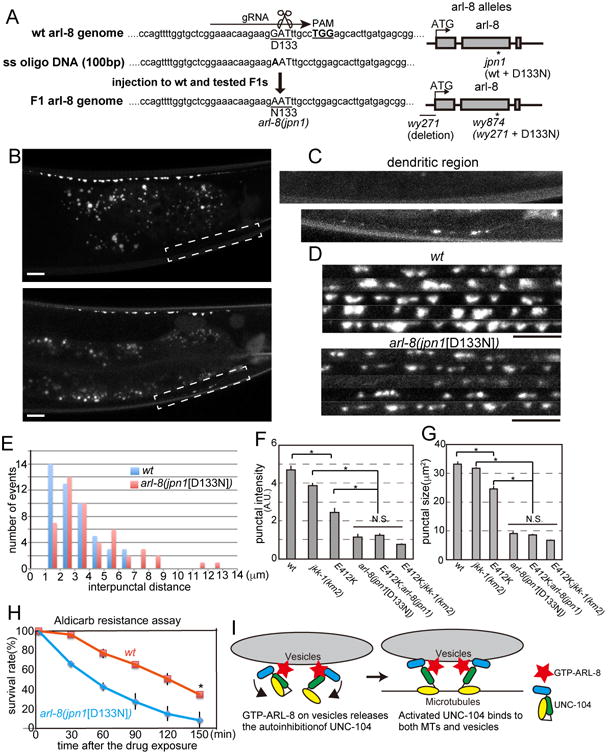
(A) Left, Genome editing using the CRISPR/CAS9 method. The D133N mutation was introduced into the wild type background to generate an arl-8 allele, arl-8(jpn1). Right, a schema showing the genomic structure of arl-8(jpn1), arl-8(wy271) and arl-8(wy874) (B-D) Representative confocal images of wild type and arl-8 (jpn1[D133N]). (B) Gross phenotype. (C) The dendritic mislocalization. and (D) Image montages of part of the synaptic region in WT and arl-8(jpn1[D133N]). Scale bars, 10 μm.
(E) The distribution of the interpunctal distance in wild type (WT) and arl-8(jpn1). In arl-8(jpn1), the mean intersynapse distance is significantly longer compared to wild type. (p < 0.01, Mann-Whitney U test, n = 50 intersynapses from 3 animals).
(F and G) Characterization of synaptic boutons in wild type, jkk-1, unc-104(wy798), arl-8(jpn1), unc-104(wy798);arl-8(jpn1) double mutant and unc-104(wy798); jkk-1 double mutant visualized by RAB-3∷GFP. Punctal intensity and size were measured as described in Figure 4. Mean ± SEM, p < 0.01, Dunnett test, n = 60 synaptic boutons.
(H) The aldicarb resistance assay. arl-8(jpn1) is resistant to the aldicarb treatment compared to wild type. Mean ± SD. *, p < 0.05, compared to wt at 150 min, t-test. Three independent experiments. 25 worms were tested in each experiment.
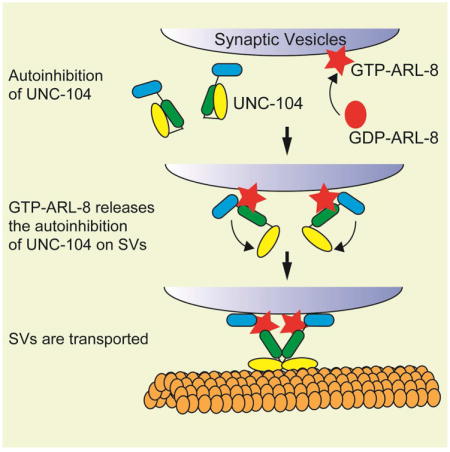
(I) A model showing how UNC-104 is activated. GTP-ARL-8 on vesicles strongly binds to the stalk domain of UNC-104 and releases the autoinhibition. Activated UNC-104 can bind to both MTs and SVs. See also Figure S5.
Finally, we found that arl-8(jpn1[D133N]) worms showed aldicarb resistance, similar to the unc-104(gf) (Fig. 7H). Together, these results argue that constitutive activation of ARL-8 leads to overactivation of UNC-104, an effect that can also be achieved by the autoinhibition mutations.
