

Abstract
Shank is a post-synaptic scaffolding protein that has many binding partners. Shank mutations and copy number variations (CNVs) are linked to several psychiatric disorders, and to synaptic and behavioral defects in mice. It is not known which Shank binding partners are responsible for these defects. Here we show that the C. elegans SHN-1/Shank binds L-type calcium channels and that increased and decreased shn-1 gene dosage alter L-channel current and activity-induced expression of a CRH-1/CREB transcriptional target (gem-4 Copine), which parallels the effects of human Shank copy number variations (CNVs) on Autism spectrum disorders and schizophrenia. These results suggest that an important function of Shank proteins is to regulate L-channel current and activity induced gene expression.
DOI: http://dx.doi.org/10.7554/eLife.18931.001
Research Organism: C. elegans
Introduction
Shank is a synaptic scaffolding protein (containing SH3, Ankyrin, PDZ, proline-rich and SAM domains) (Grabrucker et al., 2011). Because Shank is highly enriched in the post-synaptic densities of excitatory synapses, prior studies have focused on the idea that Shank proteins regulate some aspect of synapse formation or function. Through its various domains, Shank proteins bind hundreds of other synaptic proteins (Lee et al., 2011; Sakai et al., 2011) thereby potentially altering diverse cellular functions. Shank proteins have been implicated in synaptic transmission, synapse formation, synaptic plasticity, and cytoskeletal remodeling (Jiang and Ehlers, 2013).
Mammals have three Shank genes, each encoding multiple isoforms (Jiang and Ehlers, 2013). Several mouse Shank knockouts have been described but these mutants exhibit inconsistent (often contradictory) synaptic and behavioral defects (Jiang and Ehlers, 2013), most likely resulting from differences in which Shank isoforms are impacted by each mutation. The biochemical mechanism by which Shank mutations alter synaptic function and behavior has not been determined.
In humans, Shank mutations and CNVs are linked to Autism Spectrum Disorders (ASD), schizophrenia, and mania (Durand et al., 2007; Peça et al., 2011). Haploinsufficiency for 22q13 (which spans the Shank3 locus) occurs in Phelan-McDermid syndrome (PMS), a syndromic form of ASD (Phelan and McDermid, 2012). PMS patients exhibit autistic behaviors accompanied by hypotonia, delayed speech, and intellectual disability (ID) (Bonaglia et al., 2011). Heterozygous inactivating Shank3 mutations are found in sporadic ASD and schizophrenia (Durand et al., 2007; Peça et al., 2011). These genetic studies suggest that decreased Shank3 function likely plays an important role in the pathophysiology of these psychiatric disorders.
A parallel set of genetic studies suggest that increased Shank3 function also contributes to psychiatric diseases. 22q13 duplications spanning Shank3 are found in ASD, schizophrenia, ADHD, and bipolar disorder (Durand et al., 2007; Failla et al., 2007; Han et al., 2013). These 22q13 duplications involve multiple genes; consequently, the contribution of increased Shank3 to these psychiatric disorders was uncertain. To address this issue, a transgenic mouse model was developed that selectively over-expresses Shank3 (Han et al., 2013). This transgenic mouse exhibited hyperactive behavior and susceptibility to seizures. Taken together, these studies suggest that too little or too much Shank3 is associated with several psychiatric disorders.
If Shank3 mutations and CNVs are causally associated with these psychiatric disorders, cellular and circuit phenotypes should also be sensitive to Shank3 copy number. Consistent with this idea, several defects have been reported in Shank3+/- heterozygotes, including: decreased mEPSC frequency and spine density (Zhou et al., 2016), decreased Ih current density (Yi et al., 2016), decreased TRPV1 current density (Han et al., 2016), increased tactile sensitivity (Orefice et al., 2016), and decreased post-synaptic Homer abundance (Wang et al., 2016). Increased Shank expression was associated with increased spine density in hippocampus and decreased inhibitory synapses (Han et al., 2013). In many cases (Han et al., 2013; Orefice et al., 2016; Wang et al., 2016; Zhou et al., 2016), it was not determined if these phenotypes are a cell autonomous consequence of altered Shank3 copy number. While these studies identify cellular deficits associated with Shank3 CNVs, it remains unclear which Shank binding partners and cellular functions are responsible for psychiatric traits, nor why these traits are sensitive to both increased and decreased Shank gene dosage.
To further investigate how Shank proteins regulate nervous system development and function, we analyzed Shank function in an invertebrate genetic model. Here we show that C. elegans SHN-1 is a dose-sensitive regulator of Cav1 calcium current and CREB induced gene expression in C. elegans body muscles.
Results
The SHN-1 PDZ domain binds EGL-19/Cav1 channels
C. elegans has a single Shank gene, shn-1. The SHN-1 protein lacks an SH3 domain but has all other domains found in mammalian Shank proteins (Figure 1A). Many protein ligands have been identified for the Shank PDZ domain (Lee et al., 2011). Of these potential binding partners, we focused on EGL-19/Cav1 because human CACNA1C (which encodes a Cav1 α-subunit) is mutated in Timothy Syndrome (TS), a rare monogenic form of ASD (Splawski et al., 2005, 2004), and polymorphisms linked to CACNA1C are associated with multiple psychiatric disorders (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). We confirmed that SHN-1’s PDZ domain binds the EGL-19 carboxy-terminal motif (-VTTLCOOH) by both yeast two-hybrid and GST-pull down assays (Figure 1—figure supplement 1A and B). Thus, like their mammalian counterparts, SHN-1 binds to EGL-19/ Cav1 (Zhang et al., 2005).
Figure 1. SHN-1 promotes EGL-19/Cav1 channel function.
(A) The protein domains found in SHN-1 and rat Shank3A are compared. SHN-1 lacks an SH3 domain but contains all other domains found in mammalian Shank proteins. Homology between the worm and mammalian protein is shown for each domain. (B–F) Voltage-activated Ca+2 currents were recorded from adult body wall muscles of the indicated genotypes at holding potentials of −60 to +40 mV. Averaged traces (B), mean current density as a function of holding potential (C), normalized conductance as a function of holding potential (D), mean current density at 10 mV (E), and mean deactivation time constants (F) are shown. shn-1 mutants had significantly decreased Ca+2 current-density and this defect was rescued by a single copy transgene expressing SHN-1 in body muscles (nuSi26) (D). No significant differences were observed for voltage-dependence of current activation and de-activation kinetics. The number of animals analyzed is indicated for each genotype. Values that differ significantly from wild type controls are indicated (***p<0.001). Error bars indicate SEM. Mean, standard errors, sample sizes, and p values for this figure are shown in Supplementary file 1.

Figure 1—figure supplement 1. Supplemental data related to Figure 1.

EGL-19/Cav1 calcium currents are diminished in shn-1 mutants
The impact of Shank binding on Cav1.3 function has been assessed by over-expression in Xenopus oocytes and in cultured neurons (Stanika et al., 2016; Zhang et al., 2005) but has not been tested in mutant or transgenic animals. Because shn-1 is expressed in muscles (Stefanakis et al., 2015), we assayed EGL-19/Cav1 channel function by recording voltage-activated calcium currents in body muscle, using salines containing potassium channel blockers (TEA and 4AP). In these conditions, the remaining voltage-activated inward current is entirely blocked by the EGL-19 antagonist nemadipine (Lainé et al., 2011). In shn-1(tm488) null mutants, calcium current density was significantly decreased (Figure 1B,C,E). Neither the voltage-dependence of calcium current activation (Figure 1D) nor the deactivation kinetics (Figure 1F) were altered in shn-1(tm488) null mutants. The shn-1 calcium current defect was rescued by a transgene restoring SHN-1 expression in body muscles (Figure 1E), confirming that SHN-1 has a cell autonomous effect on EGL-19/Cav1 currrent. To determine if SHN-1’s effects on calcium currents were specific, we measured voltage-activated potassium currents in body muscles (Figure 1—figure supplement 1C). Neither the voltage-dependence nor the current density of fast and slow potassium currents were significantly altered in shn-1 mutants. Collectively, these results suggest that SHN-1 specifically regulates the expression or function of EGL-19/Cav1 channels.
