

ABSTRACT
Tomato is a major vegetable crop that has tremendous popularity. However, viral disease is still a major factor limiting tomato production. Here, we report the tomato virome identified through sequencing small RNAs of 170 field-grown samples collected in China. A total of 22 viruses were identified, including both well-documented and newly detected viruses. The tomato viral community is dominated by a few species, and they exhibit polymorphisms and recombination in the genomes with cold spots and hot spots. Most samples were coinfected by multiple viruses, and the majority of identified viruses are positive-sense single-stranded RNA viruses. Evolutionary analysis of one of the most dominant tomato viruses, Tomato yellow leaf curl virus (TYLCV), predicts its origin and the time back to its most recent common ancestor. The broadly sampled data have enabled us to identify several unreported viruses in tomato, including a completely new virus, which has a genome of ∼13.4 kb and groups with aphid-transmitted viruses in the genus Cytorhabdovirus. Although both DNA and RNA viruses can trigger the biogenesis of virus-derived small interfering RNAs (vsiRNAs), we show that features such as length distribution, paired distance, and base selection bias of vsiRNA sequences reflect different plant Dicer-like proteins and Argonautes involved in vsiRNA biogenesis. Collectively, this study offers insights into host-virus interaction in tomato and provides valuable information to facilitate the management of viral diseases.
IMPORTANCE Tomato is an important source of micronutrients in the human diet and is extensively consumed around the world. Virus is among the major constraints on tomato production. Categorizing virus species that are capable of infecting tomato and understanding their diversity and evolution are challenging due to difficulties in detecting such fast-evolving biological entities. Here, we report the landscape of the tomato virome in China, the leading country in tomato production. We identified dozens of viruses present in tomato, including both well-documented and completely new viruses. Some newly emerged viruses in tomato were found to spread fast, and therefore, prompt attention is needed to control them. Moreover, we show that the virus genomes exhibit considerable degree of polymorphisms and recombination, and the virus-derived small interfering RNA (vsiRNA) sequences indicate distinct vsiRNA biogenesis mechanisms for different viruses. The Chinese tomato virome that we developed provides valuable information to facilitate the management of tomato viral diseases.
KEYWORDS: small RNA sequencing, tomato virome, viral diversity and evolution
INTRODUCTION
Tomato (Solanum lycopersicum L.) is one of the most popular and extensively consumed vegetable crops. The worldwide production of tomato in 2013 was approximately 164 million tons, with a value of $60 billion (FAOSTAT, 2013; http://www.fao.org/faostat). Although the annual yield of tomato has increased over the years, the pre- and postharvest losses are still tremendous, especially in developing countries (1). Management of viral diseases, in addition to the well-appreciated fungal and bacterial diseases, is of paramount importance for tomato production. There are at least 136 characterized viral species that are capable of infecting tomato, a number greater than that for other vegetables such as pepper (n = 62), lettuce (n = 53), potato (n = 57), eggplant (n = 44), radish (n = 19), and spinach (n = 109) (2). Due to global climate changes and increased international trade, new viruses of tomato and other crops are frequently detected, and old viruses are found to be epidemic (3). Understanding the diversity and evolution of viruses across major crop production areas is critical for viral disease management.
It was not until recently that diagnosis and discovery of known and novel viruses on a global scale became feasible (4–7). Technical strides in next-generation sequencing (NGS) have made it possible to obtain the complete or near-complete genomes of novel viruses without any prior knowledge. To date, several NGS approaches have been adopted to explore viral diversity using either viral genomic RNA/DNA or small RNA (sRNA) from an infected host (reviewed in reference 8). Using these approaches, at least four viroid/viroid-like RNAs and 49 novel plant RNA and DNA viruses from 18 known or unassigned viral families have been reported during the past few years (8). Among these technologies, sequencing of virus-derived small interfering RNAs (vsiRNAs) is becoming prevalent and has been used for virus/viroid detection in different kingdoms, such as plants (9–11), fungi (12), insects (6, 13), and mammals (14). vsiRNAs are the product of the antiviral defense process that is naturally invoked by a host when perceiving viral invasion (15). Plants usually produce vsiRNAs through Dicer-like proteins (DCLs). In Arabidopsis thaliana, viral RNAs are cleaved primarily by DCL4 and secondarily by DCL2, which results in 21- and 22-nucleotide (nt) vsiRNAs, respectively (16). The Argonaute (AGO) proteins, such as AGO1 and AGO2, are essential downstream components for virus defense in Arabidopsis (17). AGOs selectively bind vsiRNAs to form the RNA-induced silencing complex (RISC) and trigger RNA interference (RNAi), and the AGO-dependent sorting on sRNAs has a strong sequence specificity (18). vsiRNAs can also be recruited by multiple host RNA-directed RNA polymerases (RdRPs) to boost the generation of vsiRNAs, thus amplifying defense signals (19, 20).
Virus detection via sRNA sequencing offers several unique advantages over other approaches. First, both DNA and RNA viruses are subjected to host antiviral immune systems (16, 21, 22); thus, they can be simultaneously detected in a single experiment. Second, unlike the sporadic distribution of host-derived sRNAs, vsiRNAs are derived from both sense and complementary strands of the viral genome. Therefore, de novo assembly of vsiRNAs is usually enough to resolve the complete viral genomes (11, 23), even though sRNA sequences are generally short (<30 bp). A recent study showed, by sequencing of both sRNAs and RNAs from viral particles, that the mutation landscapes from the two pools were very similar, implying that sRNAs are unbiased for studying the viral diversity (24).
China is the largest tomato-producing country in the world (FAOSTAT, 2013; http://www.fao.org/faostat/). Viral infection is one of the major factors limiting tomato production. To obtain an overall picture of the diversity, distribution, and evolution of tomato viruses in China, we collected 170 field-grown tomato samples across the country. Viruses in these samples were determined using sRNA sequencing. Comprehensive analysis of sequencing data revealed dozens of known and novel viruses present in tomatoes in China and provided insights into virus diversity and evolution, offering valuable information to facilitate the development of efficient strategies for tomato viral disease management.
RESULTS
Small RNA sequencing unveiled viral species present in tomato.
