

Abstract
Neuroinflammation is critically involved in numerous neurodegenerative diseases, and key signaling steps of innate immune activation hence represent promising therapeutic targets. This mini review series originated from the 4th Venusberg Meeting on Neuroinflammation held in Bonn, Germany, 7-9th May 2015, presenting updates on innate immunity in acute brain injury and chronic neurodegenerative disorders, such as traumatic brain injury and Alzheimer’s disease, on the role of astrocytes and microglia, as well as technical developments that may help elucidate neuroinflammatory mechanisms and establish clinical relevance. In this meeting report, a brief overview of physiological and pathological microglia morphology is followed by a synopsis on PGE2 receptors, insights into the role of arginine metabolism and further relevant aspects of neuroinflammation in various clinical settings, concluded by a presentation of technical challenges and solutions when working with microglia and astrocyte cultures. Microglial ontogeny and induced pluripotent stem cell derived microglia, advances of TREM2 signaling, and the cytokine paradox in Alzheimer’s disease are further contributions to this series.
Keywords: Alzheimer’s disease, blood-brain barrier (BBB), cytokines, microglia, innate immunity, Long-term potentiation (LTP), macrophage colony stimulating factor receptor (CSF-1R), non-steroidal anti-inflammatory drugs (NSAIDs), Venusberg Neuroinflammation Meeting Bonn 2015
Graphical abstract
Neuroinflammation is critically involved in numerous neurodegenerative diseases, and key signaling steps of innate immune activation hence represent promising therapeutic targets. This mini review series originated from the 4th Venusberg Meeting on Neuroinflammation held in Bonn, Germany, 7-9th May 2015, presenting updates on innate immunity in acute brain injury and chronic neurodegenerative disorders such as traumatic brain injury and Alzheimer's disease, on the role of astrocytes and microglia, as well as technical developments that may help elucidate neuroinflammatory mechanisms and establish clinical relevance. In this meeting report, a brief overview on physiological and pathological microglia morphology is followed by a synopsis on PGE2 receptors, insights into the role of arginine metabolism and further relevant aspects of neuroinflammation in various clinical settings, concluded by a presentation of technical challenges and solutions when working with microglia cultures. Microglial ontogeny and induced pluripotent stem cell derived microglia, advances of TREM2 signaling, and the cytokine paradox in Alzheimer’s disease are further contributions to this article.
Morphological aspects of microglia in development, aging and disease
Niva Russek-Blum & Alon Monsonego
Introduction
Microglia, the immune resident cells of the brain, were first described many decades ago as migratory phagocytic cells of the central nervous system (CNS) (Del Rio-Hortega 1932; Rezaie and Male 2002). In his seminal paper, del Rio-Hortega suggested that microglia exhibit an amoeboid morphology when they enter the brain during early development, then transform into a branched, ramified morphological phenotype. He observed that microglia are found almost evenly dispersed throughout the adult brain, and that, following injury, they undergo a transformation into an amoeboid morphology and have the capacity to migrate, proliferate and phagocytose similar to their characteristics during early development. Although these early morphological observations did not change dramatically since they were first described, they did undergo some refinements. Recently, the combination of advanced imaging techniques, such as confocal and two-photon microscopy, with the use of genetically based cell-specific markers and thinned-skull preparations, has allowed high resolution studies of microglia also in the living animal. This review summarizes our current understanding of structural-functional aspects of microglia, from development to the adult functioning brain, and to aging and age-related diseases.