SHN-1 binding to EGL-19 increases Cav1 current density
SHN-1 effects on calcium current density could result from direct binding of SHN-1 to EGL-19/Cav1 or indirectly via other SHN-1 binding partners. We did several experiments to distinguish between these possibilities. First, we utilized CRISPR to isolate two deletion alleles that alter the EGL-19 carboxy-terminus (nu495 and nu496) (Figure 2A). Both deletion mutants exhibited decreased calcium current density (similar to the defect observed in shn-1 null mutants) (Figure 2B–C). The egl-19(nu496) mutation had no effect on the voltage-dependence of calcium current activation nor on deactivation kinetics (Figure 2—figure supplement 1A and B). The egl-19(nu496) and shn-1 null mutations did not have additive effects on calcium current density in double mutants, as would be predicted if SHN-1’s effects on calcium current require direct binding to EGL-19’s carboxy-terminus (Figure 2D and E). To further examine the functional impact of the SHN-1 PDZ interaction with EGL-19, we analyzed shn-1(ok1241) mutants, which have an in-frame deletion spanning exons encoding the PDZ domain and part of the proline-rich domain (Figure 1—figure supplement 1D). The shn-1(ok1241) mutants exhibited a decrease in calcium current density similar to those observed in shn-1 null and the egl-19 carboxy-terminal deletion mutants and had no effect on voltage-dependence of current activation nor on deactivation kinetics (Figure 2—figure supplement 1C–F). Collectively, these results suggest that SHN-1 binding to EGL-19’s carboxy-terminus promotes the expression or function of L-type calcium channels.
Figure 2. SHN-1 binding to EGL-19’s carboxy-terminus promotes the expression or function of L-type calcium channels.
(A) Predicted c-terminal sequences of mutant EGL-19 proteins are shown. egl-19(nu496) is a 22 bp deletion and egl-19(nu495) is a 5 bp deletion, both resulting in frame shifts that delete the carboxy-terminal PDZ ligand of EGL-19 (-VTTLCOOH). Residues in blue represent the PDZ ligand. Residues in red represent those introduced by the frame shift mutations. (B–E) Voltage-activated Ca+2 currents were recorded from adult body wall muscles of the indicated genotypes at holding potentials of −60 to +40 mV. Representative traces (B), mean current density at 0 mV (C, E), and mean current density as a function of holding potential (D) are shown. The egl-19(nu496) and shn-1(tm488) single mutants had similar decreases in Ca+2 current-density, and additive defects were not observed in the double mutant. The number of animals analyzed is indicated for each genotype. Values that differ significantly from wild type controls are indicated (***p<0.001). Error bars indicate SEM. Mean, standard errors, sample sizes, and p values for this figure are shown in Supplementary file 1.

Figure 2—figure supplement 1. Supplemental data related to Figure 2.

SHN-1 promotes trafficking of EGL-19 channels to the cell surface
SHN-1 effects on calcium current could result from a change in EGL-19 delivery to the cell surface. We performed two further experiments to test this idea. First, we measured the gating currents of voltage-activated channels in body muscles (Figure 3A–B). The activation of Cav channels is mediated by depolarization-induced movements of positively charged residues in membrane-spanning S4 helices, which are termed gating charges. When there is no net calcium current (by holding the muscle membrane at the reversal potential), gating charge movement can be measured as a small voltage-activated current. The magnitude of gating currents can be used as a measure of Cav channel surface abundance (Fu et al., 2011; Hulme et al., 2006). The total voltage activated gating charge in body muscles was significantly reduced in shn-1 null mutants (Figure 3B). This shn-1 mutant defect in gating charge is unlikely to result from decreased surface delivery of other voltage-activated channels because voltage-activated potassium currents were unaltered in shn-1 mutants (Figure 1—figure supplement 1C). Thus, analysis of gating currents suggests that the decreased calcium current exhibited in shn-1 mutants arises from decreased trafficking of EGL-19/Cav1 channels to the cell surface.
Figure 3. SHN-1 promotes EGL-19/Cav1 delivery to the cell surface.

(A–B) Voltage-activating gating currents were significantly decreased in shn-1 null mutants. Averaged trace of gating current in wild type adult body muscles (A) and mean gating charge (normalized to capacitance) (B) are shown. (C–G) Surface delivery of the Terrier fusion protein is significantly reduced in shn-1 null mutants. (C) A schematic illustrating the structure of the Terrier fusion protein is shown. (D) A schematic illustrating the imaged region (left) and representative images of Terrier pHluorin fluorescence in the nerve ring are shown. Mean pHluorin puncta intensity (E), pHluorin puncta area (F), and total pHluorin puncta fluorescence (G) are shown. Regions of interest utilized to quantify Terrier fluorescence are indicated (D). The number of animals analyzed is indicated for each genotype. Values that differ significantly from wild type controls are indicated (**p<0.01, *p<0.05). Error bars indicate SEM.
To further investigate SHN-1’s effects on EGL-19 trafficking, we designed a fluorescent reporter construct containing a trans-membrane domain fused to EGL-19’s cytoplasmic tail domain (504 amino acids), which includes the carboxy-terminal PDZ ligand (Figure 3C). This chimeric protein (designated Terrier to indicate the presence of EGL-19’s cytoplasmic tail) was expressed in body muscles. The Terrier protein contains tagRFP in the cytoplasmic domain, and pHluorin (a pH-sensitive GFP) in the ectodomain (Figure 3C). Thus, red fluorescence reports total Terrier protein while green fluorescence reports surface Terrier molecules. When expressed in body muscles, Terrier exhibits a punctate green and red fluorescence in the nerve ring, where body muscles receive synaptic input (Figure 3C–D). In shn-1 null mutants, the intensity and size of green Terrier puncta were significantly decreased (Figure 3D–G), indicating decreased surface Terrier protein in the nerve ring. Thus, decreased calcium current density (20%) in shn-1 mutants was mirrored by similar decreases in gating charge (26%) and total surface Terrier fluorescence (36%). Collectively, these results suggest that the decreased calcium current in shn-1 null mutants arises from decreased delivery of EGL-19/Cav1 channels to the cell surface.
gem-4 is an activity-induced CRH-1/CREB target expressed in muscles
Increased cytoplasmic calcium activates expression of a large number genes, hereafter designated activity-induced gene expression. Although Cav1 channels account for a small fraction of bulk calcium entry in neurons, Cav1 channels account for the majority of activity-induced gene expression (Ma et al., 2012). This privileged ability of Cav1 channels to activate gene expression is thought to be mediated by direct physical coupling of Cav1 channels to the calcium sensors responsible for activating CREB (Deisseroth et al., 1996; Wheeler et al., 2008).
Because shn-1 mutations alter EGL-19/Cav1 current, we hypothesized that SHN-1 may also play a role in activity-induced gene expression. To test this idea, we first identified activity-induced muscle genes. We analyzed gene expression following depolarization of body muscles with a nicotinic acetylcholine (ACh) agonist (levamisole, Lev). Lev-induced genes were identified using the Affymetrix C. elegans gene chip. This analysis identified 427 genes whose expression was significantly increased following muscle depolarization (>2 fold change, FDR p<0.05) (Figure 4, Supplementary file 2). 67% (287/427) of Lev-induced genes contain binding sites for the myogenic transcription factor HLH-1 (<5 kb from the transcriptional start site, TSS) in chromatin-immunoprecipitation experiments (http://www.modencode.org), suggesting that these genes are expressed in body muscles. Of the HLH-1 binding genes, 81% (233/287) contain predicted CREB binding sites (<5 kb from the TSS). These results suggest that C. elegans body muscles (like other excitable cells) have a large number of activity-induced genes, many of which are potential CREB transcriptional targets.