We collected 170 field-grown tomato samples with virus-like disease symptoms in 2013 from major tomato cultivation areas in China (Fig. 1A). sRNA libraries of these samples were constructed and sequenced, which resulted in 84,996 to 6,321,108 cleaned reads for each sample, with a median of ∼1.25 million reads (see Table S1 in the supplemental material). The sRNA reads showed two distinguishable length distributions: host-derived reads were enriched at a length of 24 nt and to a lesser extent at 21 nt, while the remaining reads were preferentially enriched at 21 nt and less significantly at 22 nt (Fig. 1B). The bipartite read size distribution underlined the activity of the host immune defense system against viruses, which released large amounts of vsiRNAs of 21 and 22 nt. We analyzed the sRNA sequences using VirusDetect, a program that can efficiently identify both known and novel viruses from deep siRNA sequences (23). In total, we detected 22 viruses, including 21 known viruses and one newly discovered virus, as well as two viroids (Citrus exocortis viroid, genus Pospiviroid, and Potato spindle tuber viroid, genus Pospiviroid) from the 170 samples, and these viruses spanned 12 genera (Fig. 1C). The sample information, sRNA sequences, and the information on the identified viruses are available at the Chinese Tomato Virome Database (http://ted.bti.cornell.edu/CtomatoVirome/index.html). Positive-sense single-stranded RNA [(+)ssRNA] viruses were the dominant group, representing 77% of the identified viruses. Potyvirus was the most abundant subgroup in the (+)ssRNA viruses, with six species detected from the collected samples (Fig. 1C). We used the nonparametric viral discovery statistic (5, 25) to predict the bounds of the viral community in tomato and assess the completeness of our virus discovery effort. The curve indicated that the community may contain 30 viral species, and the viruses that we discovered from 170 samples represented ∼73% of all species that may be present in tomato in China (Fig. 1D). All the samples were diagnosed with at least one virus, consistent with their sampling based on virus-like symptoms. Moreover, ∼89% of samples contain two or more viruses, with a peak at three (Fig. 1E), suggesting a mixed infection in the majority of the collected samples.
FIG 1.

Virus detection in field-grown tomato samples in China using sRNA sequencing. (A) Distribution of the sampling regions in China. Color and size of the circles indicate the sampling size. Regions are shown by acronyms, and their full names are listed in Table S1 in the supplemental material. (B) Read size distribution in collected samples. (C) Classification of viruses detected in the tomato samples. (D) Viral discovery curve to assess the saturation and estimate the richness (number of viral species). The solid green line is the rarefaction curve, the dashed green line indicates the Chao2 estimation of asymptotic richness by sample number, and shading shows 95% confidence intervals. Current sampling size and estimated size for 97% sampling completeness are marked. (E) Summary of samples by the number of detected viruses per sample.
Despite the short length of sRNA sequencing reads, the complete genomes of 13 out of 22 detected viruses could be assembled and the genomes of another 5 were nearly complete (genome coverage ≥90%) (Fig. 2), affirming the efficiency of viral genome recovery from sRNA sequences (11). Viruses such as Tomato mosaic virus (ToMV, genus Tobamovirus), Tomato yellow leaf curl virus (TYLCV, genus Begomovirus), Potato virus Y (PVY, genus Potyvirus), Southern tomato virus (STV, genus Amalgavirus), Cucumber mosaic virus (CMV, genus Cucumovirus), Chilli veinal mottle virus (ChiVMV, genus Potyvirus), Tomato mottle mosaic virus (ToMMV, genus Tobamovirus), Tomato chlorosis virus (ToCV, genus Crinivirus), Tomato zonate spot virus (TZSV, genus Tospovirus), and Tomato spotted wilt virus (TSWV, genus Tospovirus) have been well documented in tomato in China and elsewhere. These viruses represent the most prevalent viruses in our sampling set, and the number of samples containing these viruses ranged from 6 to 118 (Fig. 2). In contrast, several detected viruses were reported for the first time in tomato, including Potato virus H (PVH, genus Carlavirus), Turnip yellows virus (TuYV, genus Polerovirus), Potato virus S (PVS, genus Carlavirus), Tobacco vein banding mosaic virus (TVBMV, genus Potyvirus), and Potato virus A (PVA, genus Potyvirus) (Fig. 2; see also Table S1 in the supplemental material). The presence of a subset of these viruses was further confirmed by reverse transcription-PCR (RT-PCR) using specific primers (PVH, 5′-CCGGATGCACAGTCAGCGAT-3′ and 5′-GTACCTCGGCCGTGAAGAGC-3′; TVBMV, 5′-GAATGGGATCGCGCCACAGA-3′ and 5′-TGTGCCGCTGGTTCCAACAT-3′) (Fig. 3). Meanwhile, we identified a fungal virus (Sclerotinia sclerotiorum mitovirus 4, genus Mitovirus) in one sample (T131; Table S1). A close examination of sequencing reads from this sample showed that ∼3% of total sRNAs came from the fungus S. sclerotiorum, suggesting the coexistence of a fungal pathogen in the tomato sample.
FIG 2.

Viral distribution pattern in field-grown tomato samples from China. Regions of samples are clustered according to virus occurrence profiles and displayed on the top. The full information for the region acronyms is listed in Table S1 in the supplemental material. Viruses that were first reported in tomato are in blue text.
FIG 3.
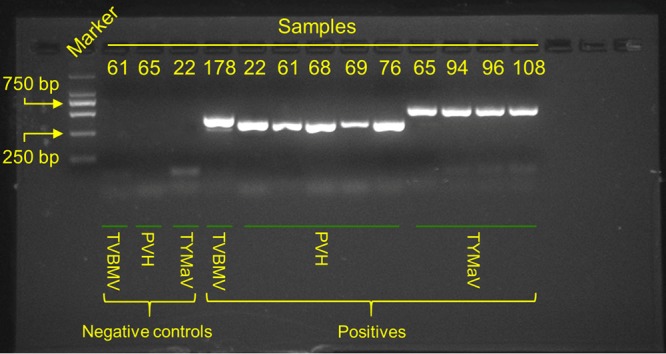
RT-PCR validation of a subset of identified tomato viruses.
