

Abstract
Aggregatibacter actinomycetemcomitans leukotoxin (LtxA) is a major virulence factor that kills leukocytes permitting it’s escape from host immune surveillance. A. actinomycetemcomitans strains can produce high or low levels of toxin. Genetic differences reside in the “so called JP2” ltxA promoter region. These hyper-leukotoxin producing strains with the 530 bp deletion have been studied in detail. However, regions contained within the 530 bp deletion that could be responsible for modulation of leukotoxin production have not been defined. Here, we report, for the first time, on regions within the 530 bp that are responsible for high-levels of ltxA expression. We constructed a deletion of 530 bps in a primate isolate of A. actinomycetemcomitans, which produced leukotoxin equivalent to the JP2 strain. We then constructed sequential deletions in regions that span the 530 bps. Results indicated that expression of the ltxA transcript was reduced by a potential transcriptional terminator in promoter region 298 to 397 with a ΔG = −7.9 kcal/mol. We also confirmed previous findings that transcriptional fusion between the orfX region and ltxC increased ltxA expression. In conclusion, we constructed a hyper-leukotoxin producing A. actinomycetemcomitans strain and identified a terminator located in the promoter region extending from 298–397 that alters ltxA expression.
Subject terms: Bacterial genes, Pathogens
Introduction
Leukotoxin (LtxA), is considered as one of the major virulence factors produced by Aggregatibacter actinomycetemcomitans. This large pore forming protein helps A. actinomycetemcomitans evade the host immune system by killing neutrophils, lymphocytes, and monocytes1, 2 and thus protects A. actinomycetemcomitans against surveillance and destruction by its native host3. Two major strains of A. actinomycetemcomitans have been reported, a minimal leukotoxin producing strain (652 type) and hyper-producing leukotoxin strain (JP2 type)4. At the genetic level the hyper-producing strain shows a deletion of 530 bp in the promoter region that appears to be responsible for increased expression of downstream ltx genes4. Regulation of ltxA expression has been studied extensively at the molecular level in naturally occurring promoter-deleted strains described as JP2-like strains4–12. Studies have shown that this deletion appears to, 1) upregulate ltxA expression, and 2) result in differential transcription that can influence ltxA expression. These studies have been performed, for the most part, in JP2-like strains that; 1) lack many characteristics seen in recently isolated clinical isolates, and 2) are devoid of the 530 bp promoter region thought to be critical for elevated leukotoxin production8. Comparison between a minimal leukotoxin producing wild-type (WT) strain, and a hyper-producing strain derived from the same WT parental strain have not been studied. It was our belief that the sequential analysis of deletions within the missing 530 bp region and the relationship of these missing regions with regulation of ltxA could be assessed in these genetically re-constructed strains. Taken together, it would seem likely that comparison between a WT parental strain containing the full-length promoter region and strains with a series of deletions in the promoter region derived from its parent strain should indicate how specific regions within the 530 bp could influence leukotoxin expression.
Over the last 30 years there have been a number of studies designed to understand the role of the promoter region in leukotoxin production12. Virtually all of the work has been described using the JP2-like strain as compared to an unrelated 652 minimal leukotoxin producing strain containing the wild-type promoter4, 8, 13, 14. Hence, most comparisons are based on speculation and abductive inference15. However, since these studies were conducted in the JP2-like strain they focused on the role of regulatory proteins that might bind to DNA upstream from the missing promoter region5, 6, 10. Regulatory proteins such as CRP, (cAMP Receptor Protein)9, IHF (Integration Host Factor)12 were recognized as either positive and/or negative regulators of ltxA expression respectively. Further, Mlc was identified as an activator of ltxA11.
Another proposal suggested that the 530 bp deletion shortens the interval between an RNA polymerase binding site and ltxA structural genes, which implies that increased transcription could occur as a result of this shortened distance15. Environmental conditions have also been shown to influence leukotoxin production. As such, increased cAMP levels, lower pH (6.0–7.0) and growth under anaerobic conditions have all been shown to increase leukotoxin transcription8, 16–18. Since the periodontal pocket is more anaerobic at its deepest point these suggestions appear to be relevant. Moreover, the oxygen tension in the pocket is known to decrease during infection in the sub-gingival region. Nevertheless, even though regulatory proteins have been identified, there are still a significant number of unanswered questions relating to ltxA expression. Chief among them is definitive proof that a specific portion of the deleted DNA promoter region affects leukotoxin expression levels4, 8–10, 12, 17.
The strains used in this study were derived from a Rhesus (Rh) monkey. Our long-term study goal was to assess a number of A. actinomycetemcomitans virulence genes and to determine their effect on A. actinomycetemcomitans colonization in the mouths of Rh monkeys. As such we deleted luxS and ltxA and studied these deletions in vivo and in vitro. The in vitro results presented herein indicate that the entire 530 bp deletion is not mandatory for excessive LtxA production. Furthermore, we found that a key determinant for expression of leukotoxin is found in a 100 bp sequence in the promoter region that contains a terminator, which when deleted permits high levels of production.
Results
Construction of a hyper LtxA producing A. actinomycetemcomitans from a minimal leukotoxin producer
Our principal goal is to study the role of different virulence factors of A. actinomycetemcomitans in vivo in a real world Rh monkey model. In this context, a previous study showed that a LtxA null producer failed to colonize the oral cavity of Rh monkeys whereas the wild-type strain RhAa3 colonized19. The initial aim of the current study was to develop a hyper LtxA producing strain from the same wild-type parental strain for testing in our monkey model. The hyper LtxA producing RhAa-ltxP530 strain was developed as described in the methods section. To assess the LtxA levels, RhAa-ltxP530 biofilm cells were grown alongside RhAa3 (wild type strain), while the RhAa-VS2 (a leukotoxin mutant strain) was used as a negative control. Both the JP2 strain and the RhAa-ltxP530 (the test strain) exhibited significantly high levels of leukotoxin activity (P < 0.0001) as compared to the RhAa3 strain (Fig. 1A). Further, qRT-PCR analysis of ltxA showed that there was a significantly high level of expression (P < 0.0001) in RhAa-ltxP530 strain as compared to minimal LtxA production in wild type RhAa3 (Fig. 1B). We also analyzed leukotoxin levels in an anaerobic cell free supernatant by SDS-PAGE SYPRO® ruby staining and western blot. Results showed an intense band in RhAa-ltxP530 strain corresponding to the molecular weight of ~113 kDa, which reacted with antibody to leukotoxin (Fig. 1C).
Figure 1.
