

ABSTRACT
Monkeypox virus (MPXV) is a human pathogen that is a member of the Orthopoxvirus genus, which includes Vaccinia virus and Variola virus (the causative agent of smallpox). Human monkeypox is considered an emerging zoonotic infectious disease. To identify host factors required for MPXV infection, we performed a genome-wide insertional mutagenesis screen in human haploid cells. The screen revealed several candidate genes, including those involved in Golgi trafficking, glycosaminoglycan biosynthesis, and glycosylphosphatidylinositol (GPI)-anchor biosynthesis. We validated the role of a set of vacuolar protein sorting (VPS) genes during infection, VPS51 to VPS54 (VPS51–54), which comprise the Golgi-associated retrograde protein (GARP) complex. The GARP complex is a tethering complex involved in retrograde transport of endosomes to the trans-Golgi apparatus. Our data demonstrate that VPS52 and VPS54 were dispensable for mature virion (MV) production but were required for extracellular virus (EV) formation. For comparison, a known antiviral compound, ST-246, was used in our experiments, demonstrating that EV titers in VPS52 and VPS54 knockout (KO) cells were comparable to levels exhibited by ST-246-treated wild-type cells. Confocal microscopy was used to examine actin tail formation, one of the viral egress mechanisms for cell-to-cell dissemination, and revealed an absence of actin tails in VPS52KO- or VPS54KO-infected cells. Further evaluation of these cells by electron microscopy demonstrated a decrease in levels of wrapped viruses (WVs) compared to those seen with the wild-type control. Collectively, our data demonstrate the role of GARP complex genes in double-membrane wrapping of MVs necessary for EV formation, implicating the host endosomal trafficking pathway in orthopoxvirus infection.
IMPORTANCE Human monkeypox is an emerging zoonotic infectious disease caused by Monkeypox virus (MPXV). Of the two MPXV clades, the Congo Basin strain is associated with severe disease, increased mortality, and increased human-to-human transmission relative to the West African strain. Monkeypox is endemic in regions of western and central Africa but was introduced into the United States in 2003 from the importation of infected animals. The threat of MPXV and other orthopoxviruses is increasing due to the absence of routine smallpox vaccination leading to a higher proportion of naive populations. In this study, we have identified and validated candidate genes that are required for MPXV infection, specifically, those associated with the Golgi-associated retrograde protein (GARP) complex. Identifying host targets required for infection that prevents extracellular virus formation such as the GARP complex or the retrograde pathway can provide a potential target for antiviral therapy.
KEYWORDS: GARP complex, HAP1 screen, poxviruses, retrograde transport
INTRODUCTION
Monkeypox virus (MPXV) is a member of the Orthopoxvirus (OPXV) genus, which also includes Variola virus (the causative agent of eradicated smallpox) and Vaccinia virus (VACV) (1). MPXV infection is an emerging zoonotic infectious disease in humans that is endemic in western and central Africa (2). Two distinct clades of MPXV have been identified, the West African and Congo Basin clades, which exhibit genetic, clinical, and geographic differences (3). Generally, the Congo Basin clade is associated with a more severe disease manifestation, with a mortality rate of up to 10% and extended human-to-human transmission compared to the West African clade (3–5). Due to the cessation of smallpox vaccination, waning immunity, and, consequently, an increased proportion of naive population members, MPXV outbreaks can pose a serious public health threat (6). For instance, the MPXV outbreak of 2003 in United States caused by the West African clade virus spread from imported infected African animals to prairie dogs and subsequently to humans (7). Although MPXV infection is preventable by preexposure vaccination, treatment options after contraction of the disease are limited due to the absence of approved therapeutics.
Orthopoxviruses are double-stranded DNA viruses that replicate exclusively in the cytoplasm of the host cell (8). Viral factories are the cytoplasmic structures engaged in viral DNA replication, gene expression, and assembly of mature virions (MVs). Although MVs are infectious, these particles remain intracellular unless they are transported out of the viral factories to the Golgi/endosomal compartment for wrapping with two additional lipid bilayers to generate extracellular viruses (EVs) (9–11). EVs are responsible for cell-to-cell and long-distance spread of virus, which is essential for pathogenicity in animals (12–14). Although poxviruses encode a majority of genes required for replication, transcription, immune evasion, and virus assembly, host proteins are also required to complete the viral life cycle.
Previous studies have utilized RNA interference (RNAi) to identify host factors required for infections by VACV, the prototype member of the poxvirus family (15–19). These studies have provided important findings of proteins and host pathways exploited by VACV, including macropinocytosis, ubiquitin-proteasome system, and nuclear pore complex pathways and a recent demonstration of the retrograde pathway (20, 21). Here, we exploited the utility of genome-scale haploid genetic screens as an alternative method to identify host factors required for MPXV infection. This approach, which generates gene knockouts (KOs) by insertional mutagenesis in a haploid human cell line (HAP1), has been successfully used to identify host factors required for infection by a variety of viral pathogens, including Ebola virus, Lassa virus, and Rift Valley fever virus (22–24).
In this study, we have identified candidate host genes necessary for infection of both clades of MPXV in haploid cells, including those involved in Golgi trafficking, glycosaminoglycan (GAG) biosynthesis, and glycosylphosphatidylinositol (GPI)-anchor biosynthesis. We confirmed the requirement of Golgi-associated retrograde protein (GARP) complex genes in MPXV infection and further demonstrate that GARP complex proteins are dispensable for MV formation but are essential for membrane wrapping to form EVs and egress.
RESULTS
A mutagenesis screen in human haploid cells identified host factors required for MPXV infection.
In order to identify host factors required for MPXV infection, we employed a large-scale gene knockout approach using a retroviral gene trap vector in human haploid HAP1 cells (22). We used two strains of MPXV, representing the two clades of virus that exhibit differences in virulence, in the haploid genetic screen, therefore providing the opportunity to identify necessary host factors required for infection by either or both strains. Mutagenized HAP1 cells were infected with MPXV (MPXV ROC [Republic of Congo] or MPXV WA [West Africa]) and screened for surviving mutant cells as described previously (22). By 3 days postinfection (dpi), infected cells exhibited cytopathic effects and regularly detached from flasks. These detached cells and secreted EVs in the media were removed every 2 to 3 days, and the culture was replenished with fresh media. By 7 dpi, most of the cells were completely detached and the few remaining adherent cells were cultured until 18 dpi and harvested for further analysis. Surviving cells were sequenced to quantify independent retroviral insertion events compared to the unselected control (see Table S1 in the supplemental material). Genes that were significantly enriched for insertions (adjusted P value < 0.05) were selected for further analysis.
