

Abstract
Background
During the development of the mammalian cerebral cortex, newborn postmitotic projection neurons are born from local neural stem cells and must undergo radial migration so as to position themselves appropriately to form functional neural circuits. The zinc finger transcriptional repressor Rp58 (also known as Znf238 or Zbtb18) is critical for coordinating corticogenesis, but its underlying molecular mechanism remains to be better characterised.
Findings
Here, we demonstrate that the co-expression of Rp58 and the cyclin dependent kinase inhibitor (CDKI) p27kip1 is important for E14.5-born cortical neurons to coordinate cell cycle exit and initiate their radial migration. Notably, we find that the impaired radial positioning of Rp58-deficient cortical neurons within the embryonic (E17.5) mouse cortex, as well as their multipolar to bipolar transition from the intermediate zone to the cortical plate can be restored by forced expression of p27kip1 in concert with suppression of Rnd2, a downstream target gene of Rp58. Furthermore, the restorative effects of p27kip1 and Rnd2 abrogation are reminiscent of suppressing RhoA signalling in Rp58-deficient cells.
Conclusions
Our findings demonstrate functional interplay between a transcriptional regulator and a CDKI to mediate neuroprogenitor cell cycle exit, as well as to promote radial migration through a molecular mechanism consistent with suppression of RhoA signalling.
Electronic supplementary material
The online version of this article (doi:10.1186/s13064-017-0084-3) contains supplementary material, which is available to authorized users.
Keywords: Cerebral Cortex, Transcription Factor, Neurodevelopment, Neurogenesis, Radial migration, Neuronal morphology
Introduction
The development of the cerebral cortex involves a precise integration of multiple molecular cues within embryonic cortical cells to coordinate the production of appropriate numbers of progenitor cells, followed by the elaboration of distinct neural cell types [1–4]. Within the developing embryonic cerebral cortex, local progenitor cells undergo multiple rounds of cell proliferation to generate neurons, astrocytes and oligodendrocytes which systematically integrate within the tissue so as to eventually form functional neural circuitry. The activities of DNA-binding transcription factors have been recognised to be crucial for guiding the developmental trajectory of embryonic cortical neurons through the initiation of cell-intrinsic programmes which specify their subtype as excitatory, glutamatergic neurons or inhibitory, GABAergic neurons [3, 5–7]. For example, the proneural bHLH transcriptional activator Neurogenin2 (Neurog2) drives a cell intrinsic programme to specify an excitatory glutamatergic neuron fate. Furthermore, Neurog2 initiates the transcription of downstream target genes, including the Rho GTPase related factor Rnd2, which specifies radial positioning of newborn neurons within the growing cortical anlagen [8, 9]. Equally, the activities of transcriptional repressors, such as the zinc finger DNA binding transcriptional repressor Rp58 (also known as Znf238 or Zbtb18), are also critical to the development of the embryonic cerebral cortex. Notably, the gene regulatory activities of Rp58 influence the timing of Neurog2 expression within the embryonic cortex to promote neurogenesis [10], and also directly regulates Rnd2 expression for the efficient radial migration of newborn cortical neurons [11]. As a corollary, loss of Rp58 expression during embryogenesis leads to neurodevelopmental defects such as premature depletion of cortical progenitors, precocious neurogenesis and gliogenesis, as well as programmed cell death [10, 12–14].
In addition to transcription factors, members of the Cip/Kip family of cyclin dependent kinase inhibitor (CDKI) proteins are also critical for coordinating neuroprogenitor cell cycle exit and differentiation within the developing cortex [12, 15–17]. Notably, the CDKI p27kip1 drives neuroprogenitor cell cycle exit and cortical neuron differentiation through its cyclin kinase inhibitor functions [15–17], while it also mediates neurite outgrowth through its capacity to suppress RhoA signalling so as to coordinate the neuronal cytoskeleton [16]. More recently, p27kip1 has also been identified to promote microtubule polymerisation to facilitate the migration of cortical cells [18]. While such findings identify critical roles for transcription factor signalling and CDKI activity during cortical neurogenesis, their cooperative functions remain less well characterised, particularly given recent evidence linking Rp58 expression and CDKI activities in the development of astrocytes [12]. Here, we report a functional relationship between Rp58 and p27kip1 to drive cell cycle exit and promote distinct phases of radial migration during cerebral cortex development.
Methods
Animals
Mice (C57BL/6 J) were housed, bred and treated within the animal facilities at Monash University. Female mice of at least 6 weeks of age were utilised for timed-matings. Rp58-knockout mice were genotyped by polymerase chain reaction. All animal procedures are approved by the Animal Ethics Committee within Monash University (Licenses MARP/08–104 and MARP/2012/068), and are compliant with guidelines provided by the National Health and Medical Research Council of Australia.
DNA plasmids and Antibodies
Mammalian (pCaggs) expression vectors encoding p27kip1, p27kip1(ck-) and RhoA(N19) have been described previously [16], while an Rp58 expression construct with a pCIG vector which lacks a GFP cassette was used [11]. RNAi for Rp58 was achieved using a pool of targeting siRNAs (Dharmacon GE Life Sciences) which was previously verified for specificity of knockdown as well as a pSilCaggs-Rnd2shRNA1 vector to induce Rnd2 RNAi [11]. Primary antibodies used for immunostaining analysis include chicken antibody to GFP (Abcam, ab13970, 1:700), mouse anti-p27kip1 (BD Biosciences, 1:400), rabbit anti-Rp58 (Proteintech Group, 1:250), rabbit anti-Ki67 (NCL-Ki67p, Leica, 1:1000), pHH3(ser10) (06–570, Merck Millipore, 1:1000), mouse anti βIII-tubulin (Covance, MMS-435P, 1:1000), mouse anti-Nestin (Millipore, MAB353, 1:300), rabbit anti-Pax6 (Covance, PRB-2788, 1:500), rabbit anti-Tbr2 antibody (Abcam, ab233345, 1:500), rabbit polyclonal antibody to GFP (Invitrogen, A6455, 1:1000). Alexa fluor secondary antibodies include goat anti- chicken IgG (Invitrogen, A11039, 1:700), goat anti-mouse (Invitrogen, A11031, 1:800), and goat anti-rabbit IgG (Invitrogen, A6455, 1:1000). The nuclei of cells were visualised with DAPI.
In Utero Electroporation
In utero electroporation experiments are performed as described [19, 20]. High quality, low endotoxin plasmid preparations (Qiagen) of DNA vectors were injected at 1 μg/μl for each plasmid, together with Fast Green (0.05%, Sigma). For RNAi experiments, Dharmacon siRNA targeting pools were injected at 10 μM concentration together with GFP expression plasmid at 1 μg/μl concentration. Following recovery from in utero electroporation, the mice were sacrificed by cervical dislocation, and the embryonic brains were harvested by dissection in cold PBS. For studies of dissociated cortical cells, dissected embryonic cortical tissue was digested to obtain a single-cell suspension and plated as per previously described [9]. For histological analysis, electroporated brains were subject to fixation in 4% paraformaldehyde solution in PBS overnight followed by three washes in PBS and permeation in 20% sucrose/PBS solution. Following tissue embedding in OCT, cryosectioning along the coronal plane (16 μm thickness) was performed followed by fluorescence immunostaining for antigens of interest. Images of brain sections were captured on an epifluorescence microscope (Olympus) equipped with a CCD camera (SPOT). Subdivisions of the embryonic cortex (VZ/SVZ, IZ and CP) were identified based on cell density as visualised with DAPI (4′6-Diamidino-2-Phenylindole) staining, as described previously [21]. Images from embryonic E17.5 cortices for cell shape analyses were acquired at ×20 magnification, as per described previously [22]. Cell counting was performed blind to the condition on representative fields of sections of electroporated brains using ImageJ software.
Cell Culture, Western blotting and immunoprecipitation
Mouse embryonic carcinoma (P19) cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco 10,313) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher HYC15–010.02), 2 mM L-glutamine (Gibco 25,030), 20 units/ml of penicillin and streptomycin (Gibco 15,140) under humidified air containing 5% CO2 at 37 °C. Transfections were performed using equal quantities of expression plasmids (pcDNA empty vector, pcDNA-Neurog2 or pcDNA-Rp58 vector) for each condition. Western blotting analysis was performed with antibodies to Rp58, actin (A5441, Sigma Aldrich) and p27kip1 together with appropriate fluorescent secondary antibodies, as described [22]. Immunoblot signals were resolved detected with an Odyssey® infrared imaging system (Li-Cor 9201–02) for analysis.