Although the tomato viral community in China was dominated by few species, the distribution of these viruses showed variations across sampling regions. ChiVMV was the major problem for tomato in the Chongqing (CQ) area, and this virus was previously shown to infect tomato crops in a nearby region (26), while CMV, ToMV, and TYLCV were more abundant in the Hengyang (HY), Shouguang (SG), and Shanghai (SH) regions, respectively (Fig. 2). Unlike the well-established ones, the newly emerged viruses showed sporadic distribution. Region CQ was the single area containing more than one aforementioned new virus, whereas in most other regions, these viruses were not detected in the studied samples (Fig. 1A and 2).
Genetic diversity of widely distributed tomato viruses.
Due to high mutation and recombination rates, viruses (particularly RNA viruses) are one of the fastest-evolving biological entities. The viral populations within the host contain many nonidentical but similar genome sequences often referred to as quasispecies (27). To evaluate the genetic complexity of viral quasispecies, we focused on the top seven widely distributed viruses, which emerged in at least seven regions. Reads from individual samples were mapped back to the viral consensus genomes to call single nucleotide polymorphisms (SNPs). The percentages of polymorphism sites in viruses fluctuated. CMV had the highest frequency of polymorphic sites (n = 296; 13.36%), which was ∼27 times higher than that of the lowest one, ToMMV (n = 32; 0.50%). The two most widely distributed viruses, ToMV and TYLCV, had relatively high frequencies of polymorphisms of 3.69% (n = 236) and 2.15% (n = 60), respectively (Fig. 4). The frequency of polymorphisms for TYLCV, a DNA virus, was even higher than that for some RNA viruses, such as PVY (n = 115; 1.18%), STV (n = 27; 0.77%), and ChiVMV (n = 117; 1.20%), concordant with a previous finding that some DNA viruses also have high evolutionary rates (28). SNPs in most viruses were scattered on the genome and did not display obvious hot spots or cold spots. However, there are two exceptions, PVY and ToMMV, which exhibited considerable numbers of SNPs in their genomes, but none of them were located in genes encoding coat proteins (CPs), indicating that negative selection might act on these regions. This finding is consistent with the fact that coat proteins usually evolve under selective constraint (28, 29).
FIG 4.

Population diversity of tomato viruses in China. Genomes of the seven most prevalent tomato viruses are shown. SNP positions in the genome of each virus are indicated as black lines in the outer circle of each plot. Colored segments close to the outer circle are coding regions. Depth for vsiRNA reads mapped on the positive strand (red) or negative strand (green) is shown in the inner circle. Orange and gray lines link the recombination sites from positive-positive and positive-negative strands, respectively. Recombination frequencies in the samples are indicated by the line width.
Recombination events in the genomes of tomato viruses were also detected. CMV and ToMV showed high frequency for both positive-to-positive-strand and positive-to-negative-strand recombination (Fig. 4). Genomic sites where recombination occurred frequently in different individual samples were suggested as hot spots. ToMV harbored the most recombination hot spots in its genome, followed by CMV. Interestingly, we found a highly frequent recombination between Nlb and other open reading frames (ORFs) in PVY and ChiVMV, both of which are potyviruses, suggesting that the Nlb region might be a recombination hot spot.
The origin and evolution of tomato viruses are critical questions in viral biology; however, answering these question is challenging for most viruses due to the limited availability of genomic information and the restricted sampling effort. Previous studies have proposed a history, origin, and worldwide spread for TYLCV (29), but how and when this virus became prevalent in China still remain unclear. Our phylogenetic analysis of whole-genome sequences from all previously reported TYLCV strains in China as well as those identified in this study revealed that all Chinese strains fell into one clade with two subgroups, indicating the parallel evolution of Chinese strains from a single origin, with their ancestor likely from the TYLCV-IL strain (Fig. 5). Chinese strains from different locations showed significant diversity. Strains from Shandong province were more divergent than those from other regions as inferred from the branch lengths of the phylogenetic tree. As Shandong is the most important vegetable-producing area in China, it might have been one of the earliest regions suffering from TYCLV infection. TYCLV was first reported in China in 2006 (30), but the exact date of its first appearance in China would undoubtedly be much earlier. To make an estimate of this date, we selected 56 representative strains and inferred the evolutionary rate and time scale using the Bayesian phylogenetic method (31). Because of the high recombination frequency in the TYCLV genome, using the whole-genome sequence would push the estimate of most recent common ancestor (MRCA) much deeper into the past (32). Therefore, a better choice would be to use the coat protein (CP) region (28, 29). The mean evolutionary rate for CP was estimated to be 9.66E−4 substitutions per site per year (95% highest posterior densities [HPDs], 5.58E−4 to 1.38E−3), which is similar to the previously reported data (29). The mean age of MRCA for TYLCV is estimated to be 83 years (95% HPDs, 31.80 to 159.81) (Fig. 6), which is in line with the time of the first description of tomato yellow leaf curl disease-like symptoms in the late 1920s (33). The Chinese strains formed a monoclade with the mean age of MRCA being 18 years (95% HPDs, 10.63 to 26.59). These data suggested that TYLCV may have been present in China as early as 1996, about 10 years before it was first reported in 2006.
FIG 5.

Phylogeny of TYLCV in China. Genome sequences of TYLCV strains identified in this study and those from China and elsewhere downloaded from GenBank (accession numbers are shown in the figure) were used for the phylogenetic analysis. A maximum likelihood (ML) tree was constructed using PhyML with the best-fitted model GTR +Γ4 and 100 bootstraps. Branches in red indicate the highly diverged strains collected from Shandong province.
FIG 6.

Maximum clade creditability tree of TYLCV coat proteins. The time scale below the tree corresponds to the mean posterior estimate of the age in years. Bars represent 95% highest posterior density (HPD) intervals for the estimated divergence time. Nodes in the tree with posterior probabilities greater than 0.5 are labeled with red circles, and those with probabilities smaller than 0.5 are labeled with green circles. Accession number, region, and sampling year for each TYLCV are included in its identifier, and samples from China are shaded with pink.
Discovery of a new virus in tomato.
