

Abstract
The UbiA superfamily is a group of intramembrane prenyltransferases that generate lipophilic compounds essential in biological membranes. These compounds, which include various quinones, hemes, chlorophylls, and vitamin E, participate in electron transport and function as antioxidants, as well as acting as structural lipids of microbial cell walls and membranes. Prenyltransferases producing these compounds are involved in important physiological processes and human diseases. These UbiA superfamily members differ significantly in their enzymatic activities and substrate selectivities. This chapter describes examples of methods that can be used to group these intramembrane enzymes, analyze their activity, and screen and crystallize homolog proteins for structure determination. Recent structures of two archaeal homologs are compared with structures of soluble prenyltransferases to show distinct mechanisms used by the UbiA superfamily to control enzymatic activity in membranes.
2. Introduction to the UbiA Superfamily
The intramembrane prenyltransferases of UbiA superfamily generate the basic skeleton of a variety of lipiophilic compounds. These prenyltransferases catalyze the fusion of an isoprenyl or phytyl chain to another molecule, often an aromatic compound from the aqueous phase of cells. Addition of the aliphatic tail confers lipid solubility on these compounds, which are recruited to cell membranes and become active in various biological pathways.
These lipophilic compounds play essential roles in all living organisms (Nowicka and Kruk, 2010). Ubiquinones and menaquinones function as electron carriers in the cellular respiration chain that generates ATP, and as antioxidants that protect membranes against lipid peroxidation. Prenylated hemes are cofactors of the terminal oxidases in the respiration chain that converts oxygen to water. In photosynthetic organisms, chlorophylls absorb energy from light, and plastoquinones transport electrons during photosynthesis. Vitamin E is a group of potent antioxidants that reduce cell damage in plants (Bonitz et al., 2011). Other prenylated compounds serve as components of the mycobacterial cell wall, as characteristic lipids of archaeal membranes, and as secondary metabolites.
The prenylation reaction that generates these compounds is generally an evolutionarily conserved and rate-limiting step of the biosynthetic pathway (Heide, 2009). The prenyltransferases require two substrates, the prenyl donor and the acceptor. The donor substrate is usually isoprenyl diphosphate (XPP; X stands for the various lengths of the isoprenyl chain). Cleavage of the diphosphate group from the XPP substrate generates a reactive carbocation intermediate at the end of the isoprenyl chain. This carbocation reacts with the acceptor substrate to form either a C–C or C–O bond to complete the prenylation reaction.
3. Biological Function and Enzymatic Activity of the Subfamilies
As the prototype of the superfamily, the UbiA enzyme from E. coli is well characterized in biochemistry. UbiA catalyzes the condensation of p-hydroxybenzoate (PHB) with XPP (Fig. 1A). The UbiA activity requires divalent metal ions, preferably magnesium (Melzer and Heide, 1994; Young et al., 1972), which are coordinated by two Asp-rich motifs in UbiA to engage the pyrophasphate group of XPP (Cheng and Li, 2014). The length of XPP can vary from 2 to 10 isoprenyl units (each unit contains 5 carbons, or C5). The E. coli UbiA enzyme appears to have low substrate affinity; the Km values for the reaction between PHB and geranyldiphosphate (GPP; C10) are 0.2 mM and 0.25 mM, respectively. A higher binding affinity is observed for longer substrates, farnesyldiphosphate (FPP; C15) and solanesyldiphosphate (SPP; C45), albeit with a lower enzymatic activity. Despite the promiscuity of XPP chain length, the prenylation occurs only at the meta-position of PHB (Wessjohann and Sontag, 1996). The control mechanism of this regiospecific reaction remains unknown.
Figure 1.
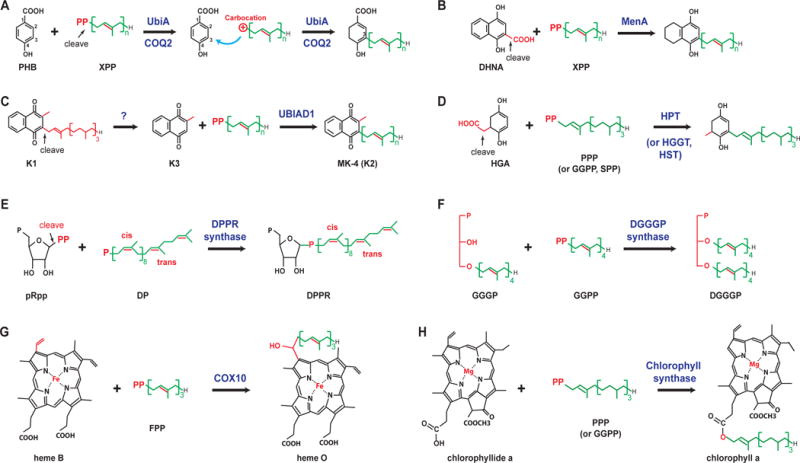
Different reactions catalyzed by UbiA superfamily prenyltransferases (A–G). Unique chemical groups are colored in red and cleavage reactions are indicated by arrows. Abbreviations: HGA, homogentisic acid; PPP, phytyl diphosphate; GGPP, geranylgeranyl diphosphate; pRpp, phosphoribose diphosphate; DP, decaprenyl phosphate; DPPR, decaprenylphosphoryl-5-phosphoribose, GGGP, geranylgeranylglyceryl phosphate; DGGGP, digeranylgeranylglycerylphosphate.
COQ2, the eukaryotic homolog of UbiA, has a very similar activity to that of PHB prenyltransferase. In fact, COQ2 or UbiA from different organisms can complement the growth of UbiA-deficient E. coli or COQ2-deficient yeast (Boehm et al., 2000; Forsgren et al., 2004; Ohara et al., 2009, 2006, 2004; Okada et al., 2004; Suzuki et al., 1994; Uchida et al., 2000). These results show that both UbiA and COQ2 are indiscriminate with respect to the chain length of XPPs, which are used in these organisms to produce ubiquinones of 6 to 10 isoprenyl units (Kainou et al., 2001; Meganathan, 2001; Okada et al., 1998, 1997, 1996). Clinically, COQ2 has been linked to infantile multisystem disease, in which COQ2 mutations result in primary ubiquinone deficiency (Quinzii et al., 2008, 2007).
The MenA enzyme synthesizes menaquinones (Shineberg and Young, 1976; Suvarna et al., 1998; Young, 1975), the quinones most often used by microbes in their respiration chains. Compared to UbiA, MenA recognizes a different aromatic substrate, 1,4-dihydroxy-2-naphthoic acid (DHNA). Decarboxylation of DHNA, which removes the 2-carboxyl group, is coupled with the condensation reaction at the same position (Fig. 1B). Recent studies of MenA (Debnath et al., 2012; Dhiman et al., 2009; Kurosu and Crick, 2009; Kurosu et al., 2007; Li et al., 2014), focused on developing new tuberculosis drugs, have observed that MenA inhibitors showed significant and specific activity against the mycobacterium at the nonreplicating stage (Debnath et al., 2012).
UBIAD1 is a prenyltransferase, found in animals, that is used for the biosynthesis of menaquinone-4 (MK4; vitamin K2), which is converted from phylloquinone (K1) obtained from food sources (Nakagawa et al., 2010). UBIAD1 prenylates menadione (K3), which is generated by the removal of the phytyl tail of K1 (Fig. 1C), either by UBIAD1 or an unknown enzyme (Nakagawa et al., 2010). UBIAD1 is involved in maintaining vascular homeostasis (Hegarty et al., 2013), preventing oxidative damage in cardiovascular tissues (Mugoni et al., 2013), and sustaining mitochondrial function (Vos et al., 2012). Mutations of UbIAD1 in humans cause Schnyder crystalline corneal dystrophy (Orr et al., 2007; Weiss et al., 2007) and urologic cancers (Fredericks et al., 2011).
Homogentisate prenyltransferases decarboxylate homogentisic acid and attach different tails. Enzymes in this group, which include homogentisate solanesyltransferases (HST), phytyltransferases (HPT), and geranylgeranyltransferases (HGGT), differ, as indicated by their names, by their specificity requirements for the prenyl donor (Fig. 1D). HST, HPT, and HGGT participate in the biosynthesis of plastoquinones, tocopherols, and tocotrienols, respectively (Soll et al., 1980), with the latter two groups of compounds collectively known as vitamin E. To improve the vitamin E content in transgenic plants, HPT and HGGT have been extensively explored for metabolic engineering (Cahoon et al., 2003; Collakova and DellaPenna, 2003; Kinney, 2006; Lee et al., 2007; Savidge et al., 2002; Schledz et al., 2001; Valentin and Qi, 2005; Venkatesh et al., 2006).
The DPPR synthase generates a precursor for arabinogalactans, which form the central layer of a highly impermeable cell wall that is required for the survival of Mycobacterium tuberculosis. DPPR synthase is a promising drug target because both the prenyl donor and the acceptor substrates, decaprenyl phosphate (with several cis- bonds) and 5-phosphoribosyl 1-pyrophosphate (Fig. 1E), are not found in humans (Huang et al., 2008, 2005). The catalysis of DPPR synthase is also unique because the prenyl acceptor contains the pyrophosphate; after its cleavage, the resulting carbocation on the ribose reacts to a monophosphate on the prenyl chain. In contrast, for all other enzymes in this superfamily, a carbocation intermediate is generated from cleaving XPP, the donor substrate (Fig. 1).
DGGGP synthase generates the skeleton of diether lipids (Zhang and Poulter, 1993) in an unsaturated form (Fig. 1F), which is the common precursor to most diether and tetraether lipids. These lipids form the core structure of unique archaeal membranes, a major evolutionary feature that distinguishes archaea from bacteria and eukaryotes (De Rosa and Gambacorta, 1988; Koga and Morii, 2007). Although most members of the superfamily use acceptors with ring structures (Fig. 1), the prenyl acceptor in DGGGP synthase is a linear compound (Hemmi et al., 2004; Zhang and Poulter, 1993). Another difference is that the prenylation by DGGGP synthase generates a C–O link on a glycerol moiety.
The heme O synthase, or protoheme IX farnesyltransferase, attaches a farnesyl tail to protoheme IX. These enzymes are also named COX10 in eukaryotes and CyoE and CtaB in E. coli and Bacillus subtilis, respectively (Fig. 1G). This universal farnesyltransferase generates heme O, which can be subsequently converted to heme A. These hemes are used in different organisms as the prosthetic group of the terminal heme-copper oxidases in the respiration chain, such as the mitochrodrial cytochrome c oxidase (complex IV) that reduces oxygen to water (Hederstedt, 2012).
As the terminal enzyme for chlorophyll biosynthesis, chlorophyll synthase esterifies chlorophyllide with phytyl or geranylgeranyl pyrophosphate (Fig. 1H) (Beale, 1999; Oster et al., 1997; Schmid et al., 2001; Willows, 2003). Modified compounds were used to deduce the orientation of the chlorophyllide binding (Rüdiger et al., 2005), which may be similar in chlorophyll synthase and COX10, because both enzymes need to accommodate a large porphyrin ring.
Overall, prenyltransferases in the UbiA superfamily are characterized by their different preferences for structurally diverse substrates. This superfamily contains a large number of intramembrane prenyltransferases that are highly diverse in sequence and function.