Figure 4. Analysis of mRNA abundance following muscle depolarization.

mRNA abundance in Lev (200 µM, 1 hr) versus mock treated synchronized L4 larvae is plotted. Fold change (x-axis) is plotted against the statistical significance (y-axis) for each probeset. Fold changes are shown in log2 scale. Adjusted P values are shown in - log10 scale. Genes with increased (red dots) and decreased (green dots) expression are indicated (>2 Fold-change, FDR p<0.05). Probe sets corresponding to gem-4 and cex-1 are indicated. All genes that are differentially expressed following Lev treatment are listed in Supplementary file 2.
Using this Lev-induced gene list, we devised a simple reporter assay for CREB induced gene expression. For this purpose, we focused on the gem-4/Copine gene, which was the top hit from our analysis of Lev-induced genes (induced ~30 fold) (Figure 4). The gem-4 promoter contains multiple CRH-1/CREB binding sites, implying that it could be a direct CRH-1 transcriptional target. Quantitative RT-PCR confirmed that Lev treatment increased gem-4 mRNA levels 8-fold (Figure 5A). We designed a transcriptional reporter (Figure 5B) that compares expression of the gem-4 promoter (expressing NLS-GFP) with a control promoter unaffected by depolarization (the myo-3 promoter, expressing NLS-mCherry) in individual muscle cells. Using this reporter, we found that Lev treatment increased gem-4 expression 8–12-fold (Figure 5C–D) while myo-3 expression was unaltered (Figure 5—figure supplement 1A). Lev-induction of the gem-4 reporter was eliminated by mutations inactivating a Lev receptor subunit (UNC-29), indicating that gem-4 induction was not mediated by an off target effect of Lev (Figure 5C). Lev-induction of gem-4 was also blocked in mutants lacking CRH-1/CREB and this defect was rescued by a transgene expressing CRH-1 in body muscles (Figure 5D). These results identify gem-4 as a CRH-1/CREB target expressed in body muscles.
Figure 5. gem-4 Copine expression in body muscle is induced by depolarization.
Induction of gem-4 expression was analyzed by qPCR (A) and using a transcriptional reporter (B–H). (A) The abundance of gem-4 mRNA (assessed by qPCR) was increased following 1 hr levamisole (Lev) exposure. The number of biological replicates is indicated. (B) A schematic diagram of the gem-4 reporter construct (left) and representative images of muscle nuclei (right) before and after a 20 min Lev exposure, and 2 hr recovery. (C–D) The mean fold induction of the gem-4 reporter (Pgem-4) after Lev treatment is shown. Lev-induced gem-4 expression was abolished in mutants lacking UNC-29, an essential subunit of the Lev receptor (C) and in mutants lacking the transcription factor CRH-1 (D). The crh-1 mutant defect in gem-4 induction was rescued by a transgene expressing CRH-1 in body muscles (D). (E) Expression of the gem-4 reporter was measured following photo-stimulation of transgenic animals that express ChR2 in cholinergic motorneurons. Expression of gem-4 was significantly increased by 2, 5, 10, and 20 Hz photo-stimulation (for 20 min). Photo-evoked gem-4 expression was not observed when animals were not cultured with ATR. (F–G) The fold induction of the gem-4 reporter following Lev exposure was significantly reduced in shn-1 null mutants (F) but not in shn-1(ok1241) mutants, which lack the PDZ domain (G). Lev-induced gem-4 expression was significantly increased in egl-19(nu496) mutants, which lack the carboxy-terminal PDZ ligand (H). (I–J) Lev induction of the cex-1 reporter is significantly reduced in shn-1 mutants. (I) Schematics of the cex-1 and myo-3 reporters are shown. (J) Expression of the cex-1 reporter (normalized to myo-3 expression in the same nucleus) was significantly increased by Lev treatment. The Lev-induced expression of the cex-1 reporter was significantly reduced in shn-1 mutants. The number of animals analyzed is indicated for each genotype. Values that differ significantly from wild type controls are indicated (***p<0.001; **p<0.01; *p<0.05). Error bars indicate SEM.

Figure 5—figure supplement 1. Supplemental data related to Figure 5.

Lev treatment is a non-physiological stimulus that produces prolonged muscle depolarization. To determine if synaptic activity induces the gem-4 reporter, we evoked excitatory synaptic input to muscles by photo-stimulating transgenic animals expressing channel rhodopsin in cholinergic motor neurons (Figure 5E). In patch clamp recordings of body muscles, spontaneous action potentials are observed at ~1 Hz (Gao and Zhen, 2011). Therefore, to mimic a realistic pattern of synaptic input, we photo-stimulated motor neurons (25 ms light pulse) at 1–20 Hz for 20 min. The gem-4 reporter was significantly induced following 2, 5, 10, and 20 Hz photo-stimulation (Figure 5E). Induction was not observed at lower frequencies (0.5 and 1 Hz) nor when animals were cultured without all trans-retinal (ATR) (Figure 5E). Photo-induction of gem-4 expression was eliminated in unc-13 mutants (Figure 5—figure supplement 1B), which have dramatically reduced synaptic vesicle exocytosis (Richmond et al., 1999). By contrast, unc-13 mutations did not prevent Lev-induced gem-4 expression (Figure 5—figure supplement 1C). Similar levels of gem-4 induction were produced by 20 Hz photo-stimulation and Lev treatment (Figure 5—figure supplement 1B–C). Thus, the gem-4 reporter was induced by both synaptic and Lev-evoked muscle depolarization. Expression of a mouse gem-4 paralog (N-copine) in hippocampal neurons is also induced by high frequency stimulation of acute brain slices (Nakayama et al., 1998); consequently, activity-induced copine expression is observed in both muscles and neurons and is conserved across phylogeny.
SHN-1 is required for gem-4 induction
Using this gem-4 reporter construct, we next asked if SHN-1 is required for CRH-1/CREB-induced gene expression. Lev-induced gem-4 expression was significantly decreased in shn-1(tm488) null mutants (Figure 5F). To determine if SHN-1 controls expression of other activity-induced genes, we developed a transcriptional reporter for a second Lev-induced gene (cex-1) (Figures 4 and 5I). Expression of the cex-1 reporter in body muscles was dramatically induced following Lev treatment (Figure 5J). Because baseline cex-1 expression in untreated muscles could not be reliably detected, we were unable to accurately measure the fold-induction of the cex-1 reporter following Lev treatment. As seen with the gem-4 reporter, we found that Lev-induced cex-1 expression in muscles was dramatically reduced in shn-1 null mutants (Figure 5J). Taken together, these results support the idea that SHN-1 promotes activity-induced gene expression in body muscles.
Next we asked if gem-4 induction requires binding of SHN-1’s PDZ domain to EGL-19’s carboxy-terminus. Deleting EGL-19’s PDZ ligand [in egl-19(nu496) mutants] significantly increased gem-4 induction while deleting SHN-1’s PDZ domain [in shn-1(ok1241) mutants] had no effect on gem-4 induction (Figure 5G–H). These results indicate that SHN-1 regulates EGL-19/Cav1 current and CRH-1/CREB activation by distinct mechanisms, since the former requires PDZ binding to EGL-19 while the latter does not.
Calcium current and gem-4 induction are sensitive to shn-1 gene dose
Deletion and duplication of human shank genes are both associated with ASD, schizophrenia, and mania (Bonaglia et al., 2006; Durand et al., 2007; Failla et al., 2007; Gauthier et al., 2010; Han et al., 2013). If Shank CNVs are causally associated with these psychiatric disorders, cellular phenotypes should be similarly sensitive to Shank copy number. To test this idea, we analyzed the effect of shn-1 gene dosage on calcium currents and Lev-induced gem-4 expression (Figure 6). We analyzed animals with 0 (tm488 homozygotes), 1 (tm488/+ heterozygotes), 2 (WT), and 4 (WT +2 single copy shn-1 transgenes) copies of shn-1. Compared to wild type controls, muscle calcium current density was significantly increased in animals containing 1 and 4 copies of shn-1 and was significantly decreased in animals containing 0 copies of shn-1 (Figure 6A–C). The kinetics of calcium current deactivation were not significantly altered by changes in shn-1 dosage (Figure 6D). Similarly, when compared to wild type controls, gem-4 induction was significantly diminished in animals containing 0, 1, and 4 shn-1 copies (Figure 6E). Thus, L-type calcium current density and gem-4 induction were both sensitive to shn-1 copy number. Interestingly, increased and decreased shn-1 gene dosage produced similar defects in calcium current and gem-4 induction.