Five samples (T065, T093, T094, T096, and T108) from the Chongqing region contained highly identical long contigs that matched viral RNA-dependent RNA polymerase (RdRp) with moderate sequence identity (<50%). The complete genome of this virus was assembled and has a size of 13,389 bp (GenBank sequence accession no. KY075646) (Fig. 7A). We have tentatively named this new virus tomato yellow mottle-associated virus (TYMaV) as the tomato leaves harboring this virus showed symptoms including epinasty of leaflet blades, yellow spots, puckering, and mottling (Fig. 7B). We confirmed the presence of TYMaV in the abovementioned samples using RT-PCR but failed to detect it in other samples without similar symptoms (primers 5′-TTTCCTCGGGGCGCTTGTTT-3′ and 5′-GAAGGCCCCCAAGGTCCCTA-3′) (Fig. 3). However, further investigation is required to determine whether TYMaV is the causal factor for the observed symptom. TYMaV has a relatively low frequency of polymorphism (n = 151; 1.1%) and recombination, implying that it is a recently emerged tomato virus in China. The closely related counterparts of TYMaV fall in the genus Cytorhabdovirus, a group of the negative-stranded RNA viruses, and their genomes usually encode six or seven proteins (Fig. 7A). Plants are the natural host for Cytorhabdovirus, but currently, only nine viral species belonging to Cytorhabdovirus are recorded in the ICTV database (http://www.ictvonline.org), although 11 described viruses have genome information (including those currently not categorized in ICTV). TYMaV showed genome structure similar to the Alfalfa dwarf virus (ADV). The abundance of sRNAs generated from the negative strand was comparable to those from the positive strand, indicating that the double-stranded RNA (dsRNA) replicative intermediates could be the primary target for siRNA biogenesis for this virus (Fig. 7A).
FIG 7.

Genome and phylogeny of tomato yellow mottle-associated virus (TYMaV). (A) Genome structure of two representative plant viruses in the genus Cytorhabdovirus and TYMaV. sRNA read distribution on the genome of TYMaV is shown at the bottom. (B) Symptoms of TYMaV-infected tomato plants. The image in the inset is the magnified tomato leaf from the indicated yellow-circled region. (C) Sequence features of the TYMaV genome. The upper panel shows the near-complementary structure of the 5′ and 3′ termini in the genome. The lower panel shows the conserved motif sequences in the intergenic regions. (D) Phylogenic tree of 11 available genomes of members in the genus Cytorhabdovirus constructed with five conserved proteins (N, P, M, G, and L).
As is typical for cytorhabdoviruses, the coding region of TYMaV is flanked by untranslated 3′ leader and 5′ trailer regions, which are nearly complementary to each other with a 1-nt overhang at the 3′ terminus (Fig. 7C). Analysis of intergenic sequences highlighted a conserved motif with the consensus sequence 3′-UAGUUUUAUUAAAN1–11CG-5′. This motif appeared in all intergenic regions, therefore assisting the prediction of the boundaries of ORFs (Fig. 7C). Phylogenetic analysis using five conserved proteins from each of the 11 viral species showed that TYMaV was evolutionarily close to an insect virus recently isolated from an aphid (34). The related Barley yellow striate mosaic virus (BYSMV) and Northern cereal mosaic virus (NCMV) are transmitted by planthoppers, and Alfalfa dwarf virus (ADV) and Lettuce necrotic yellow virus (LNYV) are transmitted by aphids (35–37). As the three Wuhan insect viruses phylogenetically clustered within the plant viruses, it is highly possible that these three viruses carried by the insect were from the host plants. Although the presence of TYMaV sequence in any aphid or aphid transmission of this virus has not been proven, the close phylogenetic relationship to the aphid-transmitted ADV suggests that TYMaV could be transmitted by aphid (Fig. 7D). The conserved intergenic motif has evolved among the 11 viral species, and many viruses (e.g., ADV, Persimmon virus A, and BYSMV) harbor similar but not identical motifs within themselves, indicating genomic recombination between different viruses (Fig. 7D).
Small RNA features underlined differences in viral biology.
Key interesting questions following the discovery of various categories of viral genomes [i.e., dsRNA, ssDNA, (−)ssRNA, and (+)ssRNA] include (i) how the sRNA core machinery differentiates vsiRNA biogenesis among different categories of viruses and (ii) how these differences are reflected in vsiRNA characteristics. To address these questions, we mapped the sRNA reads to the viral genomes in each sample and summarized the sequence-based features of vsiRNAs. We restricted our analysis to the viruses whose reconstructed genomes were no less than 95% of the complete sequences, which turned out to be 17 viruses in total. sRNA read length analysis showed that the sizes of siRNAs from all viruses were centered at 21 and 22 nt. The general read size distribution was in line with previous reports (11, 24), except that siRNAs from the Tomato chlorosis virus (ToCV) were preferentially enriched with those of 22 nt (Fig. 8A). DNA viruses may have different vsiRNA biogenesis mechanisms from RNA viruses, since they lack a dsRNA stage during their infection cycle. Pioneering studies have demonstrated that all four DCLs (DCL1, DCL2, DCL3, and DCL4) are involved in vsiRNA biogenesis for DNA viruses in Arabidopsis, instead of the predominant two (DCL2 and DCL4) for some RNA viruses, and three classes (21, 22, and 24 nt) of vsiRNAs can be observed (21, 38). Indeed, this is true for DNA virus TYLCV, as we found that siRNAs of TYLCV were significantly enriched with those of both 21- and 22-nt size classes, and the 24-nt siRNAs were less abundant but still much more prevalent than those of other sizes (Fig. 8A). This siRNA abundance pattern was not apparent for RNA viruses. The polarity of siRNA reads from different viruses displayed huge variations, although most viruses, except TYMaV, TZSV, and TYLCV, had more reads mapped to the positive strand than to the negative strand. Six viruses had a ratio of reads mapped to the positive strand to those mapped to the negative strand of less than 1.2, suggesting similar siRNA generation efficiencies for the two strands. However, viruses such as TMV, ToMMV, CMV, and ToMV were obviously biased for the positive strand. The extreme case was CMV, for which the number of vsiRNA reads mapped to the positive strand was ∼10 times those mapped to the negative strand (Fig. 8A).
FIG 8.

