

Abstract
Neutropenia is a common dose-limiting toxicity associated with irinotecan treatment. While UGT1A1 variants have been associated with neutropenia, a fraction of neutropenia risk remains unaccounted for. To identify additional genetic markers contributing to variability in irinotecan pharmacokinetics and neutropenia, a regression analysis was performed in 78 irinotecan-treated patients to comprehensively analyze three hepatic efflux transporter genes (ABCB1, ABCC1 and ABCG2). rs6498588 (ABCC1) and rs12720066 (ABCB1) were associated with increased SN-38 exposure, and rs17501331 (ABCC1) and rs12720066 were associated with lower absolute neutrophil count nadir. rs6498588 and a variant in high linkage disequilibrium are located in transcriptionally active regions or are predicted to alter transcription factor binding sites. While enhancer activity was not evident in vitro for genomic regions containing these SNPs, rs6498588 was significantly associated with ABCC1 expression in human liver. These results suggest that genetic variation in ABCC1 and ABCB1 may contribute to irinotecan-induced neutropenia by altering expression of transporters involved in irinotecan metabolite disposition.
Introduction
Irinotecan is an antineoplastic prodrug approved for first-line therapy in combination with 5-fluorouracil and leucovorin for patients with metastatic carcinoma of the colon or rectum and as a single agent for second-line therapy of fluorouracil refractory metastatic colorectal cancer. Common dose-limiting toxicities are diarrhea and neutropenia1, with up to 34% of patients experiencing grade 3–4 neutropenia2. Interindividual variation in irinotecan toxicity has prompted the search for genetic biomarkers to guide safer irinotecan treatment.
Irinotecan is activated to form the more potent topoisomerase I inhibiting active metabolite, SN-383. Elevated SN-38 plasma concentrations have been associated with neutropenia during irinotecan treatment4–7. SN-38 is subsequently inactivated to SN-38 glucuronide (SN-38G) by members of the uridine diphosphate glucuronosyl transferase family, primarily UGT1A18 (Figure 1).
Figure 1. Role of ABC transporters and UGT in irinotecan pharmacokinetics and toxicity.
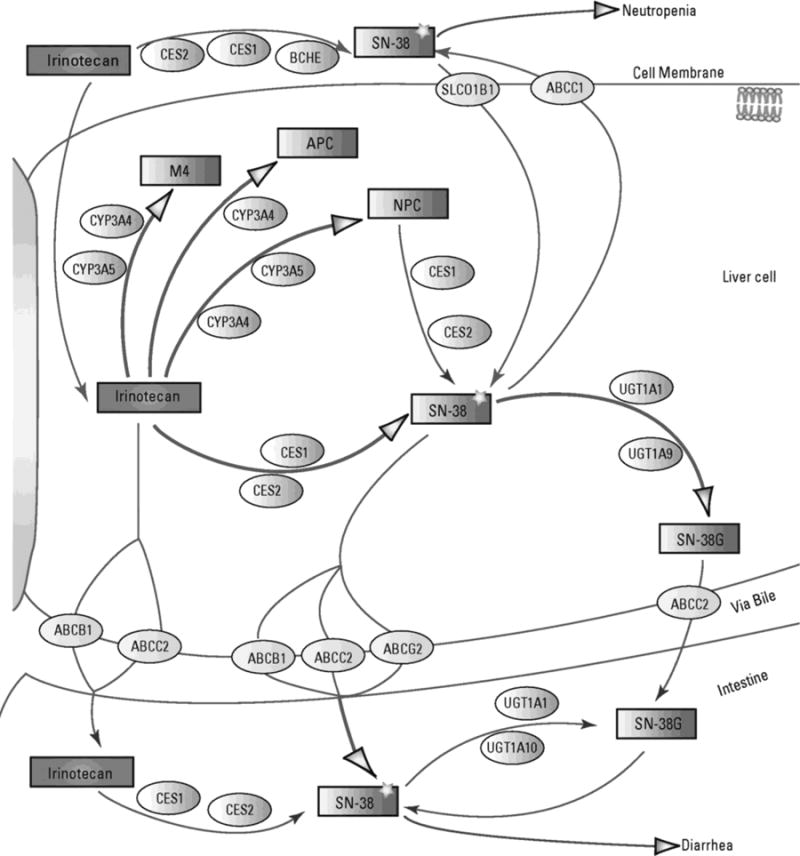
The pathway is taken from PharmGKB. Copyright PharmGKB. Permission has been given by PharmGKB and Stanford University. An original version is available online at https://www.pharmgkb.org/pathway/PA2001.
UGT1A1*28 has been associated with increased risk of neutropenia during irinotecan treatment5,9–12. The TA repeat polymorphism is located in the proximal promoter region of UGT1A1 and results in reduced expression relative to the UGT1A1*1 reference allele. Patients homozygous for UGT1A1*28 glucuronidate SN-38 less efficiently than patients who have one or more reference alleles10; subsequently, homozygous patients are especially susceptible to toxicities resulting from increased SN-38 systemic exposure (as measured by area under the plasma concentration-time curve (AUC)). UGT1A1*93 (rs10929302), a promoter polymorphism in high linkage disequilibrium (LD) with UGT1A1*28, has also been proposed as a predictor of neutropenia, with the variant being strongly associated with increased hematologic toxicity13, increased SN-38 exposure, and lower absolute neutrophil count (ANC) nadir14–16.
While UGT1A1*28-guided irinotecan dosing does reduce dose-limiting toxicities17, variability in SN-38 AUC within genotype-normalized doses suggests that UGT1A1*28 does not account for all the toxicity observed during irinotecan therapy. Other studies have demonstrated more modest effects of UGT1A1 variants and propose that other metabolizing enzymes and drug transporters may be involved18–20.