The top 20 most significant genes were highlighted and represented according to the P value and corresponding number of independent gene trap events for the MPXV ROC-resistant population (Fig. 1A) and the MPXV WA-resistant population (Fig. 1B). A total of 48 candidate host genes were significantly enriched. Eight genes were exclusively identified in the MPXV ROC screen, 26 were exclusively identified in the MPXV WA screen, and 14 were identified in both screens (Fig. 2A). These genes were subjected to functional annotation analysis using Cytoscape in combination with the ClueGO plugin. Functional annotation terms enriched in our data set included a “GARP complex” associated with vacuolar protein sorting (VPS) genes VPS51, VPS53, and VPS54; a “Golgi transport complex” associated with conserved oligomeric Golgi (COG) complex genes COG3, COG4, COG7, and COG8; “glycosaminoglycan biosynthesis” associated with B3GALT6, B3GAT3, B4GALT7, EXT1, and XYLT2; and “GPI-anchor biosynthesis” associated with phosphatidyl inositol glycan (PIG) genes PIGL, PIGP, PIGU, DPM3, and MPDU1 (Fig. 2A). Several genes were identified in multiple functional groups; therefore, functional grouping of related terms was performed, with results demonstrating that the majority of the genes identified corresponded to “glycosaminoglycan biosynthesis” and “Golgi apparatus part” (Fig. 2B). Of particular interest were genes that were part of a complex and were known to function as a unit in association with a biological process, such as the GARP complex. The GARP complex is composed of four proteins, VPS51, VPS52, VPS53, and VPS54, all of which were identified in our screen (Fig. 2A).
FIG 1.
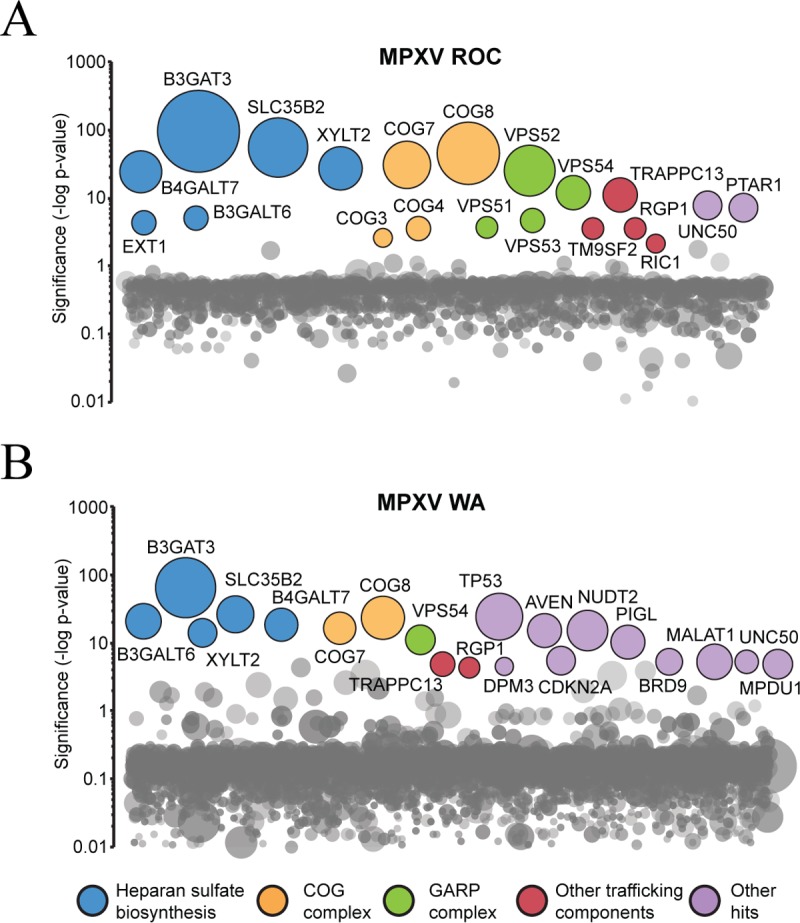
Haploid genetic screen identifies host factors required for MPXV infection in HAP1 cells. (A) Significance of enrichment of gene-trap insertions for MPXV ROC. (B) Significance of enrichment of gene-trap insertions for MPXV WA. Each circle represents a gene, and the size of the circle represents the number of independent gene-trap events identified in the selected resistant population. The y axis shows the significance of enrichments of gene-trap insertions between selected and unselected cell populations calculated using Fisher's exact test. The top 20 genes with the highest significance are labeled according to the functional annotation group.
FIG 2.

Functional gene annotation analyses of significant MPXV host factors. (A) MPXV ROC and WA enriched genes with an adjusted P value of <0.05. Genes listed in the middle were identified in both screens. (B) Overview of functional annotation ClueGO analysis. Similar terms were grouped into one representative term. The group sections represent the relative numbers of terms in the groups. The significance value for each group is indicated. **, P ≤ 0.001.
GARP complex genes required for MPXV EV production.
The GARP complex is a heteromeric multisubunit tethering complex whose members are best known for their roles in the retrograde transport of endosome-derived vesicles to the trans-Golgi network (25). In order to validate these genes, HEK293FF6 GARP complex knockout (KO) cells (VPS51KO, VPS52KO, VPS53KO, or VPS54KO) (26), along with wild-type (WT) control cells, were infected with MPXV for 24 h. GARP KO cell lines were previously generated using the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 system, and each KO cell line was evaluated for loss of expression of the target gene by DNA sequencing (26). The absence of protein expression was confirmed by immunoblotting (26) (data not shown). The yields of intracellular (MV) and extracellular virus released in the medium were determined. Tecovirimat (ST-246), an antiviral compound previously reported to target wrapping of MVs and thereby prevent the production of EV (27), was used as a control for EV release.
EV yield was significantly reduced in VPS52KO and VPS54KO cells infected with MPXV ROC compared to those infected with a VPS WT control (Fig. 3A), with no appreciable reduction in MV yield seen. The percentage of EV released was dramatically decreased in VPS52KO- and VPS54KO-infected cells and slightly decreased in VPS53KO-infected cells. Although these values did not meet statistical significance based on our cutoff P value of 0.05, the trend is consistent with the EV yield. Similarly, MPXV WA EV yields were significantly decreased in VPS52KO and VPS54KO cells (Fig. 3B). The percentage of EV released was also decreased in VPS52KO cells, in VPS54KO cells, and, to some extent, in VPS51KO cells but not in VPS53KO cells.
FIG 3.

GARP complex genes are required for MPXV EV production. (A and B) HEK293FF6 cell lines containing VPS WT, VPS51KO, VPS52KO, VPS53KO, or VPS54KO genes were infected with MPXV ROC (A) or WA (B) at a multiplicity of infection (MOI) of 1 in the absence and presence of 2 μM ST-246. MV and EV production was measured 24 h postinfection (hpi) in each cell line. The percentage of EV released was determined by calculating the proportion of EV represented in the total number of virions (MV plus EV) for each replicate. Data shown represent average percent EV released calculated from each individual replicate set. (C) GARP KO cell lines were infected with MPXV WA. After 72 h, cells were fixed and stained with MPXV reactive anti-VACV antibody followed by HRP-conjugated secondary antibody. The monolayer was developed using o-dianisidine solution. Data are shown as means (n = 3), and error bars represent standard deviations. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Extracellular viruses are required for cell-cell spread and hence determine the size of plaques. Since EV production is significantly decreased in the absence of VPS52 and VPS54, plaque formation was analyzed in KO cells. Plaque formation in the HEK293FF6 cell line was more difficult to evaluate than in BSC-40 cells, in which plaque formation is clear and distinguishable by crystal violet staining. Therefore, we used an immunostaining method for analysis of HEK293 cells infected with MPXV WA as an alternative to evaluate plaque formation. All VPS KO cells exhibited plaques that were smaller than those seen with the WT, with the greatest amount of reduction observed in VPS52KO and VPS54KO cells (Fig. 3C). Interestingly, even though the EV yields were similar in the WT and VPS53KO cells, plaque sizes were diminished in the absence of VPS53. This suggests that the plaque size is not always representative of EV yield in the medium or that the plaque assay is potentially a more sensitive assay because it relies on multiple rounds of infection, which could amplify smaller differences.