Characteristics of tomato virus siRNA sequences. (A) Length distribution, polarity profiles, and paired distance (21- and 22-nt reads) of siRNA reads mapped to viral genomes. Columns labeled with stars are positions of 2 nt for the 3′ overhang and 19 nt (for 21-nt vsiRNA) or 20 nt (for 22-nt vsiRNA) for the 5′ overhang, respectively. (B) Paired vsiRNA distance of siRNA reads in ToMV. (C) Examples of three scenarios (perfectly matched, 5′ overhang, and 3′ overhang) of paired vsiRNAs and normalized nucleotide percentage at each position of siRNAs. Normalized nucleotide percentage is calculated by dividing the base percentage at each position with the mean percentage of the respective base between positions 2 and 18. Only paired reads with a 2-nt 3′ overhang were used for calculation. (D) Clusters of tomato viruses based on the correlation coefficient of the normalized nucleotide percentage. Viruses were grouped into the same clusters if their correlation coefficient was ≥0.6. The base composition at the 5′-most position on vsiRNA reads for each cluster is indicated.
vsiRNAs generated by DCLs are intrinsically structured with a 2-nt overhang at the 3′ terminus (39). The cleaved double-stranded siRNAs are further bound and sorted by AGOs depending on the 5′-most base (18). To investigate if these patterns were incorporated into the vsiRNAs in the tomato samples, we assessed the paired vsiRNA distance for all the tomato viruses using a previously described approach (40). The distance profiles showed a significant enrichment at position 2 on 21-nt and 22-nt reads for the 3′ overhang in nearly all viruses (Fig. 8A). These data confirmed that most vsiRNAs in the tomato samples were derived from typical DCL processing. However, some viruses showed an additional peak at position 19 on 21-nt reads or 20 on 22-nt reads for the 5′ overhang (Fig. 8A). This pattern is particularly clear for high-coverage viruses, such as ToMV (Fig. 8B), suggesting that these vsiRNAs were phased. To assess the base preference on the 5′ terminus, we calculated the base percentage for each position, which was then normalized by the average percentage of each base along the selected positions on the reads. As shown for two particular viruses (ToCV and pepper mottle virus [PepMoV]) in Fig. 8C, base composition at positions 1 and 19 of 21-nt reads or 1 and 20 of 22-nt reads displayed striking variations, and the pattern was significantly different from those at other sites (permutation test, P < 0.0001 for both viruses). This observation coincided with the enrichment of a 2-nt 3′ overhang of vsiRNAs and indicated a preferential base selection at the 5′ terminus. Indeed, base G was the least favored at the 5′ terminus for nearly all viruses (Fig. 8C). Many viruses showed a distinguishable preference for the remaining three bases at the 5′ terminus: for example, U was enriched in both 21- and 22-nt vsiRNAs for ToCV, while C was enriched in PepMoV (Fig. 8C). Base compositions may somehow reflect the specific viral biology and could be similar in biologically related species. We therefore calculated the correlation coefficient (Pearson's r) of base composition for all viruses. When the r was set to be ≥0.6, the viruses could be categorized into four groups, among which nine preferred base C and another eight preferred U at the 5′ terminus (Fig. 8D). Since the 5′-terminal base composition has remarkable influence on vsiRNA sorting by AGO proteins (18), our data imply that different viruses within the same host might undergo distinct processes during vsiRNA biogenesis.
DISCUSSION
It has been suggested that tomato is more susceptible to viruses than other vegetable crops (2). However, important questions such as how many viruses actually exist in tomato and how prevalent these viruses are are still challenging to answer, because viruses are highly dynamic, as they can easily evolve and adapt to the environmental pressures, and traditional approaches for virus detection, such as RT-PCR and enzyme-linked immunosorbent assay (ELISA), often fail to discover highly diverged or new viruses. Here, we used deep sRNA sequencing to disclose the tomato viral diversity in China, the leading country in tomato production. To our knowledge, this is the first comprehensive viral landscape in crops revealed from large amounts of field samples.
Our analysis discovered dozens of viruses from 170 tomato samples collected across the country. The majority of the discovered viruses were previously characterized, but many of them are described in tomato for the first time, confirming the rapidly adaptable nature of viruses. ToMV and TYLCV, two notorious viruses found worldwide, are the top two prevalent tomato viruses in China. However, the dominances of the two viruses in different areas are dissimilar. TYLCV was first reported in China in 2006 (30), but through Bayesian phylogenic analysis, we suggested that it might have been present in China as early as 10 years before it was discovered (Fig. 6). STV, another important tomato virus, was initially discovered in Mexico and the United States in 2009 (41) and later reported in France (42), Spain (43), and China (44). Although this virus was discovered only recently and reported from a limited number of countries, we showed that it was detected in 70% of sampling areas in China and ranked as the fourth most prevalent virus in Chinese tomatoes. STV is seed transmitted, the distribution of this virus seems very fast, and the infectious risk is rapidly increasing; therefore, monitoring this virus in the field should be taken seriously in the future.
It has been well established that plants preferentially recruit DCL4 to generate abundant 21-nt vsiRNAs for most RNA viruses. However, in Turnip crinkle virus (TCV), DCL2 instead of DCL4 is the major contributor for vsiRNA biogenesis, which is likely due to the suppression of DCL4 by TCV (45). In this study, although most viruses in tomato generated 21-nt siRNAs, Tomato chlorosis virus (ToCV) generated many more 22-nt siRNAs than 21-nt siRNAs, suggesting that ToCV may inhibit the tomato RNA silencing machinery, consistent with a previous study that demonstrated that the RNA1-encoded p22 protein in ToCV can serve as an effective silencing suppressor in tobacco (46). Differently from RNA viruses, which are generally restricted in the cytoplasm, DNA viruses transcribe their genomes in the nucleus. Accumulation of vsiRNAs from DNA viruses requires the cooperation of all four DCL enzymes in a coordinated and hierarchical manner (21, 38). Indeed, TYLCV showed three types of vsiRNAs, although the 21- and 22-nt sequences are apparently more numerous than 24-nt reads, supporting different roles of DCL proteins in DNA virus processing (Fig. 8A). Once viral RNAs are cleaved, AGO proteins are then recruited for vsiRNA sorting. The Arabidopsis genome encodes 10 AGO proteins, which have diversified functions. AGO1 and AGO2 are the main antiviral AGOs against RNA viruses, with AGO5, AGO7, and AGO10 playing minor roles in some cases. AGO4 is the major antiviral AGO against DNA viruses (47). Diversification of AGO proteins is specifically reflected by their recognition of the 5′-most base of vsiRNAs (18); therefore, different viral species with the same enrichment patterns are possibly processed by the same AGO protein in plants (47). Similarly, we show that all viruses identified in this study have siRNAs enriched with one or more bases of A, U, or C at the 5′ terminus (Fig. 8C and D). We note that for some well-studied viruses, the enrichment patterns are consistent with those reported with experimental evidence (17, 48), implying that our analysis can provide reliable insights into viral biology.