Irinotecan toxicity and increased SN-38 exposure have been previously associated with several hepatic transporter polymorphisms14,21–23. The ATP-binding cassette (ABC) family includes proteins that transport irinotecan and its metabolites out of the cell24. ABCB1 variants have been associated with increased exposure to irinotecan and SN-3821, reduced irinotecan and SN-38 clearance21,25, and other toxic events15,26. ABCC1 variants were previously associated with ANC nadir, SN-38 AUC, and the ratio of SN-38G AUC to SN-38 AUC14, and a common ABCG2 haplotype was also associated with neutropenia27. However, these associations have yet to be replicated and validated. Compared to UGT1A1, hepatic transporters are relatively unexplored yet could play an important role in the distribution and accumulation of irinotecan and its metabolites.
In the current study, a candidate gene and functional analysis were performed to examine associations of ABCB1, ABCC1, and ABCG2 polymorphisms with irinotecan pharmacokinetics and neutropenia. An earlier pharmacogenetic analysis in this cohort included only a small number of polymorphisms in these three genes14. The present study covers the entire length and flanking regions of these genes for a more comprehensive analysis and tests the hypothesis that ABC transporter variants can contribute to additional variability in neutropenia observed during irinotecan treatment.
Subjects and Methods
Subjects
Human investigations were performed after review and approval by the Biological Sciences Division/University of Chicago Hospitals Institutional Review Board and in accordance with Federalwide Assurance for the protection of human subjects. Informed consent was obtained from all subjects. The study cohort consisted of 85 advanced cancer patients treated with single-agent irinotecan (300 mg/m2 or 350 mg/m2) every 3 weeks. Pharmacokinetic and clinical data, including race, sex, age, body surface area (BSA), SN-38 AUC, ANC nadir, and baseline ANC, were collected. This patient population was examined in previous studies5,14, and thus the sample size was not based on a priori calculation.
Genotyping
Tag single nucleotide polymorphisms (SNP) were selected from genomic regions covering 25 kb upstream of the transcription start site to 5 kb downstream of the 3′-UTR of each candidate gene (ABCB1, ABCC1, ABCG2). Additional SNPs were chosen to directly interrogate nonsynonymous SNPs in the UCSF Pharmacogenetics of Membrane Transporters database (http://pharmacogenetics.ucsf.edu/), SNPs with published clinical associations, and SNPs in LD with clinically associated SNPs. Fifty ABCB1 SNPs, 93 ABCC1 SNPs, and 38 ABCG2 tag SNPs were assayed. Genotyping was performed by the Mayo Clinic Genotyping Shared Resource using the Illumina VeraCode (BeadXpress) platform. Following quality control measures to eliminate suboptimal samples and markers and filtering for minor allele frequency ≥ 0.05 using PLINK v1.0728, 85 individuals and 123 SNPs were retained for analysis (Supplementary Table 1).
Statistical analysis
SNPs were tested for deviation from Hardy-Weinberg equilibrium using an exact test. Genotype-phenotype associations were analyzed using a two-stage regression analysis in R v3.2.229. Dose-adjusted SN-38 AUC and ANC nadir values were log10-transformed for the analysis. In the univariate analysis, simple regressions were first performed on each variant and clinical covariate separately, and those with a significance level of P < 0.15 were kept for further analysis. Step-wise backward selection of this subset of genotypes using the rms package30 for R led to the final multivariable models, and model fit was assessed by examining R2 values. An additive genetic model was first assumed for all genotype associations. To eliminate the driving effect of any sparse homozygous variants, when a particular variant had n ≤3 observations in the homozygous genotype group and was significant, these genotypes were grouped with the heterozygous genotypes; the variant was kept in the model if still significant after combining genotypes.
Other statistical analyses were performed using GraphPad Prism v6.0 (GraphPad Software, La Jolla, CA, USA). Comparisons for the risk alleles analysis and luciferase reporter assays were analyzed using a Mann-Whitney test or, if there were more than two groups, a Kruskal-Wallis test followed by Dunn’s multiple comparisons test. Variance was assumed to be similar between compared groups, and two-sided P-values < 0.05 were considered statistically significant.
Bioinformatic analysis
Significant SNPs and SNPs in high LD (r2 ≥ 0.8) were examined for evidence of changes in function and regulatory activity. HaploReg31 and RegulomeDB32 were used to determine whether the SNPs overlap with experimentally predicted functional elements (chromatin state segmentation33, DNase hypersensitivity peaks, ChIP-seq peaks) from the ENCODE Project34 and the Roadmap Epigenomics Project35. HaploReg31 and TRANSFAC Match36 were used to identify and predict changes in transcription factor (TF) binding.
Measurement of enhancer activity in vitro
Primers were designed with the aid of Primer337,38 to target 500 bp genomic regions flanking each significant SNP (Supplementary Table 2). These regions were PCR-amplified using PfuTurbo (Agilent, Santa Clara, CA, USA) from human genomic DNA and cloned into the pGL4.23[luc2/minP] luciferase reporter vector (Promega, Madison, WI, USA). The QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) was used to create the desired point mutation. Correct sequences were verified by Sanger sequencing. Reporter plasmids containing reference or variant sequences were transfected into Hep G2 (ATCC® HB-8065™, human hepatocellular carcinoma, passage 2–6) cells using Lipofectamine LTX and PLUS reagents (Invitrogen, Carlsbad, CA, USA). The Renilla luciferase construct pGL4.73 (Promega) was co-transfected to correct for transfection efficiency. Empty pGL4.23 vector and a known apolipoprotein E liver enhancer39 were also transfected as negative and positive controls, respectively. Cells were cultured in Eagle’s Minimum Essential Medium with EBSS, non-essential amino acids, 2 mM L-glutamine, 1 mM sodium pyruvate, 1500 mg/L sodium bicarbonate, and 10% fetal bovine serum. Twenty-four hours after transfection, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) and quantified using the GloMax-96 Microplate Luminometer (Promega). Background-subtracted firefly luciferase activity was normalized with respect to Renilla luciferase activity and expressed as a mean value relative to empty pGL4.23 vector.
Code availability
The R code for the univariate and multivariate regression analyses is available at https://github.com/kroetzlab/irinotecan-ABC.