Discussion
Activation mechanism of UNC-104/KIF1A
Previous studies showed that KIF1A is regulated by autoinhibition in vitro (Hammond et al., 2009). The similar effects by the mutations on both worm and mammalian kinesins argue that the autoinhibition is a conserved mechanism to regulate UNC-104/KIF1A. While previous studies have focused on the activation of the kinesin motor domain (Blasius et al., 2007; Cho et al., 2009; Fu and Holzbaur, 2014; Sun et al., 2011; van der Vaart et al., 2013; Yamada et al., 2007), we found that cargo binding is also regulated by an autoinhibitory mechanism (Figs 2 and 6). Introduction of gain-of-function mutations, or co-expression of ARL8B are able to activate KIF1A and drives KIF1A onto both MTs and internal membranes.
How are kinesin activation and cargo availability coordinated? Our data suggest that the arl-8/ALR8B, a conserved small GTPase, activates UNC-104/KIF1A by unlocking the autoinhibition of unc-104/KIF1A (Figures 7I and S5). Interestingly, both worm ARL-8 and vertebrate ARL8 are localized on SV membrane (Takamori et al., 2006; Wu et al., 2013). It is possible that this localization ensures UNC-104/KIF1A to be activated specifically on the cargo vesicles. Unlike other small GTPases that serve as membrane receptors for motor proteins, ARL-8 is not absolutely required for the binding of UNC-104/KIF1A to SVs, since the autoinhibition defective UNC-104/KIF1A mutants retain vesicle-binding abilities in the absence of ARL-8. Together with the previous literature, these data suggested that cargo vesicle bound ARL-8 binds to UNC-104/KIF1A and releases the autoinhibition, which enables binding of the tail domain to the cargo vesicles as shown in previous studies (Niwa et al., 2008; Wagner et al., 2009). Consequently, the cargo binding would induce dimerization (Klopfenstein et al., 2002), leading to the full activation of UNC-104 (Tomishige et al., 2002) (supplementary Figure S5).
Autoinhibition of UNC-104 regulates polarized distribution of synaptic material, synapse size and synaptic density
Although previous studies have shown that kinesins are regulated by autoinhibition (Blasius et al., 2007; Cho et al., 2009; Fu and Holzbaur, 2014; Sun et al., 2011; Yamada et al., 2007), it has not been fully clarified how the mechanism affect the cargo distribution in vivo. Our genetic data suggest that the autoinhibitory mechanism of unc-104 is required for normal SV localization and synaptic functions in vivo. unc-104 mutants lacking the autoinhibition showed decreased synaptic density and smaller synaptic puncta. These mutants were aldicarb-resistant, indicative of a synaptic transmission defect (Mahoney et al., 2006). While these assays do not provide detailed characterization of electrophysiological parameters, they strongly suggest that synaptic transmission is reduced in the autoinhibition mutants.
The factors that determine synaptic localization are fundamental to the formation of neuronal networks. Previously we have found extrinsic and intrinsic factors, such as wnt signaling molecules, Dynein and two cyclin-dependent kinase pathways, regulate the synaptic localization (Ou et al., 2010; Park et al., 2011). In addition to these factors, this study revealed that the autoinhibition of UNC-104 and the nucleotide state of ARL-8 also determine synaptic localization. In both unc-104(gf) and arl-8(gf) mutants, a small but significant amount of synaptic material was mislocalized to the dendrite. We have previously shown that SV precursors are transported by both UNC-104 and dynein (Maeder et al., 2014). In dendrites, while the majority of MTs are minus-end out, there are some MTs that are plus-end out (Yan et al., 2013). These plus-end out MTs can potentially support the trafficking of SVs into dendrite by UNC-104. Indeed, while UNC-104∷GFP in which autoinhibition is disrupted were under the detectable level in DA9 dendrites, the amount of SV precursors transported to the dendrite were increased (Fig. 3, 4A and B). This result implies that even the small amount of UNC-104 might be sufficient to transport cargos when the autoinhibitory mechanism is disrupted. Furthermore, it is possible that more ARL-8 is in the GTP state in the axon compared to the dendrite and regulates the polarized transport of SVs, but further analysis is required to test this hypothesis.