Early models on the biogenesis of vsiRNAs suggested that dsRNA replication intermediates are the major source of vsiRNAs (49). Therefore, the read depths between two strands should be similar. However, as shown in Fig. 8A, many tomato viruses identified in this study have significantly biased read distribution on the two strands, and all viruses display vsiRNA hot spots. Thus, further investigations are needed to elucidate the mechanisms underlying these phenomena.
MATERIALS AND METHODS
Sample collection and small RNA sequencing.
Field-grown tomato samples were collected from China in 2013. The sampled plants were in the early stage of fruit ripening and showed virus-like symptoms, including but not limited to stunting, curling of leaves, chlorosis on leaves and flowers, dwarfing, midrib browning, distorted apical buds, and concentric ringspots. Young leaves at the top of diseased plants were collected for RNA preparation. Total RNA was purified using the TRIzol method according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). The quality of the purified total RNA was evaluated using a Bioanalyzer 2100 (Agilent Technology, Santa Clara, CA). Libraries of small RNAs (lengths of 20 to 60 nt) were constructed from the total RNA using the protocol described previously (50).
Read processing, assembly, and virus identification.
Raw sRNA reads were first processed by trimming the adaptor sequences, and trimmed reads containing an ambiguous base (N) or that were shorter than 15 nt were discarded. Virus contigs were assembled and identified from the sRNA sequences using the VirusDetect pipeline v1.6 (23). The tomato reference genome version 2.4 (51) was used to subtract the host-derived sRNA. SPAdes v3.8.0 (52) was used to reconstruct the complete genome of tomato yellow mottle-associated virus from the pooled sRNA reads.
SNP calling and recombination detection.
For SNP analysis, we first excluded samples in which the assembled viral contigs covered less than 50% of the corresponding viral genomes. sRNA reads with a low-quality base (Q < 20) or that were shorter than 20 nt or longer than 24 nt were excluded from the analysis. The resulting sRNA reads were then mapped to the viral genomes using the Burrows-Wheeler Aligner (BWA) (53), allowing no more than three mismatches. Only uniquely mapped reads were included in the downstream analysis. The resulting BAM file was marked with duplicates using Picard (version 2.2.4; https://broadinstitute.github.io/picard/) and processed by GATK (54) for indel realignment and mapping quality recalibration. The adjusted BAM file was fed to HaplotypeCaller in GATK (version 3.5) for SNP calling. Raw SNPs were further filtered, and only those with a depth of ≥10 and a minor allele frequency of ≥5% were retained. Reads not mapped to host and viral genomes were used to identify nonhomologous recombination events using ViReMa (55) with the seed length set to 11 nt. We set the criterion of two nonidentical reads, as commonly used in other studies (24), to define a recombination event. SNPs and recombination were visualized in the viral genomes using Circos v0.69-3 software (56).
Phylogeny and dating.
The whole-genome sequences or concatenated gene regions were aligned using MUSCLE (57). trimAl (58) was then employed to remove poorly aligned regions. The resulting alignment was subjected to model selection using jModelTest 2 (59). Maximum likelihood phylogenetic trees were constructed using PhyML (60) with the best-fitted model and 100 bootstraps. To get the time scale for the evolution of TYLCV, we aligned the coat protein sequences from all collected viruses. The evolution rate and time scale were estimated using BEAST v2.4.0 (31). According to previous studies (28, 29), we used a coalescent constant population size tree prior, a log-normal relaxed molecular clock, and a general time-reversible (GTR) +Γ4 substitution model for our TYLCV analysis. Each BEAST run was performed with 200 million steps in the Markov chain and sampled every 10,000 steps to produce a posterior tree distribution containing 20,000 genealogies. The maximum clade credibility tree was then built using TreeAnnotator in the BEAST package.
Accession number(s).
Small RNA reads have been deposited in the NCBI Sequence Read Archive (SRA) under accession number SRP092384. The complete genome sequence of tomato yellow mottle-associated virus has been deposited in GenBank under accession number KY075646.
Supplementary Material
ACKNOWLEDGMENTS
We thank Keith Perry and Kai-Shu Ling for critical readings of the manuscript.
This work was supported by grants from the Development and Collaborative Innovation Center of Shanghai (no. ZF1205); the Capacity Construction Project of Local Universities, Shanghai, China (no. 14390502700); the Set-Sail Plan Project supported by the Science and Technology Commission of Shanghai, China (no. 14YF1409400); the National Natural Science Foundation of China (no. 31601769); the National Science Foundation (IOS-1110080); and USDA SCRI (2012-01507-229756).
Quanhua Wang, Xuepeng Sun, Zhangjun Fei, and Quanxi Wang designed the project. Quanhua Wang, Xiaofeng Cai, Xiaoli Wang, Chenhui Ge, and Guanghui Pan collected samples. Chenxi Xu, Chen Jiao, and Yimin Xu performed the experiments. Xuepeng Sun, Chenxi Xu, Angela Taylor, and Zhangjun Fei analyzed the data. Xuepeng Sun and Zhangjun Fei wrote the paper.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00173-17.