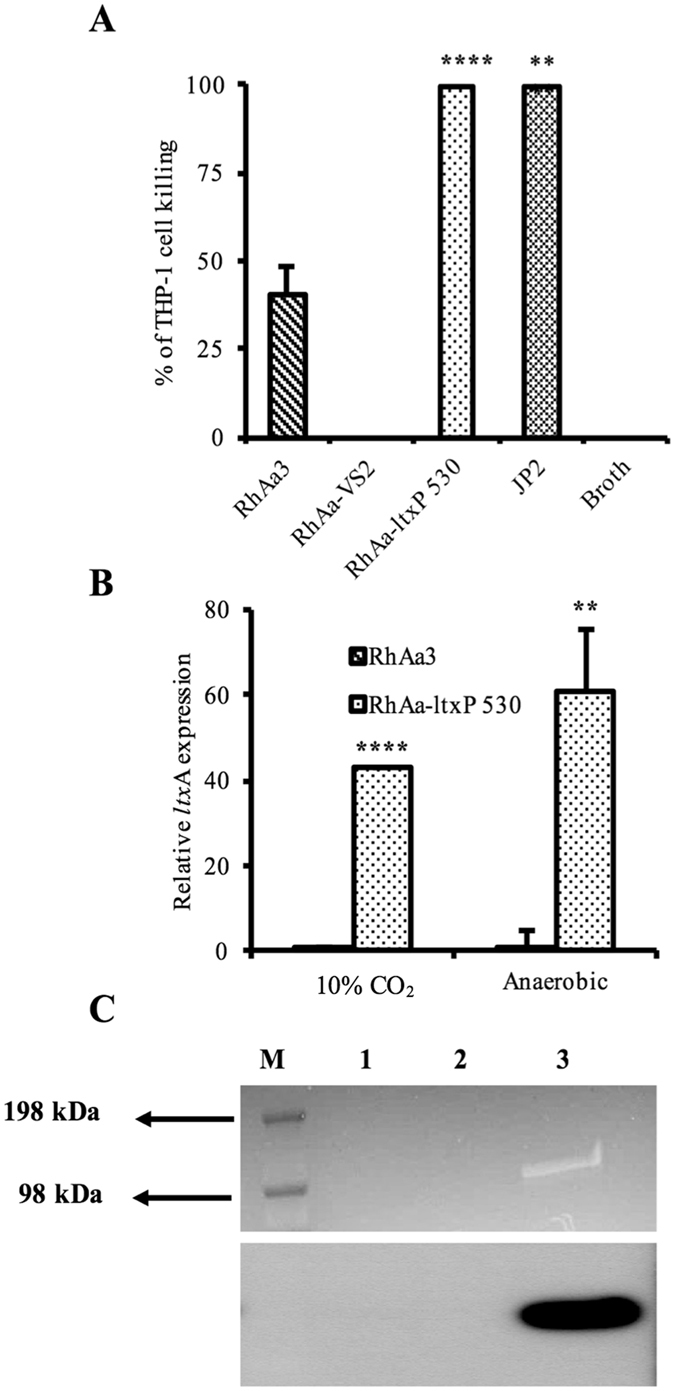
Leukotoxin expression and activity of constructed RhAa-ltxP530 strain. The leukotoxic activity of wild type RhAa3 and hyperproducer, RhAa-ltxP530 were tested in extracellular extracts against human THP-1 macrophages. The RhAa-ltxP530 strain showed significantly higher leukotoxic activity than extract from RhAa3. The JP2 strain extracellular extract used as the positive control, and uncultured growth media was used as the negative control. The significant difference in the leukotoxic activity (*P < 0.05 ± SD) between A. actinomycetemcomitans strains were calculated by One-way ANOVA test with Tukey’s multiple comparison post-hoc test. All experiments were conducted in biological triplicates (A). qRT-PCR analysis of ltxA expression in anaerobic and. The significant fold change (*P < 0.05) between RhAa3 and RhAa-ltxP530 was calculated by Student’s t-test (B). SYPRO® ruby staining (Top panel) and western blot (bottom panel) showing RhAa-ltxP530 produced high levels of leukotoxin as compared to RhAa3. The RhAa-VS2, a ltxA disrupted mutant was used as the negative control. Since the RhAa3 wild type produces minimal leukotoxin it cannot be seen in western blot (M-Marker; 1-RhAa3; 2-RhAa-VS2; 3-RhAa-ltxP530 (C).
Leukotoxin promoter region 298–397 is important in controlling leukotoxin expression
A series of deletions were made in the leukotoxin promoter region. A physical map of mutations created in the ltxA promoter region as shown in Fig. 2A. To test the leukotoxic activity due to different mutations in the promoter region, a THP-1 functional cell killing assay was performed. The results indicated that strains RhAa-ltxP98-530 (P < 0.0001), RhAa-ltxP198-530 (P < 0.0001) and RhAa-ltxP298-530 (P < 0.0001) exhibited significantly increased leukotoxic activity when compared to RhAa3 (Fig. 2B). However, RhAa-ltxP398-530 and RhAa-ltxP451-530 did not show significantly increased leukotoxin activity when compared to RhAa3 (Fig. 2B) suggesting that the region between 298–397 could be critical in controlling leukotoxin expression. Therefore, another deletion was carried out in which the region 298–397 was removed and the resultant strain RhAa-ltxP298-397 was assessed for leukotoxin activity. Interestingly, the strain RhAa-ltxP298-397 produced significantly high leukotoxic activity (P < 0.0001) compared to RhAa3 strain (Fig. 2B). Since the RhAa-ltxP298-397 strain showed significantly increased leukotoxic activity, it was proposed that this region has the potential site for a negative regulatory element binding that could alter leukotoxin expression.
Figure 2.
Physical map of the constructed ltxA promoter deletion strains and their leukotoxic activity. The physical map showing the region deleted within the 530 bp. 1 bp indicates the start of 530 bp deletion in the orfX and the respective number of base pairs deleted are indicated. 530 bp indicates the end of 530 bp deletion similar to a JP2 type strain. Bold lines indicate the region without deletion and thin line indicate the regions deleted. A red dotted vertical line represents the stop codon of orfX (A). The cell free culture supernatants were tested for the leukotoxic activity from 24 h biofilm growth of different strains. The RhAa-ltxP530, RhAa-ltxP98-530, RhAa-ltxP198-530, RhAa-ltxP298-530 and RhAa-ltxP298-397 showed significantly higher killing than the RhAa3 strain. Values indicate the mean percentages of human macrophage THP-1 cell killing performed in independent triplicate experiment. Error bars indicate standard deviation (SD). The significant killing of THP-1 cells between different A. actinomycetemcomitans strains were calculated by One-way ANOVA test. *P < 0.001 ± SD with (B).
Analysis of the mutant strains for the transcriptional fusion in orfX-ltx operon
The orfX is a gene that is located immediately upstream of ltxCABD operon. It is co-transcribed with the ltx operon4, 10, 14. In the case of the hyper-producer with the 530 bp deletion, a portion of the gene orfX was deleted leading to transcriptional fusion between orfX and ltx operon (Fig. 2A). Further analysis of the ltx promoter deletion constructs for transcriptional fusion were carried out by RT-PCR using primers orfJnF and ltxCqR. The strains RhAa-ltxP298-530, RhAa-ltxP398-530, RhAa-ltxP451-530 and RhAa-ltxP298-397 in which orfX is present were compared with RhAa-ltxP530. RT-PCR results indicated that strains RhAa-ltxP298-530, RhAa-ltxP398-530, RhAa-ltxP451-530 and RhAa-ltxP298-397 did not have transcriptional fusion between orfX and ltx operon as indicated by a lack of amplification. RhAa-ltxP530 showed amplification of an intercistronic region indicating a transcriptional fusion between orfX and ltx operon (Fig. 3A). In addition, it was also shown that RhAa-ltxP530 with transcriptional fusion had significantly increased ltxA expression (P = 0.0002) compared to RhAa3 as evidenced by qRT-PCR. Nevertheless, the strains in which transcriptional fusion had not occurred, RhAa-ltxP298-397 (P = 0.0082) and RhAa-ltxP298-530 (P = 0.0449) had a significantly higher ltxA expression compared to RhAa3 (Fig. 3B). These results suggested that the 298–397 region could contain an element that can alter ltxA expression in RhAa3 strain. We have also analyzed whether successive deletions lead to in-frame fusion of orfX to ltxC by translating the nucleotide sequence (Data not shown). The results showed that none of the mutant strains had in-frame fusions due to deletion.
Figure 3.