4. Predicting the Functional Role of Intramembrane Prenyltransferases by Sequence Clustering Analysis
Sequence clustering provides a useful tool for predicting the function of the large number of uncharacterized superfamily members, which are occasionally misannotated in databases. For example, an archaeal homolog from Archaeoglobus fulgidus, for which a crystal structure has been determined (Huang et al., 2014), is either annotated as bacteriochlorophyll synthase in NCBI or is considered to be a homolog close to MenA and UBIAD1. However, sequence clustering suggests that the function of this homolog is relatively distinct from the functions of known groups of enzymes in the superfamily (Li, 2016). The need for functional prediction is particularly compelling in the field of membrane protein crystallography, because it is often necessary to screen a large set of protein homologs to identify the very few that can grow diffracting crystals. In a large screen, activity analysis for all candidate membrane proteins is an intimidating task; sequence clustering offers a simple way to judge the candidate proteins’ functional relevance, which can be verified later if a crystallization hit is obtained.
The first step in sequence clustering is the selection of representative proteins with well-studied biochemical properties from each major subfamily of known functions (Section 3). These representative proteins, and the protein(s) to be predicted for function, are expanded through a PSI-BLAST search to a number of non-redundant proteins of considerable sequence similarity. Clustering of these sequences is performed based on pairwise sequence similarity, and the function of the target protein is predicted by its relative location on a clustered map. The clustering analysis is more accurate than conventional methods such as multiple sequence alignment and phylogenetic analysis, which can accumulate errors when analyzing thousands of proteins (Frickey and Lupas, 2004). This is the case for the numerous prenyltransferases in the UbiA superfamily, and therefore sequence clustering analysis (Fig. 2) is performed in the following steps.
Figure 2.
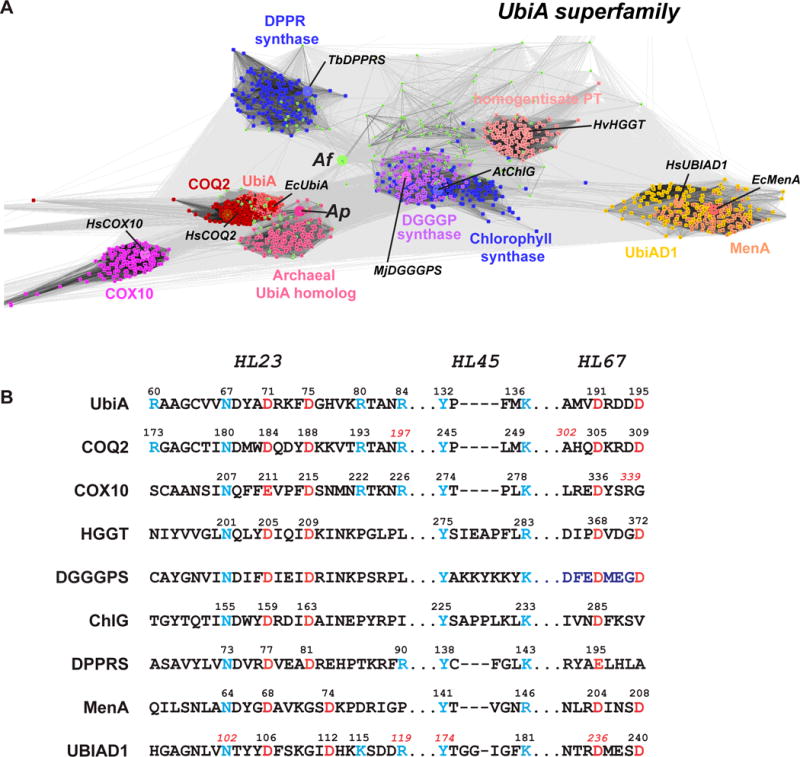
Sequence clustering and conserved regions of UbiA superfamily prenyltransferases. (A) Homology clustering. The representative proteins used in PSI-BLAST search are indicated (prefix: Hs, homo sapiens; Ec, Escherichia coli; At, Arabidopsis thaliana; Tb, Mycobacterium tuberculosis; Hv, Hordeum vulgare; Mj, Methanocaldococcus jannaschii), and their positions (large spheres) are shown within each groups (small dots). However, a cluster cannot be generated for the Af homolog (yellowgreen dots) due to its low homology to other proteins. (B) Alignment of conserved sequence motifs. The conserved Asp residues are colored in red, and other conserved residues in blue.
4.1 Selecting Representative Proteins of Each Major Subfamily
The following proteins are selected, covering most of the subfamilies of intramembrane prenyltransferases that have been identified to date (Section 3). UbiA (Melzer and Heide, 1994; Young et al., 1972) and MenA (Suvarna et al., 1998) are from Escherichia coli. COQ2 (Ashby et al., 1992; Forsgren et al., 2004), UBIAD1 (Nakagawa et al., 2010), and COX10 (Kim et al., 2012) are from Homo sapiens. Plant homologs include chlorophyll synthases (Rüdiger et al., 2005) from Arabidopsis thaliana and HGGT (Schledz et al., 2001) from Hordeum vulgare. Microbial enzymes include DPPR synthase (Huang et al., 2005) from Mycobacterium tuberculosis and DGGGP synthase (Zhang and Poulter, 1993) from Methanocaldococcus jannaschii. Sequence clustering is performed between these representative proteins and the unknown proteins to be identified for function. As examples for this functional prediction, we chose two archaeal homologs from Aeropyrum pernix and Archaeoglobus fulgidus for which crystal structures have been determined (Cheng and Li, 2014; Huang et al., 2014).
4.2 Sequence Clustering Analysis
All the following steps are conducted using the Bioinformatics Toolkit (http://toolkit.tuebingen.mpg.de/). This platform contains multiple tools, including the CLANS program (Biegert et al., 2006), that are required for clustering analysis. Use of this single platform for all steps avoids the problem of data format conversion.
Create an account in Bioinformatics Toolkit, which allows the saving of performed tasks for a period of time. The following steps will take a few hours to finish and some jobs often require rerunning; therefore, it is crucial to save the successful jobs.
Perform a PSI-BLAST search for each representative sequence (Section 4.1) to generate a number of similar sequences. Click the Search tab on top of the webpage, select for PSI-BLAST search, and input a target sequence. Under Databases, select nr70, which stands for the NCBI protein database of non-redundant proteins filtered for 70% maximum sequence identity. This relatively low level of identity is used to cover a considerable range of sequence variations. Under Search Options, input in Descriptions the number of sequences to be generated. For the 11 representative proteins being analyzed here (Section 4.1), generate 150 sequences from each PSI-BLAST search; this is to keep the total number of sequences below 2000, which is the limit of the online CLANS application (step 4). Repeat this search for each representative sequence by choosing the existing PSI-BLAST job and clicking on Submit with Same Parameters.
Click on the job number and select Alignment to view the aligned sequences. Copy the sequences from each PSI-BLAST job, and combine into a single text file; it is important to use a plain-text editor such as Notepad or Vi (instead of Word).
Input the combined sequences into the CLANS program under the Classification tab. To remain consistent with step 2, select the same options (PSI-BLAST and nr70).
Select the job number in CLANS when finished. Click on View in CLANS and Continue, which will create a new job that generates the CLANS file (filename.clans). Download this file, and also download and install the CLANS application named clans.jar (a JAVA program), following the link (ftp://ftp.tuebingen.mpg.de/pub/protevo/CLANS/) on the webpage.
Start the clans.jar application, and click File and Load Runs to load the filename.clans file obtained from step 5. Many small dots appear in the application window; each dot represents a homolog sequence. Press Initiate to randomize the location of sequence dots, and then Start Run. Homologs of significant similarity (e.g., all UbiA proteins) should start to form clusters of sequence dots. Click Stop when these clusters have converged to a relatively stable position. Click Show Connections to draw lines between dots. Each of these gray lines shows the PSI-BLAST [95] comparison of two sequences, with darker lines indicating higher similarity (lower E values). Click Show selected for a pop-up window, select similar homologs, and click OK. Choose from the menu Windows and Edit Groups to color these homologs in groups. Refer to the CLANS manual for more information.
5. Analysis of Enzymatic Activity
Several factors complicate the activity analysis of intramembrane enzymes. For example, some of these intramembrane enzymes can be active only in the lipid bilayer. For intramembrane prenyltransferases, XPPs with long aliphatic chains are less soluble, which can be problematic when supplied to an in vitro reaction system (Melzer and Heide, 1994). It is unclear whether these substrates can efficiently diffuse across the membrane-water interface or partition into detergent micelles to reach the prenyltransferases.
For UbiA and COQ2, in vitro activity assays generally use the enzymes in crude microsomal membranes, which provide a lipid bilayer environment. The activity assays often use GPP (C10), a short and artificial substrate, due to its high solubility and high yield of enzymatic product. Long substrates are less investigated, as they are difficult to obtain by chemical synthesis and are often not commercially available. Nevertheless, SPP (C45) dissolved in dimethyl sulfoxide can be used by microsomal UbiA, although the catalysis is inefficient (Vmax ~50X lower than GPP) (Melzer and Heide, 1994). For the MenA enzymes, activity analyses have been reported either using microsomes (Huang et al., 2014) or proteins purified in detergents (Li et al., 2014).
The protocol here describes an in vitro assay of E. coli UbiA activity in microsomes and a complementation assay using a quinone-deficient E. coli strain. Because the endogenous UbiA and MenA activities of E. coli interfere with the enzymatic assay, we constructed a BL21(DE3) strain with knockout of the mena gene and conditional knockout of the ubia gene. This ubia−/mena− strain maintains the BL21(DE3) overexpression system, which can be used to produce constantly high levels of wild-type or mutant proteins for activity analysis. With the engineered T7 system in BL21(DE3), pET vectors (Agilent) are used to express proteins for use in both structure determination (Section 6) and biochemical analysis. Therefore, consistency is maintained between these studies, and subcloning to different expression vectors is avoided.
For activity analysis, the ubia−/mena− strain is used to prepare microsomes for an in vitro assay free of endogenous UbiA or MenA activity. Moreover, the activity of wild-type and mutant UbiA or MenA can be conveniently evaluated by their ability to complement this quinone-deficient strain, which is otherwise prohibited from normal growth.
5.1. Generation of a ubia−/mena− Bl21(DE3) Strain
Quinones are essential electron carriers in the respiration chain of all living organisms. E. coli is a facultative anaerobic bacterium that produces both ubiquinone and menaquinone. The relative levels of these two types of quinones change with different growth conditions of E. coli. Of the total quinones generated by E. coli, under aerobic conditions, 65% is ubiquinone and 35% is menaquinone; under anaerobic conditions, this changes to 7% ubiquinone and 93% menaquinone (Wallace and Young, 1977). The key enzymes in the biosynthesis of ubiquinone and menaquinone are, respectively, UbiA and MenA; both of these genes need to be disrupted in order to make a quinone-deficient strain that can be used for the complementation assay.
We conduct the gene knockout under aerobic conditions. Since the menA gene is non-essential for the aerobic growth of E. coli, we can simply delete menA in BL21(DE3) by replacing the wild-type menA with a menA::kan allele from a JW3901 strain (Baba et al., 2006). The introduced kanamycin resistance is used as the selection marker for the knockout strain and is subsequently removed from the chromosome. Removal of the kanamycin-resistant (KanR) gene generated from the menA knockout is necessary because subsequent transduction of the deficient UbiA also needs KanR as a selection marker.
The ubiA conditional knockout is made by introducing a ΔmalF::kan…ubiA420 allele into the BL21(DE3) menA− strain. The ubiA420 allele carries a point mutation in ubiA, which was originally isolated from a quinone-deficient strain, AN384 (Wallace and Young, 1977). The mutant has a negligible UbiA activity that cannot be detected by either the complementation or microsomal activity assays (Sections 5.2 and 5.3). However, when supplied with 1 mM PHB, this mutant strain can make ~20% amount of the ubiquinone as the wild-type strain; this quinone level is sufficient for the growth of E. coli cells. Therefore, the transductants carrying ΔmalF::kan…ubiA420 are selected, as they can grow only with 1 mM PHB. The protocol of generating the ubia−/mena− Bl21(DE3) strain is as follows.