The GARP complex is required for VACV EV production.
To determine whether the GARP complex is also important for infection of other orthopoxviruses, GARP KO cells were infected with the VACV IHDJ strain, a strain that releases a higher percentage of EVs than other VACV strains (14). The yield of EV demonstrated significant reduction in all VPS KO cells, with the maximum effect in VPS52 and VPS54KO cells, similarly to MPXV (Fig. 4A). The EV yields in the absence of VPS52 and VPS54 were comparable to those seen with ST-246 treatment, demonstrating the importance of this pathway in OPXV EV release. As observed with MPXV, immunostaining of VACV in VPSKO-infected cells demonstrated the presence of smaller plaque sizes in all VPS KO cells. VPS52KO and VPS54KO cells in particular demonstrated the maximum effect on plaque formation (Fig. 4B). Thus, the results clearly demonstrate a requirement of VPS52 and VPS54 for EV production in both MPXV and VACV infections. Further investigations were focused on these two genes using VACV due to the availability of reagents and to biosafety requirements.
FIG 4.

GARP complex genes are important for VACV EV production. GARP complex KO cell lines (VPS51KO, VPS52KO, VPS53KO, and VPS54KO) were infected with VACV strain IHDJ at an MOI of 1 in the absence and presence of 2 μM ST-246. MV and EV production was measured 24 hpi in each cell line. The percentage of EV released was determined by calculating the proportion of EV represented in the total number of virions (MV plus EV). All MV and EV values were averaged first, and then percent EV released was calculated from the averages. (C) Immunostaining of VACV IHDJ-infected KO cells. After 72 h, cells were fixed and stained with anti-VACV antibody followed by HRP-conjugated secondary antibody. Data are shown as means (n = 6), and error bars represent standard deviations. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Complementation of the GARP complex in deficient cells restores viral production.
In order to determine if the defect in EV production was specific to KO genes and could be restored by ectopic expression, we performed complementation experiments in VPS52KO and VPS54KO cells. VPS52KO cells were transfected with a plasmid expressing wild-type FLAG-tagged VPS52 (pVPS52) or a control plasmid 24 h prior to infection. Immunoblotting confirmed VPS52 expression in transfected cells with pVPS52 (Fig. 5B). Transfected cells were then infected with VACV IHDJ at a multiplicity of infection (MOI) of 1, and virus yield was measured 24 h postinfection (hpi). EV yield in VPS52 transfected cells was increased compared to control results (Fig. 5A). In addition, the percentage of EV released was also increased in VPS52 transfected cells versus the control, suggesting that complementation of wild-type VPS52 gene in VPS52KO cells was able to restore EV production and release.
FIG 5.
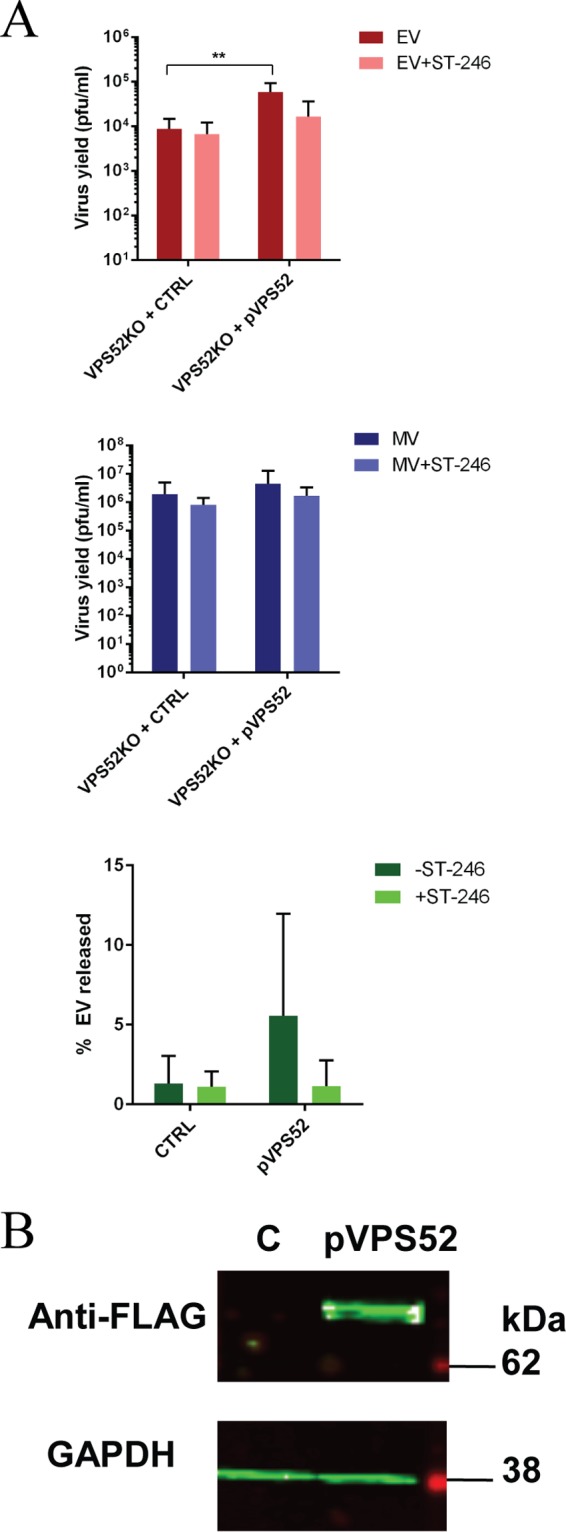
Complementation of GARP complex in KO cells restores viral production. (A) VPS52KO cells were transfected with pLenti-VPS52-FLAG plasmid (pVPS52) or control plasmid (CTRL). Cells were then infected with VACV strain IHDJ in the presence or absence of ST-246. MV and EV production was measured 24 hpi. The percentage of EV released was determined by calculating the proportion of EV represented in the total number of virions (MV plus EV). Data are shown as means (n = 7), and error bars represent standard deviations. (B) VPS52KO cells were transfected with a FLAG-tagged VPS52-expressing plasmid (pVPS52) or a non-VPS52-expressing plasmid (CTRL). After 24 h, cells were harvested and processed for Western blot analysis using anti-FLAG antibody followed by IRDye secondary antibodies and were visualized using direct infrared fluorescence. GAPDH was used as a control.