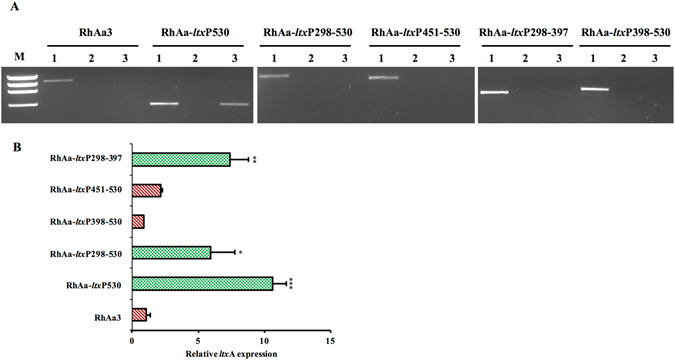
Transcriptional fusion of orfX with ltx operon due to promoter region deletion. A representative RT-PCR gel picture showing the transcriptional fusion in RhAa-ltxP530, but not in RhAa3 and RhAa-ltxP298-397 is seen. 1 – Genomic DNA template (+ve control); 2 – RNA template with no RT (−ve control); 3 – cDNA template (A). qRT-PCR analysis was carried out to assess the expression level of ltxA due to promoter deletion. There was a 10.7-fold increase in ltxA expression in RhAa-ltxP530, 5.9 fold increase in RhAa-ltxP298-530 and 7.4 fold increase in RhAa-ltxP298-397 compared to RhAa3. Values are means from a triplicate experiment. Error bars indicate ± SD. The significant fold changes (*P < 0.05 ± SD) between different A. actinomycetemcomitans strains were calculated by One-way ANOVA test with Tukey-Kramer test (B).
Analysis of the leukotoxin promoter region 298–397 for NagC binding site
Based on previous studies and in silico analysis, we predicted a NagC (a transcriptional regulator) binding consensus sequence within the ltx promoter region 298–397 (Fig. 4A)20. Further analysis of the whole genome sequence database of A. actinomycetemcomitans (strain D7S NCBI accession number CP003496) showed that the homologous genes responsible for the metabolism of N-acetyl glucosamine (GlcNAc) are nagA: D7S_0040 and nagB: D7S_00401. To show that NagC is a negative regulator and that GlcNAc can repress ltxA expression in RhAa3 strain, the cells were grown in media containing either dextrose or GlcNAc. GlcNAc is the repressor for NagC and the binding of NagC to the ltxA promoter should be blocked in the presence of GlcNAc. Thus it was expected that ltxA expression would be increased in the presence of GlcNAc. It was interesting to note that ltxA expression by RhAa3 strain in the presence of GlcNAc was significantly increased (P = 0.001) when compared to the cells grown in dextrose containing media (Fig. 4B). These results suggest that NagC might be involved in ltxA expression by interaction in the promoter region between 298–397. To confirm the above result, qRT-PCR analysis and SDS-PAGE analysis were carried out in nagC-disrupted strain RhAa-VS5. Western blot analysis did not show increased leukotoxin production in RhAa-VS5 strain as compared to RhAa3 (Fig. 4C). Further analysis of ltxA expression by qRT-PCR also showed that there was no statistical difference (P = 0.216) between RhAa-VS5 and RhAa3 (Fig. 4D). To further evaluate the influence of the 298–397 region on leukotoxin production, western blot was carried out in comparison with strains RhAa3 and RhAa-ltxP530. It was observed that there was an increased leukotoxin production as demonstrated by THP-1 cell killing assay in RhAa-ltxP530 and RhAa-ltxP298-397 whereas a decreased expression was observed in RhAa3 (Fig. 4E).
Figure 4.
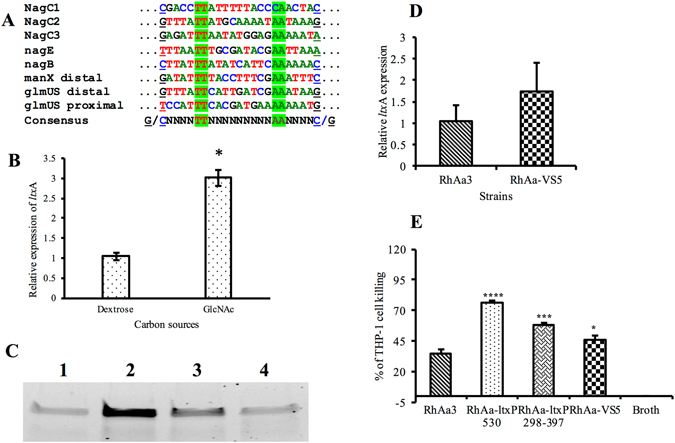
Prediction of NagC in leukotoxin regulation. In silico prediction of the NagC consensus binding site within the 530 bp of ltx operon promoter region. NagC site 2 and NagC site 3 are the predicted sites in the leukotoxin promoter region (See Supplement Figure S1). NagC site 2 is present within the region 298–397 in the 530 bp region (A). The relative ltxA expression upon supplementation of GlcNAc by RhAa3 was assessed by qRT-PCR in RhAa3 strain. Values are the mean of three independent replicate experiments. Significant difference between the group was calculated by Student’s t test P > 0.05 was considered as significance (B). Western blot analysis of the mutant strains showing the signals reacting to LtxA antibody; 1. RhAa3, 2. RhAa-ltxP530, 3. RhAa-ltxP298-397 and 4. RhAa-VS5 (C). qPCR analysis showed that there was no significant increase in ltxA expression in RhAa-VS5 compared to RhAa3 (D). The significant killing of THP-1 cells (P < 0.05) between RhAa3 and the strains were calculated by One-way ANOVA test. *indicates P < 0.001 ± SD with Tukey-Kramer test (E).
Presence of transcriptional terminator in the 298–397 region
Semi-quantitative RT-PCR was carried out using the primers orfJnF-ltxCqR to show the presence of a weak terminator in the 298–397 region that could possibly decrease the transcription in the RhAa3 strain as compared to RhAa-ltxP530 and RhAa-ltxP298-397 (Fig. 5A). This result also implies that orfX is co-transcribed in the ltx operon as it is seen that with increasing cDNA concentrations. Amplification of the intervening region between orfX and ltxC occurred even in RhAa3. In addition, qRT-PCR analysis was performed to assess and compare the expression levels of orfX with ltxC and ltxA in the three strains, RhAa3, RhAa-ltxP530 and RhAa-ltxP298-397. Results indicated that the expression levels of ltxC was significantly increased in RhAa-ltxP298-397 (P = 0.0053) when compared to expression levels of orfX further indicating the presence of a terminator in the 298–397 region which slows down the transcription in the RhAa3 strain (Fig. 5B).
Figure 5.
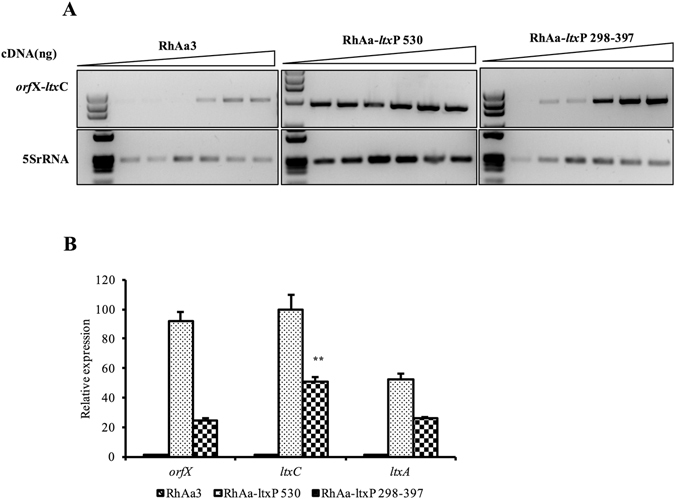
Assessment of a transcriptional terminator is present in the region 298–397. RT-PCR amplification of inter-cistronic region between orfX and ltxC in RhAa3, RhAa-ltxP530 and RhAa-ltxP298-397 with increasing concentrations of cDNA are shown as follows. Lane 1. 1 ng; Lane 2. 2 ng; Lane 3. 4 ng; Lane. 4 5 ng; Lane. 5 7.5 ng and Lane. 6 10 ng. Note that a clear signal was obtained from the 5 ng template in RhAa-ltxP298-397 (A). qRT-PCR shows significant increase in ltxC expression as compared to orfX expression in RhAa-ltxP298-397 further indicating a potential weak terminator, a negative regulatory element, within the region 298–397. Results are means ± SD for triplicate cultures normalized to 5S rRNA. The significant fold change was calculated by One-way ANOVA test. * indicates P < 0.001 ± SD with Tukey’s multiple comparison post-hoc test (B).