The synapse density is reduced in autoinhibition mutants (Fig 4). It is conceivable that the decreased synaptic density in the autoinhibition mutants might be the result of reduced pause duration or increased movement events, (Fig 5) both of which could lead to more robust anterograde movement and better separation of en passant synapses during synaptogenesis. Furthermore, the size of synapses appears to be reduced in these autoinhibition mutants. This is consistent with our previous findings that overexpression of ARL-8 reduces the size of synapses (Wu et al., 2013) and further suggest that UNC-104 mediated trafficking and SV aggregation are antagonistic aspects of synapse formation. The balance of these events is required to achieve the appropriate size and density of synapses in vivo. Our data suggests that the balance would be regulated by the autoinhibition of UNC-104 and the nucleotide state of ARL-8.
Experimental Procedures
Full Experimental procedures are described in the supplemental text.
Genetics, Cell Biology and biochemistry
C. elegans was maintained on OP50 feeder bacteria at 20 °C. Genetic screening was performed using ethyl methanesulfonate (SIGMA-ALDRICH, St Louis, MO, USA). Extrachromosomal arrays were generated by plasmid injection. Genome editing was performed as described (Arribere et al., 2014). COS cells were maintained in 5 % CO2 incubator at 37 °C. Transfection was performed using Lipofectamine 2000(Life Technologies, Waltham, MA, USA).
Microscopy
DA9 neurons were observed by LSM710 confocal microscope system (Carl Zeiss, Jena, Germany) equipped with PlanApochromat (Carl Zeiss, x63, NA1.4) as described (Wu et al., 2013). Time lapse microscopy was performed using spinning disk confocal microscope (Yokogawa, Tokyo, Japan) equipped with PlanApochromat (Carl Zeiss, x63, NA1.4) as described (Maeder et al., 2014).
Statistics
Statistical methods and the sample size are described in each figure legends. t-test and Chi-square tests and Bonferroni correction were performed using Excel (Microsoft Corporation, Redmond, WA, USA), Dunnet method and Mann–Whitney U test were performed using Excel Toukei (SSRI, Tokyo, Japan) and Kruskal-Wallis test was performed using MATLAB (MathWorks, Natick, MA, USA).
Supplementary Material
Highlights.
We describe an autoinhibitory regulation of UNC-104/KIF1A.
ARL-8 activates UNC-104 by releasing autoinhibition.
The autoinhibition of UNC-104 determines synaptic size and density
The nucleotide state of ARL-8 determines synaptic size and density
Acknowledgments
The authors wish to thank Dr. Emily Wu, Cen Gao and other members in the Shen lab at Stanford University. Some strains were provided by the CGC, which is funded by the NIH Program (P40 OD010440). Research in the Shen lab was supported by both the National Institutes of Health (R01 NS048392) and the Howard Hughes Medical Institute. Research in the Hirokawa lab was supported by a Grant-in Aid for Specially Promoted Research from MEXT to N.H.. Shinsuke Niwa's research was supported by fellowships from the JSPS, Naito Foundation and Kanae Foundation and JSPS KAKENHI (15H06033).
Footnotes
Authors contribution: Conceptualization, S.N. and K.S.; Methodology, D.M.L, C.Z., and H.L.; Investigation, S. N., D.M.L, M.M, and C.Z.; Resources, N.H., H.L. and K.S.; Writing - Original Draft, S.N. and K.S.; Writing - Review and Editing, S.N., D.M.L and K.S.; Funding Acquisition; S.N., N.H. and K.S.; Supervision, K.S..