Similarly, to assess whether VPS54 expression in VPS54KO cells could restore virus yield, we infected VPS54KO cells that were stably transfected with wild-type VPS54-expressing vector (VPS54KO-VPS54) or an empty vector control (VPS54KO-empty) (26). Expression of VPS54 was evaluated in the two cell lines by immunoblotting (Fig. 6B), with results demonstrating expression of V5-tagged VPS54 vector in VPS54KO-VPS54 cells and the absence of expression in VPS54-empty cells. EV yield and MV yield were measured 24 hpi, and the results demonstrated significantly higher EV levels in VPS54-expressing cells than were seen with the empty vector control (Fig. 6A). The higher percentage of EV released in VPS54-expressing stable cells than in the empty vector control confirms the requirement of VPS54 expression in these KO cells. Thus, the results further demonstrate the specific requirement and complementation of VPS52 and VPS54 for OPXV EV release in the respective KO cells.
FIG 6.

(A) Stable VPS54KO cell lines expressing empty vector or VPS54 were infected with VACV strain IHDJ in the presence or absence of 2 μM ST-246. MV and EV production was measured 24 hpi. The percentage of EV released was determined by calculating the proportion of EV represented in the total number of virions (MV plus EV). Data are shown as means (n = 3), and error bars represent standard deviations. (B) VPS54KO-empty and VPS54KO-VPS54 (V5-tagged) cells were harvested and processed for Western blot analysis using anti-V5 antibody, followed by HRP-conjugated secondary antibody. The blot was developed using enhanced chemiluminescent substrate. (C) Stable VPS54KO cell lines expressing empty vector or VPS54 were infected with VACV strain IHDJ. MV and EV production was measured at 0, 24, 48, 72, and 96 hpi. Data are shown as means (n = 3), and error bars represent standard deviations. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Compared to the single-step growth curve, a multistep infection also demonstrated a role for EV yields and hence for viral spread. For this experiment, VPS54KO empty and VPS54-complementing cell lines were infected with VACV IHDJ at a low MOI (0.01) and virus yields were determined at several time points. Multistep growth curve analysis demonstrated increased viral yield (and hence increased virus spread) in VPS54KO-VPS54-infected cells for both EV and MV compared to VPS54-empty infected cells over a 96-h period (Fig. 6C). Although the MV yield was slightly increased in the VPS54KO-empty cells, the EV yield was maintained at a level close to the input levels. Thus, these results further demonstrate that there was reduced or no EV release in VPS54 KO cells and that EV release was able to be restored with wild-type VPS54 expression.
Virus-induced actin tail formation is deficient in GARP complex knockout cells.
Since EV levels but not MV levels were affected in KO cells, VPS52 and VPS54 may be required for a crucial step after MV assembly. A proportion of the MVs are transported out of the viral factories via microtubules to a site for additional membrane wrapping to form wrapped virus (WV). The outer membrane then fuses with the plasma membrane to form cell-associated EVs (CEVs). Extracellular virus is formed by release of CEVs, which can occur by exocytosis, budding, or actin tail formation, which can extend CEVs toward neighboring cells (28–31). We focused on virus release mediated by actin tail formation and used confocal microscopy to evaluate actin tail projections in VPS52KO- and VPS54KO-infected cells.
VPS KO cells were infected with VACV WR A4-YFP (A4-yellow fluorescent protein) containing YFP fused to A4 core protein for virus visualization. Infected cells were then fixed and stained with phalloidin for cellular actin visualization. As shown in representative images, actin tails with fluorescent punctate viral particles on the tip were present on the surface of WT cells (Fig. 7, upper left panel). As expected, in the absence of EV formation, actin tails were absent in ST-246-treated cells (Fig. 7, upper right panel). However, accumulation of virions inside the cell (MV) demonstrated by the presence of green punctate viral particles was evident in the ST-246-treated cells. Consistent with previous results, confocal microscopy of VPS52KO-infected cells (Fig. 7, lower left panel) and VPS54KO-infected cells (Fig. 7, lower right panel) revealed a defect in actin tail formation and the absence of EV on the cell surface. Thus, the results demonstrate a requirement of VPS52 and VPS54 for virus egress and cell-to-cell spread.
FIG 7.

Actin tail deficiency in VACV-infected VPS KO cells. Cell lines were infected with VACV WRA4-YFP virus (green) and stained with Alexa Fluor 647 phalloidin (red, to visualize F-actin) and DAPI (blue, to visualize DNA) after 24 h. Representative images of infection in VPS WT (upper left), VPS WT in the presence of 2 μM ST-246 (upper right), VPS52KO (bottom left), and VPS54KO (bottom right) cells are shown. White arrowheads point to a representative actin tail with loaded virus. Original magnification: ×40.
Viral membrane wrapping is impaired in GARP complex-deficient cells.
Membrane wrapping was investigated using electron microscopy (EM) in VACV IHDJ-infected VPS52KO and VPS54KO cells. WT cells treated with ST-246 were used as a control, given that ST-246 blocks extracellular virus formation. Electron microscopic images were examined for the presence of MV, WV, and EV particles. In the absence of membrane continuity, extracellular cell-associated virus could not be differentiated on the basis of egress or the presence of superinfecting MVs. Hence, WVs and cell-associated EVs were quantified together. To obtain quantifiable information, around 20 to 25 unique fields for each condition were examined. Upon infection, both WT and KO (VPS52 and VPS54) cells exhibited different morphological forms of VACV, namely, crescents (C), immature virus (IV), and MVs. However, WVs and EVs were observed only in WT cells and were almost completely absent upon treatment with ST-246 as represented in Fig. 8A. More importantly, electron micrographs representing VPS52 and VPS54KO cells demonstrated a considerable reduction in levels of WVs, corroborating other results showing decreases in virus release (Fig. 8A). Quantification yielded 320 MVs and 36 EVs in WT cells, 340 MVs and 3 EVs in WT cells treated with ST-246, 452 MVs and 7 EVs in VPS52KO cells, and 508 MVs and 13 EVs in VPS54 KO cells. The results were used to calculate percentages of WV (Fig. 8B), and the data clearly demonstrated a reduction in membrane wrapping in VPS52 and VPS54KO cells. Therefore, VPS52 and VPS54 proteins, as part of the GARP complex, are required for membrane wrapping of MV particles.
FIG 8.

Transmission EM of VACV-infected VPS KO cells. (A) Representative images of VPS WT cells, VPS WT cells in the presence of 2 μM ST-246, VPS52KO cells, and VPS54KO cells infected with VACV IHDJ strain. An area representing the selected VPS WT is shown at higher magnification. Bar, 500 nm. The representative virion forms are labeled as follows: C, crescents; IV, immature virus; MV, mature virus; EV, extracellular virus; Nu, nucleus. (B) Quantification of percent WV in infected KO cells. A total of 20 to 25 electron microscopic images were examined for quantities of MV, WV, and EV. Comparisons of numbers of wrapped and extracellular virus to the total number of virions determined the percentage of WV present under each set of conditions.
DISCUSSION
Viruses are obligate intracellular pathogens that require and exploit host cell proteins and functions for replication. Hence, identification of host factors required for viral infection will contribute to understanding host-pathogen interactions and potential targets for antiviral therapy. By performing high-throughput genetic screens in human haploid cells, we identified candidate host genes required for MPXV infection. Since the screen was performed using two clades of MPXV, we increased the confidence levels for identifying genes specific to either clade or common to the two clades. Here, we provide evidence that VPS52 and VPS54, components of the GARP complex, are required for virus wrapping and therefore for virus egress from infected cells.