Assessment of terminator strength
In silico computational analysis of 298–397 region showed the presence of rho independent terminator loop structure with ΔG = −7.9 kcal/mol (Fig. 6A). Terminator strength (TS) was assessed as described previously21. The assay compared the expression of two fluorescent reporters, green fluorescent protein (GFP) and red fluorescent protein (RFP). The fluorescence data of the plasmid with no terminator, rrnB sequence (used as positive control) and sequences of interest are represented in Fig. 6B. Based on the TS calculation, we found that rrnB is a strong terminator with TS of 230.4 ± 21.1 and the 286 bp was found to have a weak terminator with a TS of 5.3 ± 0.43 (Fig. 6C). However, it is not clear if the region has a Rho-independent or a Rho-dependent terminator.
Figure 6.
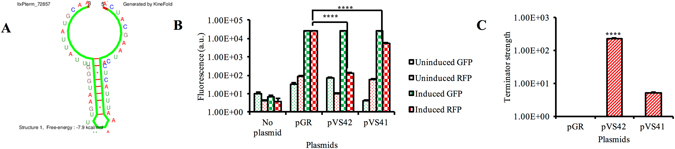
Transcriptional terminator in ltx promoter region. Putative terminator structure was predicted using KineFold software in the 298–530 bp region (A). The sequences were cloned in between GFP and RFP in a reporter plasmid, pGR. The expression of GFP and RFP fluorescence were measured before and after induction with arabinose. pGR without any cloned terminator sequence served as the negative control. The significant fluorescence (P < 0.05) between the samples was calculated by One-way ANOVA test, * indicates P < 0.001 ± SD with Tukey’s multiple comparison post-hoc test (B). The different terminator strengths were measured as described in results section. There was a presence of week terminator demonstrated in pVS41 (C).
Mlc is an activator for ltxA expression in RhAa3
It has been previously shown that the Mlc binds upstream to the leukotoxin promoter and activates ltxA transcription. These results were described in a JP2 strain and it was shown that the mlc mutant resulted in decreased ltxA expression both under aerobic and anaerobic growth conditions. To test that a similar mechanism in RhAa3 strain, we created a mlc disruption strain RhAa-VS6 from RhAa3 strain and compared the leukotoxin production. We found that leukotoxin activity was significantly reduced in RhAa-VS6 strain under both 10% CO2 (P = 0.0004) and anaerobic conditions (P < 0.0001) (Fig. 7) indicating that the Mlc positively regulate ltxA expression.
Figure 7.
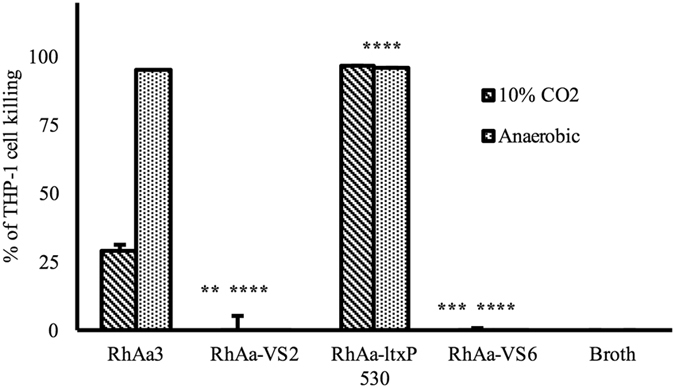
Leukotoxic activity of mlc disrupted strain. The comparison of leukotoxin production by RhAa3, RhAa-ltxP530 and RhAa-VS6 were evaluated using THP-1 human macrophage cell killing assay. Filter-sterilized culture supernatant from 24 h biofilm of RhAa3 showed significantly better killing than extract from RhAa-VS6. Uncultured growth media was used as negative control. The significant cell killing was calculated by One-way ANOVA test between different A. actinomycetemcomitans strains. Values indicate the mean percentage of THP-1 cell killing in a triplicate experiment. Error bars indicate SD. * indicates P < 0.001 ± SD with Tukey’s multiple comparaison test.
Discussion
The main aim of our ongoing studies was to assess the role of different A. actinomycetemcomitans virulence factors following implantation of these strains into the mouths of Rh monkeys. To this end, we have recently reported that a luxS deficient strain had a minimal impact on A. actinomycetemcomitans colonization while a leukotoxin null producer did not colonize supragingivally in the oral cavities of Rh monkeys19 (Unpublished data). The role of leukotoxin during periodontal disease progression and its contribution to the subgingival environment appears to be critical with respect to A. actinomycetemcomitans survival15, 22–25. After creating a leukotoxin knock-out strain our next goal was to create a hyper-producing A. actinomycetemcomitans strain by deleting the 530 bp promoter region for both in vitro and in vivo testing. Once accomplished, we wished to identify the specific region(s) within the 530 bp responsible for high levels of leukotoxin production. As a result, we created several strains that expressed differing levels of ltxA to assess regions critical for leukotoxin production.
A great deal of effort has been expended in attempts to understand increased leukotoxin production by using the JP2 strain of A. actinomycetemcomitans. This strain and those with similarly high levels of LtxA production have shown a naturally occurring deletion of 530 bp in its promoter region15. However, to date, no one has created a 530 bp deletion mutant and sequential deletions for comparison to a parental strain containing the wild type promoter. Our initial results showed that deletion of the 530 bp region led to leukotoxin production similar to that seen in the JP2 strains. As such, strains RhAa-ltxP98-530, RhAa-ltxP198-530, RhAa-ltxP298-350 and RhAa-ltxP298-397 all produced significantly increased levels of leukotoxin activity as compared to their wild type parental stain, RhAa3.
In order to understand the increased expression seen in RhAa-ltxP298-397 and possible regulatory mechanisms within that region that might be involved in leukotoxin production, region 298–530 was analyzed. It is known that an orfX gene is present upstream of ltxC in the 652 type promoter15 (Fig. 8). Examination showed that RhAa-ltxP298-530 and RhAa-ltxP298-397 deletion mutants demonstrated increased ltxA expression without transcriptional fusion of orfX. These results demonstrated the presence of regulatory elements within the 298–397 region. Initial RT-PCR experiments showed that transcriptional fusion between orfX and ltx operon was observed in RhAa-ltxP530, but not in RhAa3 and RhAa-ltxP298-397. However, when the same experiment was carried out with increasing concentrations of cDNA, the results showed that orfX was co-transcribed with the ltx operon in RhAa3 as previously reported in the JP2 strain4, 10, 14. The level of mRNA typically coincides with increased concentrations of cDNA. As seen in Fig. 5A, it is clear that the RT-PCR product appears in 2 ng of cDNA in RhAa-ltxP298-397 strain but is absent in the RT-PCR product of the WT RhAa3 strain. These results suggest that a repressor is found in the region 298–397 that causes decreased expression of ltxA in the wild-type strain. One of the limitations of this study is that occasionally we found there was no correlation found between the leukotoxicity and gene expressions analysis between the mutant strains.
Figure 8.