Results
We focussed our attention on early-mid gestation mouse embryos at embryonic day 14.5 (E14.5), a stage of neurodevelopment defined by a peak period of neurogenesis when maximal numbers of cortical neurons are born from local ventricular zone (VZ) and subventricular zone (SVZ) progenitor cells [23]. We performed immunostaining and detected Rp58 in neuroprogenitor cells of the ventricular zone (VZ) and subventricular zone (SVZ), as well as in postmitotic neurons of the intermediate zone (IZ) and cortical plate (CP) (Fig. 1a). We also characterised the immunostaining pattern for Rp58 together with p27kip1, the predominant cyclin dependent kinase inhibitor in the E14.5 embryonic cortex [16] and detected their co-expression in scattered cells within the germinal VZ/SVZ, and in virtually all cells of the IZ and CP (Fig. 1a). Since it is known that disruptions to Rp58 or p27kip1 lead to defective neuronal differentiation and radial migration [10, 11, 13, 16], we reasoned that the combined activities of both Rp58 and p27kip1 might also be relevant for coordinating the cellular activities of cortical progenitors, such as their cell cycle exit and radial migration.
Fig. 1.
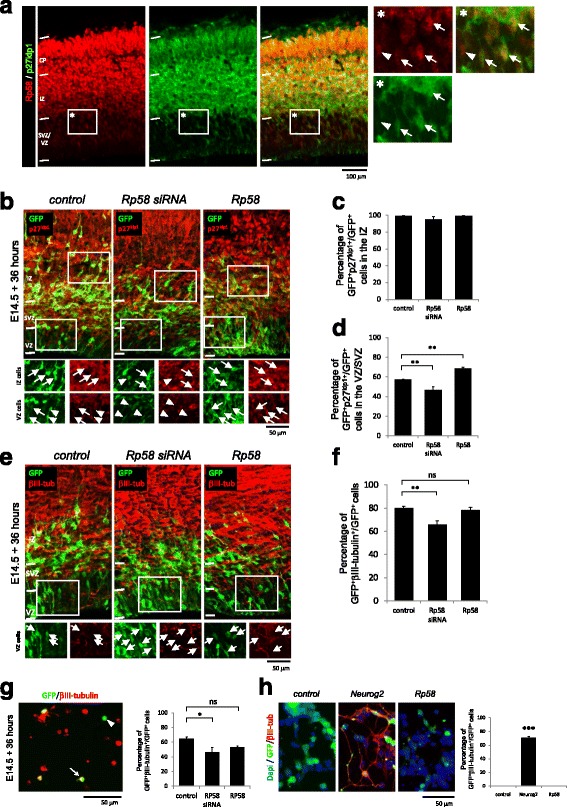
The effects of Rp58 disruption on p27kip1 within the embryonic cortex. a Immunostaining of Rp58 and p27kip1 to reveal their co-presence in cells of the VZ/SVZ, IZ and CP. Arrows point to double-positive cells, while arrowheads point to Rp58+ cells. b–d Knockdown or overexpression of Rp58 did not significantly disrupt p27kip1 co-immunostaining of GFP+ cells in the IZ (F2,6 = 3.1 p = 0.11, One-way ANOVA, >300 cells counted from 3 independent brains per condition) (c), while there was a significant effect in the VZ/SVZ (F2,6 = 45, p < 0.0005, One-way ANOVA, >400 cells counted from 3 independent brains per condition). Arrows point to double-positive cells, while arrow heads point to GFP+ cells. e, f Knockdown of Rp58 leads to a decrease in the proportion of GFP electroporated cells which co-label with βIII-tubulin, indicative of impaired neurogenesis (F2,6 = 18 p = 0.003, One-way ANOVA, >600 cells counted from 3 independent brains per condition). Arrows point to GFP+ cells which do not co-label with βIII-tubulin. g GFP labelled cells were dissociated from successfully electroporated brain tissue and plated for 2 h before fixation, immunostaining and data collection. Knockdown of Rp58 leads to a reduction in neurogenesis, while forced expression does not significantly disrupt p27kip1 expression (F2,6 = 9 p = 0.011, One-way ANOVA, >450 cells counted from 3 independent experiments per condition). Arrow points to double-positive cells, while arrow head points to GFP+ cells.. (H) Forced expression of Neurog2 but not Rp58 in P19 embyrocarcinoma cells induces neurogenesis, as evaluated by immunostaining for βIII-tubulin. *p < 0.05, **p < 0.01, ***p < 0.001; “ns” denotes not significant. Scale bar represents 50 μm
We performed Western blotting with lysates collected from whole E14.5 embryonic cortices of Rp58 wildtype, heterozygote and nullizygous embryos to find that steady-state levels of p27kip1 immunoblotted signal were not significantly different between genotypes (Additional file 1: Figure S1A-B; F2,7 = 0.26, p = 0.77 One-way ANOVA). While this result is consistent with the findings of Hirai and colleagues [12], we were intrigued by the possibility that cell-type specific fluctuations in p27kip1 within the cortical tissue might underlie Rp58 deficiency. Thus, we performed a series of experiments using E14.5 mouse embryos in which we introduced small interfering RNAs (siRNAs) by in utero electroporation to efficiently suppress Rp58 in a cell autonomous manner within the embryonic cortex, as previously reported [11]. We also performed control experiments in which cortices were electroporated with a non-targeting siRNA pool. In these experiments, a Green Fluorescent Protein (GFP) expression vector was co-electroporated to identify siRNA-treated cells. In parallel, we also electroporated brains with a bicistronic GFP expression construct encoding RP58 (together with non-targeting siRNAs) to investigate the consequence of its forced expression within the E14.5 cortex. Thirty-six hours after electroporation, successfully electroporated brains were analysed for potential effects on p27kip1 expression. As shown in Fig. 1b, we found that neither knockdown of Rp58 nor its forced expression led to a significant effect in the proportion of GFP-labelled cells which co-stain with p27kip1 in the IZ (Fig. 1c; F2,6 = 3.1, p = 0.11, One-way ANOVA, >300 cells counted from 3 independent brains per condition). We also analysed the intensity of p27kip1-immunofluorescence signal within IZ cells and found no significant difference between treatment groups (Additional file 1: Figure S1C-D; F2,222 = 2.9, p = 0.0557, One-way ANOVA, >75 cells counted from 3 independent brains per condition). However, we observed a significant reduction in the proportion of p27kip1-expressing cells within the VZ and SVZ following Rp58 knockdown, while overexpression led to a significant elevation (Fig. 1b, d; F2,6 = 45, p < 0.0005, One-way ANOVA, >400 cells counted from 3 brains per condition). Furthermore, the intensity of p27kip1-immunofluorescence signal within VZ/SVZ cells was significantly reduced upon knockdown, while overexpression did not have a significant effect (Additional file 1: Figure S1E; F2,222 = 95, p < 0.0001, One-way ANOVA, >75 cells counted from 3 independent brains per condition). These observations suggest that acute suppression of Rp58 results in a transient reduction in p27kip1 levels in VZ/SVZ cells, with potential effects on neurodifferentiation. Consistent with this finding, Rp58 knockdown led to a significant reduction in neuronal differentiation as marked by immunostaining for βIII-tubulin, but forced expression of Rp58 did not have such an effect (Fig. 1e, f, F2,6 = 18, p = 0.003, One-way ANOVA, >600 cells counted from 3 brains per condition). Parallel experiments in which electroporated tissue was dissociated and cells plated before immunostaining for βIII-tubulin further confirmed this finding (Fig. 1g, F2,6 = 9, p = 0.011, One-way ANOVA, >450 cells counted from at least 3 brains per condition). While our results could also be interpreted to indicate that Rp58 drives neurodifferentiation, we addressed this directly in an experiment using mouse embryocarcinoma P19 cells to find that forced expression of Rp58 does not promote the production of βIII-tubulin expressing neurons in vitro (Fig. 1h, F2,6 = 1212, p < 0.001, One-way ANOVA, >1000 cells counted from 3 independent experiments per condition). As a control, we transfected cells with the neurogenic transcription factor Neurog2 which robustly induced βIII-tubulin expression. Altogether, these results show that disruptions to Rp58 affect p27kip1 expression in embryonic cortical cells of the germinal VZ/SVZ, but Rp58 lacks the capacity to promote neurodifferentiation.