-
Strains and phage.
BL21(DE3) and Phage P1 vir are from the lab collection. NK319 (ΔmalF::kan…ubiA420) is a gift from Dr. Jon Beckwith. JW3901 with ΔmenA789::kan is from the Keio collection (Baba et al., 2006).
- Preparation of phage P1 lysate from donor strains.
- Grow the donor strain JW3901 in 5 mL rich media (dissolve 10 g Tryptone, 5 g yeast extract, and 5 g NaCl in 1 L H2O, adjust to pH 7.2, and autoclave). Grow the donor strain NK319 at 37°C in 5 mL rich media with 1 mM PHB to late log phage (OD600 = 0.8).
- Add 250 μL of 0.5 M CaCl2 to the 5 mL cultures. Continue to grow the cells for 10 min. Mix 1 mL aliquot of the culture with 100 μL serial dilutions of P1 lysate stock (100, 10−1, 10−2 and 10−3). Incubate at 37°C for 20 min without shaking.
- During the incubation, warm up agar plates prepared with rich media (rich plates) in a 37°C incubator.
- Add 20 μL 20% glucose, 20 μL 0.5 M CaCl2, and 2 mL melted rich top agar (0.7% agar in rich media) to the mixture of cells and phages. Vortex 10 s and pour onto pre-warmed rich plates. Allow the soft agar to solidify. Incubate the plate at 37°C for at least 8 h.
- Choose the plate with evenly distributed individual plaques. Add 2 mL rich media to the plate and scrape off the top agar. Transfer top agar and liquid media into a 50 mL centrifuge tube. Centrifuge the tube at 10,000 rpm for 10 min to remove debris. Collect the supernatant. Add 20 μL of chloroform and vortex for 10 s. Store the P1 lysates from JW3901 and NK319 at 4°C.
- P1 vir transduction.
- Grow the recipient strain BL21(DE3) at 37°C overnight in 5 mL rich media.
- Spin down 0.5 mL of the culture, discard the supernatant and resuspend the pellet into 0.5 mL MC solution (100 mM CaCl2 and 10 mM MgCl2).
- Mix 100 μL of cells with 100 μL of serial dilutions (100, 10−1 and 10−2) of P1 lysate made from JW3901. Incubate at 37°C for 20 min without shaking.
- Add 200 μL 1 M sodium citrate and 0.5 mL rich media. Shake the tube at 37°C for 1 h.
- Spin the cells and phage mixture at 13,000 rpm for 5 min on a tabletop centrifuge. Discard the supernatant and resuspend the pellet in 0.2 mL 1 M sodium citrate.
- Plate on a rich plate with 30 mg/L kanamycin to select for colonies that carry the menA::kan allele. Incubate the plate overnight at 37°C.
- Pick 3–4 colonies from the kanamycin plate and streak onto a new kanamycin plate. Grow at 37°C overnight. Repeat the colony streaking at least twice to purify the colonies.
- Confirm the P1 transduction. PCR-amplify individual colonies from the kanamycin plate with the menA forward primer AAAGGCCCCATTTTTATTGG and reverse primer TTTGTCAGTTATGCTGCCCA. The colony that has the incorporated menA::kan allele gives a 1,300 bp PCR product. For the PCR analysis, include colonies from JW3901 and BL21(DE3) as positive and negative controls, respectively. The BL21(DE3) strain carrying the wild type menA gives a 970 bp PCR product.
-
Excision of the kanamycin resistance cassette.
In the menA::kan allele, the KanR gene is flanked by a short direct repeats (FRT sites). This KanR gene can be excised in vivo from the chromosome by a yeast Flp recombinase. A temperature-sensitive plasmid pCP20 (Cherepanov and Wackernagel, 1995), which expresses Flp, is transformed into the transductant strains to release the KanR gene.- Prepare chemical competent cells of BL21(DE3) menA− following a standard protocol.
- Mix 1 μL pCP20 plasmid DNA with 50 μL competent cells and incubate on ice for 30 min. Heat-shock the competent cells by incubating the tube at 42°C for 30 s. Move the tube to ice and let it sit for 5 min. Add 1 mL SOC media to the tube and recover the cells by shaking at 30°C for 1 h. Plate 100 μL cells onto rich plate with 100 mg/L ampicillin. Incubate the plate at 30°C overnight.
- Pick 3–4 single colonies, restreak them on rich plate, and incubate the plates at 42°C.
- Pick one colony from each of the restreaked colonies, and patch them on rich plate, ampicillin plate, and kanamycin plate. Incubate the patches at 30°C overnight. Those that grow only on the rich plate, but not on the ampicillin or kanamycin plate, have lost the pCP20 plasmid (ampicillin resistance) and have the kanR flipped out.
- Confirm the removal of KanR by colony PCR with menA forward and reverse primers. Colonies carrying KanR generate a 1,300 bp PCR product, and those lost KanR give a 180 bp PCR product.
- P1 transduction of ΔmalF::kan…ubiA420 to BL21(DE3) menA−
- Perform P1 transduction of ΔmalF::kan…ubiA420 to BL21(DE3) menA−, using the same protocol as described in step 3. The ΔmalF::kan is used because malF and ubiA genes are located only 9,000 bp apart on the E. coli chromosome, and therefore the transduction of ΔmalF::kan and ubiA420 are tightly linked.
- To select for the transductants, plate the cells on kanamycin rich plate supplied with 1 mM PHB and incubate the plate at 37°C overnight. Pick 8–10 colonies and patch them on kanamycin rich plates supplied with and without 1 mM PHB. The colonies that cannot grow without 1 mM PHB carry the ΔmalF::kan…ubiA420 allele.
5.2. Complementation Assay of UbiA Activity with the ubia−mena− BL21(DE3) Strain
Prepare calcium competent cells using a standard protocol, but with the addition of PHB. Inoculate a single colony of the BL21(DE3) ubia−/mena− strain in 5 mL LB with 1mM PHB (Sigma). Grow overnight at 37°C. Take 1 mL aliquot to inoculate 100 mL of the same media and grow until the OD600 reaches 0.2–0.4. Leave cells on ice for 10 min. Collect cells by centrifugation at 4°C and resuspend the cell pellet in 12.5 mL ice-cold 0.1 M MgCl2. Centrifuge again, resuspend cells in 5 mL ice-cold 0.1 M CaCl2, and leave on ice for 20 min. Centrifuge and resuspend cells on ice in 4 mL 0.1 M CaCl2, 10% glycerol, 1 mM PHB. Flash-freeze in aliquots.
Prepare LB ampicillin plates with 1mM PHB (PHB+ plates) and without PHB (PHB− plates).
Transform wild-type and mutant UbiA in pET15b into the calcium competent BL21(DE3) ubia−/mena− cells. Split the transformed cells in half and spread on PHB− and PHB+ plates for overnight growth.
Wild-type E. coli UbiA is able to complement the growth of double-knockout cells on PHB− plates. In contrast, inactive mutants of E. coli UbiA, such as Arg60Ala and Asn67Ala, can grow only on PHB+ plates. This complementation assay is a stringent test of activity; mutants that cannot rescue the growth on plates may still have a low activity, which can be detected by the in vitro microsomal assay described below.
5.3. Assay of Enzymatic Activity in Microsomes
Determination of the UbiA prenyltransferase activity has been described before (Bräuer et al., 2008; Melzer and Heide, 1994). For each mutant or wild-type E. coli UbiA, a 100μl reaction is performed. We generally start with 1 mM GPP (Echelon Biosciences) and 1 mM PHB, which is at a high concentration to ensure that, for mutants of low activity, the reaction product is detectable by monitoring the UV absorption on HPLC (Section 5.3.3). For the analysis of enzymatic kinetics, different concentrations of GPP and PHB need to be tested. The typical concentration range of these substrates is 0.01–10 mM for E. coli UbiA.
5.3.1 Preparing Microsomes from the Bl21(DE3) Ubia−Mena− Strain
Transform wild-type or mutant UbiA into the double-knockout cell. Grow the cells on PHB+ plates overnight.
Transfer all colonies on the plate to 1 L LB media supplemented with 1 mM PHB. Grow the cells at 37°C in a 2 L flask, shaking at 225 rpm. When OD600 reaches 0.4–0.6, induce protein expression with 0.4–1 mM IPTG for 3 h.
Perform all subsequent steps at 4°C or on ice. Collect cells by centrifugation at 4,000 rpm in a JS 4.0 rotor (Beckman).
Resuspend cells in 20 mL of 50 mM Tris-HCl, pH 8.0, 0.1 M NaCl. Lyse cells immediately by sonication in presence of the protease inhibitors, 1mM phenylmethylsulfonyl fluoride (PMSF) and 2 mM benzamidine hydrochloride.
Centrifuge at 5,000 rpm for 10 min to remove unbroken cells.
Collect membrane fraction by ultracentrifugation at 50,000 rpm for 15 min in a TLA-100 rotor.
Resuspend the membrane fraction in 50mM Tris-HCl, pH 8.0, and 10 mM dithiothreitol (DTT). Centrifuge again to collect the membrane. Repeat the wash by resuspend the membrane in the same buffer and centrifuge.
Use a small glass douncer to thoroughly resuspend the pellet in 4 mL of the same buffer. Flash-freeze aliquots with liquid nitrogen. The concentration of wild-type and mutant UbiA proteins in the microsome resuspension can be matched on western blot, then quantified by comparing with dilutions of purified protein (Section 6.3) using an anti-His antibody.
5.3.2. Reaction Setup
- Prepare the following mixture for a 100 μl reaction. Multiply the volumes by the number of samples to be analyzed.
- 50 μl 100 mM Tris 7.5
- 10 μl DMSO
- 0.5 μl 1 M DTT
- 0.4 μl 1 M MgCl2
- 1 μl 100 mM GPP
- 1 μl 100 mM PHB
Mix a 60 μl aliquot of this reaction mixture with 40 μl microsome, prepared as described in Section 5.3.1. Incubate for 2 h at 37°C with nutation. At the end of the reaction, add 1 μl 25 mM 4-phenylphenol as internal control for the subsequent solvent extraction and HPLC analysis.
Immediately mix the reaction mixture with 300 μl ethyl acetate. Vortex at top speed for 1 min to extract the product, 3-geranyl-4-hydroxybenzoate. Carefully take 200 μl of the top layer and transfer to a new tube. Dry the extraction by centrifuge under vacuum or by opening the tube to air overnight.
5.3.3. HPLC Analysis of Enzymatic Activity
Dissolve the dried product in 100 μl methanol and pass through a 0.1 μm centrifuge filter.
Transfer the filtered sample to an HPLC vial. Load the samples on an HPLC (Hewlett-Packard) equipped with an autosampler and a mass spectrometer (Agilent 1100).
Inject 10 μl sample on the HPLC with a C18 reverse-phase column (Proto 300 C18 5 μm, 250 × 4.6 mm) pre-equilibrated in 5% acetonitrile. Run a gradient of 5–95% acetonitrile in 10 min at 1 mL/min and hold for another 2 min at 95% acetonitrile. The geranylated PHB elutes at ~5.16 min and the internal control 4-phenylphenol elutes at ~4.70 min (Fig. 3A). Quantify the product by peak areas on HPLC and normalize with the internal control.
Confirm the reaction product by mass spectrometry (Fig. 3B). The geranylated PHB has an m/z of 275.3.
Analyze the enzyme kinetics from multiple samples using the Prism6 software (GraphPad).
Figure 3.