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Bassam J, Cui Y, Klopfenstein D, Carragher BO, Vale RD, Milligan RA. Distinct conformations of the kinesin Unc104 neck regulate a monomer to dimer motor transition. J Cell Biol. 2003;163:743–753. doi: 10.1083/jcb.200308020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 2014;198:837–846. doi: 10.1534/genetics.114.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkus RV, Klyachko O, Horiuchi D, Dickson BJ, Saxton WM. Identification of an axonal kinesin-3 motor for fast anterograde vesicle transport that facilitates retrograde transport of neuropeptides. Mol Biol Cell. 2008;19:274–283. doi: 10.1091/mbc.E07-03-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius TL, Cai D, Jih GT, Toret CP, Verhey KJ. Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol. 2007;176:11–17. doi: 10.1083/jcb.200605099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Desai J, Miranda CJ, Duncan JS, Qiu W, Nugent AA, Kolpak AL, Wu CC, Drokhlyansky E, Delisle MM, et al. Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron. 2014;82:334–349. doi: 10.1016/j.neuron.2014.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KI, Yi H, Desai R, Hand AR, Haas AL, Ferreira PA. RANBP2 is an allosteric activator of the conventional kinesin-1 motor protein, KIF5B, in a minimal cell-free system. EMBO Rep. 2009;10:480–486. doi: 10.1038/embor.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coy DL, Hancock WO, Wagenbach M, Howard J. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat Cell Biol. 1999;1:288–292. doi: 10.1038/13001. [DOI] [PubMed] [Google Scholar]
- Crane MM, Stirman JN, Ou CY, Kurshan PT, Rehg JM, Shen K, Lu H. Autonomous screening of C. elegans identifies genes implicated in synaptogenesis. Nat Methods. 2012;9:977–980. doi: 10.1038/nmeth.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DS, Vale RD. Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat Cell Biol. 1999;1:293–297. doi: 10.1038/13008. [DOI] [PubMed] [Google Scholar]
- Fu MM, Holzbaur EL. Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends Cell Biol. 2014;24:564–574. doi: 10.1016/j.tcb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD. Highly processive microtubule-stimulated ATP hydrolysis by dimeric kinesin head domains. Nature. 1995;377:448–450. doi: 10.1038/377448a0. [DOI] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Cai D, Blasius TL, Li Z, Jiang Y, Jih GT, Meyhofer E, Verhey KJ. Mammalian Kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS Biol. 2009;7:e72. doi: 10.1371/journal.pbio.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Holzbaur EL, Scherer SS. Microtubules, axonal transport, and neuropathy. N Engl J Med. 2011;365:2330–2332. doi: 10.1056/NEJMcibr1112481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L, Yue Y, Ren J, Yu J, Liu J, Yu Y, Ye F, Xu T, Zhang M, Feng W. The CC1-FHA tandem as a central hub for controlling the dimerization and activation of kinesin-3 KIF1A. Structure. 2012;20:1550–1561. doi: 10.1016/j.str.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Klassen MP, Shen K. Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell. 2007;130:704–716. doi: 10.1016/j.cell.2007.06.046. [DOI] [PubMed] [Google Scholar]
- Klassen MP, Wu YE, Maeder CI, Nakae I, Cueva JG, Lehrman EK, Tada M, Gengyo-Ando K, Wang GJ, Goodman M, et al. An Arf-like small G protein, ARL-8, promotes the axonal transport of presynaptic cargoes by suppressing vesicle aggregation. Neuron. 2010;66:710–723. doi: 10.1016/j.neuron.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein DR, Tomishige M, Stuurman N, Vale RD. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell. 2002;109:347–358. doi: 10.1016/s0092-8674(02)00708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JR, Shin H, Choi J, Ko J, Kim S, Lee HW, Kim K, Rho SH, Lee JH, Song HE, et al. An intramolecular interaction between the FHA domain and a coiled coil negatively regulates the kinesin motor KIF1A. EMBO J. 2004;23:1506–1515. doi: 10.1038/sj.emboj.7600164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Taylor DW, Krementsova EB, Trybus KM, Taylor KA. Three-dimensional structure of the myosin V inhibited state by cryoelectron tomography. Nature. 2006;442:208–211. doi: 10.1038/nature04719. [DOI] [PubMed] [Google Scholar]
- Maeder CI, San-Miguel A, Wu EY, Lu H, Shen K. In vivo neuron-wide analysis of synaptic vesicle precursor trafficking. Traffic. 2014;15:273–291. doi: 10.1111/tra.12142. [DOI] [PubMed] [Google Scholar]