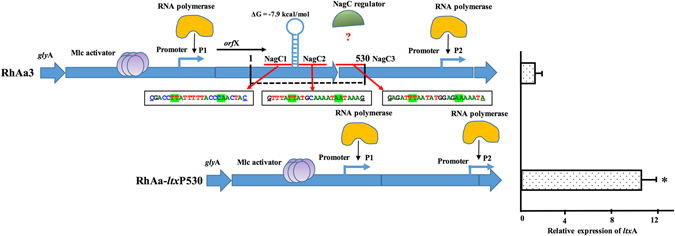
Schematic diagram showing the proposed model of transcriptional regulation of ltxA expression in RhAa3 and in RhAa-ltxP530 strains. The Mlc binding site is located preceding to orfX and the transcription of ltx operon appears to be under the direct influence of Mlc in the RhAa-ltxP530 strain. The expression level of Ltx is higher, which is shown in right side panel by graphical representation. In the RhAa3 strain due to the presence of a stem loop structure the transcription is reduced. There are three possible NagC binding sites which are predicted and shown in the red line. The sequences are shown below the RhAa3 panel. The green highlighted sequence shows TT or AA consensus bases. In the RhAa-ltxP530 strain the P1 promoter came closer due to the deletion and is one of the other mechanisms proposed for the high level of ltxA expression. *indicates P < 0.001 ± SD calculated by Student’s t test.
At one point in time it was thought that there were two promoters, P1 and P2, in the JP2 strains that controlled ltxA expression4. Later, it was shown that only one promoter P1 was present and that there was no second initiation site for leukotoxin near ltxC in JP2 as well as a Y4 strain10. Hence, it was proposed that the regulatory elements controlling P1 in the JP2 strain may modulate ltxA expression in the 652 type promoter as well.
It has been shown that CRP and Mlc are regulatory proteins that activate leukotoxin expression in the JP2 strain8. In our experiments deletion of mlc also resulted in decreased leukotoxin expression. Further, the THP-1 cell killing assay showed significantly reduced killing by RhAa-VS6 strain (the nagC deletion mutant). These results suggested that the Mlc did regulate leukotoxin expression in the monkey derived WT strain RhAa3 confirming previous results11. We also suggest that the Mlc activates ltxA expression in the RhAa3 strain with a similar mechanism as seen in the JP2-like strains. The operator region for Mlc appears to be similar in both these strains. Furthermore, this is the first time this has been reported in a wild-type strain.
Based on the in silico prediction results, NagC was identified as another possible modulator of ltxA expression (Fig. 4A). NagC is a dual-function, activator-repressor, which acts by binding to two operators forming a DNA loop26. GlcNAc-6-P, the product of GlcNAc transport by PTS system is the specific inducer of NagC. Further, NagC and Mlc have almost identical amino acid sequences in their helix-turn-helix (H-T-H), DNA binding motif. In addition, the consensus binding sites are also very similar20. In agreement with previous results we also conclude that the Mlc protein acts as an activator for the expression of ltxA11. In silico computational analysis suggested the presence of a binding site for the NagC, a potential transcriptional modulator. Analysis of the nagC mutant RhAa-VS5, revealed that no significant changes were found in ltxA expression or production as evidenced by RT-PCR and western blot. A simple mechanism of gene regulation is seen in the competition between repressors and activators for common or overlapping DNA binding sites at the promoter regions27. In this case, it is possible that the condition we tested may favor binding of the activator, Mlc in its’ binding at, the position 298–397, while neglecting binding of NagC. It is also possible that the increased expression of ltxA suggests that NagC may have a role in activation in an indirect manner within the three predicted binding sites as demonstrated in Fig. 8. Therefore, at this point we speculate that NagC is not directly involved in ltxA expression. It is also possible that NagC can trigger other factors to act on ltxA expression. Further study is required to trace the protein-protein or protein DNA interactions that could lead to the over expression of ltxA.
Next, the region between 245–530 in the 530 bp region was analyzed for other regulatory elements. This region represents the intervening area between 3′ end of orfX and the end of the 530 bp deletion. The presence of a transcriptional terminator within the region was shown by in silico analysis using KineFold software (Supplementary Fig. S1). It was also shown that a stem-loop structure was found with a ΔG = −7.9 kcal/mol in the 298–397 region. The expression levels of orfX and ltxC were assessed for RhAa3, RhAa-ltxP530 and RhAa-ltxP298-397 by qRT-PCR. These results showed an increased expression of ltxC in RhAa-ltxP298-397 suggesting a negative regulatory element between orfX and ltxC. Our results suggest a potentially weak terminator, in this region. Terminator strength assessment was studied within this region. It was shown that a weak terminator was present within the 298–530 bp region.
It is known that transcription terminators can be present at the end of genes or in the upstream regulatory regions28. Terminators at the end of the genes prevent unintended transcription in downstream genes29. Terminators in the regulatory regions are known to regulate the expression of the structural gene in response to mechanical and environmental stimuli29. Based on these findings, we speculate that there is a potential negative regulator, which acts as a weak terminator, in the region between 298–530 in the 530 bp region. The terminator could be a Rho-dependent or a Rho-independent terminator30, 31. It is highly likely that multiple factors control leukotoxin expression.
In conclusion, we confirmed the fact that deletion of 530 bp in the structural leukotoxin promoter region increased ltxA expression. We also confirm that the structure of this regulatory region within the promoter of RhAa-ltxP530 and RhAa3 strains are alike and can be regulated in a different manner. Further, in our study deletion of fewer than 530 bp (deletion of 298–397 bases) resulted in increased leukotoxin expression. These results suggest the presence of regulatory elements within specific sites in the 530 bp region control ltxA expression. Analysis of terminators and terminator strength showed the presence of a possible weak terminator in the region between 298–530 that can influence ltxA expression. Moreover, our results confirmed the observation that Mlc controls leukotoxin expression in our wild type RhAa3 strain as has been shown in the JP2 strain11. Further analysis of the exact region of the terminator and other possible regulatory elements in the leukotoxin promoter region is required. This newly developed strain will make it possible to study various strains of A. actinomycetemcomitans that differentially expresses ltxA in vivo in the oral cavity of monkeys.
Methods
Bacterial strains, growth conditions and plasmids
The strains and plasmids used in this study are listed in Table 1. A minimally leukotoxic 652 type A. actinomycetemcomitans (RhAa3) was used in this study19, 32. A. actinomycetemcomitans strains were routinely grown on Brain Heart Infusion (BHI) agar (BD company, NJ) supplemented with 0.6% yeast extract, 0.8% dextrose and 0.4% sodium bicarbonate. When necessary 0.8% dextrose was replaced with N-Acetyl-D-glucosamine (GlcNAc) (Sigma, St. Louis, MO). A. actinomycetemcomitans strains were incubated at 37 °C in a 10% CO2 or in a, anaerobic incubator for 16–48 h. Escherichia coli strains were routinely grown on LB broth or LB agar media supplemented with appropriate antibiotics in a 37 °C aerobic chamber.
Table 1.
Plasmids and bacterial strains used in this study.