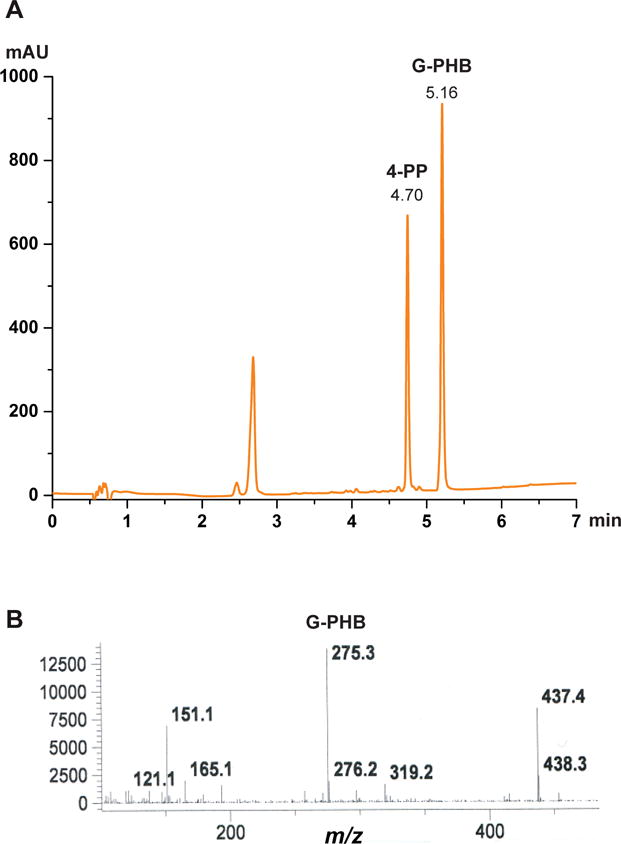
Activity analysis of wild-type EcUbiA prepared in microsomes. (A) HPLC profile (254 nm) shows the product, 3-geranyl-4-hydroxybenzoate (G-PHB), and the internal control, 4-phenylphenol (4-PP). (B) Confirmation of the G-PHB peak (from A) by mass spectrometry.
6. Structural Studies of the UbiA Superfamily
6.1 Cloning, Expression, and Selection of Protein Constructs
Most of the homologs that we have tested in this superfamily are of microbial origin, for which heterogeneous expression in E. coli is the primary choice. The genes of these homologs were either PCR amplified from the genomic DNA, if available from ATCC, or obtained from gene synthesis services. The genes were subcloned into either the PET15b or the PET20b vector, which adds either an N- or C-terminal His tag to the protein, respectively.
We screen the expression levels of recombinant homolog proteins for the combination of N- or C-His tags, at 37°C or 22°C, and in three E. coli strains, BL21(DE3), C41(DE3) and C43(DE3). The C41(DE3) and C43(DE3) strains carry mutations on the T7 RNA polymerase (Miroux and Walker, 1996) that hinder the protein expression; slowly expressed proteins tend to fold better in membranes, presumably because the folding machinery in E. coli cells is less overwhelmed. To slow down the protein expression, we also perform expression tests at a lower temperature, i.e., 22°C (sometimes 16°C). Western blot towards the His affinity tag is used as a quick readout for the protein expression levels.
Transform different homologs with N- or C- His tags into BL21(DE3), C41(DE3) and C43(DE3). Grow each of these constructs on an LB ampicillin plate at 37°C overnight.
Inoculate a single colony from each plate to 5 mL LB ampicillin media and grow at 37°C. Add 1 mM IPTG at OD600 0.6–0.8. Immediately split 2.5 mL culture to another tube and induce at 22°C overnight. Keep the rest of the 2.5 mL culture at 37°C and induce for 3–6 h.
Centrifuge to collect 50–100 μl of cells at OD600 = 1 or an equivalent volume. Match the volume of cell samples by their cell density (OD600) to ensure that the same number of cells are compared for expression levels.
Resuspend the cell pellet with 50 μl 2X SDS-PAGE sample buffer. Apply to a 12% SDS-PAGE with 1.5 mm thick wells. Heating the cells at 95°C in sample buffer gives a less viscous solution, but may change the gel mobility of some membrane proteins. Therefore we generally skip the heating step (or heat at 60°C in some cases), but use a small amount of cells to lower the viscosity.
Detect the expression level of His-tagged protein by western blot. Transfer protein bands on the SDS-PAGE gel to a nitrocellulose membrane at 100 V for 1 h using a wet-blotting system (BIORAD). Block the membrane at 22°C for 1 h in 10 mL TBS-T (50 mM Tris pH 7.4, 150 mM NaCl, and 0.1% v/v Tween 20) with 5% milk. Incubate the membrane at 22°C for 1 h with 5 μL mouse anti-His antibody (Genscript) in 10 mL TBS-T. Wash the membrane three times with 10 mL TBS-T for 10 min each time. Incubate the membrane at 22°C for 1 h with 2 μL of goat-anti-mouse IgG peroxidase (Sigma) in 10 mL of TBS-T. Wash the membrane three times with 10 mL of fresh TBS-T for 10 min.
-
Immediately add ECL western blotting substrate (Pierce). Develop the film in a short exposure time (10–30 s). A strong band on the film indicates a well-expressed protein (Fig. 4A). Constructs showing weak bands are difficult to pursue further because the protein usually cannot be purified to crystallographic quality in the end; the purification protocol described here relies primarily on an effective His-affinity chromatography step (Section 6.3).
This first round of western blot analysis significantly narrows down the protein targets: only those that are sufficiently expressed are pursued further (Fig. 4A). However, this preliminary test cannot exclude misfolded and aggregated proteins, such as those in the inclusion body. To overcome this problem, we take the assumption (Sonoda et al., 2010) that proteins that are properly folded in membranes can be efficiently extracted by n-Dodecyl-β-D-Maltopyranoside (DDM), the most commonly used detergent in membrane protein crystallography.
Freshly transform selected constructs with high level of protein expression. Grow and induce cells with the best conditions identified above.
Resuspend and sonicate the same amount of cells (matched by OD600) from ~10 ml culture in 500 μl of 20 mM Tris-HCl 7.5, 0.1 M NaCl. Add powder of DDM to 1% final concentration, and incubate for 1 h with nutation for membrane protein extraction. For comparison, save an aliquot of cells without DDM extraction.
Centrifuge and run the supernatant from DDM extraction on western blot. Run the unextracted cells side-by-side. The protein constructs that can be effectively solubilized by DDM are pursued further.
Figure 4.
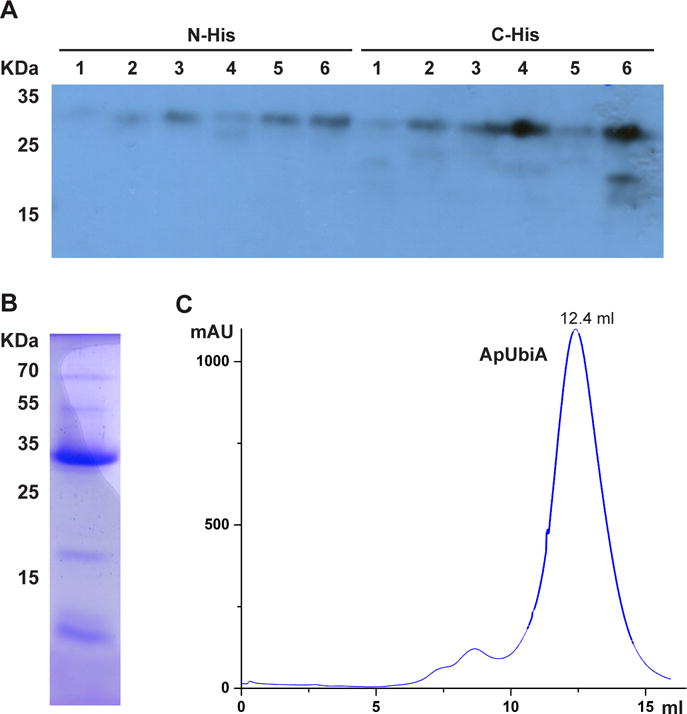
Selection of protein constructs for crystallographic studies. (A) Western blot of six UbiA homologs with N- or C-His tags. As examples, homolog 1 is not worth pursuing further, whereas homolog 4 expresses well only with a C-His tag. (B) Test purification of a well-expressed homolog from a 2 L culture. The sample is from the elution of Ni chromatography. (C) Profile of ApUbiA on size-exclusion chromatography shows a sharp and symmetrical peak.
For each selected protein, a medium scale of purification is conducted from a 2 L culture. We generally purify 6 proteins (i.e., a total of 12 L culture) at the same time, following a protocol described in Section 6.3. The proteins purified after Ni chromatography can be directly compared on SDS-PAGE for amount and purity. Proteins to be pursued further should be relatively pure at this stage (Fig. 4B), and the amount purified from the 2L culture is generally sufficient for the subsequent analysis of the elution profile by size exclusion chromatography. Protein constructs that give sharp and symmetrical peaks at the proper elution volume (Fig. 4C) are selected for large-scale purification and crystallization trials (Sections 6.2–6.4).
An alternative approach, which is more efficient than the western blot method described here, is to clone proteins with GFP tags and use fluorescence size exclusion chromatography (FSEC) to screen for the expression level and elution profile at the same time (Kawate and Gouaux, 2006). The FSEC method requires cloning into the vectors with the GFP sequence engineered, in addition to cloning into the PET vectors. Conversely, if the GFP vectors are directly used for expression and purification, cleavage of the GFP tag is normally required at a later step. In some cases, although the GFP-tagged protein gives a sharp and symmetrical peak at the initial FSEC test, removing the GFP tag by protease digestion may destabilize the target protein and lead to aggregation. Although our lab routinely uses FSEC for membrane proteins expressed in eukaryotic systems, for E. Coli expression, our experience is that the western blot method is as effective as FSEC in selecting the constructs suitable for crystallographic studies.
After screening large numbers of UbiA and MenA homologs, we identified an archaeal UbiA homolog from Aeropyrum pernix (ApUbiA) that is well expressed and shows a good profile on size exclusion chromatography (Fig. 4C). Interestingly, one of the few homologs that behave well in our hands, the homolog from Archaeoglobus fulgidus (Af), was also independently identified by another group (Huang et al., 2014); this homolog was initially found during a structural genomics search, and also succeeded in structure determination. This coincidence suggests that there are only a small number of protein homologs in this large superfamily that behave well enough to be readily pursued for crystallographic studies. Other protein homologs in this family remain possible targets for structural determination, but to be successful, these may require more optimization efforts to find better expression conditions, suitable detergents, and proper protein constructs, such as truncations that remove flexible or degradable regions.
The ApUbiA homolog is best expressed in C43(DE3) cells at 22°C. In addition, minimal media gives a significantly higher level of protein expression compared to LB media. This growth condition was found during the process of crystal structure determination, in which the minimal media is routinely used for selenomethonine (SeMet) labeling to obtain experimental phasing. The improvement of protein expression in SeMet media has also been occasionally observed for other membrane proteins (Zimmer et al., 2008). Therefore, SeMet labeling should probably be always tested for new membrane proteins under investigation, with the improvement of expression level as a bonus to obtaining experimental phasing. The following protocol describes the expression and purification of the ApUbiA protein with SeMet labeling, which can serve as a model for producing other microbial proteins in the UbiA superfamily for crystallographic studies.
6.2 Expression of the ApUbiA Protein in a Minimal Media Used for SeMet Labeling
The following steps are modified from a published protocol (Doublié, 2007). Six amino acids (Lys, Thr, Phe, Leu, Ile, and Val) are added before induction to inhibit Met biosynthesis, which in turn allows efficient SeMet incorporation.
- Prepare the following.
- 1.5 L of 10X M9 medium. Dissolve 132.9 g Na2HPO4•7H2O, 58.5 g KH2PO4, 9.75 g NaCl, and 19.5 g NH4Cl in 1.5 L H2O, and transfer to a 2 L flask.