We previously reported that Rp58 cell autonomously regulates progenitor cell cycle exit and initiation of radial migration within the E14.5 embryonic cortex [10, 11]. Given our findings that Rp58 is co-labelled with p27kip1 within the embryonic E14.5 cortex (Fig. 1a), and that disruptions to Rp58 lead to changes in p27kip1 expression within the VZ/SVZ (Fig. 1b–d; Additional file 1: Figure S1C-E), we hypothesised that Rp58 mediated neuroprogenitor proliferation via a p27kip1-mediated process. Should this be the case, we would thus predict that forced expression of p27kip1 might abrogate Rp58 deficiency in neural progenitor cells. To test this, we co-delivered expression constructs encoding p27kip1 or a variant which lacks its cyclin kinase function but retains its capacity for signalling cell migration (termed p27kip1ck-) in Rp58-deficient cortical cells of the E14.5 cortex and examined their potential restorative effects on cell proliferation and radial migration. It is known that p27kip1 promotes cell cycle exit through its activities involving cyclin dependent kinases as well as cell migration through a mechanism independent of its cyclin kinase activity [16, 24], and we confirmed this effect in the E14.5 cortex (Additional file 2: Figure S2). As shown in Fig. 2, while Rp58 siRNA treatment leads to a significant increase in the proportion of GFP-labelled cells which co-label with the proliferation markers Ki67 (Fig. 2a–d, and Fig. 2e, f3,8 = 73, p < 0.001, One-way ANOVA, >700 cells counted from 3 independent brains per condition) or pHH3 (Fig. 2k; F2,8 = 20, p = 0.004, One-way ANOVA, >700 cells counted from 3 independent brains per condition), co-delivery of p27kip1 led to a significant restoration of these markers, while co-delivery of a p27kip1ck- expression construct did not have a restorative effect. Notably, p27kip1 suppressed cell proliferation to an extent beyond control levels (Fig. 2e, Ki67 16.6% ± 2.1% in control vs 4.9% ± 0.6% in Rp58 siRNA restored with p27kip1 expression; p < 0.01), suggesting that p27kip1 and Rp58 have different potencies for mediating Ki67 and pHH3 expression in cortical cells. We also found a significant interaction between non-surface (SVZ) divisions in treated cells identified as mitoses marked by pHH3 expression away from the ventricular surface (Additional file 3: Figure S3A; F3,9 = 7, p < 0.0102, One-way ANOVA, 3 independent brains per condition), which could suggest an effect of Rp58 disruption on neuroprogenitor cells. To investigate this further, we analysed the proportion of Pax6-expressing radial glial progenitors to find that this was not significantly affected between treatment groups (Additional file 3: Figure S3B-F; F3,9 = 0.89, p = 0.4818, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). We also did not find a significant difference in surface versus non-surface positioning of GFP + Pax6+ cells (data not shown). In contrast, treatment with Rp58 siRNAs led to a significant reduction in the proportion of Tbr2-expressing intermediate progenitors which could not be augmented by co-delivery of p27kip1 or p27kip1ck- expression constructs (Additional file 3: Figure S3G-K; F3,11 = 6.5, p = 0.0085, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). Overexpression of p27kip1 or p27kip1ck- alone did not influence Pax6-immunoreactive radial glial progenitors, while only p27kip1 overexpression resulted in a reduction in Tbr2-immunopositive cells (Additional file 2: Figure S2E-F). Thus, the effect of Rp58 knockdown on intermediate progenitors cannot be restored by forced expression of p27kip1 or p27kip1ck-.
Fig. 2.
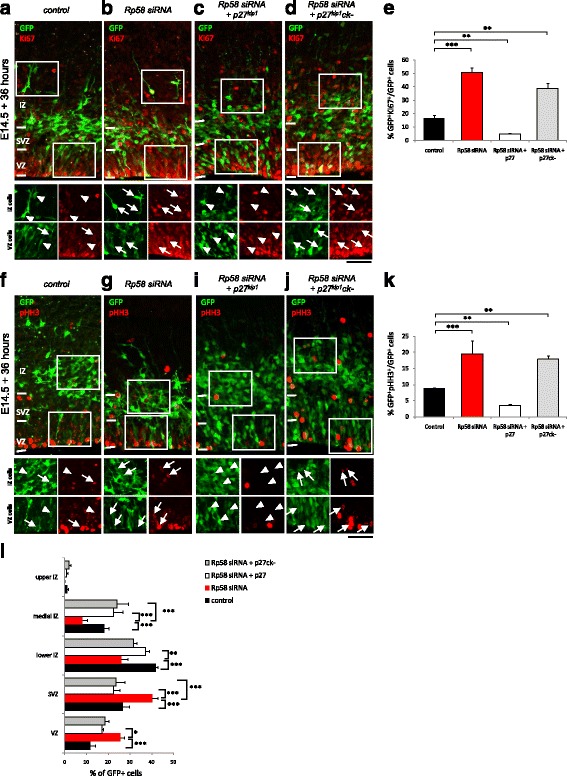
p27kip1 restores the defective cell proliferation and radial migration of Rp58 siRNA-treated cortical progenitors. Knockdown of Rp58 leads to significant reductions in the expression of the cell proliferation marker Ki67. a–d The defective expression of Ki67 in Rp58 siRNA-treated cells could be restored with p27kip1, but not p27kip1(ck-) which is incapable of signalling cell cycle exit owing to a mutation which impairs its cyclin kinase function (e) (F3,8 = 73, p < 0.001, One-way ANOVA, >700 cells counted from 3 independent brains per condition). Similar effects on the co-detection of pHH3, a marker of cell mitosis, were observed (f–k, F2,8 = 20, p = 0.004, One-way ANOVA, >700 cells counted from 3 independent brains per condition). l In addition, suppression of Rp58 by siRNA treatment impaired the migration of GFP-labelled cells, while treatment with either p27kip1 or p27kip1(ck-) promoted the radial migration of Rp58-siRNA treated cells from the VZ/SVZ to the IZ (F2,8 = 12, p < 0.0001, One-way ANOVA, >550 cells counted from 3 independent brains per condition). Scale bar represents 50 μm
During cortical development, immature neurons undertake distinct migratory behaviours as they leave the germinal VZ to migrate through the IZ so as to position themselves appropriately within the CP. We previously reported that Rp58-deficient cells are impaired in their capacity for radial migration within the embryonic cortex [10, 11]. However, p27kip1 is known to promote the migration of neurons into the embryonic cortical plate ([16] and Additional file 2: Figure S2G), thus we hypothesised that both factors could signal cooperatively to promote radial migration. Curiously, in addition to the restorative effects of p27kip1 over-expression on cell cycle exit in Rp58-deficient cortical cells (Fig. 2a–k), we also observed that co-delivery of either p27kip1 or p27kip1ck- augmented their migration from the VZ/SVZ to the IZ (Fig. 2l; F2,8 = 12, p < 0.0001, One-way ANOVA, >550 cells counted from 3 independent brains per condition). Guided by this finding, we explored whether forced expression of p27kip1 could restore the defective migration of Rp58-deficient neurons into the CP of the embryonic mouse cortex. We thus performed in utero electroporation experiments with E14.5 embryos in which we delivered Rp58 siRNAs together with p27kip1 and examined treated brains 72 h later. Previously, we reported that suppression of Rp58 expression using siRNAs in E14.5-born cortical cells impaired their capacity to migrate into the CP of the embryonic E17.5 cortex, and that this defect was restored by co-delivery of an Rp58 expression construct which was refractory to silencing [11]. As shown in Fig. 3, while Rp58-siRNA treated cells were defective in their migration into the CP, co-delivery of p27kip1 led only to a modest increase in the proportion of cells in the CP (Fig. 3a–c, f). Furthermore, we examined the distribution of GFP-labelled cells within the CP to find that co-delivery of p27kip1 did not correct the intracortical positioning of Rp58-deficient cells (Fig. 3a–g). However, we have also previously reported that Rp58 regulates radial migration through transcriptional regulation of downstream target genes such as Rnd2, and that the defective migration of Rp58-deficient cells could be partially corrected by abrogating Rnd2 expression [10, 11]. Therefore, we reasoned that the mechanistic actions for Rp58 to signal radial migration within the cortex could involve both p27kip1, as well as transcriptional regulation of Rnd2. When we directly tested this hypothesis by co-delivering Rp58 siRNAs with p27kip1 as well as a short hairpin RNA expression vector with moderate silencing activity for Rnd2 (known as Rnd2shRNA1, see [11]), we found that the radial migration of these cells was significantly augmented, such that their migration into the CP (Fig. 3f, 44.66% ± 2.79% in the CP for control treatment versus 46.01% ± 2.59% in the CP for Rp58 siRNA + p27kip1 + Rnd2shRNA1 treatment, p > 0.05 Bonferroni posthoc t-test) as well as their intracortical positioning (Fig. 3g, 17.99% ± 1.01% in the upper CP for control treatment versus 16.09% ± 0.81% in the upper CP for Rp58 siRNA + p27kip1 + Rnd2shRNA1 treatment, p > 0.05 Bonferroni posthoc t-test) was restored to levels not significantly different to control profile. This was further confirmed when we analysed the phases of cell migration within each embryonic cortical subcompartment, namely the VZ to IZ migration (Fig. 3h, F4,12 = 39, p < 0.001 One-WAY ANOVA), IZ to CP migration (Fig. 3i, F4,12 = 99, p < 0.001 One-WAY ANOVA) and lower CP to upper CP migration (Fig. 3j, F4,12 = 19, p < 0.001 One-WAY ANOVA). As shown, while Rp58 siRNA-treatment impaired migration across all compartments, co-delivery of both p27kip1 and Rnd2 shRNA led to a significant improvement in all phases of migration by Rp58-deficient cells to levels not significantly different to control profile (Fig. 3h–j).
Fig. 3.