The GARP complex is categorized as one of several multisubunit tethering complexes involved in vesicle recognition for membrane fusion (32, 33). In combination with other molecular machinery, the GARP complex promotes fusion between endosomally derived transport carriers and the trans-Golgi network as part of a retrograde transport pathway (25). The GARP complex is composed of four distinct proteins, VPS51, VPS52, VPS53, and VPS54, present at a 1:1:1:1 ratio (34). Our data show significant reduction of EV production in VPS52 and VPS54KO cells infected with MPXV ROC and MPXV WA, without significant differences in MV production. However, EV yield of MPXV WA was lower than that of the WT in VPS51KO cells but not that of MPXV ROC, probably due to genetic differences in the viral genomes of the two strains. Additional studies are required to determine if the genes identified in the WA clade that are not present in the ROC clade provide the differential requirements exhibited by these two different MPXV clades (Fig. 2A). We extended our findings to VACV infection, specifically with the IHDJ strain, which is known to release more EVs than other strains (14). EV production, and not MV production, was also significantly reduced in VPS52KO and VPS54KO cells, similarly to MPXV, suggesting that orthopoxviruses exploit the GARP complex for EV production. This effect was also evident in examining plaque formation in VPS KO cells, showing decreased plaque size in VPS51KO- and VPS53KO-infected cells and no plaque formation in VPS52KO- and VPS54KO-infected cells. Therefore, we hypothesized that these genes were important for EV formation or virus egress, which is required for virus dissemination within a host. Confocal microscopy further revealed that one mechanism of viral egress, actin tail formation, was deficient in VPS52KO and VPS54KO cells infected with VACV. EV production was further investigated by EM, which demonstrated a decrease in the percentage of WVs in VPS52KO- and VPS54KO-infected cells. Collectively, these data demonstrate that components of the GARP complex and, potentially, the retrograde transport pathway are required for EV formation and cell-to-cell spread. In our survival-based genetic knockout screen, mutant cells that are deficient in producing new infectious viral particles are selected, whereas susceptible cells detach and are removed from the culture. However, GARP complex genes were enriched in the recovered surviving cells even though formation of infectious intracellular MV was not affected. Although MVs are infectious, they require an additional membrane-wrapping process in order to generate EVs for subsequent infection and cell-to-cell spread. We speculate that completion of the virus life cycle, including MV wrapping and EV egress, leads to rapid detachment of infected cells. Thus, cells with deficiencies in these steps of the viral life cycle harbor an advantage during the selection process. However, further studies are needed to fully determine how the absence of the GARP complex promotes survival in the genetic screen.
GARP complex genes were identified previously as part of an RNAi screen performed using VACV (16), and recent follow-up studies revealed that the retrograde transport pathway is required for virus wrapping (20, 21). In addition, these studies also demonstrated inhibition of viral egress by Retro-2, an inhibitor of the retrograde pathway. These results further validate our finding that VPS52 and VPS54 are required for virus egress of orthopoxviruses, including MPXV, an emerging human pathogen. Here, we identify additional candidate genes and pathways required for MPXV infection. For instance, our screen also revealed genes associated with the “Golgi transport complex,” such as COG complex genes COG3, COG4, COG7, and COG8. Similarly to the GARP complex, the COG complex is a multisubunit tethering complex important for intra-Golgi retrograde trafficking (35). Other genes identified in our screen, such as RGP1 and RIC1, have also been reported to be involved in the retrograde trafficking pathway (36), highlighting the importance of this host pathway in viral infection.
In addition to the Golgi trafficking proteins, levels of members of the “glycosaminoglycan (GAG) biosynthesis” and “GPI-anchor biosynthesis” functional annotation groups were also significantly increased in our screen. GAG biosynthesis pathway genes included the B3GAT3, B3GALT6, B4GALT7, XYLT2, EXT1, and PTAR1 genes. Collectively, these genes encode GAG linker region enzymes and glycosyltransferases, which carry out enzymatic reactions necessary for GAG synthesis such that mutations in these genes can lead to severe disorders such as linkeropathy syndrome and connective tissue disorder (37–39). In addition, these specific genes were previously identified in a haploid screen as factors required for Rift Valley fever virus infection, demonstrating the importance of GAGs in virus entry and binding (24). GAGs have also previously been shown to be important for VACV host cell entry (40). It is postulated that the outer EV membrane is disrupted prior to plasma membrane fusion, allowing the MV to interact with GAGs at the cell surface, thereby facilitating viral entry into the host cell (41). However, since other entry mechanisms and binding factors, such as laminin (42) and macropinocytosis (43, 44), have been previously described, the specific role of GAG biosynthesis genes requires further investigation.
Furthermore, our screen also identified genes encoding GPI-anchor biosynthesis pathway proteins such as PIGL, PIGP, PIGU, DPM3, and MPDU1. PIG complementation group genes participate in GPI precursor biosynthesis in the endoplasmic reticulum and play an important role in membrane anchoring of around 150 host proteins (45). Some of these genes encode proteins that catalyze enzymatic reactions in the GPI-anchor biosynthesis process as part of the GPI transamidase component (46, 47). Mutations in GPI-anchor biosynthesis pathway genes have been shown to lead to inherited GPI deficiencies associated with hyperphosphatasia mental retardation syndrome patients (45, 48). However, studies to determine the role of GPI-anchor-related genes in OPXV infection are needed. Analysis of the roles of GAG and GPI-anchor biosynthetic genes identified in our genetic screen was not part of previously published VACV RNAi screens. Similarly, some of the genes involved in ubiquitin proteasome system pathways, nuclear pore complex pathways, and other essential cellular pathways identified through RNAi were not significantly enriched in our genetic screen. Considering that HAP1 cells are nearly haploid (49), mutant cells that lack essential genes are not viable and would not be identified in our study. Hence, RNAi and genetic screen methods might complement the identification of host factors required for OPXV infection.
In summary, our data demonstrate the role of the GARP complex in OPXV infection and implicate the trans-Golgi retrograde trafficking system in this process. Importantly, three independent screens, including our current study, using two different approaches (RNAi and genome-wide genetic screening), identified these genes as important for poxvirus infection. Further studies are required to determine the mechanism and differential requirements of GARP subunit proteins for OPXV EV formation. Although EVs represent 1% to 10% of total infectious virus, prevention of EV formation was as effective as postexposure treatment in animals (50, 51). Small-molecule inhibitors of the retrograde pathway, such as Retro-2, were shown to inhibit EV production both in vitro and in vivo (20, 21) and to inhibit polyomavirus entry in vitro (52, 53). Hence, such inhibitors represent members of a class of small-molecule inhibitors targeting the host rather than the virus, which could potentially be used for antiviral therapy.
MATERIALS AND METHODS
Cell lines.