Figure 6. shn-1 gene dosage regulates calcium current density and gem-4 induction.
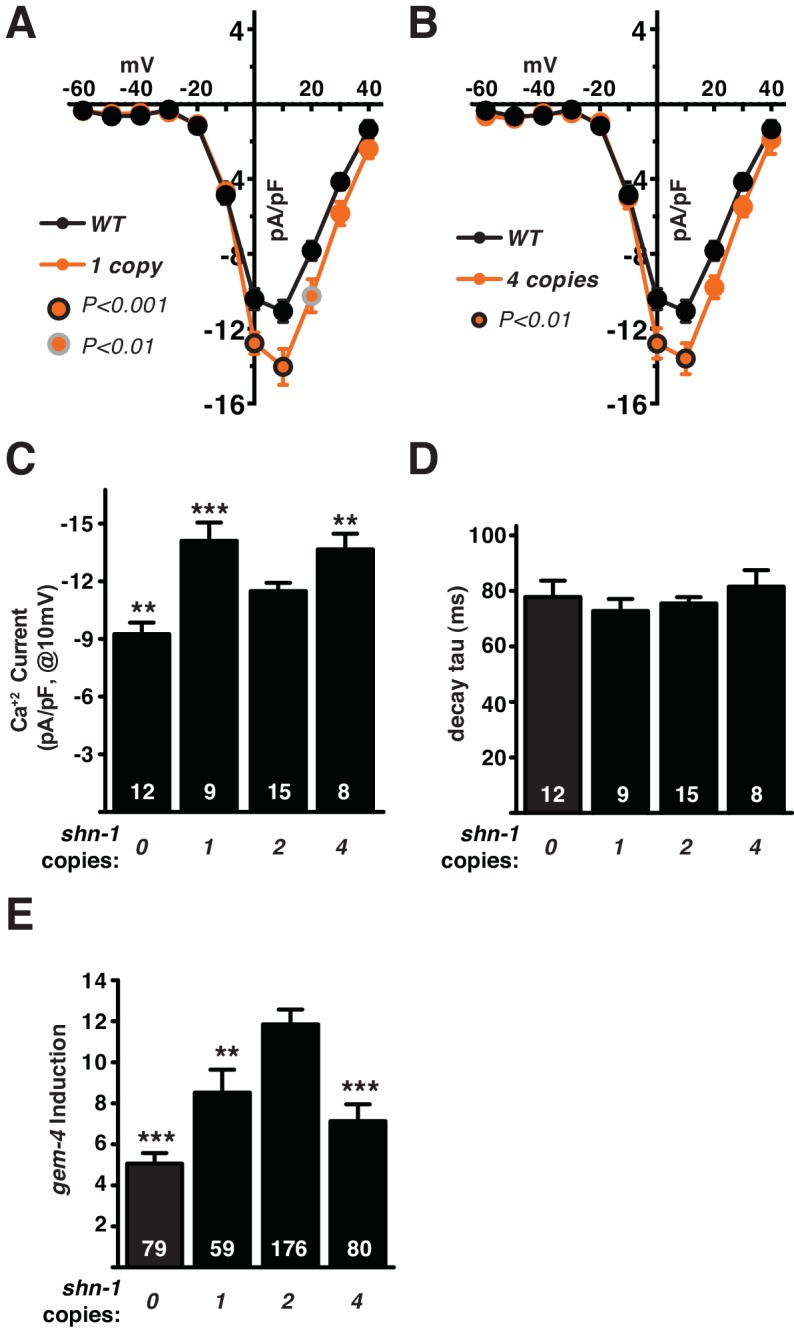
The effect of varying shn-1 gene dosage on calcium current density (A–B) and gem-4 induction (C) was analyzed. The following genotypes were analyzed: 0 copies of shn-1 [shn-1(tm488) homozygotes], 1 copy of shn-1 [shn-1(tm488)/+ heterozygotes], 2 copies of shn-1 (wild-type) and 4 copies of shn-1 (nuSi26 homozygotes in wild-type). (A–B) Muscle Ca+2 current was sensitive to changes in shn-1 gene dose, with decreased (0 shn-1 copies) and increased (1 and 4 shn-1 copies) current density observed in the indicated genotypes. Mean current density as a function of holding potential (A), mean current density at 10 mV (B), and mean current deactivation time constants (C) are shown. (D) Lev-induced gem-4 expression was significantly reduced in animals with 0, 1, and 4 copies of shn-1. The number of animals analyzed is indicated for each genotype. Values that differ significantly from wild type controls are indicated (***p<0.001; **p<0.01). Error bars indicate SEM. Mean, standard errors, sample sizes, and p values for panels A-C are shown in Supplementary file 1.
Cholinergic transmission is not sensitive to shn-1 gene dosage
To determine SHN-1’s role in synaptic transmission, we recorded excitatory post-synaptic currents (EPSCs) from body muscles. Evoked responses were significantly larger in shn-1 null mutants (Figure 7A,C). The shape of evoked responses in wild type and shn-1 mutants were indistinguishable, indicating that the kinetics of evoked release was unaltered (Figure 7B). The rate and amplitude of spontaneous miniature EPSCs (mEPSCs) were unaltered in shn-1 mutants (Figure 7—figure supplement 1A–C). The change in evoked EPSC amplitude combined with unaltered mEPSC amplitudes indicates a pre-synaptic change in cholinergic transmission. The shn-1 null EPSC defect was rescued by transgenes expressing SHN-1 in body muscles, implying that SHN-1 functions post-synaptically (Figure 7B). The evoked EPSCs (Figure 7C) and mEPSCs (Figure 7—figure supplement 1D–E) observed in animals with 1, 2, and 4 shn-1 copies were not significantly different, indicating that synaptic transmission is not sensitive to shn-1 copy number. Similar results were recently reported in mice where glutamatergic transmission in the striatum was enhanced in Shank3B-/- homozygotes but this effect was not observed in Shank3B+/- heterozygotes (Peixoto et al., 2016).
Figure 7. Synaptic transmission at the NMJ is not sensitive to shn-1 gene dosage.
Stimulus-evoked EPSCs were recorded from adult body wall muscles. Representative traces of evoked responses (A), averaged shn-1 and WT evoked responses (normalized to equal amplitudes) (B), and mean evoked charge transfer (C) are shown. Evoked charge was significantly increased in shn-1 homozygotes and this defect was rescued by a single copy transgene (nuSi26) that restored SHN-1 expression in body muscles (C). Averaged peak normalized shn-1 and WT evoked responses have indistinguishable rise and decay times, indicating that rise and decay kinetics were unaltered (B). Evoked charge transfer (D) and peak amplitudes (E) did not differ significantly in animals containing 1, 2, and 4 shn-1 copies. The number of animals analyzed is indicated for each genotype. Values that differ significantly from wild type controls are indicated (**p<0.01; ns, not significant). Error bars indicate SEM. Mean, standard errors, sample sizes, and p values for this figure are shown in Supplementary file 1.

Figure 7—figure supplement 1. shn-1 mutations and gene dosage had no effect on mEPSCs.

Discussion
Because Shank proteins are highly enriched at post-synaptic densities, prior studies focused on the idea that Shank regulates synapse formation or function in some manner (Jiang and Ehlers, 2013). Here we provide evidence that Shank regulates Cav1 calcium current density and activity-induced gene expression, and that these two cellular functions are mediated by distinct dosage-sensitive mechanisms.