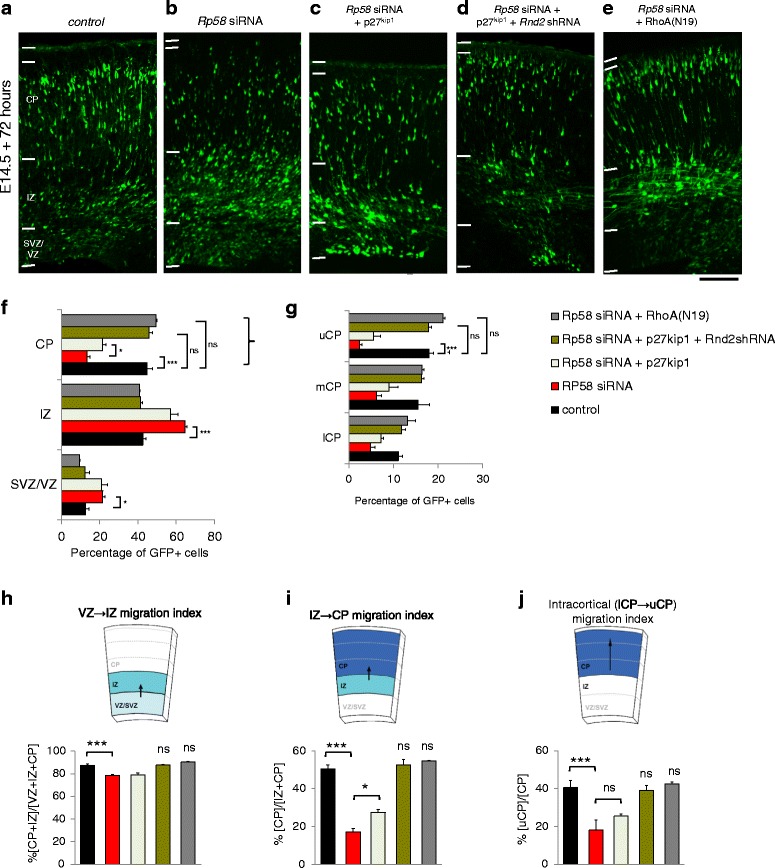
The defective migration of Rp58-deficient cells can be restored with forced expression of p27kip1 and concurrent suppression of Rnd2 by RNAi, and this phenotype is reminiscent of Rp58-deficient cells co-treated with a dominant-negative RhoA(N19) expression construct. a–e Representative images of GFP-labelled cells within the embryonic cortex following treatment with the conditions indicated. f There was a significant interaction between treatment condition and distribution of GFP-labelled cells (F8,36 = 106, p < 0.0001, Two-way ANOVA, >700 cells counted from 3 to 4 independent brains per condition). Knockdown of Rp58 with siRNAs leads to significant decrease in migration into the CP (a, b, f; p < 0.0001 Bonferroni posthoc t-test), but CP entry is only modestly restored by co-delivery with p27kip1 (b, c; f; p < 0.01 Bonferroni posthoc t-test). Forced expression of p27kip1 alone promotes radial migration into the CP (Additional file 6: Figure S5A–D). However, only co-delivery of p27kip1 and Rnd2 shRNA leads to a restoration of the migration of Rp58 siRNA-treated cells (b, d; b; p < 0.0001 Bonferroni posthoc t-test) to levels not significantly different to control CP profile. The radial migration of Rp58-deficient cells can also be restored by co-delivery of RhoA(N19) (b, e; f; p < 0.0001 Bonferroni posthoc t-test), and the CP profile is not significantly different to control. l Quantification of the proportion of GFP-expressing cells within each subcompartment of the embryonic (E17.5) cortex. g There was a significant interaction between treatment condition and distribution of GFP-expressing cells within the lower, medial and upper cortical plate (lCP, mCP and uCP, respectively) (F8,33 = 9.0, p < 0.0001, Two-way ANOVA, >700 cells counted from 3 to 4 independent brains per condition). h The VZ to IZ phase of migration calculated across conditions. i The IZ to CP phase of migration calculated across conditions. j The lower CP to upper CP phase of migration calculated across conditions. One-way ANOVA followed by Bonferroni’s posthoc t-test; *p < 0.05, **p < 0.01, ***p < 0.001 for (h) to (j). Scale bar represents 100 μm
It has been reported that both p27kip1 and Rnd2 mediate cell migration within the embryonic cortex by suppressing RhoA signalling [16, 25]. Thus, we extended our investigation to determine if the defective migration of Rp58-deficient cells could be restored by co-delivery of an expression construct encoding a dominant-negative form of RhoA, (denoted RhoA(N19) [26]). As shown, forced expression of RhoA(N19) significantly augmented the migration of Rp58 siRNA-treated cells to a profile which was not significantly different to control treatment in the CP (Fig. 3a–f; 44.66% ± 2.79% in the CP for control treatment versus 49.53% ± 0.35% in the CP for Rp58 siRNA + RhoA(N19) treatment, p > 0.05), as well as within the upper CP (Fig. 3g; 17.99% ± 1.01% in the upper CP for control treatment versus 21.15% ± 0.44% in the upper CP for Rp58 siRNA + RhoA(N19) treatment, p < 0.05 Bonferroni posthoc t-test). Furthermore, treatment with RhoA(N19) corrected all phases of migration (Fig. 3h–j). We performed immunostaining with Nestin antibody to confirm that the radial glial scaffold of the embryonic cortices between treatment groups was not significantly perturbed (Additional file 4: Figure S4), and that our tissue specimens were prepared in a consistent manner. Therefore, these studies indicate that Rp58 mediates cell migration through p27kip1 and Rnd2 via a cell autonomous mechanism which phenocopies the suppression of RhoA signalling.
Radial migration by cortical neurons involves a series of dynamic changes to their morphologies which, in turn, culminate in directional movement as multipolar-shaped neurons or as bipolar-shaped cells [2, 27]. As newborn neurons leave the germinal VZ/SVZ, they migrate as bipolar shaped neurons then transit as multipolar-shaped neurons within the IZ. Finally, these neurons undergo multipolar-to-bipolar transition, engage a radialglial fibre and then migrate to their appropriate position within the cortical plate by locomotion. We performed high power confocal microscopy on GFP-labelled cortical cells within the IZ and CP in each of our treatment conditions in order to understand how modulation of Rp58, p27kip1 and Rnd2 expression influenced their morphology. We found that knockdown of Rp58 resulted in an accumulation of multipolar-shaped cells with very short processes interspersed amongst round-shaped cells within the intermediate zone (Fig. 4a–f), consistent with previous reports [10, 11, 25]. Forced expression of p27kip1 significantly restored the morphological profiles of Rp58-deficient cells to control levels within the IZ, suggesting that p27kip1 and Rp58 may share common functions for mediating the shapes of IZ cells (Fig. 4a–f). However, within the CP compartment, forced expression of p27kip1 could not restore the elevated proportion of round cells and concomitant reduction in uni/bipolar shaped neurons as a consequence of Rp58 siRNA treatment (Fig. 4g–i, m). Furthermore, we found that while knockdown of Rp58 results in a significant reduction in the length of the leading processes of CP neurons, forced expression of p27kip1 also could not restore this feature (Fig. 4 g–i, l, n). In contrast, however, treatment with both p27kip1 and Rnd2 shRNA restored the morphological profiles of Rp58-deficient neurons to levels not significantly different to control treatment (Fig. 4f and m for IZ and CP cell shape distributions, respectively), suggesting that p27kip1 and Rp58 (through suppression of Rnd2) are both required to regulate distinct morphological characteristics of embryonic cortical neurons over the course of their radial migration. Furthermore, we observed that the morphological profiles for Rp58-deficient cells co-treated with RhoA(N19) were also restored to levels not significantly different to control treatment (Fig. 4 f–n). Thus, our findings suggest that the defective morphological properties of Rp58-deficient cells can be corrected by augmenting p27kip1 levels in combination with suppression of Rnd2, and that these restorative effects on cell shape are reminiscent of a molecular mechanism involving suppression of RhoA signalling.
Fig. 4.