REFERENCES
- 1.Bani R, Josiah M, Kra E. 2006. Postharvest losses of tomatoes in transit. Agric Mech Asia Afr Lat Am 37:84–86. [Google Scholar]
- 2.Brunt A, Crabtree K, Dallwitz M, Gibbs A, Watson L, Zurcher E (ed). 1997. Plant viruses online: descriptions and lists from the VIDE database. Version 16 January 1997 Australian National University, Canberra, Australia: http://sdb.im.ac.cn/vide/refs.htm. [Google Scholar]
- 3.Hanssen IM, Lapidot M, Thomma BP. 2010. Emerging viral diseases of tomato crops. Mol Plant Microbe Interact 23:539–548. doi: 10.1094/MPMI-23-5-0539. [DOI] [PubMed] [Google Scholar]
- 4.Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, Rubin E, Ivanova NN, Kyrpides NC. 2016. Uncovering Earth's virome. Nature 536:425–430. doi: 10.1038/nature19094. [DOI] [PubMed] [Google Scholar]
- 5.Anthony SJ, Islam A, Johnson C, Navarrete-Macias I, Liang E, Jain K, Hitchens PL, Che X, Soloyvov A, Hicks AL. 2015. Non-random patterns in viral diversity. Nat Commun 6:8147. doi: 10.1038/ncomms9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster CL, Waldron FM, Robertson S, Crowson D, Ferrari G, Quintana JF, Brouqui J-M, Bayne EH, Longdon B, Buck AH. 2015. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLoS Biol 13:e1002210. doi: 10.1371/journal.pbio.1002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R. 2009. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388:1–7. doi: 10.1016/j.virol.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 8.Wu Q, Ding SW, Zhang Y, Zhu S. 2015. Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Annu Rev Phytopathol 53:425–444. doi: 10.1146/annurev-phyto-080614-120030. [DOI] [PubMed] [Google Scholar]
- 9.Wu Q, Wang Y, Cao M, Pantaleo V, Burgyan J, Li WX, Ding SW. 2012. Homology-independent discovery of replicating pathogenic circular RNAs by deep sequencing and a new computational algorithm. Proc Natl Acad Sci U S A 109:3938–3943. doi: 10.1073/pnas.1117815109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons HE, Dunham JP, Stack JC, Dickins BJ, Pagan I, Holmes EC, Stephenson AG. 2012. Deep sequencing reveals persistence of intra- and inter-host genetic diversity in natural and greenhouse populations of zucchini yellow mosaic virus. J Gen Virol 93:1831–1840. doi: 10.1099/vir.0.042622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li R, Gao S, Hernandez AG, Wechter WP, Fei Z, Ling KS. 2012. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS One 7:e37127. doi: 10.1371/journal.pone.0037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vainio EJ, Jurvansuu J, Streng J, Rajamäki M-L, Hantula J, Valkonen JP. 2015. Diagnosis and discovery of fungal viruses using deep sequencing of small RNAs. J Gen Virol 96:714–725. doi: 10.1099/jgv.0.000003. [DOI] [PubMed] [Google Scholar]
- 13.Aguiar ER, Olmo RP, Paro S, Ferreira FV, de Faria IJ, Todjro YM, Lobo FP, Kroon EG, Meignin C, Gatherer D, Imler JL, Marques JT. 2015. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res 43:6191–6206. doi: 10.1093/nar/gkv587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schopman NC, Willemsen M, Liu YP, Bradley T, van Kampen A, Baas F, Berkhout B, Haasnoot J. 2012. Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res 40:414–427. doi: 10.1093/nar/gkr719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamilton AJ, Baulcombe DC. 1999. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 16.Diaz-Pendon JA, Li F, Li W-X, Ding S-W. 2007. Suppression of antiviral silencing by cucumber mosaic virus 2b protein in Arabidopsis is associated with drastically reduced accumulation of three classes of viral small interfering RNAs. Plant Cell 19:2053–2063. doi: 10.1105/tpc.106.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang XB, Jovel J, Udomporn P, Wang Y, Wu Q, Li WX, Gasciolli V, Vaucheret H, Ding SW. 2011. The 21-nucleotide, but not 22-nucleotide, viral secondary small interfering RNAs direct potent antiviral defense by two cooperative argonautes in Arabidopsis thaliana. Plant Cell 23:1625–1638. doi: 10.1105/tpc.110.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, Chen S, Hannon GJ, Qi Y. 2008. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133:116–127. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang XB, Wu Q, Ito T, Cillo F, Li WX, Chen X, Yu JL, Ding SW. 2010. RNAi-mediated viral immunity requires amplification of virus-derived siRNAs in Arabidopsis thaliana. Proc Natl Acad Sci U S A 107:484–489. doi: 10.1073/pnas.0904086107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Ruiz H, Takeda A, Chapman EJ, Sullivan CM, Fahlgren N, Brempelis KJ, Carrington JC. 2010. Arabidopsis RNA-dependent RNA polymerases and dicer-like proteins in antiviral defense and small interfering RNA biogenesis during turnip mosaic virus infection. Plant Cell 22:481–496. doi: 10.1105/tpc.109.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blevins T, Rajeswaran R, Shivaprasad PV, Beknazariants D, Si-Ammour A, Park H-S, Vazquez F, Robertson D, Meins F, Hohn T. 2006. Four plant Dicers mediate viral small RNA biogenesis and DNA virus induced silencing. Nucleic Acids Res 34:6233–6246. doi: 10.1093/nar/gkl886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma M, Huang Y, Gong Z, Zhuang L, Li C, Yang H, Tong Y, Liu W, Cao W. 2011. Discovery of DNA viruses in wild-caught mosquitoes using small RNA high throughput sequencing. PLoS One 6:e24758. doi: 10.1371/journal.pone.0024758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, Gao S, Padmanabhan C, Li R, Galvez M, Gutierrez D, Fuentes S, Ling K-S, Kreuze J, Fei Z. 2017. VirusDetect: an automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 500:130–138. doi: 10.1016/j.virol.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Kutnjak D, Rupar M, Gutierrez-Aguirre I, Curk T, Kreuze JF, Ravnikar M. 2015. Deep sequencing of virus-derived small interfering RNAs and RNA from viral particles shows highly similar mutational landscapes of a plant virus population. J Virol 89:4760–4769. doi: 10.1128/JVI.03685-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chao A, Gotelli NJ, Hsieh T, Sander EL, Ma K, Colwell RK, Ellison AM. 2014. Rarefaction and extrapolation with Hill numbers: a framework for sampling and estimation in species diversity studies. Ecol Monogr 84:45–67. doi: 10.1890/13-0133.1. [DOI] [Google Scholar]