- For 1 L adaptive media (below), add 50 g glucose and 5 g LB to 920 mL H2O in a 2 L flask.
- For 12X 1 L expression media (below), add 800 mL H2O and 50 g glucose to each 2 L flask.
- Autoclave media 1) to 3), along with empty centrifuge bottles and empty cylinders (used below).
- Dissolve 65g Yeast Nitrogen Base (YNB) in 1 L H2O and sterile filter (10X 6.5% YNB stock solution).
- Make 200 mL of 10 M NaOH.
- After the media is cooled down, use the sterilized cylinder to
- add 80 mL of 10X M9 medium to make 1L adaptive media;
- add 100 mL of 10X M9 medium and 100 mL of 10X YNB to make each 1L expression media.
- Grow 12 L cells to express the ApUbiA protein with SeMet labeling.
- Transform 1 μl plasmid DNA, pET15b-ApUbiA, to 30 μl calcium-competent C43(DE3) cells. Grow on LB ampicillin plate overnight.
- Scrape all colonies on the plate into the 1 L adaptive media. Grow at 37°C until OD600 reaches 0.6–0.8.
- Collect cells by centrifuge at 4,000 rpm at 4°C for 10 min. Resuspend cells in 240 mL of expression media. Add 20 mL of cells to each 1L of expression media in the 12 flasks. Grow at 37°C until OD600 reaches 1.0.
- Add 4.5 mL 10 M NaOH dropwise to each 1L of expression media. Change temperature to between 16°C and 22°C. After 20 min, add seven amino acids: Lys, Thr, and Phe at 100 mg/L; Leu, Ile, and Val at 50 mg/L; and SeMet at 60 mg/L. Resuspend the amino acids by stirring in 120 mL H2O first, and pipet 10 mL of the suspension to each 1L expression media.
- After 30 min, add 0.4–1 mM IPTG to induce protein expression. Shake at 180 rpm for 13–17 h.
- Centrifuge at 4,000 rpm at 4°C for 20 min to collect the cells. The cells can be frozen by liquid nitrogen and stored at −80°C for later use.
6.3 Protein Purification
Perform all subsequent steps in this section at 4°C or on ice. Thaw and resuspend the cells from 12 L culture (Section 6.2) in 120 mL buffer A containing 20 mM Tris-HCl, pH 8.0, 0.3 M NaCl, and 10% glycerol. Use a food processor to thoroughly resuspend the cells.
Lyse the cells by passing twice through a microfluidizer (Microfluidics M-110L) with the pressure set at 15,000 psi. After the first pass, add 1.2 mL of 100X stock solution of protease inhibitors, which is made by dissolving 0.1 M PMSF and 0.2 M benzamidine in ethanol.
Centrifuge the lysed cells at 5,000 g for 10 min to remove unbroken cells.
Pour the supernatant to 2 of 70 mL ultracentrifuge tubes, and collect the cell membrane by ultracentrifugation at 42,000 rpm in a Ti45 rotor (Beckman) for 1 h.
Remove supernatant and use a 55 mL glass douncer (attached to an electrical overhead stirrer) to homogenize the membrane in a final volume of 60 mL buffer A with 1% (w/v) DDM (Sol-Grade from Anatrace). Stir the mixture for 1 h to thoroughly solubilize the membrane in DDM.
Ultracentrifuge at 42,000 rpm for 30 min in a Ti45 rotor.
Prepare a 10 mL suspension of Ni-NTA resin (Qiagen) in a 50 mL gravity-flow column. Wash the resin with 100 mL water in steps to remove ethanol. To equilibrate the resin, wash with 100 mL Buffer A in 10 mL steps, and then 10 mL Buffer A with 0.03% DDM. Resuspend the resin by 5 mL Buffer A with 0.03% DDM.
Add the resin to the supernatant from Step 6. Stir the Ni-NTA resin with the DDM solubilized protein for 1 h for batch binding.
Collect the resin back in the 50 mL column. Carry out all subsequent washing and elution in 5–10 mL additions at a time. Thoroughly wash the resin in the following steps: 300 mL Buffer A with 0.03% DDM; 300 mL Buffer A with 0.03% DDM and 20 mM imidazole; 600 mL Buffer A with 0.06% 6-Cyclohexyl-1-Pentyl-β-D-Maltoside (CYMAL6) and 40 mM imidazole. Elute the protein in 40 mL Buffer A with 0.06% CYMAL6 and 0.3 M imidazole.
Run an SDS-PAGE gel of aliquots from each of the wash and elution steps. The samples are mixed with SDS-PAGE sample buffer without heating. Proceed only if a significant amount of protein is purified in the elution fraction.
Concentrate the eluted protein to 0.5 mL with a 50 KDa cutoff Amicon Ultra (Millipore) centrifuged at 2,000–4,000 rpm. Frequently check and mix the sample to avoid over-concentrating. Centrifuge in a 0.01 μm Ultrafree filter unit (Millipore) to clear up any precipitation in the sample.
Immediately apply the filtered sample on a Superdex 200 size exclusion column (GE Healthcare) that has been pre-equilibrated in a buffer containing 10 mM Tris-HCl, pH 8.0, 0.1 M NaCl, and 0.25% 5-Cyclohexyl-1-Pentyl-β-D-Maltoside (CYMAL5). Collect fractions and run SDS-PAGE gel.
Concentrate the protein to 17.5 mg/mL. Determine the protein concentration by OD280 using a Nanodrop spectrophotometer.
6.4 Selection of detergents
Careful detergent selection is essential for work with membrane proteins, because their structural stability strongly depends on the detergent being used. Moreover, membrane protein crystals are formed both by the protein and by associated detergent micelles; therefore, the micelle size is an important factor in improving crystal diffraction.
The choice of detergents is essential in crystallizing the ApUbiA protein and improving crystal diffraction. This protein can be crystallized in DDM and in other detergents that have a maltose head group and a similar-length aliphatic chain. Compared to crystallization in DDM, crystallization in CYMAL5 improved the diffraction from ~8 Å to 3.3 Å, which allowed the building of a reliable structural model. In addition, ApUbiA that is purified in the lauryldimethylamine-N-Oxide (LDAO) detergent, which has a relatively small micelle, can be crystallized in a different crystal form for which the diffraction spots beyond 3 Å can be observed. However, the LDAO crystal form is severely twinned and was not able to provide any more structural information than the 3.3 Å model of the CYMAL5 crystals.
The initial screen of the detergents is usually conducted at the gel filtration step. The ApUbiA protein is purified in DDM until its elution from the Ni-NTA column, with the assumption that DDM is a mild detergent that maintains stability for most of the membrane proteins (Sonoda et al., 2010). The protein is concentrated in the buffer with DDM (Section 6.3), and applied to the Superdex-200 column that has been pre-equilibrated with the buffer containing various detergents. The detergents with the maltose head group and varying aliphatic groups, such as n-Decyl-β-D-Maltoside (DM) and CYMALs, are generally tested first. In addition, we chose detergents that have been reported to be most successful in crystallization. As nicely summarized by previous reviews (Newstead et al., 2007; Sonoda et al., 2011, 2010), these detergents include the glucosides, octyl- (OG) and nonyl-glucopyranoside (NG); Octaethylene glycol monododecyl ether (C12E8); LDAO; and more recently, detergents with two maltose head groups, such as lauryl maltose neopentyl glycol (LMNG).
If a detergent other than DDM gives a sharp and symmetrical peak at the gel filtration step, this detergent can be further tested for use at an earlier step in purification, such as the initial solubilization step or the wash and elution step after the protein has bound to the Ni-NTA resin. For ApUbiA, we found that the best protocol is to solubilize the protein in DDM, wash and elute with CYMAL6 on Ni-resin, and run gel filtration in CYMAL5 (Section 6.5).
6.5 Crystallization and Structure Determination
Structures of two archaeal homologs have been determined in the UbiA superfamily. The ApUbiA homolog was crystallized in detergent and the apo crystal diffracted to 3.3Å (Cheng and Li, 2014). A 3.6 Å structure was determined with geranyl thiolodiphosphate (GSPP); this noncleavable substrate was used because UbiA was previously reported to hydrolyze GPP even in the absence of PHB (Bräuer et al., 2008). The crystals were also soaked with PHB and Mg, both of which were modeled into the structure at this moderate resolution. The Af homolog was crystallized in liquid cubic phase (LCP) and the structures with GPP or dimethylallyldiphosphate (DMAPP; C5) were determined to be 2.4–2.5 Å (Huang et al., 2014). The positions of Mg2+ ions in this structure were confirmed by anomalous signals from Cd, a heavier divalent atom in substitution for Mg.
Crystals grown in LCP generally diffract better than those grown in detergents because the protein molecules are more closely packed with interactions by membrane domains. Besides the Af homolog, we have recently obtained LCP crystals of other proteins in the superfamily (unpublished data). As crystallization in detergent is a more straightforward method, the protocols described below put more emphasis on the LCP method.
6.5.1 Crystallization in Detergents
Initial crystallization screens of ApUbiA are performed with chemical solutions from commercial screening kits (Hampton Research and Qiagen), with the aid of a Mosquito crystallization robot. Crystallization conditions are optimized for buffer conditions, protein concentrations, and additives. The protein in LDAO crystallizes at a concentration of 13 mg/mL with an initial 1:1 mix of the protein to 0.12 M Na2SO4, 20.5% (w/v) PEG400, and 0.1 M Tris-HCl, at pH 8.0. The crystals are grown by the hanging drop vapor diffusion method at 22°C, and these hexagonal plates reach final size in 2 weeks. The protein in CYMAL5 crystallizes at 17.5 mg/mL with 0.1 M Na2SO4, 26.5% (w/v) PEG400, and 0.1 M sodium cacodylate, at pH 5.0. The crystals are rod-shaped and grow to final size in 4 weeks. The crystals are soaked in 5 mM GSPP, 10 mM MgCl2 and 10 mM PHB for 1 h. Both the native and soaked crystals are directly flash-frozen in liquid nitrogen.
6.5.2 Crystallization in LCP
The LCP method has been described before (Liu and Cherezov, 2011); the following protocol is adapted to the UbiA superfamily proteins. The LCP lipid we normally use is monoacylglycerol (MAG9.9) monoolein, and short-chain MAGs can be explored to improve crystal diffraction.
- Reconstitute the protein in LCP.
- Purify a UbiA superfamily protein in a detergent solution. Concentrate the protein to ~30 mg/mL. Avoid over-concentrating the detergent during this process. To do this, choose and combine only the peak fractions from the size exclusion chromatography and concentrate the complex of protein (usually ~30 KDa) and detergent micelle in an Amicon Ultra Centrifugal Filter with a 50 KDa cutoff.
- Warm up a 70 μl monoolein aliquot (NU CHEK) at 40°C for few minutes, until the lipid melts.
- Load one of the LCP mixing syringes (Art Robbins) with the melted monoolein. Remove the plunger from the syringe. Use a 200 μl pipette mounted with a yellow pipet tip to add the lipid from the top of the syringe. This tip fills directly in the syringe, and the lipid can be pushed slowly down with the syringe slightly tilted. After adding the monoolein, put the plunger into the syringe. If air bubbles are trapped, push the plunger down slowly and then move it up quickly a few times, which generally removes the bubbles. Record the volume of the lipid in the syringe.
- Load another syringe with the protein solution through the syringe tip. The volume of the protein is generally 2/3 of the lipid volume. Take caution to avoid generating bubbles in the protein solution.
- Connect both syringes together through a syringe coupler.