| Plasmid/bacterial strain | Relevant genotype/characteristics | Source |
|---|---|---|
| E. coli Stellar™ Competent Cells | F–, endA1, supE44, thi-1, recA1, relA1, gyrA96, phoA, Φ80d lacZΔ M15, Δ (lacZYA - argF) U169, Δ (mrr - hsdRMS - mcrBC), ΔmcrA, λ− | Clone tech laboratories Inc, CA |
| RhAa3 | Wild type strain | 19 |
| JP2 | A hyper-leukotoxic strain | 40 |
| RhAa-VS2 | SpR ltxA disrupted strain | 19 |
| RhAa-VS5 | nagC disrupted RhAa3 | This study |
| RhAa-VS6 | mlc disrupted RhAa3 | This study |
| RhAa-ltxP530 | 530 bp of ltxA promoter deleted strain | This study |
| RhAa-ltxP98-530 | 432 bp of ltxA promoter deleted strain | This study |
| RhAa-ltxP198-530 | 332 bp of ltxA promoter deleted strain | This study |
| RhAa-ltxP298-530 | 232 bp of ltxA promoter deleted strain | This study |
| RhAa-ltxP398-530 | 132 bp of ltxA promoter deleted strain | This study |
| RhAa-ltxP451-530 | 79 bp of ltxA promoter deleted strain | This study |
| RhAa-ltxP298-397 | 99 bp of ltxA promoter deleted strain | This study |
| pJT1 | SpR; gene disruption plasmid for A. actinomyctemcomitans | 33 |
| pGR | Reporter plasmid for measuring terminator strength | 21 |
| pVS31 | Deletion plasmid to create RhAa-ltxP530 strain | This study |
| pVS32 | Deletion plasmid to create RhAa-ltxP98-530 strain | This study |
| pVS33 | Deletion plasmid to create RhAa-ltxP198-530 strain | This study |
| pVS34 | Deletion plasmid to create RhAa-ltxP298-530 strain | This study |
| pVS35 | Deletion plasmid to create RhAa-ltxP398-530 strain | This study |
| pVS36 | Deletion plasmid to create RhAa-ltxP451-530 strain | This study |
| pVS37 | Deletion plasmid to create RhAa-ltxP298-397 strain | This study |
| pVS41 | Promoter regions sequence from 298–530 cloned between gfp and rfp of pGR rrnB | This study |
| pVS42 | Terminator cloned between gfp and rfp of pGR | This study |
Biofilm growth conditions
A. actinomycetemcomitans strains from −80 °C freezer were streaked on to brain heart infusion agar (BHI) plates containing yeast extract (0.6%), sodium bicarbonate (0.4%), dextrose (0.75%) and 1.5% agar. After 24 hr growth in a 37 °C incubator at 10% CO2/90% air atmosphere, a single colony was picked from the well separated area of the agar plates and suspended in BHI broth (BHI) containing yeast extract (0.6%), sodium bicarbonate (0.4%) and glucose (0.75%). Single colony was picked from the well-separated area from the agar plates, suspended in BHI broth containing yeast extract (0.6%), sodium bicarbonate (0.4%) and glucose (0.75%). The aggregated cells from the colony were disrupted using a hand held homogenizer (Kimble Chase, Vineland, NJ) and the non-aggregated free cells were removed by leaving the suspension on ice for two minutes. The cell density of the top portion was adjusted to ~108 cells per ml (OD600 = 0.7–0.8). These cells were used as the inoculum for the bio film growth. Biofilms were grown on brain BHI broth for 24 hr in a 37 °C incubator at 10% CO2/90% air atmosphere or in a 37 °C anaerobic chamber containing 80% N2, 10% CO2 and 10%H2. For testing the leukotoxic activity of each strain by THP-1 cell killing assay and western blot, 2 ml of supernatant was collected and passed through a 0.2 μ filter and used immediately or stored at −80 °C freezer. The mature biofilm cells were washed with PBS, cells were collected using a cell scrapper and stored in RNA stabilizing solution (ice cold 0.9% saline supplemented with 1/10th volume of 95% ethanol and 5% citric acid saturated phenol mixture).
DNA manipulations
PCR amplifications were performed using Phusion DNA polymerase (Thermoscientific, Waltham, MA). Oligonucleotides used in this study were synthesized from Integrated DNA technologies (Coralville, IA) and are listed in Supplementary Table 1. Genomic DNA plasmid and gel extraction kits were purchased from Qiagen (Qiagen, Foster City CA) and used as per manufacturer’s instruction. Restriction enzymes were purchased from New England Biolabs (New England Biolabs, Inc. Ipswich, MA) and used as per the manufacturer’s directions. Stellar competent cells were transformed as instructed in manufacturer’s manual (Clontech®, Mountain View, CA). The gene disruption and promoter deletion plasmids were electroporated as described previously19. All plasmid constructs and mutant A. actinomycetemcomitans strains were verified by DNA sequencing (Macrogen Inc, New York, NY).
Construction of ltxA promoter deletion in A. actinomycetemcomitans
A series of deletions within the ltxA promoter was carried out in RhAa3 as described previously33. Initially, a JP2 like 530 bp ltxA promoter strain was created. The aim was to mimic the JP2-type promoter by deleting the 530 bp in the promoter region. A scarless, marker-less deletion approach was used to construct a hyper-LtxA producing strain as described previously33. Primers 530USSUpF, 530UpR and 530DnF, 530DnR were used to amplify the upstream and downstream flanking regions of the 530 bp region in the ltxA promoter region to be deleted. Both the fragments were amplified with 15 bp complementary to each other to enable fusion between them. In addition, restriction sites XmaI and SacI were introduced into the 5′ and 3′ ends of the flanking fragments respectively by PCR to enable directional cloning into pJT1. Overlap extension PCR was performed as described previously34 with the first PCR step using equimolar concentrations of the upstream and downstream flanking fragments. The end primers were added and the second PCR step using 5 µl template from the first PCR step. The fused PCR amplified fragment was then digested with XmaI and SacI and ligated into restriction digested pJT1. The resultant plasmid was designated pVS30. The plasmid pVS30 was electroporated into RhAa3 and the transformants were screened by PCR using primers 530bpscreenF and 530bpscreenR. The resultant strain was designated as RhAa-ltxP530.
The successive ltxA promoter deletion mutants were created as follows. PCR products containing different lengths of the leukotoxin promoter along with portions of upstream gene glyA and downstream ltx operon were amplified using primer sets 530USSUpF, 400UpR and 400DnF, 530DnR; 530USSUpF, 300UpR and 300DnF, 530DnR; 530USSUpF, 200UpR and 200DnF; 530DnR; 530USSUpF, 100UpR and 100DnF, 530DnR; 530USSUpF, 80UpR and 530DnF, 80DnR; 530USSUpF (Supplementary Table S1). The upstream and downstream amplified products from each set were fused by overlap extension PCR as described above. The fused fragments were digested with XmaI and SacI and ligated into pJT1. The resultant plasmids were designated pVS31-pVS35. The plasmids were transformed into RhAa3 by electroporation as described previously and the strains were designated as RhAa-ltxP98-530, RhAa-ltxP198-530, RhAa-ltxP298-530, RhAa-ltxP398-530 and RhAa-ltxP451-530 respectively (Table 1). Another leukotoxin promoter deletion was made in a similar way with minor modifications. The primer sets used for amplification of upstream and downstream fragments were 530UpFI, -nagUpR and -nagDnF, 530DnRI. Fusion of the fragments into the vector was done by In-Fusion HD cloning (Clontech®, Mountain View, CA) as per the manufacturer’s instructions. The vector pJT1 was linearized with XhoI and NotI. The gel purified fragments and the linearized vector were incubated with the 5X In-Fusion HD Enzyme premix at 37 °C for 15 min followed by 50 °C for 15 min. The mixture was then transformed into Stellar competent E. coli. The resultant plasmid was designated as pVS36 after confirmation by sequencing. The plasmid was electroporated into RhAa3 as previously described and the mutant was designated as RhAa-ltxP298-397.
Construction of nagC and mlc mutants
Scarless markerless deletion of nagC and mlc were carried out in RhAa3 strain as described previously33. Briefly, the flanking regions of nagC (Acc. No. CP003496 locus id. D7S_00428), were amplified with nagCDNFNew, NAGC3R and 3619 R, nagCURnew primer. In the case of mlc (Acc. No. CP003496 locus id. D7S_02207), the flanking regions were amplified using primer sets mlcUF, mlcUR and mlcDF, mlcDR. The PCR amplified flanking fragments were cloned at XhoI and NotI sites of pJT1 plasmid using In-Fusion HD cloning kit as per the manufacturer’s instructions. The resultant nagC and mlc knockout plasmids were designated as pVS39 and pVS40 respectively. The plasmids pVS39 and pVS40 were electroporated into RhAa3 and the nagC and mlc mutants were screened with primer sets NAGC4R, 3843 R and mlc5′, mlc3′ respectively. The nagC mutant was designated as RhAa-VS5 and mlc mutant was designated as RhAa-VS6.
THP-1 cell killing assay
The leukotoxic activity of different A. actinomycetemcomitans strains were assessed by THP-1 cell killing assay as described previously35. Cell viability was the measure of luminescence using the microplate reader (Infinite M200pro, Tecan, Austria GmbH, Austria) which was proportional to the amount of ATP released by the live cells. In all experiments, uncultured liquid media and purified leukotoxin were used as negative and positive controls respectively.