- 26.Zhao FF, Xi DH, Liu J, Deng XG, Lin HH. 2014. First report of chilli veinal mottle virus infecting tomato (Solanum lycopersicum) in China. Plant Dis 98:1589. doi: 10.1094/PDIS-11-13-1188-PDN. [DOI] [PubMed] [Google Scholar]
- 27.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol Mol Biol Rev 76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duffy S, Holmes EC. 2008. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus. J Virol 82:957–965. doi: 10.1128/JVI.01929-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefeuvre P, Martin DP, Harkins G, Lemey P, Gray AJ, Meredith S, Lakay F, Monjane A, Lett JM, Varsani A, Heydarnejad J. 2010. The spread of tomato yellow leaf curl virus from the Middle East to the world. PLoS Pathog 6:e1001164. doi: 10.1371/journal.ppat.1001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Dai F, Zhou X. 2006. First report of Tomato yellow leaf curl virus in China. Plant Dis 90:1359–1359. doi: 10.1094/PD-90-1359C. [DOI] [PubMed] [Google Scholar]
- 31.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awadalla P. 2003. The evolutionary genomics of pathogen recombination. Nat Rev Genet 4:50–60. doi: 10.1038/nrg964. [DOI] [PubMed] [Google Scholar]
- 33.Antignus Y, Cohen S. 1994. Complete nucleotide sequence of an infectious clone of a mild isolate of tomato yellow leaf curl virus (TYLCV). Phytopathology 84:707–712. doi: 10.1094/Phyto-84-707. [DOI] [Google Scholar]
- 34.Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, Qin XC, Xu J, Holmes EC, Zhang YZ. 2015. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 4:e05378. doi: 10.7554/eLife.05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Higgins CM, Bejerman N, Li M, James AP, Dietzgen RG, Pearson MN, Revill PA, Harding RM. 2016. Complete genome sequence of Colocasia bobone disease-associated virus, a putative cytorhabdovirus infecting taro. Arch Virol 161:745–748. doi: 10.1007/s00705-015-2713-7. [DOI] [PubMed] [Google Scholar]
- 36.Heim F, Lot H, Delecolle B, Bassler A, Krczal G, Wetzel T. 2008. Complete nucleotide sequence of a putative new cytorhabdovirus infecting lettuce. Arch Virol 153:81–92. doi: 10.1007/s00705-007-1071-5. [DOI] [PubMed] [Google Scholar]
- 37.Ito T, Suzaki K, Nakano M. 2013. Genetic characterization of novel putative rhabdovirus and dsRNA virus from Japanese persimmon. J Gen Virol 94:1917–1921. doi: 10.1099/vir.0.054445-0. [DOI] [PubMed] [Google Scholar]
- 38.Aregger M, Borah BK, Seguin J, Rajeswaran R, Gubaeva EG, Zvereva AS, Windels D, Vazquez F, Blevins T, Farinelli L. 2012. Primary and secondary siRNAs in geminivirus-induced gene silencing. PLoS Pathog 8:e1002941. doi: 10.1371/journal.ppat.1002941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma JB, Ye K, Patel DJ. 2004. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 429:318–322. doi: 10.1038/nature02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Lu J, Han Y, Fan X, Ding S-W. 2013. RNA interference functions as an antiviral immunity mechanism in mammals. Science 342:231–234. doi: 10.1126/science.1241911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabanadzovic S, Valverde RA, Brown JK, Martin RR, Tzanetakis IE. 2009. Southern tomato virus: the link between the families Totiviridae and Partitiviridae. Virus Res 140:130–137. doi: 10.1016/j.virusres.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 42.Candresse T, Marais A, Faure C. 2013. First report of Southern tomato virus on tomatoes in southwest France. Plant Dis 97:1124. doi: 10.1094/PDIS-01-13-0017-PDN. [DOI] [PubMed] [Google Scholar]
- 43.Verbeek M, Dullemans A, Espino A, Botella M, Alfaro-Fernández A, Font M. 2015. First report of Southern tomato virus in tomato in the Canary Islands, Spain. J Plant Pathol 97(2) doi: 10.4454/JPP.V97I2.038. [DOI] [Google Scholar]
- 44.Padmanabhan C, Zheng Y, Li R, Sun SE, Zhang D, Liu Y, Fei Z, Ling KS. 2015. Complete genome sequence of Southern tomato virus identified in China using next-generation sequencing. Genome Announc 3:e01226-15. doi: 10.1128/genomeA.01226-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouché N, Lauressergues D, Gasciolli V, Vaucheret H. 2006. An antagonistic function for Arabidopsis DCL2 in development and a new function for DCL4 in generating viral siRNAs. EMBO J 25:3347–3356. doi: 10.1038/sj.emboj.7601217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cañizares MC, Navas-Castillo J, Moriones E. 2008. Multiple suppressors of RNA silencing encoded by both genomic RNAs of the crinivirus, Tomato chlorosis virus. Virology 379:168–174. doi: 10.1016/j.virol.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 47.Carbonell A, Carrington JC. 2015. Antiviral roles of plant ARGONAUTES. Curr Opin Plant Biol 27:111–117. doi: 10.1016/j.pbi.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morel JB, Godon C, Mourrain P, Béclin C, Boutet S, Feuerbach F, Proux F, Vaucheret H. 2002. Fertile hypomorphic ARGONAUTE (ago1) mutants impaired in post-transcriptional gene silencing and virus resistance. Plant Cell 14:629–639. doi: 10.1105/tpc.010358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahlquist P. 2002. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 296:1270–1273. doi: 10.1126/science.1069132. [DOI] [PubMed] [Google Scholar]
- 50.Chen YR, Zheng Y, Liu B, Zhong S, Giovannoni J, Fei Z. 2012. A cost-effective method for Illumina small RNA-Seq library preparation using T4 RNA ligase 1 adenylated adapters. Plant Methods 8:41. doi: 10.1186/1746-4811-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomato Genome Consortium. 2012. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Routh A, Johnson JE. 2014. Discovery of functional genomic motifs in viruses with ViReMa—a Virus Recombination Mapper—for analysis of next-generation sequencing data. Nucleic Acids Res 42:e11. doi: 10.1093/nar/gkt916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.