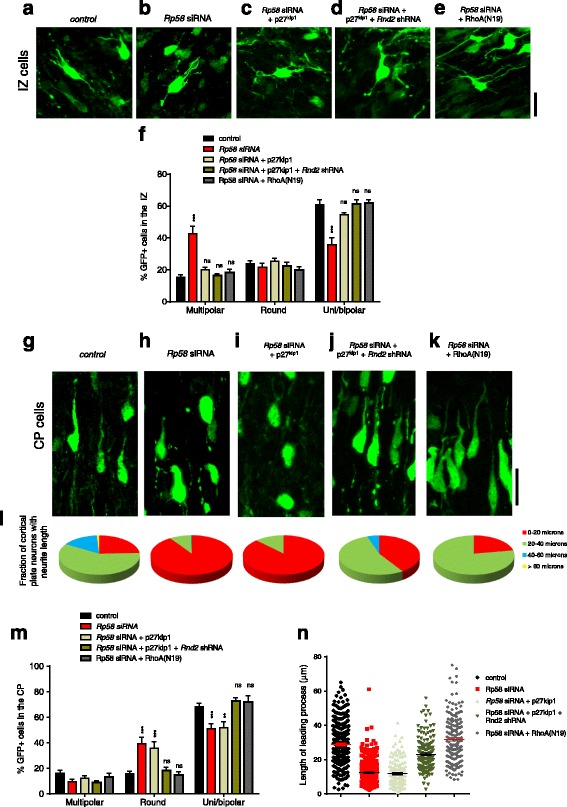
The effects of p27kip1 and Rnd2 abrogation on the morphology of Rp58 siRNA-treated embryonic cortical neurons. a–e Representative images of IZ neurons from each treatment group. f There is a significant interaction between the distribution of round, uni/bipolar and multipolar cell shapes and treatment condition (F8,60 = 20, p < 0.0001, Two-way ANOVA followed by Bonferroni’s posthoc t-test, >300 cells counted from 3 to 4 independent brains per condition), with a significant increase in the proportion of multipolar cells and a concomitant decrease in uni/bipolar shaped cells upon Rp58 siRNA treatment (p < 0.001 Bonferroni posthoc t-test). g–k Representative images of CP neurons from each treatment group. l Pie charts plotting distribution of the lengths of leading processes of uni/bipolar shaped neurons within the CP for each treatment condition. m There is a significant interaction between the distribution of round, uni/bipolar and multipolar cell shapes and treatment condition (F8,60 = 12, p < 0.0001, Two-way ANOVA followed by Bonferroni’s posthoc t-test, >300 cells counted from 3 to 4 independent brains per condition), with a significant increase in the proportion of round cells and a concomitant decrease in multipolar shaped cells upon Rp58 siRNA treatment (p < 0.001 Bonferroni posthoc t-test). n The average lengths of leading process of uni/bipolar shaped neurons is decreased upon Rp58 siRNA treatment, and this is restored by co-treatment with p27kip1 and Rnd2shRNA1 or RhoA(N19) (F4,1187 = 211, p < 0.0001, One-way ANOVA, >160 neurons analysed per condition). Scale bars represent 20 μm
Discussion
In this study, we have revealed cooperative functions for Rp58 and the cyclin dependent kinase inhibitor p27kip1 during the development of the embryonic mouse cerebral cortex. Our results indicate that suppression of Rp58 leads to a transient loss of p27kip1, and that forced expression of p27kip1 restores the defective cell cycle exit and radial migration of Rp58-deficient cortical cells from the germinal VZ/SVZ to the IZ. Furthermore, we find that the defective CP migration of Rp58-deficient cells could only be restored by p27kip1 in combination with down-regulation of Rnd2 by RNAi, and that the cellular features of these neurons are consistent with a mechanism involving down regulation of RhoA signalling. Thus, our findings support a role for Rp58 and p27kip1 to jointly coordinate cell cycle exit and the early (VZ/SVZ-to-IZ) phase of cell migration within the developing cerebral cortex, while the capacity for cortical cells to undergo the late (IZ-to-CP) phase of migration involves distinct functions mediated by p27kip1 and Rp58-Rnd2 signalling pathways (see Additional file 5: Figure S6).
Rp58 deficiency during embryogenesis leads to profound defects in cortical development, including cell proliferation, differentiation and tissue morphogenesis [10, 12–14, 28]. However, a precise understanding of its mechanistic actions on the development of distinct neural cell types is only beginning to emerge. A recent study found that loss of Rp58 led to aberrant neuroprogenitor proliferation and precocious gliogenesis through a molecular mechanism involving de-repression of Id genes which, in turn, resulted in the elevation of steady state levels of the CDKI p57kip2 in the embryonic cortex [12]. However, while Ids are known to influence the expression of multiple CDKIs [29–31], levels of p27kip1 were not significantly disrupted in Rp58 heterozygous and homozygous cortices [12]. Crucially, our findings provide evidence that Rp58 deficiency leads to a transient loss in p27kip1 expression, with consequences to cell cycle exit, neurogenesis and radial migration likely through multiple signals (see summary diagram in Additional file 5: Figure S6). In neuroprogenitors, Rp58 directly represses the cell cycle regulatory genes Id1–4 [12], and Id3 is a repressor of p27 kip1 [29]. Based on our findings, we entertain the hypothesis that Rp58 deficiency results in defective cell cycle progression attributable to de-repression of Id3 in VZ/SVZ cells (see Fig. 2 of [12]) which, in turn, leads to a transient loss of p27 kip1 within this sub-compartment of the embryonic cortex. However, the subsequent restoration of p27kip1 in IZ cells is more difficult to explain, and remains to be clarified in future investigations. Regardless, our data is consistent with the notion that Rp58 and p27kip1 signal cell cycle exit through distinct pathways, since we observed that forced expression of p27kip1 in Rp58-deficient cells resulted in the suppression of proliferation markers Ki67 and pHH3 to levels beyond control treatment.
As cortical neuroprogenitors undergo cell division, p27kip1 drives cell cycle exit and these cells subsequently express the proneural transcription factor Neurog2 so as to specify their identity as excitatory, glutamatergic neurons [4, 32]. Also, Neurog2 initiates a gene regulatory programme for cell migration which involves the transcriptional activation of downstream target genes including Rnd2 [5, 33, 34]. In parallel, the transcriptional repressor activity of Rp58 is critical for negatively regulating Neurog2 expression in cortical cells to coordinate timing of neurogenesis [10]. Crucially, p27kip1 stabilises Neurog2 protein so as to promote neurodifferentiation [16], and so our current findings suggest that the defects in cortical neurogenesis as a consequence of Rp58-deficiency is also attributable to a transient fluctuation in p27kip1 levels. This, in turn, would negatively influence Neurog2 protein levels and ultimately disrupt proneural transcriptional regulation in embryonic cortical cells. Our findings are therefore consistent with a model which reconciles the transcriptional regulatory functions for Rp58 with an epistatic mechanism for cell cycle exit involving signalling cooperativity with p27kip1 (Additional file 5: Figure S6).
In contrast to the epistatic effects of Rp58 and p27kip1 on cell proliferation, we find that treatment with Rp58 siRNAs led to a reduction in expression of the intermediate progenitor marker Tbr2, and that forced expression of p27kip1 or p27kip1ck- was not restorative for this defect. While an investigation to establish the underlying molecular mechanisms for these effects is beyond the scope of our present study, our result is in contrast to a previous finding that Rp58 nullizygous mouse cortices display elevated numbers of Tbr2-immunopositive cells [10, 13]. We believe that these observations point to distinct effects on intermediate progenitors which differ between acute loss of Rp58 expression following siRNA treatment (reported in our study and in [11]) versus chronic loss of Rp58 in homozygous mutant (Rp58−/−) mice [10, 13]. A recent study in which the authors performed in utero electroporation of E14.5 cortices to introduce cre recombinase reported no significant effect on Tbr2 immunostaining in labelled cells 24 h after treatment [10]. However, it was reported that cre electroporation did not lead to complete loss of Rp58 protein expression after 24 h, which is likely to explain the lack of effect on Tbr2 expression. Collectively, these data altogether demonstrate a temporal effect of Rp58 disruption on the development of cortical neuroprogenitors during embryonic development.
In newborn postmitotic neurons, Rp58 negatively regulates appropriate levels of Rnd2 within migrating cortical neurons so as to guide cells to their appropriate positions within the cortex [10, 11]. We previously reported that Rp58 regulates radial migration through Rnd2, but we also recognised that additional signals were at play, since abrogation of Rnd2 expression was not sufficient to fully restore the defective migration of Rp58-deficient cells [11]. Based on the results from our current study, we find that the defective, early (VZ/SVZ to IZ) phase of migration of Rp58-deficient cells can be restored by co-delivery of p27kip1, while the late (IZ to CP) phase of migration can only be restored by manipulation of both p27kip1 and Rp58-Rnd2 signalling. While we acknowledge that our results are based on gain-of-function approaches to augment p27kip1 in cells, we believe they are sufficient to draw the conclusion that Rp58 influences CDKI activity in VZ/SVZ cells to drive neuroprogenitor cell cycle exit and migration into the IZ, while parallel signals from a CDKI such as p27kip1 combine with Rp58-Rnd2 signalling for the appropriate positioning of neurons within the CP. Presently, it is recognised that forced expression of p57kip2 restores the defective proliferation of E14.5-treated cells of Rp58-nullizygous mouse cortices [12]. However, knockdown of p57kip2 impairs the migration of E14.5-born mouse cortical neurons [35], yet Nguyen and colleagues reported that forced expression of p57kip2 did not significantly enhance migration [16]. Taken together, given the potencies of both CDKIs p27kip1 and p57kip2, our study is consistent with the notion that p27kip1 is a crucial, physiologically-relevant effector for coordinating neurodifferentiation and cell migration in concert with Rp58 within the E14.5 embryonic cortex.