SHN-1 promotes EGL-1/Cav1 channel delivery to the cell surface
We find that SHN-1’s PDZ domain binds the EGL-19/Cav1 carboxy-terminus (like their mammalian counterparts) and that disrupting this interaction decreased muscle calcium current. Mutations deleting SHN-1’s PDZ domain and those deleting EGL-19’s PDZ ligand both decreased EGL-19 calcium current and additive defects were not observed in double mutants. These results suggest that SHN-1’s effects on calcium current are mediated by binding of its PDZ domain to EGL-19’s carboxy-terminus. Analysis of gating currents and Terrier reporter fluorescence suggest that the decreased muscle calcium currents in shn-1 mutants arises primarily from decreased trafficking of EGL-19/Cav1 channels to the plasma membrane.
Shank effects on Cav1 current in mammalian neurons or muscles have not been reported. The carboxy-terminus of mammalian Cav1.3 (-ITTLCOOH) and Cav1.2 (-VSSLCOOH) channels are both predicted to bind PDZ domains (Zhang et al., 2005). When mutant constructs lacking the carboxy-terminal PDZ ligands of Cav1.3 or Cav1.2 channels were expressed in hippocampal neurons, no changes in calcium current were observed; however, the Cav1.3 truncation mutant exhibited decreased synaptic localization (Weick et al., 2003; Zhang et al., 2005). The rat Shank 1 and 3 PDZ domains exhibit strong binding to Cav1.3 (but not Cav1.2). Thus, Shank proteins could promote synaptic targeting of Cav1.3; however, the PDZ protein responsible for Cav1.3 targeting has not been identified. These studies relied on over-expression of mutant Cav1 channels (rather than knockin mutations altering the endogenous genes); consequently, it remains possible that Shank proteins could also regulate Cav1 current in mammalian neurons or muscles.
SHN-1 promotes activity-induced expression of gem-4
Prior studies of mammalian neurons proposed that Cav1 channel binding to a PDZ scaffolding protein promotes activity-induced CREB phosphorylation (Weick et al., 2003; Zhang et al., 2005). These studies showed that deleting the carboxy-terminal PDZ ligand in Cav1.2 or 1.3, or over-expressing peptides containing the PDZ-ligands diminish activity-induced CREB phosphorylation and transcription of CREB targets. The PDZ protein responsible for these effects was not identified in these studies. Here we show that SHN-1 promotes expression of a CRH-1/CREB transcriptional target in muscles but that this effect does not require binding of SHN-1’s PDZ domain to EGL-19’s carboxy-terminus. In fact, deleting EGL-19’s PDZ ligand significantly enhanced depolarization induced CRH-1/CREB target expression, perhaps by disrupting EGL-19 interaction with another protein.
Our results suggest that SHN-1’s effects on calcium current density and gem-4 induction are mediated by distinct mechanisms. Increased calcium current is mediated by SHN-1’s PDZ domain binding to EGL-19’s carboxy-terminus whereas another SHN-1 domain (most likely the Ankyrin or Proline-rich domains) enhances CREB activation.
A puzzling aspect of our results is that calcium current density and induced gem-4 expression were poorly correlated. For example, reduced gem-4 induction was observed in mutants with decreased (shn-1 null mutants) and increased (shn-1/+ heterozygotes) EGL-19/Cav1 current density. Similar results were also reported in cultured mammalian neurons, where CREB phosphorylation was poorly correlated with calcium current density and instead was better correlated with Cav1 open probability (Wheeler et al., 2008). Based on these results (and others), these authors proposed that CREB phosphorylation is mediated by a calcium sensor that is spatially very close to (and possibly physically associated with) activated Cav1 channels (Deisseroth et al., 1996; Wheeler et al., 2008). In this scenario, CREB phosphorylation would be strongly correlated with calcium levels in the nanodomain of a Cav1 channel but less correlated with global cytoplasmic calcium. Thus, our results suggest that CRH-1 activation may also be mediated by tight coupling of a calcium sensor to activated EGL-19/Cav1 channels.
Implications for understanding psychiatric disorders
In humans, Shank deletions and duplications both confer risk for ASD and schizophrenia (Bonaglia et al., 2006; Durand et al., 2007; Failla et al., 2007; Gauthier et al., 2010; Han et al., 2013). Thus, too little or too much Shank is associated with psychiatric traits. If Shank CNVs are causally linked to these psychiatric disorders, cellular defects linked to ASD should exhibit similar sensitivity to Shank gene dosage. Consistent with this idea, several studies previously reported cellular and behavioral defects in Shank3m/+ heterozygotes (Han et al., 2016; Orefice et al., 2016; Wang et al., 2016; Yi et al., 2016). Increased Shank3 copy number has been analyzed in a single study (Han et al., 2013), which reported several behavioral and synaptic phenotypes. None of these studies identified phenotypes that were shared between decreased and increased Shank3 dosage. In most cases, it was not determined if cellular defects were cell autonomous consequences of altered Shank3 dosage (Han et al., 2013; Orefice et al., 2016; Wang et al., 2016; Zhou et al., 2016). Here we find that changes in shn-1 gene dosage alter two cell autonomous phenotypes in muscles: L-channel calcium current and expression of CREB target genes. For both phenotypes, similar defects were observed in muscles with decreased and increased shn-1 dosage. Thus, we identify two cellular phenotypes that exhibit the same pattern of dose sensitivity that is observed for Shank3 in schizophrenia and ASD. Based on these results, we propose that human Shank CNVs also cause cellular phenotypes, which may include altered calcium current and CREB target expression. By contrast, other shn-1 mutant phenotypes (e.g. evoked EPSCs) were not sensitive to shn-1 gene dosage. Thus, sensitivity to Shank copy number provides a useful criterion to determine which phenotypes are more likely to contribute to the psychiatric traits associated with Shank CNVs. Two recent studies showed that Shank3 binds directly to Ih and TRPV1 channels in neurons and that the corresponding currents densities are significantly reduced in Shank3+/- heterozygotes (Han et al., 2016; Yi et al., 2016). These results (together with those reported here) suggest that an important function of Shank proteins is to regulate ion channel densities.
How does shn-1 dose alter L-current and CREB target expression? In general, dosage sensitive phenotypes are thought to occur by disrupting the function of multimeric protein complexes. In such cases, under and over-expression of individual subunits alter the stoichiometry of assembled holo-complexes, leading to decreased activity. For example, increased and decreased expression of the yeast histones H2A and H2B results in similar loss of function phenotypes (chromosome loss and altered gene expression) (Clark-Adams et al., 1988; Meeks-Wagner and Hartwell, 1986). Shank binds many other synaptic proteins (Lee et al., 2011; Sakai et al., 2011) and undergoes zinc induced polymerization into large complexes (Baron et al., 2006; Hayashi et al., 2009). Thus, increased and decreased Shank abundance could disrupt the stoichiometry of post-synaptic complexes. Our results suggest that L-current and CREB activation are sensitive to subtle changes in the stoichiometry of Shank protein complexes. Our results further suggest that dynamic changes in the composition of Shank complexes provides a mechanism to regulate circuit function and plasticity (and potentially psychiatric traits).
Shank regulation of L-currents and CREB activation could both contribute to the pathophysiology of ASD. Human Shank mutations are particularly linked to ASD associated with intellectual disability (Leblond et al., 2014). CREB has long been linked to learning and memory in several model organisms (Bourtchuladze et al., 1994; Dash et al., 1990; Yin et al., 1994); consequently, Shank’s CREB activation function could directly contribute to cognitive deficits associated humans Shank CNVs. CREB-induced BDNF expression promotes development of inhibitory synapses in the cortex (Hong et al., 2008). Thus, Shank’s CREB activation function could alter synaptic inhibition, a phenotype found in several ASD models (Dani et al., 2005; Rubenstein and Merzenich, 2003). Finally, L-channels play an important role in calcium signaling in dendrites and spines (Higley and Sabatini, 2012). Thus, Shank’s Cav1 current density function could contribute to cognitive or developmental defects in ASD by adjusting dendritic calcium signaling in CNS neurons. For these reasons, we propose that Shank effects on Cav1 currents and CREB target expression could play an important role in the pathophysiology of ASD (and potentially other psychiatric disorders).