HAP1 cells were cultured in Iscove's modified Dulbecco's medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 10 units/ml penicillin, and 100 μg/ml streptomycin. HEK293FF6 VPS51-VPS54KO and VPS54KO cells stably expressing empty vector or VPS54 vector were generated and verified as previously described (26). In brief, HEK293FF6 VPS51-54KO cells were cultured in Dulbecco's minimal essential medium (DMEM) supplemented with 10% FBS, l-glutamine, penicillin, streptomycin, puromycin (1 μg/ml), and G418 (600 μg/ml). VPS54KO stable cell lines were cultured in DMEM supplemented with 10% FBS, l-glutamine, penicillin, streptomycin, puromycin, G418, and blasticidin (10 μg/ml). BSC-40 cells were cultured in DMEM supplemented with 10% FBS, l-glutamine, penicillin, and streptomycin. All virus infections were performed in DMEM supplemented with 2% FBS, l-glutamine, penicillin, and streptomycin (2% DMEM).
Viruses and infection.
A MPXV West African strain (MPXV-USA-2003-044) obtained during the 2003 U.S. outbreak (7) and a MPXV Republic of Congo (3) strain (MPXV-RCG-2003-358) from the 2003 ROC outbreak were used in this study. Experiments were conducted with MPXV in a biosafety level 3 (BSL-3) select agent laboratory according to Biosafety in Microbiological and Biomedical Laboratories (BMBL) recommendations (55). VACV strain IHDJ (14) and recombinant VACV expressing yellow fluorescent protein-fused A4 (VACV WR A4-YFP) (10) were also used in this study. For virus yield experiments, cells were infected with virus at a MOI of 1 to 3 for 24 h, during which the supernatant and the cell monolayer were collected separately. For VACV IHDJ multistep infection, using VPS54KO-empty and VPS54KO-VPS54, cells were infected at an MOI of 0.01 and supernatant and cell monolayers were collected at 0, 24, 48, 72, and 96 h postinfection (hpi). Infections involving Tecovirimat (ST-246) were performed at a 2 μM final concentration in growth medium. Virus yield was determined by plaque assay in BSC-40 cells with 2% DMEM containing a 0.05% methylcellulose overlay and visualized with crystal violet supplemented with 10% formalin at 48 hpi.
Haploid screen and functional gene annotation analysis.
HAP1 mutagenized cells were generated using the retroviral gene-trap method as previously described (22). Mutagenized cells were infected with MPXV WA or MPXV ROC at an MOI of 0.5 for 1 h at 37°C, washed, and cultured in Iscove’s modified Dulbecco’s medium supplemented with 10% FBS, with a medium change performed every 2 to 3 days. Resistant clones were cultured for 18 days after infection and harvested for genomic DNA isolation using a QIAamp DNA column. Gene-trap insertion sites were identified by linear amplification of retroviral integrations in the genomic DNA, and subsequent sequencing was performed using an Illumina NextSeq system. Reads were aligned to the hg19 human reference genome using Bowtie. Significantly enriched genes were determined by comparing independent gene-trap insertions in infected cells versus an uninfected control as previously described (22). Gene Ontology (GO) enrichment analysis was done on significantly enriched genes (adjusted P value, <0.05) using Cytoscape (version 3.2.0) software with the ClueGO plug-in (version 2.1.5). The GO term database file (20 March 2014) was used. Significance was determined using the right-sided hypergeometric test and Benjamini-Hochberg correction. Significantly overrepresented terms were defined as those having a P value of less than 0.05, a minimum of 3 genes per term, and at least 4% of the genes from the data set associated with the term. Associated GO terms were grouped into representative terms on the basis of the kappa score, and the results were summarized by functional group in an overview chart.
Plaque immunostaining.
Cells were infected with MPXV-WA or VACV IHDJ for 72 h, with a methylcellulose overlay. The infected monolayer was fixed with 1:1 acetone-methanol and incubated with rabbit anti-vaccinia virus antibody (Virostat, Inc., Portland, ME) followed by horseradish peroxidase (HRP)-conjugated secondary antibody. The foci were developed using o-dianisidine H2O2 solution.
Transfection.
Transfection of VPS52KO cells was performed using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions 24 h prior to infection. Transfected plasmid (pVPS52) was constructed by inserting FLAG-tagged VPS52 into a pLenti CMV Puro DEST backbone (Addgene catalog no. 17452). A non-VPS52-expressing plasmid was used as a control. After transfection, cells were infected with VACV IHDJ at an MOI of 1 for 24 h and the supernatant and the cell monolayer were collected for virus titer determinations. To validate expression of pVPS52, transfected VPS52KO cells were harvested with NuPAGE 1× LDS sample buffer with reducing agent and processed for Western blotting.
Western blot analysis.
VPS52 KO cells transfected with the pVPS52 gene (FLAG epitope tag) or VPS54KO cells stably expressing epitope-tagged VPS54 (V5 and His tags) were harvested and lysed by boiling in NuPAGE 1× LDS sample buffer with a reducing agent. The samples were separated on a 4% to 12% Bis-Tris polyacrylamide gel and transferred to a nitrocellulose membrane using an iBlot system (ThermoFisher). Membranes were incubated with mouse monoclonal anti-FLAG M2 antibody (Sigma-Aldrich, St. Louis, MO) for pVPS52 transfected samples, mouse monoclonal anti-V5 antibody (ThermoFisher Scientific) for VPS54KO stably transfected samples, and anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) antibody (Covance Inc., Princeton, NJ) as a control. After incubation with primary antibody, membranes were washed once with 1× phosphate-buffered saline (PBS) with 0.05% Tween 20 (PBST) and twice with 1× PBS. The blots were developed by incubation with secondary antibodies tagged to IRDye 800CW and IRDye 680RD (Li-COR Biotechnology, Lincoln, NE) (for FLAG-VPS52) or to HRP (Clarity Western ECL substrate kit; Bio-Rad, Hercules, CA) (for V5-VPS54).
Confocal microscopy.
Cells were seeded into 24-well glass-bottom chamber culture slides (Mattek Corporation). The next day, cells were infected with VACV WR A4-YFP at an MOI of 0.01 for 24 h, after which cells were rinsed with ice-cold PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. This was followed by permeabilization performed with a mixture containing 0.1% Triton X-100, 0.05% Tween 20, and PBS buffer for 30 min. Fixed cells were staining with Alexa Fluor 647 phalloidin (ThermoFisher) for analysis of actin levels and with DAPI (4′,6-diamidino-2-phenylindole) for visualization of nuclei. The cells were then washed with cold PBS three times for 3 min each time, followed by mounting performed with Prolong Antifade mounting media (Molecular Probes). The cells were examined by the use of an LSM 710 inverted confocal microscope (Zeiss, Oberkochen, Germany), and images were processed using Adobe Photoshop.
Electron microscopy.
HEK293FF6 WT, VPS52KO, and VPS54KO cells were infected with VACV IHDJ for 24 h at an MOI of 1. Cell monolayers were gently scrapped, spun down, and processed for thin-section EM. Specimens were fixed in buffered 2.5% glutaraldehyde 24 h postinoculation, postfixed in 1% osmium tetroxide, stained en bloc with 4% uranyl acetate, dehydrated through a graded alcohol and acetone series, and embedded in a mixture of Epon substitute and Araldite. Thin sections were stained with 4% uranyl acetate and Reynolds' lead citrate.
Statistical analysis.