Cell motility is orchestrated by the activities of small GTPases, including RhoA and Rac1, in order to form a leading process and to retract the trailing process, respectively, for directional movement [36]. In fibroblasts, RhoA signalling is down-regulated by p27kip1 so as to enable cells to limit the formation of stress fibres and focal adhesions which can impair their motility [24]. Similarly, p27kip1 is found to downregulate RhoA signalling in cortical neurons to facilitate their radial migration [16]. While these lines of evidence indicate that RhoA signalling is relevant to migration, appropriate levels of Rnd2 are also crucial to down regulate RhoA signalling during this process [9, 25]. In our study, we find that the defective migration of Rp58-deficient neurons could be restored by forced expression of p27kip1, while simultaneously suppressing Rnd2 expression via RNAi. Furthermore, the defective migration of Rp58-deficient cells within the embryonic cortex could be corrected by suppressing RhoA signalling by forced expression of a dominant-negative (N19) variant. Based on our results, we thus conclude that the co-presence of Rp58 and p27kip1 in embryonic cortical cells drives radial migration through parallel mechanisms which converge to suppress RhoA signalling. Notably, our results are consistent with the notion that RhoA signalling requires distinct upstream triggers [25, 36–38], since p27kip1 cannot substitute for the defective migration and cell morphological properties of Rp58-deficient neurons within the CP. Taken altogether, our results therefore raise the hypothesis for a dual-signalling cascade involving Rp58-p27kip1 and Rp58-Rnd2 pathways which, in turn, co-ordinately suppress RhoA signalling so as to modulate the neuronal cytoskeleton and drive efficient radial migration into the CP. As such, our study provides a compelling account of the molecular interplay between transcription factors and cyclin dependent kinase inhibition to promote cell cycle exit and radial migration during the development of the cerebral cortex. Future work will address the subcellular regulation of RhoA signalling which underlies the functional cooperativity of Rp58 and p27kip1 in these critical cellular processes for the assembly of functional neural circuits in the central nervous system.
Additional files
Steady state levels of p27kip1 are not significantly altered in Rp58(+/−) and Rp58(−/−) mutant mice compared to Rp58(+/+) wildtype littermate controls. (A) Western blotting of mouse brain lysates from E14.5 mouse embryonic cortices of wildtype, heterozygote and homozygote mutant Rp58-knockout mice. Quantification was conducted on biological triplicates. (B) There was no significant difference in the steady state levels of immunoblotted p27kip1 signal (F2,7 = 0.26, p = 0.77, One-way ANOVA). (C) Analysis of p27kip1 immunofluorescence signal (red) in E14.5-electroporated, GFP-expressing cortical cells collected 36 h after surgical manipulation. Boxed inserts are representative of individual cells analysed from the IZ (C′) and VZ/SVZ (C″). (D) Raw images were analysed using ImageJ software to measure the average intensity of p27kip1 signal in GFP-labelled, IZ cells, with values in Arbitrary Units defined as the signal intensity of an IZ cell divided by the average intensity of 20 CP cells which are negative for GFP immunostaining, and multiplied by 100. There was no significant interaction between treatment groups (F2,222 = 2.9, p = 0.0557, One-way ANOVA, >75 cells counted from 3 independent brains per condition). (E) The average intensity of p27kip1 signal in GFP-labelled, VZ/SVZ cells, with values in Arbitrary Units defined as the signal intensity of an IZ cell divided by the average intensity of unelectroporated CP cells, and multiplied by 100. The intensity of p27kip1-immunofluorescence signal within VZ/SVZ cells was significantly reduced upon knockdown, while overexpression did not have a significant effect (F2,222 = 95, p < 0.0001, One-way ANOVA, >75 cells counted from 3 independent brains per condition). Values in (B), (D) and (E) represent mean ± SEM. Scale bars represent 100 μm (C) and 20 μm (C″) respectively. (PDF 208 kb)
p27kip1 is a potent mediator of cell cycle exit and radial migration within the embryonic cortex. In utero electroporation of a control (GFP only), p27kip1 or p27kip1(ck-) construct into E14.5 embryonic cortices collected 36 h after surgery for analysis. (A-B) Sections were immunostained for Ki67 and counted to find that forced expression of p27kip1 but not p27kip1(ck-) led to a significant suppression in Ki67 co-staining (F2,6 = 53, p = 0.0002). (C-D) Sections were immunostained for pHH3, a marker of cell mitosis to find that forced expression of p27kip1 but not p27kip1(ck-) led to a significant suppression in pHH3 co-staining (F2,6 = 56, p = 0.0001). (E) Overexpression of p27kip1 or p27kip1ck- alone did not influence Pax6-immunoreactive radial glial progenitors (F2,7 = 0.35, p = 0.7171, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). (F) Treatment with Rp58 siRNAs led to a significant reduction in the proportion of Tbr2-expressing intermediate progenitors which could not be augmented by co-delivery of p27kip1 or p27kip1ck- expression constructs (F2,7 = 11, p = 0.0072, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). (G) The distribution of GFP-labelled cells is significantly affected by treatment with p27kip1 or p27kip1(ck-). Forced expression of p27kip1 or p27kip1(ck-) led to a significant increase in the proportion of treated cells arriving within the upper IZ (F8,30 = 18, p < 0.0001; Two-way ANOVA followed by Bonferroni’s posthoc t-test; *p < 0.05, **p < 0.01, ***p < 0.001). Values in graphs represent mean ± SEM. Scale bars represent 50 μm. (PDF 428 kb)
The effects of Rp58 siRNA treatment together with expression constructs for p27kip1 and p27kip1ck- on progenitors. (A) There was a significant interaction between non-surface (SVZ) divisions in treated cells, identified as mitoses marked by pHH3 expression away from the ventricular surface (F3,9 = 7, p < 0.0102, One-way ANOVA, 3 independent brains per condition). (B-E) Representative photomicrographs of immunostained sections from each treatment, as indicated. (F) The proportion of Pax6-expressing radial glial progenitors was not significantly affected between treatment groups (F3,9 = 0.89, p = 0.4818, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). (G-J) Representative photomicrographs of GFP and Tbr2 immunostained sections from each treatment, as indicated. (K) The proportion of Tbr2-expressing intermediate progenitors was significantly affected between treatment groups (F3,11 = 6.5, p = 0.00858, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). Values in graphs represent mean ± SEM. Scale bars represent 50 μm. (PDF 193 kb)
Immunostaining for Nestin reveals radial glial fibres within sections of electroporated (E14.5 + 72 h) mouse brains. Nestin immunostaining indicates that the radial glial scaffolds within the cortices of each treatment group are not qualitatively different. Scale bar represents 50 μm. (PDF 6040 kb)
A summary diagram highlighting the combinatorial functions for Rp58 and p27kip1 on cell cycle exit and radial migration during the development of the mouse embryonic cerebral cortex. In this scheme, the regulation of neuroprogenitor cell cycle exit and neurogenesis is mediated by Rp58. Newborn postmitotic cells within the E14.5 embryonic cortex undergo cell cycle exit and express p27kip1 and Neurog2. The timing of p27kip1 expression and Neurog2 expression is influenced by Rp58. Neurog2 stimulates the expression of Rp58 which induces a feedback loop to temper Neurog2 expression levels, as well as to abrogate Rnd2 expression in migrating cortical neurons. Meanwhile, p27kip1 stabilises Neurog2 protein levels to specify glutamatergic neuron identity as well as promote radial migration. In the context of Rp58 deficiency, cortical cells lose their capacity to transit from the IZ to the CP owing to their failure to undergo MP to BP transition (B). Neither forced expression of p27kip1 nor Rnd2 RNAi alone was capable of restoring the capacity of Rp58-deficient cells to migrate into the CP (C-D). However, the defective migration of Rp58-deficient cells was significantly augmented by the combination of p27kip1 overexpression and Rnd2 RNAi (F). This restorative capacity is reminiscent of Rp58-deficient cells co-treated with RhoA(N19), a dominant-negative form which suppresses RhoA signalling (see Fig. 4). (PDF 123 kb)
Forced expression of p27kip1 enhances radial migration and alters the morphological characteristics of embryonic cortical neurons. (A-B) Forced expression of p27kip1 leads to enhanced radial migration, observed as a significant increase in the proportion of cells arriving in the CP (F2,12 = 7.245, p = 0.0085; Two-way ANOVA followed by Bonferroni’s posthoc t-test; *p < 0.05) (C). (D) The intracortical positioning of p27kip1-overexpressing cells is also affected, observed as a significant increase in the proportion of cells within the upper CP (uCP) (F2,12 = 2.645, p = 0.118; Two-way ANOVA followed by Bonferroni’s posthoc t-test; *p < 0.05). (E-F) Representative images of IZ neurons within each treatment condition. (G) The distribution of cell shapes within the IZ are not significantly affected by p27kip1 overexpression (F2,24 = 1.8, p = 0.194; Two-way ANOVA. (H-J) Representative images of CP neurons within each treatment condition. Pie charts representing the lengths of the leading processes of uni/bipolar shaped neurons within the CP are represented for each treatment condition. (J) There is a significant interaction between the distribution of CP cell shapes and p27kip1 overexpression (F2,27 = 5.0, p = 0.0147; Two-way ANOVA), but the differences for a given cell shape was not significantly different upon posthoc t-testing. (K) The leading processes of p27kip1-overexpressing neurons were significantly shorter when compared with control (p < 0.0001 student’s t-test, two-tailed; n > 285 neurons per condition). Scale bars represent 100 μm (B) and 20 μm (F, I) respectively. (PDF 331 kb)
Acknowledgments
Funding
JI-TH was supported by a grant from the National Health and Medical Research Council of Australia (ID:1011505) as well as funding from the Victorian Government through the Operational Infrastructure Scheme. This work was also supported by the Harry Perkins Institute of Medical Research and The University of Western Australia, as well as The Australian Regenerative Medicine Institute, and Monash University.