Results
Cohort analysis
The original study cohort consisted of 85 patients, with the majority being Caucasian or African American. Seven subjects (2 Asian, 1 Filipino, 4 Hispanic) were omitted from the initial regression analyses to avoid dilution of association signals by genetic heterogeneity. Characteristics of patients included in the primary analysis are listed in Supplementary Table 3. The analyzed cohort had a median age of 57 and slightly more males (58%). The majority (76%) received a 350 mg/m2 dose of irinotecan, while 24% received a 300 mg/m2 dose. The outcomes examined in this study were SN-38 AUC (adjusted for irinotecan dose) and ANC nadir. Distributions of these phenotypes in the analyzed cohort are shown in Supplementary Figure 1. None of the clinical covariates (race, sex, age, BSA, baseline ANC) were significantly associated with SN-38 AUC or ANC nadir.
Association of ABC SNPs with SN-38 exposure
Eight SNPs met the P < 0.15 threshold in univariate regression analyses for SN-38 exposure. Of these, rs12720066, rs6498588, rs35621, and rs2373586 had P-values < 0.05 (Table 1). The genotype frequencies for these SNPs in Caucasians and African Americans are shown in Supplementary Table 4. All eight ABC SNPs were taken into the multivariate analysis along with the UGT1A1*93 genotype (rs10929302). The final log SN-38 AUC model included UGT1A1*93, an intronic ABCB1 variant (rs12720066), and an ABCC1 variant (rs6498588) in the 5′-flanking region (Table 1). The model explained 34% (R2 = 0.345) of the variation in SN-38 exposure, with all three SNPs contributing similar variances to the model (rs12720066: β = −0.337; rs6498588: β = 0.325; rs10929302: β = 0.390). The UGT1A1 and ABCC1 SNPs were associated with increased exposure of patients to SN-38, while the ABCB1 SNP was associated with decreased exposure (Figure 2).
Table 1.
Univariate and multivariate analysis results for log SN-38 AUC1
| Univariate | Multivariate3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| rsid | Gene | Function | Ref>Alt | Estimate | SE | P | Estimate | SE | P |
| rs127200662 | ABCB1 | intronic | A>C | −0.204 | 0.070 | 0.005 | −0.217 | 0.061 | 6.24 × 10−4 |
| rs6498588 | ABCC1 | 5′-gene | A>T | 0.111 | 0.042 | 0.010 | 0.123 | 0.036 | 9.50 × 10−4 |
| rs356212 | ABCC1 | intronic | C>T | 0.191 | 0.083 | 0.025 | |||
| rs2373586 | ABCB1 | intronic | A>C | 0.086 | 0.040 | 0.033 | |||
| rs8187843 | ABCC1 | intronic | G>A | −0.160 | 0.084 | 0.061 | |||
| rs37435272 | ABCC1 | 3′-UTR | C>T | 0.113 | 0.062 | 0.071 | |||
| rs2150952 | ABCC1 | intronic | G>A | 0.118 | 0.073 | 0.109 | |||
| rs212082 | ABCC1 | intronic | A>G | −0.080 | 0.054 | 0.142 | |||
| rs10929302 | UGT1A1 | promoter | G>A | 0.145 | 0.042 | 0.001 | 0.154 | 0.037 | 9.00 × 10−5 |
SN-38 AUC expressed in units of ng*hr/mL; adjusted for irinotecan dose
Homozygous alternate genotypes grouped with heterozygous genotypes
R2 = 0.345
Figure 2. Association of ABC and UGT variants with SN-38 AUC.
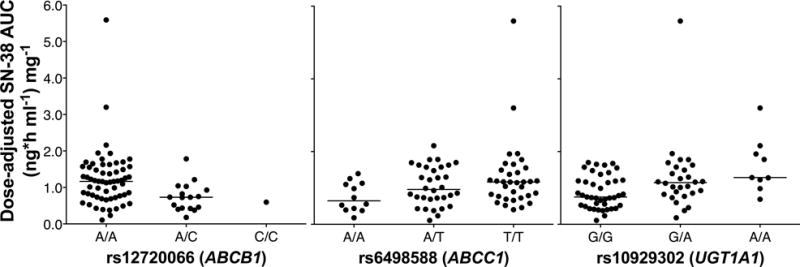
The relationship between genotype and SN-38 AUC (adjusted for irinotecan dose) is shown for all analyzed patients. Horizontal bars represent the median values for each genotype group.
Inclusion of only Caucasian genotypes in the association analyses yielded similar results as obtained with the combined population (Supplementary Table 5). The multivariable model for log SN-38 AUC also contained UGT1A1*93, rs12720066, and rs6498588, with similar effect sizes as those for the model including African Americans. Inclusion of the seven Asian, Filipino, and Hispanic subjects into the final model (Supplementary Table 6) resulted in similar coefficients and P-values.
Association of ABC SNPs with neutropenia
In univariate linear regression analyses of ABC SNPs with ANC nadir, ten SNPs met the P < 0.15 threshold and three (rs17501331, rs12720066, and rs3743527) had P-values < 0.05 (Table 2). Genotype frequencies of these SNPs are listed in Supplementary Table 4. The final model following multivariable regression and inclusion of UGT1A1*93 included an intronic SNP in ABCB1 (rs12720066) and an intronic SNP in ABCC1 (rs17501331) (Table 2). This model accounted for 39% (R2 = 0.390) of the variation in ANC nadir, with UGT1A1*93 explaining more variance in the outcome (β = −0.479) than the other two SNPs (rs17501331: β = −0.295; rs12720066: β = 0.286). The ABCB1 SNP was associated with increased ANC nadir and the UGT1A1 and ABCC1 SNPs were associated with reduced nadirs (Figure 3). As for the SN-38 AUC analysis, the final multivariable model for log ANC nadir for Caucasians only contained the same SNPs with similar effect sizes as those for the model including African Americans (Supplementary Table 7). Inclusion of the seven Asian, Filipino, and Hispanic subjects into the final model (Supplementary Table 8) also resulted in similar coefficients and P-values.