SDS-PAGE and western blot analysis of LtxA
Protein samples were processed and separated onto SDS-PAGE gel (Bio-Rad, Hercules, CA), stained with SYPRO® Ruby and the image was captured as described in the product manual (Thermoscientific, Waltham, MA). SeeBlue® Plus2 Pre-Stained standard was used to determine the protein molecular weight (Invitrogen, Carlsbad, CA). Protein samples were subjected to western blot analysis as previously described36. Briefly, protein bands were transferred from SDS-PAGE onto a nitrocellulose membrane, probed with polyclonal anti-LtxA antibody (1:2000-Monoclonal anti-mouse antibody, ProMab, Richmond, CA) and peroxidase labeled goat-anti-mouse antibody (1:10,000, Sigma, St. Louis, MO). Reactive bands were visualized by treating the membrane with Super signal West Femto substrate (Pierce, Rockford, IL) and exposing the membrane to FluorChem Q system (San Jose, CA).
RNA extraction from biofilm, RT-PCR and qRT-PCR analysis
RNA was isolated as previously described19. The RNA was purified using Micro Bio-Spin P-30 Gel Columns (Bio-Rad, Hercules, CA) and treated with DNaseI and RNA purification kit (Zymo Research, Irvine) to remove genomic DNA contamination. PCR using ltxA primers was used to confirm the elimination of genomic DNA contamination before proceeding to qPCR.
Transcriptional fusion analysis was performed to demonstrate whether the difference in leukotoxin expression was as a result of transcriptional fusion between orfX and ltxA operon in the created mutant strains as described previously37. To do this we carried out two step RT-PCR. The total RNA was extracted from the strains and 2 μg of total RNA was converted into cDNA in the first step as described in manufacturer’s instruction (Applied Biosystems, Foster City, CA). A second PCR step was carried out with primers OrfXJnF and ltxCqR using the cDNA template. The forward primer was designed to prime at the 5′ end of orfX gene and the reverse primer was used in the case of ltxC. The DNA and RNA were used as positive and negative controls in the second PCR step reactions respectively. To further analyze if there was a low level of transcript in the inter cistronic region, increasing concentrations of cDNA template was used for second RT-PCR step.
For qRT-PCR, LightCycler480 system was used using Roche SYBR green master mix. cDNA was used in the dilution of 1:100 for reference gene 5SrRNA and 1:25 was used for target genes as described previously19. The cycling condition was followed as described in the product manual (Roche Life Science, Indianapolis, IN). Melting curve analysis was done to assess product specificity. Data analysis was done using LightCycler 480 software (Version 1.2.9.11).
Transcriptional terminator assessment
The stem loop structure in the RNA secondary structure was predicted using KineFold web server38. The plasmid pGR (A generous gift from Christopher Voigt-Addgene plasmid #46002) was used for terminator assessment assays. The terminator sequence of interest was amplified with primers pGR153F and pGR286R using the RhAa3 DNA as template. As a control, rrnB terminator was amplified from pJAK16 using primers pGRrrnBF and pGRrrnBR. All the sequences were cloned at EcoRI and SpeI sites of pGR plasmid using an In-Fusion HD cloning kit as described in the product manual. The resultant plasmids were designated as pVS41 and pVS42 respectively. The transcriptional terminator strength assays were performed in Stellar E. coli as described previously21. The cell growth and collection of cells were carried out as described previously39. LSRFortessa X-20 flow cytometer (BD Biosciences, CA) was used for flow cytometry measurements as described previously21. GFP data was extracted from FITC and RFP data from PE-CF594 channels respectively. GFP and RFP fluorescence was the measure of the geometric means of the data obtained from FITC and PE-CF594 channels respectively. The data was analyzed using FlowJo 9.9.4. TS and was calculated by applying the equation; TS = 1/1 − TE = [(GFP)Term/(RFP)Term] [(GFP)0/(RFP)0]−1, where TE refers to terminator efficiency, (GFP)Term and (RFP)Term is the averages of populations measured by flow cytometry when terminator is present and (GFP)0 and (RFP)0 refers to the measurements of the control plasmid with no terminator present. TS = 1 refers to no termination. The data presented are as means ± SD. *P < 0.05.
Statistical Analysis
In all cases where two samples were compared we used a Students-t-test to determine statistically significant differences using a level of P < 0.05 as our end point determinant. In any case where three of more comparisons were being assessed we used ANOVA and relied on Tukey-Kramer test to discriminate between means that were significantly different setting the level at P < 0.05.
Electronic supplementary material
Acknowledgements
D.H.F. would like to acknowledge support derived from grant 1R21DE021172-01A1 from the National Institute of Dental and Craniofacial Research (NIDCR) to support the work performed in this study. Authors would like to thank Dr. Donald R. Demuth for providing plasmids; Sukhwinder Singh, Technical Director of NJMS Flow Cytometry And Immunology Core Laboratory, Newark, N.J. We like to thank Dr. Scott Kachlany for providing the primary leukotoxin antibody.
Author Contributions
Conceptualization D.H.F., V.S.K. and V.S. Funding acquisition D.H.F. Writing the original draft, review & Editing D.H.F., V.S. and S.K.V. D.G. helped in performing cell killing assays and biofilm growth. V.S., S.K.V. & D.H.F., performed statistical analyses. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Vandana Sampathkumar and Senthil Kumar Velusamy contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01692-6
References
- 1.Menestrina G, Mackman N, Holland IB, Bhakdi S. Escherichia coli haemolysin forms voltage-dependent ion channels in lipid membranes. Biochim Biophys Acta. 1987;905:109–117. doi: 10.1016/0005-2736(87)90014-9. [DOI] [PubMed] [Google Scholar]
- 2.Benz R, Schmid A, Wagner W, Goebel W. Pore formation by the Escherichia coli hemolysin: evidence for an association-dissociation equilibrium of the pore-forming aggregates. Infect Immun. 1989;57:887–895. doi: 10.1128/iai.57.3.887-895.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henderson B, Ward JM, Ready D. Aggregatibacter (Actinobacillus) actinomycetemcomitans: a triple A* periodontopathogen? Periodontol 2000. 2010;54:78–105. doi: 10.1111/j.1600-0757.2009.00331.x. [DOI] [PubMed] [Google Scholar]
- 4.Brogan JM, Lally ET, Poulsen K, Kilian M, Demuth DR. Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect Immun. 1994;62:501–508. doi: 10.1128/iai.62.2.501-508.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spitznagel J, Jr., Kraig E, Kolodrubetz D. The regulation of leukotoxin production in Actinobacillus actinomycetemcomitans strain JP2. Adv Dent Res. 1995;9:48–54. doi: 10.1177/08959374950090010901. [DOI] [PubMed] [Google Scholar]
- 6.Spitznagel J, Jr., Kraig E, Kolodrubetz D. Regulation of leukotoxin in leukotoxic and nonleukotoxic strains of Actinobacillus actinomycetemcomitans. Infect Immun. 1991;59:1394–1401. doi: 10.1128/iai.59.4.1394-1401.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aberg CH, Kwamin F, Claesson R, Johansson A, Haubek D. Presence of JP2 and Non-JP2 Genotypes of Aggregatibacter actinomycetemcomitans and attachment loss in adolescents in Ghana. J Periodontol. 2012;83:1520–1528. doi: 10.1902/jop.2012.110699. [DOI] [PubMed] [Google Scholar]