Availability of data and materials
Available upon request.
Authors’ contributions
JI-TH conceived the project and performed animal experiments with OC, IAH, IEG-N and ZQ. IAH, SSL and IEG-N performed molecular biology and cell culture, with input from MP. JI-TH wrote the manuscript with OC and IAH, with all authors providing comment. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Our animal research is governed by project licenses approved by the Animal Ethics Committee within Monash University (Licenses MARP/08–104 and MARP/2012/068), and are compliant with guidelines provided by the National Health and Medical Research Council of Australia.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- CDKI
Cyclin dependent kinase inhibitor
- CP
Cortical plate
- GFP
Green Fluorescent Protein
- IZ
Intermediate zone
- lCP
Lower cortical plate
- mCP
Medial cortical plate
- Rp58
Repressor Protein of 58 kDa
- siRNA
Small inhibitory RNA
- SVZ
Subventricular zone
- uCP
Upper cortical plate
- VZ
Ventricular zone
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13064-017-0084-3) contains supplementary material, which is available to authorized users.
Contributor Information
Olivier Clément, Email: olivier.clement@perkins.uwa.edu.au.
Isabel Anne Hemming, Email: isabel.hemming@research.uwa.edu.au.
Ivan Enghian Gladwyn-Ng, Email: ivanng.eh@gmail.com.
Zhengdong Qu, Email: zhengdongqu@gmail.com.
Shan Shan Li, Email: shanshan.li@hotmail.com.
Michael Piper, Email: m.piper@uq.edu.au.
Julian Ik-Tsen Heng, Phone: (61)-413-509-384, Email: Julian.Heng@curtin.edu.au.
References
- 1.Gupta A, Tsai LH, Wynshaw-Boris A. Life is a journey: a genetic look at neocortical development. Nat Rev Genet. 2002;3(5):342–355. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- 2.Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27(7):392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8(6):427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 4.Schuurmans C, Guillemot F. Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr Opin Neurobiol. 2002;12(1):26–34. doi: 10.1016/S0959-4388(02)00286-6. [DOI] [PubMed] [Google Scholar]
- 5.Heng J, Guillemot F. Proneural proteins and the development of the cerebral cortex. In: Kageyama R, editor. Cortical Development: Neural Diversity and Neocortical Organization. Springer Global; 2013. p. In press.
- 6.Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- 7.Molyneaux BJ, Arlotta P, Macklis JD. Molecular development of corticospinal motor neuron circuitry. Novartis Found Symp. 2007;288:3–15. doi: 10.1002/9780470994030.ch2. [DOI] [PubMed] [Google Scholar]
- 8.Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, et al. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron. 2005;48(1):45–62. doi: 10.1016/j.neuron.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 9.Heng JI, Nguyen L, Castro DS, Zimmer C, Wildner H, Armant O, et al. Neurogenin 2 controls cortical neuron migration through regulation of Rnd2. Nature. 2008;455(7209):114–118. doi: 10.1038/nature07198. [DOI] [PubMed] [Google Scholar]
- 10.Ohtaka-Maruyama C, Hirai S, Miwa A, Heng JI, Shitara H, Ishii R, et al. RP58 regulates the multipolar-bipolar transition of newborn neurons in the developing cerebral cortex. Cell Rep. 2013;3(2):458–471. doi: 10.1016/j.celrep.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Heng JI, Qu Z, Ohtaka-Maruyama C, Okado H, Kasai M, Castro D et al. The zinc finger transcription factor RP58 negatively regulates Rnd2 for the control of neuronal migration during cortical development. Cereb Cortex. 2013. doi:bht277. [DOI] [PubMed]
- 12.Hirai S, Miwa A, Ohtaka-Maruyama C, Kasai M, Okabe S, Hata Y, et al. RP58 controls neuron and astrocyte differentiation by downregulating the expression of Id1-4 genes in the developing cortex. EMBO J. 2012;31(5):1190–1202. doi: 10.1038/emboj.2011.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okado H, Ohtaka-Maruyama C, Sugitani Y, Fukuda Y, Ishida R, Hirai S, et al. The transcriptional repressor RP58 is crucial for cell-division patterning and neuronal survival in the developing cortex. Dev Biol. 2009;331(2):140–151. doi: 10.1016/j.ydbio.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 14.Xiang C, Baubet V, Pal S, Holderbaum L, Tatard V, Jiang P et al. RP58/ZNF238 directly modulates proneurogenic gene levels and is required for neuronal differentiation and brain expansion. Cell Death Differ. 2011. doi:cdd2011144. [DOI] [PMC free article] [PubMed]
- 15.Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol. 2006;8(1):17–26. doi: 10.1038/ncb1338. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, et al. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20(11):1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS. Overexpression of p27 Kip 1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex. 2005;15(9):1343–1355. doi: 10.1093/cercor/bhi017. [DOI] [PubMed] [Google Scholar]
- 18.Godin JD, Thomas N, Laguesse S, Malinouskaya L, Close P, Malaise O, et al. p27(Kip1) is a microtubule-associated protein that promotes microtubule polymerization during neuron migration. Dev Cell. 2012;23(4):729–744. doi: 10.1016/j.devcel.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Breuss M, Heng JI, Poirier K, Tian G, Jaglin XH, Qu Z, et al. Mutations in the beta-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012;2(6):1554–1562. doi: 10.1016/j.celrep.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ngo L, Haas M, Qu Z, Li SS, Zenker J, Teng KS, et al. TUBB5 and its disease-associated mutations influence the terminal differentiation and dendritic spine densities of cerebral cortical neurons. Hum Mol Genet. 2014;23(19):5147–5158. doi: 10.1093/hmg/ddu238. [DOI] [PubMed] [Google Scholar]
- 21.Haas MA, Ngo L, Li SS, Schleich S, Qu Z, Vanyai HK, et al. De Novo Mutations in DENR Disrupt Neuronal Development and Link Congenital Neurological Disorders to Faulty mRNA Translation Re-initiation. Cell Rep. 2016;15(10):2251–2265. doi: 10.1016/j.celrep.2016.04.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gladwyn-Ng IE, Li SS, Qu Z, Davis JM, Ngo L, Haas M, et al. Bacurd2 is a novel interacting partner to Rnd2 which controls radial migration within the developing mammalian cerebral cortex. Neural Dev. 2015;10:9. doi: 10.1186/s13064-015-0032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi T, Nowakowski RS, Caviness VS., Jr The leaving or Q fraction of the murine cerebral proliferative epithelium: a general model of neocortical neuronogenesis. J Neurosci. 1996;16(19):6183–6196. doi: 10.1523/JNEUROSCI.16-19-06183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18(8):862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pacary E, Heng J, Azzarelli R, Riou P, Castro D, Lebel-Potter M et al. Proneural transcription factors regulate different steps of cortical neuron migration through Rnd-mediated inhibition of RhoA signaling. Neuron. 2011;69(6):1069-1084. doi:S0896-6273(11)00116-4. [DOI] [PMC free article] [PubMed]
- 26.Wennerberg K, Forget MA, Ellerbroek SM, Arthur WT, Burridge K, Settleman J, et al. Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr Biol. 2003;13(13):1106–1115. doi: 10.1016/S0960-9822(03)00418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29(7):407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Yokoyama S, Ito Y, Ueno-Kudoh H, Shimizu H, Uchibe K, Albini S, et al. A systems approach reveals that the myogenesis genome network is regulated by the transcriptional repressor RP58. Dev Cell. 2009;17(6):836–848. doi: 10.1016/j.devcel.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrett-Engele CM, Tasch MA, Hwang HC, Fero ML, Perlmutter RM, Clurman BE, et al. A mechanism misregulating p27 in tumors discovered in a functional genomic screen. PLoS Genet. 2007;3(12):e219. doi: 10.1371/journal.pgen.0030219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lasorella A, Benezra R, Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer. 2014;14(2):77–91. doi: 10.1038/nrc3638. [DOI] [PubMed] [Google Scholar]
- 31.Pagliuca A, Gallo P, De Luca P, Lania L. Class A helix-loop-helix proteins are positive regulators of several cyclin-dependent kinase inhibitors’ promoter activity and negatively affect cell growth. Cancer Res. 2000;60(5):1376–1382. [PubMed] [Google Scholar]
- 32.Guillemot F, Hassan BA. Beyond proneural: emerging functions and regulations of proneural proteins. Curr Opin Neurobiol. 2016;42:93–101. doi: 10.1016/j.conb.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Heng JI, Chariot A, Nguyen L. Molecular layers underlying cytoskeletal remodelling during cortical development. Trends Neurosci. 2010;33(1):38-47. doi:S0166-2236(09)00169-6. [DOI] [PubMed]