- Push the two syringe plungers back and forth slowly to mix the lipid and protein through the needle part of the coupler. This mixing usually takes more than 100 times. The protein and lipid mixture is initially a white color, and it becomes transparent after thorough mixing. This indicates that a homogeneous lipid mesophase has been obtained through mechanical mixing, and that the protein has been reconstituted in the lipid bilayer of LCP.
- Push all mixture to one syringe. Take off the other syringe and the coupler. Add on a needle and a metal ring to the syringe in use.
- Set up LCP crystallization with a Gryphon LCP robot (Art Robbins).
- Clamp the syringe in position on the dispensing arm of the robot. Place an LCP plate (Molecular Dimensions or custom-made glass plates) and a 96-well block containing the precipitant solutions on the designated decks of the robot.
- Load the 96-tip head of the robot with precipitant solutions. Commercial screening kits, such as MB Class Suite and MB Class Suite II (Qiagen) should be diluted to 70% to lower the precipitant concentration, because high precipitant concentration may disrupt the LCP mesophase (Johansson et al., 2009).
- Dispense the LCP mixture and precipitants with the robot programmed as follows: sequentially dispense 100 nL of the mesophase protein from the syringe onto the 96 wells of LCP plate; dispense 800 nL of precipitant solutions on top of the mesophase with the 96-tip head of the robot. The entire dispensing process takes about 2 min to finish.
- Remove the LCP plate from the platform of the robot and seal with a cover glass. Ensure that all 96 wells are centered and fully covered. Label and place the plate in incubator (usually at 22°C) for crystal growth.
- Remove the precipitant block from the deck of the robot, seal it tightly, and put it back into storage. Remove the syringe from the dispensing arm of the robot. Dismantle the syringe and wash the syringe, needle, and ferrule with methanol. Air-dry these parts for future use.
- Harvest LCP crystals.
- Place an LCP plate on the stage of a light microscope. Use a glass-cutting tool (Hampton Research) to draw two concentric circles on the cover glass around the well that contains the LCP crystals. Break up the glass along the circles to release the central piece of cover glass. Remove the dust of broken glass with moistened Kimwipes. Grip the cover glass with a fine tipped tweezer, tilt the glass piece away, and take it off the well.
- The mesophase bolus can be either on the base plate or the cover glass. Zoom in the microscope to get a clear view of the LCP crystals. Use a cryo-loop to take the crystals out from the freshly exposed mesophase lipids. Plunge the crystals into liquid nitrogen to freeze. To avoid crystal damage, harvest the LCP crystals as fast as possible after breaking the cover glass.
6.6 Structure Comparison of Intramembrane and Soluble Prenyltransferases
Besides intramembrane prenyltransferases, several classes of soluble enzymes catalyze either the fusion of two molecules, a prenyl donor with an acceptor, or similar fusion reactions within one molecule (Brandt et al., 2009). Central to these reactions is a resonance-stabilized carbocation intermediate that is generated after the diphosphate group of XPPs is cleaved off. Terpene synthases join this carbocation intramolecularly, often with a C–C double bond, to form a large variety of circlized molecules. Isoprenyl diphosphate synthases catalyze the elongation of linear XPPs by the intermolecular reaction of the carbocation from the allylic XPP to the unsaturated terminal of a homoallylic substrate, isopentenyl diphosphate (IPP). Protein prenyltransferases add the carbocation to the sulfur atom of a cysteine residue in a protein. Soluble aromatic prenyltransferases catalyze the carbocation reaction to aromatic compounds, with the formation of C–C, C–O, or C–N bonds. Here we will focus on the structural comparison of these enzymes to explore their substrate specificity and their different strategies of catalysis.
Two structures of intramembrane prenyltransferases have been determined (Fig. 5A). The Ap (Cheng and Li, 2014) and Af homologs (Huang et al., 2014), both of archaeal origin, contain nine transmembrane helices (TM) that arrange counterclockwise to form a U-shaped architecture. An extramembrane cap domain forms over the membrane domain and contains most of the conserved residues, including the two Asp-rich motifs. The Asp residues coordinate Mg2+ ions to engage the pyrophosphate group of XPP. These well-conserved Asp residues are essential for the activity of various enzymes in the superfamily (Bräuer et al., 2004; Cheng and Li, 2014; Huang et al., 2014, 2008; Ohara et al., 2013, 2009; Saiki et al., 1993; Stec and Li, 2012).
Figure 5.
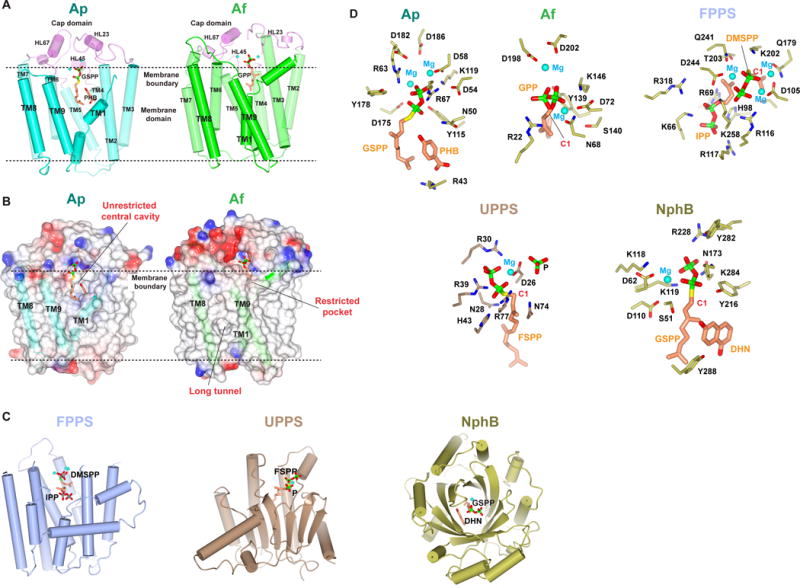
Structural comparison of intramembrane and soluble prenyltransferases. (A) Crystal structures of intramembrane prenyltransferases. Structures of the Ap (PDB: 4OD5) and Af (PDB: 4TQ3) homologs are shown in the same orientation. The cap domains are colored in pink, and TM domains of Ap and Af in cyan and green, respectively. The Mg2+ ions are shown as cyan spheres. (B) Comparison of the Ap and Af substrate-binding pockets. (C) Overall structures of soluble prenyltransferases with substrates. Trans-, cis-, and aromatic prenyltransferases are each shown with a representative structure, farnesyl pyrophosphate synthase (FPPS; PDB 1RQI), undecaprenyl pyrophosphate synthase (UPPS; PDB 1X06), and NphB (PDB 1ZB6), respectively. (D) Comparison of the active site residues and Mg positions in all these structures. The carbocation is generated on the C1 atom (red). Abbreviations: DMSPP and FSPP, uncleavable thiophosphate derivative of DMAPP and FPP, respectively; P, a phosphate in substitute of IPP in the UPPS structure; DHN, dihydroxynaphthalene, an analog of the aromatic substrate in NphB structure.
The Ap and Af structures differ significantly in the two substrate pockets that bind the prenyl donor and acceptor (Fig. 5B). ApUbiA has a large central cavity that opens laterally to the lipid bilayer, thereby creating a unique passage to the active site that may facilitate the binding of long-chain XPP substrates and the release of their prenylated products in membranes. This unrestricted binding chamber also explains the chain-length promiscuity of UbiA and COQ2; these enzymes accept a variety of IPP lengths to generate ubiquinones with 6–10 isoprenyl units, which are characteristic of different species. The central cavity contains a hydrophobic bottom wall and a small basic pocket, which have been proposed to bind the isoprenyl chain of XPP and the aromatic substrate, respectively (Cheng and Li, 2014).
In contrast, the central cavity of the Af homolog is not directly exposed to lipid. Instead, a long empty tunnel comes off the active site, with a small opening to the membrane. This long tunnel is shaped to bind a linear substrate, which is presumably the prenyl acceptor. Conversely, the prenyl donor, GPP or DMAPP, is observed to bind to another pocket in the structure. This pocket is restricted by the protein backbone (Fig. 5B) and therefore is not capable of accommodating longer XPPs.
The different substrate binding pockets in the two structures are generated by the different spacings between TM1, TM8 and TM9. The large separation between TM1 and TM9 in the Ap structure generates the open lateral portal. In contrast, TM1 and TM9 in the Af structure are close together, thereby blocking the direct opening of the Af central cavity to lipids. Because TM9 is positioned away from TM8, a larger space is generated between these helices, thereby creating the long tunnel. The variation between the two structures implies that the relative positions of these three TMs largely defines the substrate specificity, and this may be a general mechanism that is used also by other enzymes in the superfamily to determine their substrate preferences.
The 9-TM structure of intramembrane prenyltransferases is surprisingly similar to the isoprenyl synthase fold shared by trans-prenyltransferases and terpene synthases (Fig. 5C). A DALI search (Holm and Rosenström, 2010) indicates that the closest match to intramembrane prenyltransferases is the farnesyl pyrophosphate synthase (Hosfield et al., 2004; Kellogg and Poulter, 1997; Wallrapp et al., 2013), which has seven helices superimposable to the TM 1–7 in ApUbiA (Cheng and Li, 2014). Similar to enzymes in the UbiA superfamily, trans-prenyltransferases and terpene synthases also contain two Asp-rich motifs that bind two or three Mg2+ ions (Fig. 5D). In contrast, cis-prenyltransferases, although they have a similar function as the trans- type, adopt a different structural fold, using a single Asp or Glu residue to coordinate one Mg2+ ion for enzymatic activity (Fig. 5C, D).
Soluble aromatic prenyltransferases share a similar function to the intramembrane prenyltransferases, but there is no sequence or structural similarity. All soluble aromatic prenyltransferases have a PT-barrel fold made of tandem ααββ structural repeats (Fig. 5C). The α-helices surround a β-barrel, inside of which are the substrate binding pockets (Jost et al., 2010; Kuzuyama et al., 2005; Metzger et al., 2010, 2009; Saleh et al., 2009). Soluble aromatic prenyltransferases do not contain Asp-rich motifs, and the divalent metal ion is dispensable in some cases (e.g., indole prenyltransferases). Instead, an aromatic side chain in these enzymes may stabilize the carbocation by a cation–π interaction (Fig. 5D). For the prenylation of aromatic compounds, the reactive carbocation needs to be shielded from water (Bonitz et al., 2011; Metzger et al., 2009). In soluble APTs, the shielding is achieved by the β-barrel, whereas in intramembrane prenyltransferases, the TMs and surrounding lipids may play a similar role.
Overall, the prenyltransferases known to date show large variation in their structures and catalytic mechanisms. Structural and biochemical analyses of the intermembrane prenyltransferases reveal new strategies of catalysis that have been adapted to the membrane environment. The active site of these enzymes is opened to the lipid to allow ready access to long-chain substrates, as well as the efficient dissociation of prenylated products of long tails. Catalysis is permitted with this lipid opening because the reactive carbocation is protected from water by the surrounding lipids. Future studies, using the methods described in this chapter, will reveal detailed catalytic mechanisms, such as the mechanism of generating and stabilizing the carbocation, and the condensation reaction that joins the prenyl donor and acceptor together.
Acknowledgments
W. L. is supported by a R01 (HL121718) and a R00 grant (HL097083) from National Heart, Lung, and Blood Institute, a grant-in-aid (14GRNT20310017) from American Heart Association, and a scholar award from the American Society of Hematology.