Table 2.
Univariate and multivariate analysis results for log ANC nadir1
| Univariate | Multivariate3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| rsid | Gene | Function | Ref>Alt | Estimate | SE | P | Estimate | SE | P |
| rs17501331 | ABCC1 | intronic | A>G | −0.255 | 0.106 | 0.019 | −0.281 | 0.088 | 0.002 |
| rs127200662 | ABCB1 | intronic | A>C | 0.227 | 0.102 | 0.030 | 0.261 | 0.084 | 0.003 |
| rs37435272 | ABCC1 | 3′-UTR | C>T | −0.194 | 0.089 | 0.033 | |||
| rs212090 | ABCC1 | 3′-UTR | T>A | 0.102 | 0.059 | 0.089 | |||
| rs1128503 | ABCB1 | synonymous | A>G | −0.097 | 0.056 | 0.090 | |||
| rs8050881 | ABCC1 | 5′-gene | A>G | −0.098 | 0.061 | 0.110 | |||
| rs2725264 | ABCG2 | intronic | C>T | −0.127 | 0.080 | 0.118 | |||
| rs35626 | ABCC1 | intronic | G>T | −0.107 | 0.069 | 0.123 | |||
| rs1967120 | ABCC1 | intronic | G>A | −0.108 | 0.070 | 0.124 | |||
| rs152023 | ABCC1 | intronic | C>T | −0.093 | 0.063 | 0.144 | |||
| rs10929302 | UGT1A1 | promoter | G>A | −0.262 | 0.057 | 1.98 × 10−5 | −0.268 | 0.052 | 1.99 × 10−6 |
ANC nadir expressed as cells/μL
Homozygous alternate genotypes grouped with heterozygous genotypes
R2 = 0.390
Figure 3. Association of ABC and UGT variants with ANC nadir.
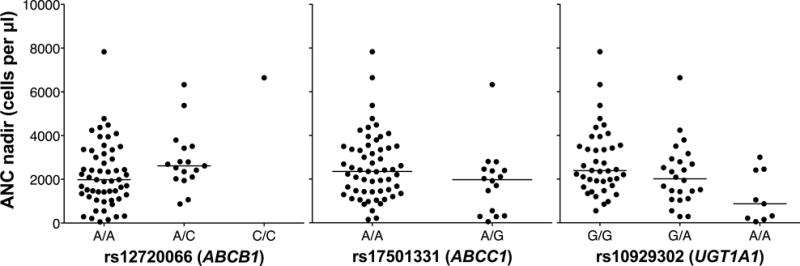
The relationship between genotype and absolute neutrophil count nadir is shown for all analyzed patients. Horizontal bars represent the median values for each genotype group.
Contribution of multiple risk alleles
To examine the combined effects of risk alleles, composite genotypes were constructed for each outcome. Patients carrying more risk alleles for rs12720066, rs6498588, and UGT1A1*93 had significantly higher SN-38 AUC (P < 0.0001), while patients carrying more risk alleles for rs12720066, rs17501331, and UGT1A1*93 had significantly lower ANC values (P = 0.0006) (Supplementary Figure 2). Almost all (91%) patients with grade 3–4 neutropenia (< 1000 cells/μl) carried at least four risk alleles among all three ABC SNPs and UGT1A1*93, compared to 53% of patients with no or less severe neutropenia (P = 0.001). Of the patients with grade 3–4 neutropenia, 91% are homozygous for rs12720066 and 46% are homozygous for UGT1A1*93, while of patients with no or less severe neutropenia, 73% are homozygous for rs12720066 and 7% are homozygous for UGT1A1*93. Dose-adjusted SN-38 AUC was also confirmed to be higher in patients with grade 3–4 neutropenia (Supplementary Figure 3; P = 0.0001).
Bioinformatic analysis
To investigate the putative function of the SNPs in the final models, overlapping regulatory elements and protein binding sites were examined. The intronic ABCC1 SNP rs17501331 is in a predicted region of transcriptional transition in Hep G2 cells and is predicted to alter two regulatory motifs, DMRT1 and SOX6. rs6498588, located 11 kb upstream of ABCC1, is predicted to alter three regulatory motifs (GR, Mef2, YY1). rs4148330, a SNP in high LD (r2 = 0.8) with rs6498588, resides 1.7 kb upstream of ABCC1 in a region with H3K4 methylation in Hep G2 cells and DNase hypersensitivity in Hep G2 cells and hepatocytes, and is predicted to alter E2F and NRSF TF binding. rs12720066 and other SNPs in high LD with rs6498588 were also predicted to alter regulatory motifs but were not associated with an enrichment in enhancer or DNase activity in liver or liver-derived cells.
Enhancer assays
Genomic regions containing the ABCB1 and ABCC1 SNPs in the final models for SN-38 AUC and ANC nadir plus additional SNPs in high LD with these variants (Supplementary Table 2) were tested in vitro for enhancer activity and the effect of genetic variation on this activity. None of the tested genomic regions exhibited significant enhancer activity and the introduction of the indicated variants did not change the transcriptional activity (Figure 4).
Figure 4. Analysis of genomic regions and genetic variants for enhancer activity.
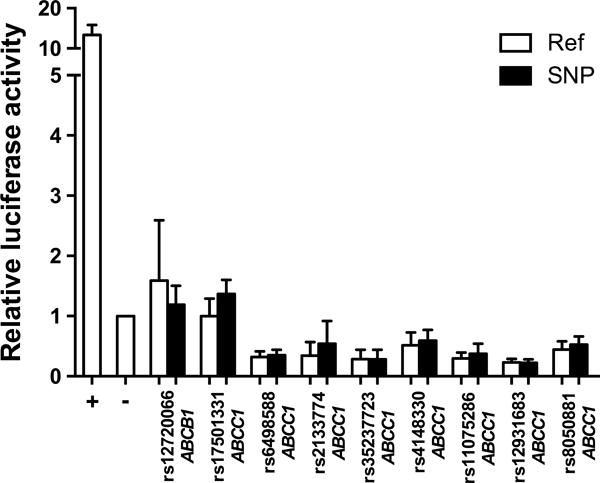
Enhancer activity was measured in Hep G2 cells using a luciferase reporter gene assay. The values plotted represent mean ± SD of the relative luciferase activity in cells transfected with the indicated genomic region without (Ref) and with (SNP) the variant compared to empty pGL4.23 construct (−). A known liver enhancer of the APOE gene was used as the positive (+) control. Data are representative of at least three independent experiments of six biological replicates each.