Results from replicate experiments are presented as means ± standard deviations. Data were analyzed in GraphPad Prism using Student's t test to calculate P values and were corrected for multiple comparisons using the Holm-Sidak method. Statistical significance was defined as represented by P values of <0.05. No statistics were reported for ST-246 since it served as a control.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bernard Moss, Juan Bonifacino, Rama Amara, and Michael Flint for reagents.
The study was supported by the CDC Intramural Research and Biomedical Advanced Research and Developmental Authority (BARDA), NIH AI109662 (to J.E.C.), the David and Lucile Packard Foundation (J.E.C.), an ASM/CDC Postdoctoral Fellowship (S.R.), a Stanford Graduate Fellowship (A.S.P.), and Boehringer Ingelheim Fonds (A.S.P.).
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00011-17.
REFERENCES
- 1.Damon IK. 2013. Poxviruses, p 2160–2184. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Di Giulio DB, Eckburg PB. 2004. Human monkeypox: an emerging zoonosis. Lancet Infect Dis 4:15–25. doi: 10.1016/S1473-3099(03)00856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Likos AM, Sammons SA, Olson VA, Frace AM, Li Y, Olsen-Rasmussen M, Davidson W, Galloway R, Khristova ML, Reynolds MG, Zhao H, Carroll DS, Curns A, Formenty P, Esposito JJ, Regnery RL, Damon IK. 2005. A tale of two clades: monkeypox viruses. J Gen Virol 86:2661–2672. doi: 10.1099/vir.0.81215-0. [DOI] [PubMed] [Google Scholar]
- 4.Saijo M, Ami Y, Suzaki Y, Nagata N, Iwata N, Hasegawa H, Iizuka I, Shiota T, Sakai K, Ogata M, Fukushi S, Mizutani T, Sata T, Kurata T, Kurane I, Morikawa S. 2009. Virulence and pathophysiology of the Congo Basin and West African strains of monkeypox virus in non-human primates. J Gen Virol 90:2266–2271. doi: 10.1099/vir.0.010207-0. [DOI] [PubMed] [Google Scholar]
- 5.Learned LA, Reynolds MG, Wassa DW, Li Y, Olson VA, Karem K, Stempora LL, Braden ZH, Kline R, Likos A, Libama F, Moudzeo H, Bolanda JD, Tarangonia P, Boumandoki P, Formenty P, Harvey JM, Damon IK. 2005. Extended interhuman transmission of monkeypox in a hospital community in the Republic of the Congo, 2003. Am J Trop Med Hyg 73:428–434. [PubMed] [Google Scholar]
- 6.Reynolds MG, Damon IK. 2012. Outbreaks of human monkeypox after cessation of smallpox vaccination. Trends Microbiol 20:80–87. doi: 10.1016/j.tim.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Reed KD, Melski JW, Graham MB, Regnery RL, Sotir MJ, Wegner MV, Kazmierczak JJ, Stratman EJ, Li Y, Fairley JA, Swain GR, Olson VA, Sargent EK, Kehl SC, Frace MA, Kline R, Foldy SL, Davis JP, Damon IK. 2004. The detection of monkeypox in humans in the Western Hemisphere. N Engl J Med 350:342–350. doi: 10.1056/NEJMoa032299. [DOI] [PubMed] [Google Scholar]
- 8.Moss B. 2013. Poxviridae, p 2129–2159. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 9.Sanderson CM, Hollinshead M, Smith GL. 2000. The vaccinia virus A27L protein is needed for the microtubule-dependent transport of intracellular mature virus particles. J Gen Virol 81:47–58. doi: 10.1099/0022-1317-81-1-47. [DOI] [PubMed] [Google Scholar]
- 10.Ward BM. 2005. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J Virol 79:4755–4763. doi: 10.1128/JVI.79.8.4755-4763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blasco R, Moss B. 1991. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J Virol 65:5910–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blasco R, Moss B. 1992. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol 66:4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelstad M, Smith GL. 1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 194:627–637. doi: 10.1006/viro.1993.1302. [DOI] [PubMed] [Google Scholar]
- 14.Payne LG. 1980. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J Gen Virol 50:89–100. doi: 10.1099/0022-1317-50-1-89. [DOI] [PubMed] [Google Scholar]
- 15.Mercer J, Snijder B, Sacher R, Burkard C, Bleck CK, Stahlberg H, Pelkmans L, Helenius A. 2012. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep 2:1036–1047. doi: 10.1016/j.celrep.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Sivan G, Martin SE, Myers TG, Buehler E, Szymczyk KH, Ormanoglu P, Moss B. 2013. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc Natl Acad Sci U S A 110:3519–3524. doi: 10.1073/pnas.1300708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filone CM, Caballero IS, Dower K, Mendillo ML, Cowley GS, Santagata S, Rozelle DK, Yen J, Rubins KH, Hacohen N, Root DE, Hensley LE, Connor J. 2014. The master regulator of the cellular stress response (HSF1) is critical for orthopoxvirus infection. PLoS Pathog 10:e1003904. doi: 10.1371/journal.ppat.1003904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beard PM, Griffiths SJ, Gonzalez O, Haga IR, Pechenick Jowers T, Reynolds DK, Wildenhain J, Tekotte H, Auer M, Tyers M, Ghazal P, Zimmer R, Haas J. 2014. A loss of function analysis of host factors influencing vaccinia virus replication by RNA interference. PLoS One 9:e98431. doi: 10.1371/journal.pone.0098431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moser TS, Sabin LR, Cherry S. 25 August 2010. RNAi screening for host factors involved in vaccinia virus infection using Drosophila cells. J Vis Exp doi: 10.3791/2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sivan G, Weisberg AS, Americo JL, Moss B. 12 September 2016. Retrograde transport from early endosomes to the trans-Golgi network enables membrane wrapping and egress of vaccinia virions. J Virol doi: 10.1128/JVI.01114-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison K, Haga IR, Pechenick Jowers T, Jasim S, Cintrat JC, Gillet D, Schmitt-John T, Digard P, Beard PM. 31 August 2016. Vaccinia virus uses retromer-independent cellular retrograde transport pathways to facilitate the wrapping of intracellular mature virions during viral morphogenesis. J Virol doi: 10.1128/JVI.01464-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P, Wessels MW, Lefeber DJ, Whelan SP, van Bokhoven H, Brummelkamp TR. 2013. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riblett AM, Blomen VA, Jae LT, Altamura LA, Doms RW, Brummelkamp TR, Wojcechowskyj JA. 2015. A haploid genetic screen identifies heparan sulfate proteoglycans supporting Rift Valley fever virus infection. J Virol 90:1414–1423. doi: 10.1128/JVI.02055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pérez-Victoria FJ, Bonifacino JS. 2009. Dual roles of the mammalian GARP complex in tethering and SNARE complex assembly at the trans-Golgi network. Mol Cell Biol 29:5251–5263. doi: 10.1128/MCB.00495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirata T, Fujita M, Nakamura S, Gotoh K, Motooka D, Murakami Y, Maeda Y, Kinoshita T. 2015. Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol Biol Cell 26:3071–3084. doi: 10.1091/mbc.E14-11-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duraffour S, Lorenzo MM, Zoller G, Topalis D, Grosenbach D, Hruby DE, Andrei G, Blasco R, Meyer H, Snoeck R. 2015. ST-246 is a key antiviral to inhibit the viral F13L phospholipase, one of the essential proteins for orthopoxvirus wrapping. J Antimicrob Chemother 70:1367–1380. doi: 10.1093/jac/dku545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meiser A, Sancho C, Krijnse Locker J. 2003. Plasma membrane budding as an alternative release mechanism of the extracellular enveloped form of vaccinia virus from HeLa cells. J Virol 77:9931–9942. doi: 10.1128/JVI.77.18.9931-9942.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsutsui K. 1983. Release of vaccinia virus from FL cells infected with the IHD-W strain. J Electron Microsc (Tokyo) 32:125–140. [PubMed] [Google Scholar]