References
- Ashby MN, Kutsunai SY, Ackerman S, Tzagoloff A, Edwards PA. COQ2 is a candidate for the structural gene encoding para-hydroxybenzoate:polyprenyltransferase. J Biol Chem. 1992;267:4128–36. [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale SI. Enzymes of chlorophyll biosynthesis. Photosynth Res. 1999;60:43–73. doi: 10.1023/A:1006297731456. [DOI] [Google Scholar]
- Biegert A, Mayer C, Remmert M, Söding J, Lupas AN. The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 2006;34:W335–W339. doi: 10.1093/nar/gkl217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm R, Sommer S, Severin K, Li SM, Heide L. Active expression of the ubiA gene from E. coli in tobacco: Influence of plant ER-specific signal peptides on the expression of a membrane-bound prenyltransferase in plant cells. Transgenic Res. 2000;9:477–486. doi: 10.1023/A:1026507803067. [DOI] [PubMed] [Google Scholar]
- Bonitz T, Alva V, Saleh O, Lupas AN, Heide L. Evolutionary relationships of microbial aromatic prenyltransferases. PLoS One. 2011;6:e27336. doi: 10.1371/journal.pone.0027336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt W, Bräuer L, Günnewich N, Kufka J, Rausch F, Schulze D, Schulze E, Weber R, Zakharova S, Wessjohann L. Molecular and structural basis of metabolic diversity mediated by prenyldiphosphate converting enzymes. Phytochemistry. 2009;70:1758–75. doi: 10.1016/j.phytochem.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Bräuer L, Brandt W, Schulze D, Zakharova S, Wessjohann L. A structural model of the membrane-bound aromatic prenyltransferase UbiA from E. coli. Chembiochem. 2008;9:982–92. doi: 10.1002/cbic.200700575. [DOI] [PubMed] [Google Scholar]
- Bräuer L, Brandt W, Wessjohann LA. Modeling the E. coli 4-hydroxybenzoic acid oligoprenyltransferase (ubiA transferase) and characterization of potential active sites. J Mol Model. 2004;10:317–27. doi: 10.1007/s00894-004-0197-6. [DOI] [PubMed] [Google Scholar]
- Cahoon EB, Hall SE, Ripp KG, Ganzke TS, Hitz WD, Coughlan SJ. Metabolic redesign of vitamin E biosynthesis in plants for tocotrienol production and increased antioxidant content. Nat Biotechnol. 2003;21:1082–7. doi: 10.1038/nbt853. [DOI] [PubMed] [Google Scholar]
- Cheng W, Li W. Structural Insights into Ubiquinone Biosynthesis in Membranes. Science. 2014;343:878–81. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- Collakova E, DellaPenna D. Homogentisate phytyltransferase activity is limiting for tocopherol biosynthesis in Arabidopsis. Plant Physiol. 2003;131:632–642. doi: 10.1104/pp.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa M, Gambacorta A. The lipids of archaebacteria. Prog Lipid Res. 1988;27:153–175. doi: 10.1016/0163-7827(88)90011-2. [DOI] [PubMed] [Google Scholar]
- Debnath J, Siricilla S, Wan B, Crick DC, Lenaerts AJ, Franzblau SG, Kurosu M. Discovery of selective menaquinone biosynthesis inhibitors against Mycobacterium tuberculosis. J Med Chem. 2012;55:3739–55. doi: 10.1021/jm201608g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhiman RK, Mahapatra S, Slayden RA, Boyne ME, Lenaerts A, Hinshaw JC, Angala SK, Chatterjee D, Biswas K, Narayanasamy P, Kurosu M, Crick DC. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol Microbiol. 2009;72:85–97. doi: 10.1111/j.1365-2958.2009.06625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S. Production of selenomethionyl proteins in prokaryotic and eukaryotic expression systems. Methods Mol Biol. 2007;363:91–108. doi: 10.1007/978-1-59745-209-0_5. [DOI] [PubMed] [Google Scholar]
- Forsgren M, Attersand A, Lake S, Grünler J, Swiezewska E, Dallner G, Climent I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem J. 2004;382:519–26. doi: 10.1042/BJ20040261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericks WJ, McGarvey T, Wang H, Lal P, Puthiyaveettil R, Tomaszewski J, Sepulveda J, Labelle E, Weiss JS, Nickerson ML, Kruth HS, Brandt W, Wessjohann LA, Malkowicz SB. The bladder tumor suppressor protein TERE1 (UBIAD1) modulates cell cholesterol: implications for tumor progression. DNA Cell Biol. 2011;30:851–64. doi: 10.1089/dna.2011.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frickey T, Lupas A. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004;20:3702–3704. doi: 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]
- Hederstedt L. Heme A biosynthesis. Biochim Biophys Acta. 2012;1817:920–927. doi: 10.1016/j.bbabio.2012.03.025. [DOI] [PubMed] [Google Scholar]
- Hegarty J, Yang H, Chi NCN. UBIAD1-mediated vitamin K2 synthesis is required for vascular endothelial cell survival and development. Development. 2013;140:1713–9. doi: 10.1242/dev.093112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide L. Prenyl transfer to aromatic substrates: genetics and enzymology. Curr Opin Chem Biol. 2009;13:171–9. doi: 10.1016/j.cbpa.2009.02.020. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Shibuya K, Takahashi Y, Nakayama T, Nishino T. (S)-2,3-Di-O-geranylgeranylglyceryl phosphate synthase from the thermoacidophilic archaeon Sulfolobus solfataricus. Molecular cloning and characterization of a membrane-intrinsic prenyltransferase involved in the biosynthesis of archaeal ether-linked memb. J Biol Chem. 2004;279:50197–50203. doi: 10.1074/jbc.M409207200. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279:8526–9. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- Huang H, Berg S, Spencer JS, Vereecke D, D’Haeze W, Holsters M, McNeil MR. Identification of amino acids and domains required for catalytic activity of DPPR synthase, a cell wall biosynthetic enzyme of Mycobacterium tuberculosis. Microbiology. 2008;154:736–43. doi: 10.1099/mic.0.2007/013532-0. [DOI] [PubMed] [Google Scholar]
- Huang H, Levin EJ, Liu S, Bai Y, Lockless SW, Zhou M. Structure of a membrane-embedded prenyltransferase homologous to UBIAD1. PLoS Biol. 2014;12:e1001911. doi: 10.1371/journal.pbio.1001911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Scherman MS, D’Haeze W, Vereecke D, Holsters M, Crick DC, McNeil MR. Identification and active expression of the Mycobacterium tuberculosis gene encoding 5-phospho-{alpha}-d-ribose-1-diphosphate: decaprenyl-phosphate 5-phosphoribosyltransferase, the first enzyme committed to decaprenylphosphoryl-d-arabinose synthesis. J Biol Chem. 2005;280:24539–43. doi: 10.1074/jbc.M504068200. [DOI] [PubMed] [Google Scholar]
- Johansson LC, Wöhri AB, Katona G, Engström S, Neutze R. Membrane protein crystallization from lipidic phases. Curr Opin Struct Biol. 2009;19:372–8. doi: 10.1016/j.sbi.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Jost M, Zocher G, Tarcz S, Matuschek M, Xie X, Li SM, Stehle T. Structure-function analysis of an enzymatic prenyl transfer reaction identifies a reaction chamber with modifiable specificity. J Am Chem Soc. 2010;132:17849–17858. doi: 10.1021/ja106817c. [DOI] [PubMed] [Google Scholar]
- Kainou T, Okada K, Suzuki K, Nakagawa T, Matsuda H, Kawamukai M. Dimer formation of octaprenyl-diphosphate synthase (IspB) is essential for chain length determination of ubiquinone. J Biol Chem. 2001;276:7876–7883. doi: 10.1074/jbc.M007472200. [DOI] [PubMed] [Google Scholar]
- Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14:673–81. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Kellogg BA, Poulter CD. Chain elongation in the isoprenoid biosynthetic pathway. Curr Opin Chem Biol. 1997;1:570–578. doi: 10.1016/S1367-5931(97)80054-3. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Khalimonchuk O, Smith PM, Winge DR. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim Biophys Acta. 2012;1823:1604–1616. doi: 10.1016/j.bbamcr.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney AJ. Metabolic engineering in plants for human health and nutrition. Curr Opin Biotechnol. 2006 doi: 10.1016/j.copbio.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Koga Y, Morii H. Biosynthesis of ether-type polar lipids in archaea and evolutionary considerations. Microbiol Mol Biol Rev. 2007;71:97–120. doi: 10.1128/MMBR.00033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu M, Crick DC. MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Med Chem. 2009;5:197–207. doi: 10.2174/157340609787582882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu M, Narayanasamy P, Biswas K, Dhiman R, Crick DC. Discovery of 1,4-didydroxy-2-naphthoate prenyltransferase inhibitors: New drug leads for multidrug-resistant gram-positive pathogens. J Med Chem. 2007;50:3973–3975. doi: 10.1021/jm070638m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuyama T, Noel JP, Richard SB. Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature. 2005;435:983–7. doi: 10.1038/nature03668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Lee SM, Park SR, Jung J, Moon JK, Cheong JJ, Kim M. Overexpression of Arabidopsis homogentisate phytyltransferase or tocopherol cyclase elevates vitamin E content by increasing gamma-tocopherol level in lettuce (Lactuca sativa L.) Mol Cells. 2007;24:301–306. [PubMed] [Google Scholar]
- Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, Gulati A, No JH, Cintra G, Bogue S, Liu YL, Molohon K, Orlean P, Mitchell DA, Freitas-Junior L, Ren F, Sun H, Jiang T, Li Y, Guo RT, Cole ST, Gennis RB, Crick DC, Oldfield E. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem. 2014;57:3126–3129. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Bringing Bioactive Compounds into Membranes: The UbiA Superfamily of Intramembrane Aromatic Prenyltransferases. Trends Biochem Sci. 2016 doi: 10.1016/j.tibs.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Cherezov V. Crystallization of membrane proteins in lipidic mesophases. J Vis Exp. 2011 doi: 10.3791/2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meganathan R. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol Lett. 2001;203:131–139. doi: 10.1016/S0378-1097(01)00330-5. [DOI] [PubMed] [Google Scholar]
- Melzer M, Heide L. Characterization of polyprenyldiphosphate: 4-hydroxybenzoate polyprenyltransferase from Escherichia coli. Biochim Biophys Acta. 1994;1212:93–102. doi: 10.1016/0005-2760(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Metzger U, Keller S, Stevenson CEM, Heide L, Lawson DM. Structure and mechanism of the magnesium-independent aromatic prenyltransferase CloQ from the clorobiocin biosynthetic pathway. J Mol Biol. 2010;404:611–26. doi: 10.1016/j.jmb.2010.09.067. [DOI] [PubMed] [Google Scholar]
- Metzger U, Schall C, Zocher G, Unsöld I, Stec E, Li SM, Heide L, Stehle T. The structure of dimethylallyl tryptophan synthase reveals a common architecture of aromatic prenyltransferases in fungi and bacteria. Proc Natl Acad Sci U S A. 2009;106:14309–14. doi: 10.1073/pnas.0904897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Mugoni V, Postel R, Catanzaro V, De Luca E, Turco E, Digilio G, Silengo L, Murphy MP, Medana C, Stainier DYR, Bakkers J, Santoro MM. Ubiad1 Is an Antioxidant Enzyme that Regulates eNOS Activity by CoQ10 Synthesis. Cell. 2013;152:504–18. doi: 10.1016/j.cell.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K, Hirota Y, Sawada N, Yuge N, Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y, Okano T. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature. 2010;468:117–21. doi: 10.1038/nature09464. [DOI] [PubMed] [Google Scholar]