Discussion
This study provides a comprehensive analysis of the genetic variants spanning three hepatic ABC transporter genes of the irinotecan pharmacokinetic pathway. Three SNPs (rs12720066, rs6498588, rs17501331) were significantly associated with changes in SN-38 AUC or ANC nadir. These variants could potentially predict neutropenia during irinotecan treatment and provide additional insight into the molecular mechanism of the toxicity.
UGT1A1*93 (rs10929302) was confirmed as a strong predictor of irinotecan-induced neutropenia, reflected by associations with higher SN-38 AUC and lower ANC nadirs. The effect sizes of UGT1A1*93 for associations with SN-38 AUC or ANC nadir were similar or slightly more significant than those of UGT1A1*28. UGT1A1*93 was previously associated with increased hematologic toxicity, increased SN-38 exposure, and lower ANC nadir13–15. A recent study confirmed UGT1A1*93 as a more robust marker for neutropenia than UGT1A1*2816.
The three ABC transporter SNPs in the final multivariable models are in noncoding regions, either intronic or upstream of their respective genes, and are not in high linkage disequilibrium with any coding SNPs. This was expected, considering the low number of high-frequency coding SNPs in these genes. Previous studies of the effects of nonsynonymous SNPs on the expression and activity of these ABC transporters have yielded mostly negative or inconclusive results40–42. Of the SNPs with P < 0.15 in the univariate analyses, only one (rs1128503) is in a coding region and one intronic SNP (rs2373586) is in high LD with a common missense SNP, rs2032582. rs1128503 and rs2032582 are both in ABCB1 and, together with rs1045642, make up the ABCB1*2 haplotype. This haplotype has been previously associated with early toxicity during irinotecan treatment26. However, these SNPs dropped out of significance during the selection of the multivariable models. These coding SNPs have been frequently studied together and individually for association with transporter activity and changes in exposure and response to numerous drugs, but with discordant results40.
In contrast to coding SNPs, due to lower selective pressures and more modest effects, variants in the regulatory regions of membrane transporter genes are more abundant and exist at higher allele frequencies43,44. Functional studies of variants in the proximal promoter44–47, untranslated48,49 and intronic50,51 regions of ABC and SLCO transporter genes have demonstrated changes in mRNA or protein expression levels and interindividual variation in pharmacokinetics52,53. In light of this, it is likely that noncoding genetic variation has a more widespread impact on transporter function. Our results highlight the importance of including noncoding SNPs in pharmacogenetic studies and the need to further characterize regulatory regions in pharmacogenes.
The significant variants identified in this study may contribute to irinotecan-induced neutropenia by altering expression of ABCB1 or ABCC1. ABCB1 encodes P-glycoprotein, which is involved in irinotecan and SN-38 biliary excretion54,55. An increase in ABCB1 expression could result in increased biliary secretion of SN-38 and a corresponding reduction in plasma SN-38 levels, which are inversely correlated with ANC nadir. ABCC1 encodes MRP1, a hepatic basolateral efflux transporter of SN-3856. Increased ABCC1 expression is hypothesized to result in increased systemic levels of SN-38, which is consistent with the decreased ANC nadir associated with this variant. It should be noted that ABCC1/MRP1 expression in the liver is relatively low57, so increased expression may have a significant impact on basolateral efflux of SN-38 back into the blood.
Bioinformatic analyses predicted 5′-flanking ABCC1 SNP rs6498588 to alter TF binding and rs4148330 (in LD with rs6498588) to have both enhancer activity in hepatic cells and effects on TF binding. A preliminary eQTL meta-analysis of human liver samples found rs6498588 to be significantly associated (meta-adjusted P = 0.05) with increased ABCC1 expression58. These data are consistent with the association of rs6498588 with increased SN-38 AUC, as higher ABCC1 expression would result in increased transport of SN-38 into the blood. However, our in vitro enhancer assays did not confirm these predictions for either rs6498588 or rs4148330. Enhancer assays conducted in additional hepatic cell lines or with different-sized genomic regions are warranted.
A combined allele analysis showed that a higher number of risk alleles from our models is correlated with irinotecan-induced toxicity. A significantly greater proportion of patients with grade 3–4 neutropenia carried at least four risk alleles for both phenotypes when compared to the group with no or less severe neutropenia. rs12720066 and UGT1A1*93 appear to be driving this effect, with the directions of their effects consistent with the toxicity in both pharmacokinetic and pharmacodynamic outcomes. While rs6498588 was only in the final SN-38 AUC model, a SNP in high LD (rs8050881) had a weaker univariate association with ANC nadir; the variant alleles of these SNPs are also associated with the toxicity in both outcomes. However, the contribution of rs17501331 is less clear; this variant was only associated with ANC nadir and not at all with SN-38 AUC, suggesting a mechanism that may affect neutrophil count independent of SN-38 exposure. Given the complexity of the many metabolizing enzymes and transporters acting on SN-38, further eQTL and functional characterization studies are needed to validate the effect of these SNPs before proposing a combined PK/PD model.
The addition of rs6498588 to our previously published model14 of log SN-38 AUC containing rs35605 (ABCC1), rs10276036 (ABCB1) and UGT1A1*93 improved the percent variation of SN-38 AUC explained by genetic determinants (adjusted R2 increased from 0.221 to 0.284, P = 0.009, ANOVA), with all four variants in the model remaining significant. When rs12720066 and rs17501331 were added to our previous model of log ANC nadir14 containing rs3765129 (ABCC1), rs2306283 (SLCO1B1), and UGT1A1*93, only rs17501331 had marginal significance (P = 0.05) and a smaller effect than the other variants in the initial model. The previously identified SNPs, especially UGT1A1*93, may explain more of the genetic variation in ANC nadir. Again, a better mechanistic understanding of this toxicity is needed before proposing a predictive multivariable model for irinotecan-induced neutropenia.