- 30.Stokes GV. 1976. High-voltage electron microscope study of the release of vaccinia virus from whole cells. J Virol 18:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang WH, Wilcock D, Smith GL. 2000. Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J Virol 74:11654–11662. doi: 10.1128/JVI.74.24.11654-11662.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou HT, Dukovski D, Chambers MG, Reinisch KM, Walz T. 2016. CATCHR, HOPS and CORVET tethering complexes share a similar architecture. Nat Struct Mol Biol 23:761–763. doi: 10.1038/nsmb.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonifacino JS, Hierro A. 2011. Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol 21:159–167. doi: 10.1016/j.tcb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liewen H, Meinhold-Heerlein I, Oliveira V, Schwarzenbacher R, Luo G, Wadle A, Jung M, Pfreundschuh M, Stenner-Liewen F. 2005. Characterization of the human GARP (Golgi associated retrograde protein) complex. Exp Cell Res 306:24–34. doi: 10.1016/j.yexcr.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 35.Vasile E, Oka T, Ericsson M, Nakamura N, Krieger M. 2006. IntraGolgi distribution of the Conserved Oligomeric Golgi (COG) complex. Exp Cell Res 312:3132–3141. doi: 10.1016/j.yexcr.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Pusapati GV, Luchetti G, Pfeffer SR. 2012. Ric1-Rgp1 complex is a guanine nucleotide exchange factor for the late Golgi Rab6A GTPase and an effector of the medial Golgi Rab33B GTPase. J Biol Chem 287:42129–42137. doi: 10.1074/jbc.M112.414565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones KL, Schwarze U, Adam MP, Byers PH, Mefford HC. 2015. A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am J Med Genet A 167A:2691–2696. doi: 10.1002/ajmg.a.37209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakajima M, Mizumoto S, Miyake N, Kogawa R, Iida A, Ito H, Kitoh H, Hirayama A, Mitsubuchi H, Miyazaki O, Kosaki R, Horikawa R, Lai A, Mendoza-Londono R, Dupuis L, Chitayat D, Howard A, Leal GF, Cavalcanti D, Tsurusaki Y, Saitsu H, Watanabe S, Lausch E, Unger S, Bonafe L, Ohashi H, Superti-Furga A, Matsumoto N, Sugahara K, Nishimura G, Ikegawa S. 2013. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am J Hum Genet 92:927–934. doi: 10.1016/j.ajhg.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang C, Zhou J, Wu S, Shan Y, Teng S, Yu L. 2004. Cloning and tissue distribution of the human B3GALT7 gene, a member of the beta1,3-glycosyltransferase family. Glycoconj J 21:267–273. doi: 10.1023/B:GLYC.0000045098.78968.4c. [DOI] [PubMed] [Google Scholar]
- 40.Carter GC, Law M, Hollinshead M, Smith GL. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol 86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- 41.Law M, Carter GC, Roberts KL, Hollinshead M, Smith GL. 2006. Ligand-induced and nonfusogenic dissolution of a viral membrane. Proc Natl Acad Sci U S A 103:5989–5994. doi: 10.1073/pnas.0601025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiu WL, Lin CL, Yang MH, Tzou DL, Chang W. 2007. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J Virol 81:2149–2157. doi: 10.1128/JVI.02302-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandgren KJ, Wilkinson J, Miranda-Saksena M, McInerney GM, Byth-Wilson K, Robinson PJ, Cunningham AL. 2010. A differential role for macropinocytosis in mediating entry of the two forms of vaccinia virus into dendritic cells. PLoS Pathog 6:e1000866. doi: 10.1371/journal.ppat.1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmidt FI, Bleck CK, Helenius A, Mercer J. 2011. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J 30:3647–3661. doi: 10.1038/emboj.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinoshita T. 2014. Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proc Jpn Acad Ser B Phys Biol Sci 90:130–143. doi: 10.2183/pjab.90.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watanabe R, Murakami Y, Marmor MD, Inoue N, Maeda Y, Hino J, Kangawa K, Julius M, Kinoshita T. 2000. Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2. EMBO J 19:4402–4411. doi: 10.1093/emboj/19.16.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong Y, Ohishi K, Kang JY, Tanaka S, Inoue N, Nishimura J, Maeda Y, Kinoshita T. 2003. Human PIG-U and yeast Cdc91p are the fifth subunit of GPI transamidase that attaches GPI-anchors to proteins. Mol Biol Cell 14:1780–1789. doi: 10.1091/mbc.E02-12-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujiwara I, Murakami Y, Niihori T, Kanno J, Hakoda A, Sakamoto O, Okamoto N, Funayama R, Nagashima T, Nakayama K, Kinoshita T, Kure S, Matsubara Y, Aoki Y. 2015. Mutations in PIGL in a patient with Mabry syndrome. Am J Med Genet A 167A:777–785. doi: 10.1002/ajmg.a.36987. [DOI] [PubMed] [Google Scholar]
- 49.Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, Marceau C, Janssen H, Carette JE, Bennett KL, Colinge J, Superti-Furga G, Brummelkamp TR. 2015. Gene essentiality and synthetic lethality in haploid human cells. Science 350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 50.Zaitseva M, Shotwell E, Scott J, Cruz S, King LR, Manischewitz J, Diaz CG, Jordan RA, Grosenbach DW, Golding H. 2013. Effects of postchallenge administration of ST-246 on dissemination of IHD-J-Luc vaccinia virus in normal mice and in immune-deficient mice reconstituted with T cells. J Virol 87:5564–5576. doi: 10.1128/JVI.03426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huggins J, Goff A, Hensley L, Mucker E, Shamblin J, Wlazlowski C, Johnson W, Chapman J, Larsen T, Twenhafel N, Karem K, Damon IK, Byrd CM, Bolken TC, Jordan R, Hruby D. 2009. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother 53:2620–2625. doi: 10.1128/AAC.00021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, DiMaio D. 2013. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A 110:7452–7457. doi: 10.1073/pnas.1302164110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nelson CD, Carney DW, Derdowski A, Lipovsky A, Gee GV, O'Hara B, Williard P, DiMaio D, Sello JK, Atwood WJ. 2013. A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. mBio 4:e00729-13. doi: 10.1128/mBio.00729-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reference deleted.
- 55.Richmond JY, Nesby-O'Dell SL. 2002. Laboratory security and emergency response guidance for laboratories working with select agents. Centers for Disease Control and Prevention. MMWR Recomm Rep 51(RR-19):1–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.