- Newstead S, Kim H, von Heijne G, Iwata S, Drew D. High-throughput fluorescent-based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2007;104:13936–41. doi: 10.1073/pnas.0704546104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka B, Kruk J. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim Biophys Acta. 2010;1797:1587–605. doi: 10.1016/j.bbabio.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Ohara K, Kokado Y, Yamamoto H, Sato F, Yazaki K. Engineering of ubiquinone biosynthesis using the yeast coq2 gene confers oxidative stress tolerance in transgenic tobacco. Plant J. 2004;40:734–43. doi: 10.1111/j.1365-313X.2004.02246.x. [DOI] [PubMed] [Google Scholar]
- Ohara K, Mito K, Yazaki K. Homogeneous purification and characterization of LePGT1 – A membrane-bound aromatic substrate prenyltransferase involved in secondary metabolism of Lithospermum erythrorhizon. FEBS J. 2013;280:2572–2580. doi: 10.1111/febs.12239. [DOI] [PubMed] [Google Scholar]
- Ohara K, Muroya A, Fukushima N, Yazaki K. Functional characterization of LePGT1, a membrane-bound prenyltransferase involved in the geranylation of p-hydroxybenzoic acid. Biochem J. 2009;421:231–41. doi: 10.1042/BJ20081968. [DOI] [PubMed] [Google Scholar]
- Ohara K, Yamamoto K, Hamamoto M, Sasaki K, Yazaki K. Functional characterization of OsPPT1, which encodes p-hydroxybenzoate polyprenyltransferase involved in ubiquinone biosynthesis in Oryza sativa. Plant Cell Physiol. 2006;47:581–90. doi: 10.1093/pcp/pcj025. [DOI] [PubMed] [Google Scholar]
- Okada K, Kainou T, Matsuda H, Kawamukai M. Biological significance of the side chain length of ubiquinone in Saccharomyces cerevisiae. FEBS Lett. 1998;431:241–244. doi: 10.1016/S0014-5793(98)00753-4. [DOI] [PubMed] [Google Scholar]
- Okada K, Minehira M, Zhu X, Suzuki K, Nakagawa T, Matsuda H, Kawamukai M. The ispB gene encoding octaprenyl diphosphate synthase is essential for growth of Escherichia coli. J Bacteriol. 1997;179:3058–3060. doi: 10.1128/jb.179.9.3058-3060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Ohara K, Yazaki K, Nozaki K, Uchida N, Kawamukai M, Nojiri H, Yamane H. The AtPPT1 gene encoding 4-hydroxybenzoate polyprenyl diphosphate transferase in ubiquinone biosynthesis is required for embryo development in Arabidopsis thaliana. Plant Mol Biol. 2004;55:567–77. doi: 10.1007/s11103-004-1298-4. [DOI] [PubMed] [Google Scholar]
- Okada K, Suzuki K, Kamiya Y, Zhu X, Fujisaki S, Nishimura Y, Nishino T, Nakagawa T, Kawamukai M, Matsuda H. Polyprenyl diphosphate synthase essentially defines the length of the side chain of ubiquinone. Biochim Biophys Acta – Lipids Lipid Metab. 1996;1302:217–223. doi: 10.1016/0005-2760(96)00064-1. [DOI] [PubMed] [Google Scholar]
- Orr A, Dubé MP, Marcadier J, Jiang H, Federico A, George S, Seamone C, Andrews D, Dubord P, Holland S, Provost S, Mongrain V, Evans S, Higgins B, Bowman S, Guernsey D, Samuels M. Mutations in the UBIAD1 gene, encoding a potential prenyltransferase, are causal for Schnyder crystalline corneal dystrophy. PLoS One. 2007;2:e685. doi: 10.1371/journal.pone.0000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster U, Bauer CE, Rüdiger W. Characterization of chlorophyll a and bacteriochlorophyll a synthases by heterologous expression in Escherichia coli. J Biol Chem. 1997;272:9671–9676. doi: 10.1074/jbc.272.15.9671. [DOI] [PubMed] [Google Scholar]
- Quinzii CM, Hirano M, DiMauro S. CoQ10 deficiency diseases in adults. Mitochondrion. 2007;7(Suppl):S122–6. doi: 10.1016/j.mito.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinzii CM, López LC, Naini A, DiMauro S, Hirano M. Human CoQ10 deficiencies. Biofactors. 2008;32:113–118. doi: 10.1002/biof.5520320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüdiger W, Böhm S, Helfrich M, Schulz S, Schoch S. Enzymes of the last steps of chlorophyll biosynthesis: Modification of the substrate structure helps to understand the topology of the active centers. Biochemistry. 2005;44:10864–10872. doi: 10.1021/bi0504198. [DOI] [PubMed] [Google Scholar]
- Saiki K, Mogi T, Hori H, Tsubaki M, Anraku Y. Identification of the functional domains in heme O synthase. Site-directed mutagenesis studies on the cyoE gene of the cytochrome bo operon in Escherichia coli. J Biol Chem. 1993;268:26927–26934. [PubMed] [Google Scholar]
- Saleh O, Haagen Y, Seeger K, Heide L. Prenyl transfer to aromatic substrates in the biosynthesis of aminocoumarins, meroterpenoids and phenazines: the ABBA prenyltransferase family. Phytochemistry. 2009;70:1728–38. doi: 10.1016/j.phytochem.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Savidge B, Weiss JD, Wong YHH, Lassner MW, Mitsky TA, Shewmaker CK, Post-Beittenmiller D, Valentin HE. Isolation and characterization of homogentisate phytyltransferase genes from Synechocystis sp PCC 6803 and Arabidopsis. Plant Physiol. 2002;129:321–332. doi: 10.1104/pp.010747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schledz M, Seidler A, Beyer P, Neuhaus G. A novel phytyltransferase from Synechocystis sp PCC 6803 involved in tocopherol biosynthesis. FEBS Lett. 2001;499:15–20. doi: 10.1016/S0014-5793(01)02508-X. [DOI] [PubMed] [Google Scholar]
- Schmid HC, Oster U, Kögel J, Lenz S, Rüdiger W. Cloning and characterisation of chlorophyll synthase from Avena sativa. Biol Chem. 2001;382:903–911. doi: 10.1515/BC.2001.112. [DOI] [PubMed] [Google Scholar]
- Shineberg B, Young IG. Biosynthesis of bacterial menaquinones: the membrane-associated 1,4-dihydroxy-2-naphthoate octaprenyltransferase of Escherichia coli. Biochemistry. 1976;15:2754–2758. doi: 10.1021/bi00658a007. [DOI] [PubMed] [Google Scholar]
- Soll J, Kemmerling M, Schultz G. Tocopherol and plastoquinone synthesis in spinach chloroplasts subfractions. Arch Biochem Biophys. 1980;204:544–50. doi: 10.1016/0003-9861(80)90066-1. [DOI] [PubMed] [Google Scholar]
- Sonoda Y, Cameron A, Newstead S, Omote H, Moriyama Y, Kasahara M, Iwata S, Drew D. Tricks of the trade used to accelerate high-resolution structure determination of membrane proteins. FEBS Lett. 2010 doi: 10.1016/j.febslet.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Sonoda Y, Newstead S, Hu NJ, Alguel Y, Nji E, Beis K, Yashiro S, Lee C, Leung J, Cameron AD, Byrne B, Iwata S, Drew D. Benchmarking membrane protein detergent stability for improving throughput of high-resolution x-ray structures. Structure. 2011;19:17–25. doi: 10.1016/j.str.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec E, Li SM. Mutagenesis and biochemical studies on AuaA confirmed the importance of the two conserved aspartate-rich motifs and suggested difference in the amino acids for substrate binding in membrane-bound prenyltransferases. Arch Microbiol. 2012;194:589–595. doi: 10.1007/s00203-012-0795-0. [DOI] [PubMed] [Google Scholar]
- Suvarna K, Stevenson D, Meganathan R, Hudspeth ME. Menaquinone (vitamin K2) biosynthesis: localization and characterization of the menA gene from Escherichia coli. J Bacteriol. 1998;180:2782–7. doi: 10.1128/jb.180.10.2782-2787.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Ueda M, Yuasa M, Nakagawa T, Kawamukai M, Matsuda H. Evidence that Escherichia coli ubiA product is a functional homolog of yeast COQ2, and the regulation of ubiA gene expression. Biosci Biotechnol Biochem. 1994;58:1814–9. doi: 10.1271/bbb.58.1814. [DOI] [PubMed] [Google Scholar]
- Uchida N, Suzuki K, Saiki R, Kainou T, Tanaka K, Matsuda H, Kawamukai M. Phenotypes of fission yeast defective in ubiquinone production due to disruption of the gene for p-hydroxybenzoate polyprenyl diphosphate transferase. J Bacteriol. 2000;182:6933–9. doi: 10.1128/jb.182.24.6933-6939.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin HE, Qi Q. Biotechnological production and application of vitamin E: Current state and prospects. Appl Microbiol Biotechnol. 2005;68:436–444. doi: 10.1007/s00253-005-0017-7. [DOI] [PubMed] [Google Scholar]
- Venkatesh TV, Karunanandaa B, Free DL, Rottnek JM, Baszis SR, Valentin HE. Identification and characterization of an Arabidopsis homogentisate phytyltransferase paralog. Planta. 2006;223:1134–1144. doi: 10.1007/s00425-005-0180-1. [DOI] [PubMed] [Google Scholar]
- Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B, Meganathan R, Morais VA, Verstreken P. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306–10. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- Wallace BJ, Young IG. Role of quinones in electron transport to oxygen and nitrate in Escherichia coli. Studies with a ubiA- menA- double quinone mutant. Biochim Biophys Acta. 1977;461:84–100. doi: 10.1016/0005-2728(77)90071-8. [DOI] [PubMed] [Google Scholar]
- Wallrapp FH, Pan JJJ, Ramamoorthy G, Almonacid DE, Hillerich BS, Seidel R, Patskovsky Y, Babbitt PC, Almo SC, Jacobson MP, Poulter CD. Prediction of function for the polyprenyl transferase subgroup in the isoprenoid synthase superfamily. Proc Natl Acad Sci U S A. 2013;110:E1196–202. doi: 10.1073/pnas.1300632110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JS, Kruth HS, Kuivaniemi H, Tromp G, White PS, Winters RS, Lisch W, Henn W, Denninger E, Krause M, Wasson P, Ebenezer N, Mahurkar S, Nickerson ML. Mutations in the UBIAD1 gene on chromosome short arm 1, region 36, cause Schnyder crystalline corneal dystrophy. Invest Ophthalmol Vis Sci. 2007;48:5007–12. doi: 10.1167/iovs.07-0845. [DOI] [PubMed] [Google Scholar]
- Wessjohann L, Sontag B. Prenylation of Benzoic Acid Derivatives Catalyzed by a Transferase from Escherichia coli Overproduction: Method Development and Substrate Specificity. Angew Chemie Int Ed English. 1996;35:1697–1699. doi: 10.1002/anie.199616971. [DOI] [Google Scholar]
- Willows RD. Biosynthesis of chlorophylls from protoporphyrin IX. Nat Prod Rep. 2003;20:327–341. doi: 10.1039/b110549n. [DOI] [PubMed] [Google Scholar]
- Young IG. Biosynthesis of bacterial menaquinones. Menaquinone mutants of Escherichia coli. Biochemistry. 1975;14:399–406. doi: 10.1021/bi00673a029. [DOI] [PubMed] [Google Scholar]
- Young IG, Leppik RA, Hamilton JA, Gibson F. Biochemical and genetic studies on ubiquinone biosynthesis in Escherichia coli K-12:4-hydroxybenzoate octaprenyltransferase. J Bacteriol. 1972;110:18–25. doi: 10.1128/jb.110.1.18-25.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Poulter CD. Biosynthesis of archaebacterial ether lipids. Formation of ether linkages by prenyltransferases. J Am Chem Soc. 1993;115:1270–1277. doi: 10.1021/ja00057a008. [DOI] [Google Scholar]
- Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–43. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]