There are no known clinical associations of the variants identified in the current association studies. However, there is still enough compelling functional evidence to suggest that genetic variants in ABCB1 and ABCC1 are relevant to irinotecan toxicity. MRP1-overexpressing KB-3–1 cells showed increased SN-38 resistance compared to normal cells56, affirming that expression of the transporter does affect SN-38 transport. In ABCB1 knockout mice, irinotecan and SN-38 plasma levels were increased relative to the wildtype mice, consistent with a role for P-gp in biliary secretion of this drug/metabolite pair55. Cell lines overexpressing BCRP (encoded by ABCG2) are also resistant to irinotecan59 and SN-3860, and ABCG2 421C>A reduced gene expression and conferred irinotecan resistance in cancer cell lines61. In the current study, there was no association with any of the tested ABCG2 variants and SN-38 exposure or ANC nadir.
Other genes in the irinotecan pharmacokinetic pathway including ABCC2, SLCO1B1, CES1, CES2, CYP3A4 and additional variants in UGT1A1 and other UGT genes may also contribute to SN-38 exposure and neutropenia and warrant further study62–64. In contrast, genetic variation in genes responsible for irinotecan pharmacodynamics do not seem to play a role65. More comprehensive genotyping of pharmacokinetic genes has the potential to identify additional genetic variants more strongly associated with irinotecan exposure and toxicity. Discovery of additional genetic and non-genetic determinants of irinotecan-induced neutropenia will improve the predictive power of a clinical pharmacogenetic algorithm over using a single UGT1A1*28 test for drug selection and dosing. Replication and prospective studies in large independent cohorts as well as in other ethnic groups are necessary to validate and expand these findings.
A current gap in the field of pharmacogenetics is understanding the clinical value of variants. Our study used a pharmacokinetic/pharmacodynamic phenotyping approach to discover new variants associated with neutropenia during irinotecan treatment. Further investigation of these variants and others in ABC transporter genes may not only potentially contribute to the improvement of individualized treatment with irinotecan, but may also have broader implications for other substrates of ABC transporters.
Supplementary Material
Acknowledgments
This work was supported by NIH/NIGMS U01GM061390 (Pharmacogenetics of Membrane Transporters), NIH/NIGMS U01GM0061393 (PAAR-Pharmacogenomics of Anticancer Agents Research Group), NIH/NIGMS T32 GM007175, NIH/NIDDK R21DK081157, and NIH/NCI K07CA140390-01.
Footnotes
Conflict of Interest
Drs. Innocenti and Ratain are patent holders on the UGT1A1 testing for irinotecan neutropenia. Dr. Ratain also receives intermittent royalties on multiple patents related to irinotecan pharmacogenetics and is an inventor on a pending patent application for a genomic prescribing system. The remaining authors declare no conflicts of interest.
Supplementary information is available at The Pharmacogenomics Journal’s website.
References
- 1.de Forni M, Bugat R, Chabot GG, Culine S, Extra JM, Gouyette A, et al. Phase I and pharmacokinetic study of the camptothecin derivative irinotecan, administered on a weekly schedule in cancer patients. Cancer Res. 1994;54:4347–4354. [PubMed] [Google Scholar]
- 2.Fuchs CS, Moore MR, Harker G, Villa L, Rinaldi D, Hecht JR. Phase III comparison of two irinotecan dosing regimens in second-line therapy of metastatic colorectal cancer. J Clin Oncol. 2003;21:807–814. doi: 10.1200/JCO.2003.08.058. [DOI] [PubMed] [Google Scholar]
- 3.Slatter JG, Su P, Sams JP, Schaaf LJ, Wienkers LC. Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions. Drug Metab Dispos. 1997;25:1157–1164. [PubMed] [Google Scholar]
- 4.Ramchandani RP, Wang Y, Booth BP, Ibrahim A, Johnson JR, Rahman A, et al. The role of SN-38 exposure, UGT1A1*28 polymorphism, and baseline bilirubin level in predicting severe irinotecan toxicity. J Clin Pharmacol. 2007;47:78–86. doi: 10.1177/0091270006295060. [DOI] [PubMed] [Google Scholar]
- 5.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 6.Minami H, Sai K, Saeki M, Saito Y, Ozawa S, Suzuki K, et al. Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese: roles of UGT1A1*6 and *28. Pharmacogenet Genomics. 2007;17:497–504. doi: 10.1097/FPC.0b013e328014341f. [DOI] [PubMed] [Google Scholar]
- 7.Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, et al. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol. 2006;24:2237–2244. doi: 10.1200/JCO.2005.03.0239. [DOI] [PubMed] [Google Scholar]
- 8.Iyer L, King CD, Whitington PF, Green MD, Roy SK, Tephly TR, et al. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest. 1998;101:847–854. doi: 10.1172/JCI915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, et al. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 2000;60:6921–6926. [PubMed] [Google Scholar]
- 10.Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 11.Rouits E, Boisdron-Celle M, Dumont A, Guerin O, Morel A, Gamelin E. Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin Cancer Res. 2004;10:5151–5159. doi: 10.1158/1078-0432.CCR-03-0548. [DOI] [PubMed] [Google Scholar]
- 12.McLeod HL, Parodi L, Sargent DJ, Marsh S, Green E, Abreu P, et al. UGT1A1*28, toxicity and outcome in advanced colorectal cancer: results from trial N9741. J Clin Oncol. 2006;24(Suppl 18) Abstract 3520. [Google Scholar]
- 13.Côté JF, Kirzin S, Kramar A, Mosnier JF, Diebold MD, Soubeyran I, et al. UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin Cancer Res. 2007;13:3269–3275. doi: 10.1158/1078-0432.CCR-06-2290. [DOI] [PubMed] [Google Scholar]