- 8.Hritz M, Fisher E, Demuth DR. Differential regulation of the leukotoxin operon in highly leukotoxic and minimally leukotoxic strains of Actinobacillus actinomycetemcomitans. Infect Immun. 1996;64:2724–2729. doi: 10.1128/iai.64.7.2724-2729.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feuerbacher LA, Burgum A, Kolodrubetz D. The cyclic-AMP receptor protein (CRP) regulon in Aggregatibacter actinomycetemcomitans includes leukotoxin. Microb Pathog. 2011;51:133–141. doi: 10.1016/j.micpath.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolodrubetz D, et al. cis Elements and trans factors are both important in strain-specific regulation of the leukotoxin gene in Actinobacillus actinomycetemcomitans. Infect Immun. 1996;64:3451–3460. doi: 10.1128/iai.64.9.3451-3460.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Childress C, Feuerbacher LA, Phillips L, Burgum A, Kolodrubetz D. Mlc is a transcriptional activator with a key role in integrating cyclic AMP receptor protein and integration host factor regulation of leukotoxin RNA synthesis in Aggregatibacter actinomycetemcomitans. J Bacteriol. 2013;195:2284–2297. doi: 10.1128/jb.02144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolodrubetz D, Phillips L, Burgum A. Repression of aerobic leukotoxin transcription by integration host factor in Aggregatibacter actinomycetemcomitans. Res Microbiol. 2010;161:541–548. doi: 10.1016/j.resmic.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He T, Nishihara T, Demuth DR, Ishikawa I. A novel insertion sequence increases the expression of leukotoxicity in Actinobacillus actinomycetemcomitans clinical isolates. J Periodontol. 1999;70:1261–1268. doi: 10.1902/jop.1999.70.11.1261. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell C, Gao L, Demuth DR. Positive and negative cis-acting regulatory sequences control expression of leukotoxin in Actinobacillus actinomycetemcomitans 652. Infect Immun. 2003;71:5640–5649. doi: 10.1128/IAI.71.10.5640-5649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kachlany SC. Aggregatibacter actinomycetemcomitans leukotoxin: from threat to therapy. J Dent Res. 2010;89:561–570. doi: 10.1177/0022034510363682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue T, et al. Fermentable-sugar-level-dependent regulation of leukotoxin synthesis in a variably toxic strain of Actinobacillus actinomycetemcomitans. Microbiology. 2001;147:2749–2756. doi: 10.1099/00221287-147-10-2749. [DOI] [PubMed] [Google Scholar]
- 17.Kolodrubetz D, Phillips L, Jacobs C, Burgum A, Kraig E. Anaerobic regulation of Actinobacillus actinomycetemcomitans leukotoxin transcription is ArcA/FnrA-independent and requires a novel promoter element. Res Microbiol. 2003;154:645–653. doi: 10.1016/j.resmic.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Ohta, H., Miyagi, A., Kato, K. & Fukui, K. The relationships between leukotoxin production, growth rate and the bicarbonate concentration in a toxin-production-variable strain of Actinobacillus actinomycetemcomitans. Microbiology142 (Pt 4), 963–970, doi:10.1099/00221287-142-4-963 (1996). [DOI] [PubMed]
- 19.Velusamy SK, Sampathkumar V, Godboley D, Fine DH. Profound Effects of Aggregatibacter actinomycetemcomitans Leukotoxin Mutation on Adherence Properties Are Clarified in in vitro Experiments. PLoS One. 2016;11:e0151361. doi: 10.1371/journal.pone.0151361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plumbridge J. DNA binding sites for the Mlc and NagC proteins: regulation of nagE, encoding the N-acetylglucosamine-specific transporter in Escherichia coli. Nucleic Acids Res. 2001;29:506–514. doi: 10.1093/nar/29.2.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen, Y.-J. et al. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat Meth10, 659–664, doi:10.1038/nmeth.2515, http://www.nature.com/nmeth/journal/v10/n7/abs/nmeth.2515.html (2013). [DOI] [PubMed]
- 22.Aberg CH, Kelk P, Johansson A. Aggregatibacter actinomycetemcomitans: virulence of its leukotoxin and association with aggressive periodontitis. Virulence. 2015;6:188–195. doi: 10.4161/21505594.2014.982428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fine DH, et al. A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J Clin Microbiol. 2013;51:2850–2861. doi: 10.1128/jcm.00729-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herbert, B. A., Novince, C. M. & Kirkwood, K. L. Aggregatibacter actinomycetemcomitans, a potent immunoregulator of the periodontal host defense system and alveolar bone homeostasis. Mol Oral Microbiol, doi:10.1111/omi.12119 (2015). [DOI] [PMC free article] [PubMed]
- 25.Johansson A. Aggregatibacter actinomycetemcomitans leukotoxin: a powerful tool with capacity to cause imbalance in the host inflammatory response. Toxins (Basel) 2011;3:242–259. doi: 10.3390/toxins3030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plumbridge J. Regulation of PTS gene expression by the homologous transcriptional regulators, Mlc and NagC, in Escherichia coli (or how two similar repressors can behave differently) J Mol Microbiol Biotechnol. 2001;3:371–380. [PubMed] [Google Scholar]
- 27.Browning DF, Busby SJ. The regulation of bacterial transcription initiation. Nat Rev Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 28.Peters JM, Vangeloff AD, Landick R. Bacterial transcription terminators: the RNA 3′-end chronicles. J Mol Biol. 2011;412:793–813. doi: 10.1016/j.jmb.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santangelo TJ, Artsimovitch I. Termination and antitermination: RNA polymerase runs a stop sign. Nat Rev Microbiol. 2011;9:319–329. doi: 10.1038/nrmicro2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ermolaeva MD, Khalak HG, White O, Smith HO, Salzberg SL. Prediction of transcription terminators in bacterial genomes. J Mol Biol. 2000;301:27–33. doi: 10.1006/jmbi.2000.3836. [DOI] [PubMed] [Google Scholar]
- 31.Lesnik EA, et al. Prediction of rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 2001;29:3583–3594. doi: 10.1093/nar/29.17.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fine, D. H. et al. Colonization and Persistence of Labeled and “Foreign” Strains of Inoculated into the Mouths of Rhesus Monkeys. J Oral Biol (Northborough) 2, 10.13188/2377-987X.1000005 (2015). [DOI] [PMC free article] [PubMed]
- 33.Juarez-Rodriguez MD, Torres-Escobar A, Demuth DR. Construction of new cloning, lacZ reporter and scarless-markerless suicide vectors for genetic studies in Aggregatibacter actinomycetemcomitans. Plasmid. 2013;69:211–222. doi: 10.1016/j.plasmid.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J, Lee HJ, Shin MK, Ryu WS. Versatile PCR-mediated insertion or deletion mutagenesis. Biotechniques. 2004;36:398–400. doi: 10.2144/04363BM04. [DOI] [PubMed] [Google Scholar]
- 35.DiFranco KM, et al. Leukotoxin (Leukothera(R)) targets active leukocyte function antigen-1 (LFA-1) protein and triggers a lysosomal mediated cell death pathway. J Biol Chem. 2012;287:17618–17627. doi: 10.1074/jbc.M111.314674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diaz R, et al. Characterization of leukotoxin from a clinical strain of Actinobacillus actinomycetemcomitans. Microb Pathog. 2006;40:48–55. doi: 10.1016/j.micpath.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Phongsisay V, Fry BN. Bidirectional transcription of lipooligosaccharide synthesis genes from Campylobacter jejuni. Int J Med Microbiol. 2007;297:431–441. doi: 10.1016/j.ijmm.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Xayaphoummine A, Bucher T, Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005;33:W605–610. doi: 10.1093/nar/gki447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen YJ, et al. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat Methods. 2013;10:659–664. doi: 10.1038/nmeth.2515. [DOI] [PubMed] [Google Scholar]
- 40.Balashova NV, Diaz R, Balashov SV, Crosby JA, Kachlany SC. Regulation of Aggregatibacter (Actinobacillus) actinomycetemcomitans leukotoxin secretion by iron. J Bacteriol. 2006;188:8658–8661. doi: 10.1128/jb.01253-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.