- 34.Merot Y, Retaux S, Heng JI. Molecular mechanisms of projection neuron production and maturation in the developing cerebral cortex. Semin Cell Dev Biol. 2009; S1084-9521(09)00084-6. [DOI] [PubMed]
- 35.Itoh Y, Masuyama N, Nakayama K, Nakayama KI, Gotoh Y. The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J Biol Chem. 2007;282(1):390–396. doi: 10.1074/jbc.M609944200. [DOI] [PubMed] [Google Scholar]
- 36.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 37.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4(6):446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 38.Xu C, Funahashi Y, Watanabe T, Takano T, Nakamuta S, Namba T, et al. Radial Glial Cell-Neuron Interaction Directs Axon Formation at the Opposite Side of the Neuron from the Contact Site. J Neurosci. 2015;35(43):14517–14532. doi: 10.1523/JNEUROSCI.1266-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Steady state levels of p27kip1 are not significantly altered in Rp58(+/−) and Rp58(−/−) mutant mice compared to Rp58(+/+) wildtype littermate controls. (A) Western blotting of mouse brain lysates from E14.5 mouse embryonic cortices of wildtype, heterozygote and homozygote mutant Rp58-knockout mice. Quantification was conducted on biological triplicates. (B) There was no significant difference in the steady state levels of immunoblotted p27kip1 signal (F2,7 = 0.26, p = 0.77, One-way ANOVA). (C) Analysis of p27kip1 immunofluorescence signal (red) in E14.5-electroporated, GFP-expressing cortical cells collected 36 h after surgical manipulation. Boxed inserts are representative of individual cells analysed from the IZ (C′) and VZ/SVZ (C″). (D) Raw images were analysed using ImageJ software to measure the average intensity of p27kip1 signal in GFP-labelled, IZ cells, with values in Arbitrary Units defined as the signal intensity of an IZ cell divided by the average intensity of 20 CP cells which are negative for GFP immunostaining, and multiplied by 100. There was no significant interaction between treatment groups (F2,222 = 2.9, p = 0.0557, One-way ANOVA, >75 cells counted from 3 independent brains per condition). (E) The average intensity of p27kip1 signal in GFP-labelled, VZ/SVZ cells, with values in Arbitrary Units defined as the signal intensity of an IZ cell divided by the average intensity of unelectroporated CP cells, and multiplied by 100. The intensity of p27kip1-immunofluorescence signal within VZ/SVZ cells was significantly reduced upon knockdown, while overexpression did not have a significant effect (F2,222 = 95, p < 0.0001, One-way ANOVA, >75 cells counted from 3 independent brains per condition). Values in (B), (D) and (E) represent mean ± SEM. Scale bars represent 100 μm (C) and 20 μm (C″) respectively. (PDF 208 kb)
p27kip1 is a potent mediator of cell cycle exit and radial migration within the embryonic cortex. In utero electroporation of a control (GFP only), p27kip1 or p27kip1(ck-) construct into E14.5 embryonic cortices collected 36 h after surgery for analysis. (A-B) Sections were immunostained for Ki67 and counted to find that forced expression of p27kip1 but not p27kip1(ck-) led to a significant suppression in Ki67 co-staining (F2,6 = 53, p = 0.0002). (C-D) Sections were immunostained for pHH3, a marker of cell mitosis to find that forced expression of p27kip1 but not p27kip1(ck-) led to a significant suppression in pHH3 co-staining (F2,6 = 56, p = 0.0001). (E) Overexpression of p27kip1 or p27kip1ck- alone did not influence Pax6-immunoreactive radial glial progenitors (F2,7 = 0.35, p = 0.7171, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). (F) Treatment with Rp58 siRNAs led to a significant reduction in the proportion of Tbr2-expressing intermediate progenitors which could not be augmented by co-delivery of p27kip1 or p27kip1ck- expression constructs (F2,7 = 11, p = 0.0072, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). (G) The distribution of GFP-labelled cells is significantly affected by treatment with p27kip1 or p27kip1(ck-). Forced expression of p27kip1 or p27kip1(ck-) led to a significant increase in the proportion of treated cells arriving within the upper IZ (F8,30 = 18, p < 0.0001; Two-way ANOVA followed by Bonferroni’s posthoc t-test; *p < 0.05, **p < 0.01, ***p < 0.001). Values in graphs represent mean ± SEM. Scale bars represent 50 μm. (PDF 428 kb)
The effects of Rp58 siRNA treatment together with expression constructs for p27kip1 and p27kip1ck- on progenitors. (A) There was a significant interaction between non-surface (SVZ) divisions in treated cells, identified as mitoses marked by pHH3 expression away from the ventricular surface (F3,9 = 7, p < 0.0102, One-way ANOVA, 3 independent brains per condition). (B-E) Representative photomicrographs of immunostained sections from each treatment, as indicated. (F) The proportion of Pax6-expressing radial glial progenitors was not significantly affected between treatment groups (F3,9 = 0.89, p = 0.4818, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). (G-J) Representative photomicrographs of GFP and Tbr2 immunostained sections from each treatment, as indicated. (K) The proportion of Tbr2-expressing intermediate progenitors was significantly affected between treatment groups (F3,11 = 6.5, p = 0.00858, One-way ANOVA, >600 cells counted from 3 to 4 independent brains per condition). Values in graphs represent mean ± SEM. Scale bars represent 50 μm. (PDF 193 kb)
Immunostaining for Nestin reveals radial glial fibres within sections of electroporated (E14.5 + 72 h) mouse brains. Nestin immunostaining indicates that the radial glial scaffolds within the cortices of each treatment group are not qualitatively different. Scale bar represents 50 μm. (PDF 6040 kb)
A summary diagram highlighting the combinatorial functions for Rp58 and p27kip1 on cell cycle exit and radial migration during the development of the mouse embryonic cerebral cortex. In this scheme, the regulation of neuroprogenitor cell cycle exit and neurogenesis is mediated by Rp58. Newborn postmitotic cells within the E14.5 embryonic cortex undergo cell cycle exit and express p27kip1 and Neurog2. The timing of p27kip1 expression and Neurog2 expression is influenced by Rp58. Neurog2 stimulates the expression of Rp58 which induces a feedback loop to temper Neurog2 expression levels, as well as to abrogate Rnd2 expression in migrating cortical neurons. Meanwhile, p27kip1 stabilises Neurog2 protein levels to specify glutamatergic neuron identity as well as promote radial migration. In the context of Rp58 deficiency, cortical cells lose their capacity to transit from the IZ to the CP owing to their failure to undergo MP to BP transition (B). Neither forced expression of p27kip1 nor Rnd2 RNAi alone was capable of restoring the capacity of Rp58-deficient cells to migrate into the CP (C-D). However, the defective migration of Rp58-deficient cells was significantly augmented by the combination of p27kip1 overexpression and Rnd2 RNAi (F). This restorative capacity is reminiscent of Rp58-deficient cells co-treated with RhoA(N19), a dominant-negative form which suppresses RhoA signalling (see Fig. 4). (PDF 123 kb)
Forced expression of p27kip1 enhances radial migration and alters the morphological characteristics of embryonic cortical neurons. (A-B) Forced expression of p27kip1 leads to enhanced radial migration, observed as a significant increase in the proportion of cells arriving in the CP (F2,12 = 7.245, p = 0.0085; Two-way ANOVA followed by Bonferroni’s posthoc t-test; *p < 0.05) (C). (D) The intracortical positioning of p27kip1-overexpressing cells is also affected, observed as a significant increase in the proportion of cells within the upper CP (uCP) (F2,12 = 2.645, p = 0.118; Two-way ANOVA followed by Bonferroni’s posthoc t-test; *p < 0.05). (E-F) Representative images of IZ neurons within each treatment condition. (G) The distribution of cell shapes within the IZ are not significantly affected by p27kip1 overexpression (F2,24 = 1.8, p = 0.194; Two-way ANOVA. (H-J) Representative images of CP neurons within each treatment condition. Pie charts representing the lengths of the leading processes of uni/bipolar shaped neurons within the CP are represented for each treatment condition. (J) There is a significant interaction between the distribution of CP cell shapes and p27kip1 overexpression (F2,27 = 5.0, p = 0.0147; Two-way ANOVA), but the differences for a given cell shape was not significantly different upon posthoc t-testing. (K) The leading processes of p27kip1-overexpressing neurons were significantly shorter when compared with control (p < 0.0001 student’s t-test, two-tailed; n > 285 neurons per condition). Scale bars represent 100 μm (B) and 20 μm (F, I) respectively. (PDF 331 kb)
Data Availability Statement
Available upon request.