- 14.Innocenti F, Kroetz DL, Schuetz E, Dolan ME, Ramírez J, Relling M, et al. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol. 2009;27:2604–2614. doi: 10.1200/JCO.2008.20.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lara PN, Jr, Natale R, Crowley J, Lenz HJ, Redman MW, Carleton JE, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530–2535. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crona DJ, Ramirez J, Qiao W, de Graan AJ, Ratain MJ, van Schaik RH, et al. Clinical validity of new genetic biomarkers of irinotecan neutropenia: an independent replication study. Pharmacogenomics J. 2015 doi: 10.1038/tpj.2015.23. e-pub ahead of print 14 April 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Innocenti F, Schilsky RL, Ramírez J, Janisch L, Undevia S, House LK, et al. Dose-finding and pharmacokinetic study to optimize the dosing of irinotecan according to the UGT1A1 genotype of patients with cancer. J Clin Oncol. 2014;32:2328–2334. doi: 10.1200/JCO.2014.55.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcuello E, Altés A, Menoyo A, Del Rio E, Gómez-Pardo M, Baiget M. UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br J Cancer. 2004;91:678–682. doi: 10.1038/sj.bjc.6602042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braun MS, Richman SD, Thompson L, Daly CL, Meade AM, Adlard JW, et al. Association of molecular markers with toxicity outcomes in a randomized trial of chemotherapy for advanced colorectal cancer: the FOCUS trial. J Clin Oncol. 2009;27:5519–5528. doi: 10.1200/JCO.2008.21.6283. [DOI] [PubMed] [Google Scholar]
- 20.Schulz C, Heinemann V, Schalhorn A, Moosmann N, Zwingers T, Boeck S, et al. UGT1A1 gene polymorphism: impact on toxicity and efficacy of irinotecan-based regimens in metastatic colorectal cancer. World J Gastroenterol. 2009;15:5058–5066. doi: 10.3748/wjg.15.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathijssen RH, Marsh S, Karlsson MO, Xie R, Baker SD, Verweij J, et al. Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin Cancer Res. 2003;9:3246–3253. [PubMed] [Google Scholar]
- 22.Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY, et al. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer. 2007;110:138–147. doi: 10.1002/cncr.22760. [DOI] [PubMed] [Google Scholar]
- 23.Han JY, Lim HS, Park YH, Lee SY, Lee JS. Integrated pharmacogenetic prediction of irinotecan pharmacokinetics and toxicity in patients with advanced non-small cell lung cancer. Lung Cancer. 2009;63:115–120. doi: 10.1016/j.lungcan.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat. 2003;6:71–84. doi: 10.1016/s1368-7646(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 25.Sai K, Kaniwa N, Itoda M, Saito Y, Hasegawa R, Komamura K, et al. Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecan. Pharmacogenetics. 2003;13:741–757. doi: 10.1097/00008571-200312000-00005. [DOI] [PubMed] [Google Scholar]
- 26.Glimelius B, Garmo H, Berglund A, Fredriksson LA, Berglund M, Kohnke H, et al. Prediction of irinotecan and 5-fluorouracil toxicity and response in patients with advanced colorectal cancer. Pharmacogenomics J. 2011;11:61–71. doi: 10.1038/tpj.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sai K, Saito Y, Maekawa K, Kim SR, Kaniwa N, Nishimaki-Mogami T, et al. Additive effects of drug transporter genetic polymorphisms on irinotecan pharmacokinetics/pharmacodynamics in Japanese cancer patients. Cancer Chemother Pharmacol. 2010;66:95–105. doi: 10.1007/s00280-009-1138-y. [DOI] [PubMed] [Google Scholar]
- 28.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. https://www.R-project.org/ [Google Scholar]
- 30.Harrell FE. rms: Regression Modeling Strategies. R package version 4.4-0. 2015 http://CRAN.R-project.org/package=rms.
- 31.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roadmap Epigenomics Consortium. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kel AE, Gössling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 39.Simonet WS, Bucay N, Lauer SJ, Taylor JM. A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J Biol Chem. 1993;268:8221–8229. [PubMed] [Google Scholar]
- 40.Hodges LM, Markova SM, Chinn LW, Gow JM, Kroetz DL, Klein TE, et al. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein) Pharmacogenet Genomics. 2011;21:152–161. doi: 10.1097/FPC.0b013e3283385a1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Létourneau IJ, Deeley RG, Cole SP. Functional characterization of non-synonymous single nucleotide polymorphisms in the gene encoding human multidrug resistance protein 1 (MRP1/ABCC1) Pharmacogenet Genomics. 2005;15:647–657. doi: 10.1097/01.fpc.0000173484.51807.48. [DOI] [PubMed] [Google Scholar]
- 42.Conseil G, Cole SP. Two polymorphic variants of ABCC1 selectively alter drug resistance and inhibitor sensitivity of the multidrug and organic anion transporter multidrug resistance protein 1. Drug Metab Dispos. 2013;41:2187–2196. doi: 10.1124/dmd.113.054213. [DOI] [PubMed] [Google Scholar]