Materials and methods
Experimental procedures
Strains
Strain maintenance and genetic manipulation were performed as described (Brenner, 1974). Animals were cultivated at room temperature (~22°C) on agar nematode growth media seeded with OP50 bacteria. The following strains were used in this study:
KP7624 nuIs525[Pgem-4::NLS-GFP; Pmyo-3::NLS-mCherry] V
KP7598 unc-29(x29) I; nuIs525 V
KP7583 crh-1(tz2) III; nuIs525 V
KP7601 oxSi91[Punc17::ChIEF::mCherry] II; nuIs525 V
KP7618 unc-13(s69) I; nuIs525 V
KP7896 unc-13(s69) I; oxSi91 II; nuIs525 V
KP7032 shn-1(tm488) II
KP7272 shn-1(ok1241) II
KP7461 nuSi26[Pmyo-3::shn-1] shn-1(tm488) II
KP7573 shn-1(tm488) II; nuIs525 V
KP7574 bli-2(e768) shn-1(tm488) II; nuIs525 V
KP7567 nuSi26 II; nuIs525 V
KP7493 nuSi26 II
KP7212 bli-2(e768) shn-1(tm488) II
KP7992 egl-19(nu496) IV
KP7997 egl-19(nu496) IV; nuIs525 V
KP8046 shn-1(tm488) II; egl-19(nu496) IV
KP8047 shn-1(tm488) II; egl-19(nu496) IV; nuIs525 V
KP7991 egl-19(nu495) IV
KP8303 nuSi74 [Pmyo-3::Terrier]
KP8304 nuSi74; shn-1(tm488)
KP8274 nuSi66 [Pmyo-3::NLS::TagBFP2]; nuSi67 [Pcex-1::NLS::mNeonGreen]; nuSi70[Pgem-4::NLS::tagRFPt]
KP8301 nuSi66;nuSi67;nuSi70;shn-1(tm488)
Transgenic animals were prepared by microinjection, and integrated transgenes were isolated following UV irradiation, as described (Dittman and Kaplan, 2006). Single copy transgenes were isolated by the MoSCI and miniMoS techniques (Frøkjaer-Jensen et al., 2008; Frøkjær-Jensen et al., 2014).
shn-1 dosage experiments
Animals with different shn-1 copy numbers were constructed as follows: 0 copies, shn-1(tm488) homozygotes; 1 copy, WT males were crossed with bli-2 shn-1(tm488) homozygotes [KP7212 (for electrophysiology) and KP7574 (for gem-4 induction)] and non-Blister F1 hermaphrodites were analyzed; 2 copies, WT males were crossed with bli-2 homozygotes and non-Blister F1 hermaphrodites were analyzed; 4 copies, WT animals homozygous for the single copy transgene nuSi26. gem-4 reporter expression in bli-2/+ heterozygotes and WT controls did not differ significantly and were pooled for 2 copies of shn-1.
Constructs and transgenes
Pgem-4 reporter
2 kb of 5’ non-coding sequences from the gem-4 gene was cloned into a vector expressing NLS-GFP (pPD122.56). A myo-3 promoter region (2.3 kb) was sub-cloned into a vector expressing NLS-mCherry. Both were injected into WT animals at 5 ng/µl and stable arrays picked. A single extrachromosomal array was integrated by UV irradiation and outcrossed six times (nuIs525).
Pcex-1 reporter
KP#3310 has the cex-1 promoter (1038 bp), a single SV40 NLS, mNeonGreen (Shaner et al., 2013) (codon optimized for C. elegans) followed by the EGL-13 NLS and the cex-1 3’ UTR (2097bp) inserted between the SacII and PstI sites of pCFJ1662. A single hygromycin resistant integrant, nuSi67 was obtained as described (Frøkjær-Jensen et al., 2014). cex-1 reporter expression in each muscle nucleus was normalized to BFP expressed in the same nucleus (using the myo-3 promoter).
myo-3 expression of BFP
KP#3309 has the myo-3 promoter (2386 bp), a single SV40 NLS, mTagBFP2 (Subach et al., 2011) (codon optimized for C. elegans) followed by the EGL-13 NLS and the unc-54 3’ UTR inserted between the SbfI and SnaBI sites of pCFJ901. A single G418 resistant integrant, nuSi66 was obtained and mapped within the abch-1 gene on Chromosome II as described (Frøkjær-Jensen et al., 2014).
Terrier
KP#3308 has the myo-3 promoter, PAT-4 signal sequence (from pPD122.36 Addgene), super-ecliptic pHluorin (Dittman and Kaplan, 2006), PAT-3 transmembrane domain (pPD122.36) followed by the cDNA of EGL-19B coding for residues 1374–1872, tagRFP-T (codon optimized for C. elegans), and the cDNA of EGL-19B coding for residues 1873–1877, followed by the let-858 3’ UTR (pPD122.36) inserted between the HindIII and AflII sites in the polylinker of the miniMOS vector pCFJ910 (Addgene).
Single copy insertions of Terrier were obtained using the miniMOS technique as described (Frøkjær-Jensen et al., 2014).
shn-1 rescue
A C33B4.3a cDNA was cloned using gateway into DONR221. Multisite gateway was used to assemble Pmyo-3::shn-1::unc54UTR into pCFJ150 and a single copy transgene (nuSi26) was obtained by injecting EG6699. The unc-119(ed3) allele was outcrossed from nuSi26 prior to analysis.
Fluorescence imaging
Confocal imaging was performed using an Olympus 60x objective (NA 1.45) on an Olympus FV-1000 confocal microscope at 5x digital zoom. For Pgem-4 imaging, ~15 worms were exposed to 1 mM Lev for 20 mins. Two hours after Lev stimulation, worms were immobilized on 10% agarose pads with 0.3 µl of 0.1 µm diameter polystyrene microspheres (Polysciences 00876–15, 2.5% w/v suspension). Individual muscle nuclei were imaged next to the terminal bulb of the pharynx and analysed using FIJI (https://fiji.sc). The ratio was obtained of green (Pgem-4) to red (Pmyo-3) for each nucleus. A same day WT control (+/- Lev) was analyzed for all genotypes. For terrier imaging ~6–10 worms were immobilized on 10% agarose pads with 0.3 µl of 0.1 µm diameter polystyrene microspheres and for each worm the closest neuromuscular junction to the surface was imaged. Worms were only accepted for analysis if they had clearly identifiable red and green puncta. Both intensity and area were analysed using FIJI.
Retinal plates
4 µl of all trans-retinal (ATR, 100 mM dissolved in ethanol) was mixed with 250 µl of OP50 E. coli and spread on 60 mm NGM plates. Plates were allowed to dry for 24 hr and approx. 40 L4 animals were added and allowed to grow in the dark for 16–24 hr before the assay. Control plates used 4 µl of ethanol mixed with 250 µl of OP50.
Optogenetic stimulation
ACh release at NMJs was evoked in animals expressing the Channelrhodopsin variant ChIEF in cholinergic neurons (oxSi93) (Watanabe et al., 2013). Animals were photo-stimulated with seven 470 nm Rebel LEDs mounted on a 40 mm SinkPadII fitted with a Round Concentrator Lens and powered with a 700 mA DC Driver (Luxeon Star LEDs). 20 min of 25 ms light pulses were generated at the indicated frequencies using an Arduino Uno. Pulse frequency and duration were confirmed using a photodiode and oscilloscope.
qPCR
Total RNA was purified from a synchronized population of young adult worms treated with 200 μM Levamisole for 1 hr and mock-treated samples. RNA was isolated using standard Trizol-bromochloropropane extraction methods in combination with the Qiagen RNeasy Kit. RNA was DNase treated using the Qiagen on-column RNase free DNase set. Samples were prepared from the following genotypes: wild-type (N2 Bristol) and shn-1 (tm488) on two separate days. One micrograms of total RNA was used to synthesize cDNA using RETROscript (Ambion). Real-time PCR was performed using iTaq Universal SYBR Green Supermix (BioRad) and a 7500 Fast Real-Time PCR System (Applied Biosystems). All reactions were run in triplicate and on at least two biological replicates. All the values are normalized to rpl-32 as internal control as well as to the transcript levels in untreated samples. Statistical significance was determined using the two-tailed Student’s t test.