- Mahoney TR, Luo S, Nonet ML. Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat Protoc. 2006;1:1772–1777. doi: 10.1038/nprot.2006.281. [DOI] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Brown H, Tiwari A, Hayward L, et al. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J Cell Biol. 2003;162:1045–1055. doi: 10.1083/jcb.200302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa S, Takahashi H, Hirokawa N. β-Tubulin mutations that cause severe neuropathies disrupt axonal transport. EMBO J. 2013;32:1352–1364. doi: 10.1038/emboj.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa S, Tanaka Y, Hirokawa N. KIF1Bbeta- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat Cell Biol. 2008;10:1269–1279. doi: 10.1038/ncb1785. [DOI] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Otsuka AJ, Jeyaprakash A, García-Añoveros J, Tang LZ, Fisk G, Hartshorne T, Franco R, Born T. The C. elegans unc-104 gene encodes a putative kinesin heavy chain-like protein. Neuron. 1991;6:113–122. doi: 10.1016/0896-6273(91)90126-k. [DOI] [PubMed] [Google Scholar]
- Ou CY, Poon VY, Maeder CI, Watanabe S, Lehrman EK, Fu AK, Park M, Fu WY, Jorgensen EM, Ip NY, Shen K. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell. 2010;141:846–858. doi: 10.1016/j.cell.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack-Chung E, Kurshan PT, Dickman DK, Schwarz TL. A Drosophila kinesin required for synaptic bouton formation and synaptic vesicle transport. Nat Neurosci. 2007;10:980–989. doi: 10.1038/nn1936. [DOI] [PubMed] [Google Scholar]
- Park M, Watanabe S, Poon VY, Ou CY, Jorgensen EM, Shen K. CYY-1/cyclin Y and CDK-5 differentially regulate synapse elimination and formation for rewiring neural circuits. Neuron. 2011;70:742–757. doi: 10.1016/j.neuron.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce DW, Hom-Booher N, Otsuka AJ, Vale RD. Single-molecule behavior of monomeric and heteromeric kinesins. Biochemistry. 1999;38:5412–5421. doi: 10.1021/bi9830009. [DOI] [PubMed] [Google Scholar]
- Sun F, Zhu C, Dixit R, Cavalli V. Sunday Driver/JIP3 binds kinesin heavy chain directly and enhances its motility. EMBO J. 2011;30:3416–3429. doi: 10.1038/emboj.2011.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sönnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Tomishige M, Klopfenstein DR, Vale RD. Conversion of Unc104/KIF1A kinesin into a processive motor after dimerization. Science. 2002;297:2263–2267. doi: 10.1126/science.1073386. [DOI] [PubMed] [Google Scholar]
- Torisawa T, Ichikawa M, Furuta A, Saito K, Oiwa K, Kojima H, Toyoshima YY, Furuta K. Autoinhibition and cooperative activation mechanisms of cytoplasmic dynein. Nat Cell Biol. 2014;16:1118–1124. doi: 10.1038/ncb3048. [DOI] [PubMed] [Google Scholar]
- van der Vaart B, van Riel WE, Doodhi H, Kevenaar JT, Katrukha EA, Gumy L, Bouchet BP, Grigoriev I, Spangler SA, Yu KL, et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev Cell. 2013;27:145–160. doi: 10.1016/j.devcel.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Wagner OI, Esposito A, Köhler B, Chen CW, Shen CP, Wu GH, Butkevich E, Mandalapu S, Wenzel D, Wouters FS, Klopfenstein DR. Synaptic scaffolding protein SYD-2 clusters and activates kinesin-3 UNC-104 in C. elegans. Proc Natl Acad Sci U S A. 2009;106:19605–19610. doi: 10.1073/pnas.0902949106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Huo L, Maeder CI, Feng W, Shen K. The balance between capture and dissociation of presynaptic proteins controls the spatial distribution of synapses. Neuron. 2013;78:994–1011. doi: 10.1016/j.neuron.2013.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KH, Hanada T, Chishti AH. The effector domain of human Dlg tumor suppressor acts as a switch that relieves autoinhibition of kinesin-3 motor GAKIN/KIF13B. Biochemistry. 2007;46:10039–10045. doi: 10.1021/bi701169w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Chao DL, Toba S, Koyasako K, Yasunaga T, Hirotsune S, Shen K. Kinesin-1 regulates dendrite microtubule polarity in Caenorhabditis elegans. Elife. 2013;2:e00133. doi: 10.7554/eLife.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Sheng Y, Zhang HN, Yu Y, Huo L, Feng W, Xu T. The CC1-FHA dimer is essential for KIF1A-mediated axonal transport of synaptic vesicles in C. elegans. Biochem Biophys Res Commun. 2013;435:441–446. doi: 10.1016/j.bbrc.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.