- 43.Yee SW, Chen L, Giacomini KM. Pharmacogenomics of membrane transporters: past, present and future. Pharmacogenomics. 2010;11:475–479. doi: 10.2217/pgs.10.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hesselson SE, Matsson P, Shima JE, Fukushima H, Yee SW, Kobayashi Y, et al. Genetic variation in the proximal promoter of ABC and SLC superfamilies: liver and kidney specific expression and promoter activity predict variation. PLoS ONE. 2009;4:e6942. doi: 10.1371/journal.pone.0006942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi JH, Yee SW, Kim MJ, Nguyen L, Lee JH, Kang JO, et al. Identification and characterization of novel polymorphisms in the basal promoter of the human transporter, MATE1. Pharmacogenet Genomics. 2009;19:770–780. doi: 10.1097/FPC.0b013e328330eeca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tahara H, Yee SW, Urban TJ, Hesselson S, Castro RA, Kawamoto M, et al. Functional genetic variation in the basal promoter of the organic cation/carnitine transporters OCTN1 (SLC22A4) and OCTN2 (SLC22A5) J Pharmacol Exp Ther. 2009;329:262–271. doi: 10.1124/jpet.108.146449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yee SW, Shima JE, Hesselson S, Nguyen L, De Val S, Lafond RJ, et al. Identification and characterization of proximal promoter polymorphisms in the human concentrative nucleoside transporter 2 (SLC28A2) J Pharmacol Exp Ther. 2009;328:699–707. doi: 10.1124/jpet.108.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.To KK, Zhan Z, Litman T, Bates SE. Regulation of ABCG2 expression at the 3′ untranslated region of its mRNA through modulation of transcript stability and protein translation by a putative microRNA in the S1 colon cancer cell line. Mol Cell Biol. 2008;28:5147–5161. doi: 10.1128/MCB.00331-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan YZ, Morris ME, Yu AM. MicroRNA-328 negatively regulates the expression of breast cancer resistance protein (BCRP/ABCG2) in human cancer cells. Mol Pharmacol. 2009;75:1374–1379. doi: 10.1124/mol.108.054163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim MJ, Skewes-Cox P, Fukushima H, Hesselson S, Yee SW, Ramsey LB, et al. Functional characterization of liver enhancers that regulate drug-associated transporters. Clin Pharmacol Ther. 2011;89:571–578. doi: 10.1038/clpt.2010.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Brien VP, Bokelmann K, Ramírez J, Jobst K, Ratain MJ, Brockmöller J, et al. Hepatocyte nuclear factor 1 regulates the expression of the organic cation transporter 1 via binding to an evolutionary conserved region in intron 1 of the OCT1 gene. J Pharmacol Exp Ther. 2013;347:181–192. doi: 10.1124/jpet.113.206359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poonkuzhali B, Lamba J, Strom S, Sparreboom A, Thummel K, Watkins P, et al. Association of breast cancer resistance protein/ABCG2 phenotypes and novel promoter and intron 1 single nucleotide polymorphisms. Drug Metab Dispos. 2008;36:780–795. doi: 10.1124/dmd.107.018366. [DOI] [PubMed] [Google Scholar]
- 53.Zhou Q, Sparreboom A, Tan EH, Cheung YB, Lee A, Poon D, et al. Pharmacogenetic profiling across the irinotecan pathway in Asian patients with cancer. Br J Clin Pharmacol. 2005;59:415–424. doi: 10.1111/j.1365-2125.2004.02330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iyer L, Ramírez J, Shepard DR, Bingham CM, Hossfeld DK, Ratain MJ, et al. Biliary transport of irinotecan and metabolites in normal and P-glycoprotein-deficient mice. Cancer Chemother Pharmacol. 2002;49:336–341. doi: 10.1007/s00280-001-0420-4. [DOI] [PubMed] [Google Scholar]
- 55.Tagen M, Zhuang Y, Zhang F, Harstead KE, Shen J, Schaiquevich P, et al. P-glycoprotein, but not multidrug resistance protein 4, plays a role in the systemic clearance of irinotecan and SN-38 in mice. Drug Metab Lett. 2010;4:195–201. doi: 10.2174/187231210792928251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen ZS, Furukawa T, Sumizawa T, Ono K, Ueda K, Seto K, et al. ATP-Dependent efflux of CPT-11 and SN-38 by the multidrug resistance protein (MRP) and its inhibition by PAK-104P. Mol Pharmacol. 1999;55:921–928. [PubMed] [Google Scholar]
- 57.Roelofsen H, Vos TA, Schippers IJ, Kuipers F, Koning H, Moshage H, et al. Increased levels of the multidrug resistance protein in lateral membranes of proliferating hepatocyte-derived cells. Gastroenterology. 1997;112:511–521. doi: 10.1053/gast.1997.v112.pm9024305. [DOI] [PubMed] [Google Scholar]
- 58.Seiser EL, Xia K, Wright FA, Innocenti F. Meta-analysis of liver eQTL studies and cross-tissue eQTL comparison using GTEx data (Abstract #667S). Presented at the 64th Annual Meeting of The American Society of Human Genetics; October 20, 2014; San Diego, CA. [Google Scholar]
- 59.Schellens JH, Maliepaard M, Scheper RJ, Scheffer GL, Jonker JW, Smit JW, et al. Transport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implications. Ann N Y Acad Sci. 2000;922:188–194. doi: 10.1111/j.1749-6632.2000.tb07037.x. [DOI] [PubMed] [Google Scholar]
- 60.Candeil L, Gourdier I, Peyron D, Vezzio N, Copois V, Bibeau F, et al. ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan-treated metastases. Int J Cancer. 2004;109:858–854. doi: 10.1002/ijc.20032. [DOI] [PubMed] [Google Scholar]
- 61.Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002;1:611–616. [PubMed] [Google Scholar]
- 62.Phelps MA, Sparreboom A. Irinotecan pharmacogenetics: a finished puzzle? J Clin Oncol. 2014;32:2287–2289. doi: 10.1200/JCO.2014.56.3387. [DOI] [PubMed] [Google Scholar]
- 63.Fujiwara Y, Minami H. An overview of the recent progress in irinotecan pharmacogenetics. Pharmacogenomics. 2010;11:391–406. doi: 10.2217/pgs.10.19. [DOI] [PubMed] [Google Scholar]
- 64.Marsh S, Hoskins JM. Irinotecan pharmacogenomics. Pharmacogenomics. 2010;11:1003–1010. doi: 10.2217/pgs.10.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoskins JM, Rosner GL, Ratain MJ, McLeod HL, Innocenti F. Pharmacodynamic genes do not influence risk of neutropenia in cancer patients treated with moderately high-dose irinotecan. Pharmacogenomics. 2009;10:1139–1146. doi: 10.2217/pgs.09.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.