Microarray analysis
RNA isolation and cDNA synthesis was performed as described for qPCR. 6 Affymetrix C. elegans GeneChip were used (3 × 1 hr 200 μM Levamisole exposure and 3x mock-treated samples). Expression values were determined using the Robust Multi-chip Average (RMA) method. Probe sets that showed a > 2 fold change between mock and levamisole treated, with an unadjusted p-value of <0.0001 were considered to be Levamisole responsive. HLH-1 chip-seq data was taken from modencode (http://www.modencode.org). The PWM used for analysis of crh-1 binding sites was from Homer (http://homer.ucsd.edu/homer/).
Electrophysiology
Whole-cell patch-clamp measurements were performed using a Axopatch 200B amplifier with pClamp 10 software (Molecular Devices). The data were sampled at 10 kHz and filtered at 5 kHz. All recordings were performed at room temperature (~19–21°C)
Evoked EPSCs
Worms were superfused in an extracellular solution containing (in mM) 127 NaCl, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 20 glucose, 1 CaCl2 and 4 MgCl2, bubbled with 5% CO2, 95% O2 at 20°C. Whole cell recordings were carried out at –60 mV using an internal solution containing 105 mM CH3O3SCs, 10 mM CsCl, 15 mM CsF, 4 mM MgCl2, 5 mM EGTA, 0.25 mM CaCl2, 10 mM HEPES and 4 mM Na2ATP, adjusted to pH 7.2 using CsOH. Under these conditions, we only observed endogenous acetylcholine EPSCs. For endogenous GABA IPSC recordings the holding potential was 0 mV. All recording conditions were as described (McEwen et al., 2006). Stimulus-evoked EPSCs were stimulated by placing a borosilicate pipette (5–10 µm) near the ventral nerve cord (one muscle distance from the recording pipette) and applying a 0.4 ms, 30 µA square pulse using a stimulus current generator (WPI).
Ca+2 current recordings
The pipette solution contained (in mM): 140 CsCl; 10 TEA-Cl; 5 MgCl2; 5 EGTA; 10 Hepes, pH 7.2, with ~320 mosM CsOH. The extracellular solution contained (in mM): 140 TEA-Cl; 5 CaCl2; 1 MgCl2; 3 4-aminopyridine; 10 glucose; five sucrose; 15 Hepes, pH 7.4, with ~330 mosM CsOH. The voltage-clamp protocol consisted of −60 mV for 50 ms, −90 mV for 50 ms, test voltage (from −60 mV to +4 mV) 200 ms. Access resistance was continuously monitored, and ranged between 7 and 15 MΩ. Series resistance was not compensated. The voltage dependence of the Ca+2 current density were fitted with the equation: I(V) = Gmax(V – Vrev) ⁄ ({1 + exp[(V0.5 – V) ⁄ k]}), where I(V) is the density of the current measured, V is the test pulse, Gmax is the maximum conductance, Vrev is the apparent reversal potential, V0.5 is the half-activation voltage, and k is a steepness factor. The decay tau was well fit by a single exponential function and calculated from a test potential of 0 mV fitting the curve from the peak of the current till the end of the pulse.
Gating currents
The pipet and bath solutions were as described for the calcium current recordings. To resolve gating currents leak and capacitive transients were subtracted using a p/4 protocol, and measured by applying a series of test pulses at 5s intervals from the holding potential of −90 mV to potentials between +40 mV and +50 mV in 2 mV increments and integrating the gating charge movement at the reversal potential for the ionic current.
K+ current recordings
The bath solution contained (in mM): NaCl 140, KCl 5, CaCl2 5, MgCl2 5, dextrose 11 and HEPES 5 (pH 7.2, 320 mOsm); and the pipette solution contained (in mM): KCl 120, KOH 20, Tris 5, CaCl2 0.25, MgCl2 4, sucrose 36, EGTA five and Na2ATP 4 (pH 7.2, 323 mOsm). The voltage-clamp protocol consisted of −60 mV for 50 ms, −90 mV for 50 ms, test voltage (from −60 mV to +60 mV) 1000 ms. IKfast was defined as the peak current after the capacitance transients, and IKslow was defined as the average current of the last 100 ms of each voltage step.
Statistics
Data was assessed for a normal distribution using the D'Agostino-Pearson normality test and equality of variances using the F-Test. For comparisons of normally distributed data with equal variances a two-tailed Student’s t-test was used. For all other comparisons the Mann–Whitney U test was used. For analysis of calcium current-voltage relationships, two-way ANOVA with Sidak’s correction for multiple comparisons was utilized. All statistics were performed in GraphPad Prism 6 and significant differences are indicated as follows: *p<0.05, **p<0.01, and ***p<0.001.
Acknowledgements
We thank the following for strains, advice, reagents, and comments on the manuscript: C. elegans stock center, S Mitani, and members of the Kaplan lab. This work was supported by a postdoctoral fellowship from the Nancy Lurie Marks Family Foundation (EP), and by research grants to JK from the NIH (NS32196) and from the Simons Foundation for Autism Research (SF273555). Additional data described in the manuscript are presented in the supporting online material.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Funding Information
This paper was supported by the following grants:
National Institute of Neurological Disorders and Stroke NS32196 to Joshua M Kaplan.
Simons Foundation SF273555 to Joshua M Kaplan.
Nancy Lurie Marks Family Foundation to Edward Pym.
Additional information
Competing interests
The authors declare that no competing interests exist.
Author contributions
EP, Conceptualization, Investigation, Writing—original draft, Writing—review and editing.
NS, Investigation, Methodology.
KLT-P, Investigation.
DJS, Investigation, Methodology.
AA, Formal analysis, microarray data analysis.
RS, Formal analysis, microarray data analysis.
QH, Investigation, Methodology.
SN, Resources, Investigation.
JMK, Conceptualization, Supervision, Writing—original draft, Writing—review and editing.
Additional files
References
- Baron MK, Boeckers TM, Vaida B, Faham S, Gingery M, Sawaya MR, Salyer D, Gundelfinger ED, Bowie JU. An architectural framework that may lie at the core of the postsynaptic density. Science. 2006;311:531–535. doi: 10.1126/science.1118995. [DOI] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Mani E, Aceti G, Anderlid BM, Baroncini A, Pramparo T, Zuffardi O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. Journal of Medical Genetics. 2006;43:822–828. doi: 10.1136/jmg.2005.038604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Beri S, De Agostini C, Novara F, Fichera M, Grillo L, Galesi O, Vetro A, Ciccone R, Bonati MT, Giglio S, Guerrini R, Osimani S, Marelli S, Zucca C, Grasso R, Borgatti R, Mani E, Motta C, Molteni M, Romano C, Greco D, Reitano S, Baroncini A, Lapi E, Cecconi A, Arrigo G, Patricelli MG, Pantaleoni C, D'Arrigo S, Riva D, Sciacca F, Dalla Bernardina B, Zoccante L, Darra F, Termine C, Maserati E, Bigoni S, Priolo E, Bottani A, Gimelli S, Bena F, Brusco A, di Gregorio E, Bagnasco I, Giussani U, Nitsch L, Politi P, Martinez-Frias ML, Martínez-Fernández ML, Martínez Guardia N, Bremer A, Anderlid BM, Zuffardi O. Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genetics. 2011;7:e1002173. doi: 10.1371/journal.pgen.1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Adams CD, Norris D, Osley MA, Fassler JS, Winston F. Changes in histone gene dosage alter transcription in yeast. Genes & Development. 1988;2:150–159. doi: 10.1101/gad.2.2.150. [DOI] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of rett syndrome. PNAS. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/S0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Kaplan JM. Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. PNAS. 2006;103:11399–11404. doi: 10.1073/pnas.0600784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature Genetics. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failla P, Romano C, Alberti A, Vasta A, Buono S, Castiglia L, Luciano D, Di Benedetto D, Fichera M, Galesi O. Schizophrenia in a patient with subtelomeric duplication of chromosome 22q. Clinical Genetics. 2007;71:599–601. doi: 10.1111/j.1399-0004.2007.00819.x. [DOI] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nature Genetics. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Sarov M, Taylor J, Flibotte S, LaBella M, Pozniakovsky A, Moerman DG, Jorgensen EM. Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nature Methods. 2014;11:529–534. doi: 10.1038/nmeth.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Westenbroek RE, Yu FH, Clark JP, Marshall MR, Scheuer T, Catterall WA. Deletion of the distal C terminus of CaV1.2 channels leads to loss of beta-adrenergic regulation and heart failure in vivo. Journal of Biological Chemistry. 2011;286:12617–12626. doi: 10.1074/jbc.M110.175307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Zhen M. Action potentials drive body wall muscle contractions in Caenorhabditis elegans. PNAS. 2011;108:2557–2562. doi: 10.1073/pnas.1012346108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Champagne N, Lafrenière RG, Xiong L, Spiegelman D, Brustein E, Lapointe M, Peng H, Côté M, Noreau A, Hamdan FF, Addington AM, Rapoport JL, Delisi LE, Krebs MO, Joober R, Fathalli F, Mouaffak F, Haghighi AP, Néri C, Dubé MP, Samuels ME, Marineau C, Stone EA, Awadalla P, Barker PA, Carbonetto S, Drapeau P, Rouleau GA, S2D Team De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. PNAS. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabrucker AM, Schmeisser MJ, Schoen M, Boeckers TM. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends in Cell Biology. 2011;21:594–603. doi: 10.1016/j.tcb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Han K, Holder JL, Schaaf CP, Lu H, Chen H, Kang H, Tang J, Wu Z, Hao S, Cheung SW, Yu P, Sun H, Breman AM, Patel A, Lu HC, Zoghbi HY. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503:72–77. doi: 10.1038/nature12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Kim YH, Wang X, Liu D, Zhang ZJ, Bey AL, Lay M, Chang W, Berta T, Zhang Y, Jiang YH, Ji RR. SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron. 2016;92:1279–1293. doi: 10.1016/j.neuron.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi MK, Tang C, Verpelli C, Narayanan R, Stearns MH, Xu RM, Li H, Sala C, Hayashi Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell. 2009;137:159–171. doi: 10.1016/j.cell.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Calcium signaling in dendritic spines. Cold Spring Harbor Perspectives in Biology. 2012;4:a005686. doi: 10.1101/cshperspect.a005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. The Journal of Physiology. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainé V, Frøkjær-Jensen C, Couchoux H, Jospin M. The alpha1 subunit EGL-19, the alpha2/delta subunit UNC-36, and the beta subunit CCB-1 underlie voltage-dependent calcium currents in Caenorhabditis elegans striated muscle. Journal of Biological Chemistry. 2011;286:36180–36187. doi: 10.1074/jbc.M111.256149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, Giuliano F, Stordeur C, Depienne C, Mouzat K, Pinto D, Howe J, Lemière N, Durand CM, Guibert J, Ey E, Toro R, Peyre H, Mathieu A, Amsellem F, Rastam M, Gillberg IC, Rappold GA, Holt R, Monaco AP, Maestrini E, Galan P, Heron D, Jacquette A, Afenjar A, Rastetter A, Brice A, Devillard F, Assouline B, Laffargue F, Lespinasse J, Chiesa J, Rivier F, Bonneau D, Regnault B, Zelenika D, Delepine M, Lathrop M, Sanlaville D, Schluth-Bolard C, Edery P, Perrin L, Tabet AC, Schmeisser MJ, Boeckers TM, Coleman M, Sato D, Szatmari P, Scherer SW, Rouleau GA, Betancur C, Leboyer M, Gillberg C, Delorme R, Bourgeron T. Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genetics. 2014;10:e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Park H, Park SJ, Kim HJ, Eom SH. The structural flexibility of the shank1 PDZ domain is important for its binding to different ligands. Biochemical and Biophysical Research Communications. 2011;407:207–212. doi: 10.1016/j.bbrc.2011.02.141. [DOI] [PubMed] [Google Scholar]
- Ma H, Cohen S, Li B, Tsien RW. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Bioscience Reports. 2012;33:97–101. doi: 10.1042/BSR20120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen JM, Madison JM, Dybbs M, Kaplan JM. Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC-13. Neuron. 2006;51:303–315. doi: 10.1016/j.neuron.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Meeks-Wagner D, Hartwell LH. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell. 1986;44:43–52. doi: 10.1016/0092-8674(86)90483-6. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Yaoi T, Yasui M, Kuwajima G. N-copine: a novel two C2-domain-containing protein with neuronal activity-regulated expression. FEBS Letters. 1998;428:80–84. doi: 10.1016/S0014-5793(98)00497-9. [DOI] [PubMed] [Google Scholar]
- Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, Ginty DD. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell. 2016;166:299–313. doi: 10.1016/j.cell.2016.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(-/-) mice. Nature Neuroscience. 2016;19:716–724. doi: 10.1038/nn.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome) Molecular Syndromology. 2012;2:186–201. doi: 10.1159/000334260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nature Neuroscience. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain and Behavior. 2003;2:255–267. doi: 10.1034/j.1601-183X.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, Zoghbi HY. Protein interactome reveals converging molecular pathways among autism disorders. Science Translational Medicine. 2011;3:ra49. doi: 10.1126/scitranslmed.3002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature Methods. 2013;10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. PNAS. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika R, Campiglio M, Pinggera A, Lee A, Striessnig J, Flucher BE, Obermair GJ. Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Scientific Reports. 2016;6:34528. doi: 10.1038/srep34528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanakis N, Carrera I, Hobert O. Regulatory logic of Pan-Neuronal gene expression in C. elegans. Neuron. 2015;87:733–750. doi: 10.1016/j.neuron.2015.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Cranfill PJ, Davidson MW, Verkhusha VV. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One. 2011;6:e28674. doi: 10.1371/journal.pone.0028674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, Dutta N, Hayrapetyan V, Yu C, Gaidis E, Zhao S, Ding JD, Xu Q, Chung L, Rodriguiz RM, Wang F, Weinberg RJ, Wetsel WC, Dzirasa K, Yin H, Jiang YH. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nature Communications. 2016;7:11459. doi: 10.1038/ncomms11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Liu Q, Davis MW, Hollopeter G, Thomas N, Jorgensen NB, Jorgensen EM. Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. eLife. 2013;2:e00723. doi: 10.7554/eLife.00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick JP, Groth RD, Isaksen AL, Mermelstein PG. Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. Journal of Neuroscience. 2003;23:3446–3456. doi: 10.1523/JNEUROSCI.23-08-03446.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. The Journal of Cell Biology. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, Südhof TC. Autism-associated SHANK3 haploinsufficiency causes ih channelopathy in human neurons. Science. 2016;352:aaf2669. doi: 10.1126/science.aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of CaV1.3 L-type calcium channels with Shank. Journal of Neuroscience. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, Barak B, Zeng M, Li C, Lu C, Wells M, Amaya A, Nguyen S, Lewis M, Sanjana N, Zhou Y, Zhang M, Zhang F, Fu Z, Feng G. Mice with Shank3 mutations associated with ASD and Schizophrenia display both shared and distinct defects. Neuron. 2016;89:147–162. doi: 10.1016/j.neuron.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]