Microglia morphology in the developing CNS
Developmental studies show that microglia cells originate from yolk sac-derived precursors in a Myb-independent manner via PU.1 and interferon regulatory factor (IRF8)-dependent pathways (Figure 1). This lineage is primarily regulated by macrophage colony stimulating factor receptor (CSF-1R), its ligand IL-34, and transforming growth factor-β (TGF-β). Microglia population is established during prenatal development, but how these cells are maintained throughout the organism’s lifespan remains an ongoing debate (Eglitis and Mezey 1997; Herbomel, Thisse et al. 2001; Ginhoux, Greter et al. 2010; Schulz, Gomez Perdiguero et al. 2012; Varvel, Grathwohl et al. 2012; Kierdorf, Erny et al. 2013; Shiau, Monk et al. 2013; Elmore, Najafi et al. 2014 Bruttger, Karram et al. 2015). After invading the brain’s parenchyma, these precursor cells acquire a ramified and quiescent phenotype, unique to the CNS milieu. The mechanisms underlying the differentiation of yolk sac precursors to “resting”, ramified microglia phenotype are mostly unknown. Cell-culture studies have suggested a few candidates that affect the ramification process such as cytokines, namely, TGF-β, macrophage colony-stimulating factor (M-CSF), and granulocyte/macrophage colony stimulating factor (GM-CSF) (Schilling, Nitsch et al. 2001; Abutbul, Shapiro et al. 2012). TGF-β, M-CSF and GM-CSF were shown to induce ramification and up-regulation of delayed K+ channels in newborn mice-derived primary microglia. Transformation from amoeboid into ramified morphology induced in microglia by exposure to astrocyte-conditioned medium (ACM) was inhibited by neutralizing antibodies against TGF-β, M-CSF or GM-CSF, while ACM-induced delayed rectifier (DR) K+ channel expression was exclusively inhibited by antibodies against TGF-β (Schilling, Nitsch et al. 2001). The role of TGF-β in acquiring a microglia-like quiescent phenotype along with ramified morphology was studied in our laboratory by using co-cultures of hematopoietic progenitors or bone marrow derived dendritic cells (BMDCs) with organotypic hippocampal slices or primary glia. TGF-β signaling via SMAD2/3 phosphorylation was found to be essential for myeloid-derived cells to upregulate CX3CR1 and to acquire a microglia-like ramified morphology and a quiescent phenotype (Abutbul, Shapiro et al. 2012). These findings were further supported by the identification of a unique microglial molecular signature, which depends on TGF-β signaling (Butovsky, Jedrychowski et al. 2014). Furthermore, study of transcriptomes and enhancer landscapes of microglia indicated specificity of the SMAD motif, which collaborates with PU.1 to establish microglia-specific enhancers (Gosselin, Link et al. 2014). The cellular and molecular mechanisms that mediate the specification, migration, and differentiation of developing microglia remain mostly unknown and raise fundamental questions regarding the cellular source and signaling pathways of TGF-β in differentiating microglia, and how it is orchestrated with other signaling cues in the developing brain.
Figure 1. Microglial differentiation.

Rounded yolk sac derived precursors invade the brain parenchyma in PU.1- and IRF8- dependent pathways and are regulated by CSF-1R, its ligand IL-34, and TGF-β to become fully differentiated microglial cells. During embryonic development, these cells progressively develop ramified morphology and constantly scan the brain microenvironment, eliminating neurons and pruning excessive synapses. In the mature healthy brain, surveilling microglia assist in maintaining brain homeostasis and function by regulating neuronal activity using special morphological features- bulbous endings, controlling synaptic plasticity by finger-like protrusions that wrap around dendritic spines, and engulfing cells undergoing apoptosis. These cells are highly ramified expressing CX3CR1, Iba-1 and P2YR12, are highly dynamic and dense to cover the tissue uniformly. During aging, microglia exhibit chronic mildly activated phenotype with increased expression of proinflammatory mediators, such as TNF-α, IL-1β, and IL-6 and exhibit reduced ramification with process shortening and thickening and some aberrant morphological features resembling dystrophy. Their coverage of the tissue is impaired. In Alzheimer’s disease (AD), microglia proliferate and accumulate with activated and dystrophic phenotypes around plaques of amyloid β (Aβ). Microglia cells surround the plaque with distinct morphological phenotypes. The first layer of more amoeboid cells are found in close proximity to the plaque and resemble activated microglia and/or infiltrating monocytes. Additional layers of microglial cells are located on the plaque edges, exhibiting decreased morphological complexity compared to the healthy mature microglia. Microglia cell coverage is impaired in APP-Tg mice, in which tissue space is left devoid of microglia processes. Considering the dynamic nature of the microglial processes, such a robust loss of branches during aging and disease may significantly impair the overall sensing capacity of microglia.
Confocal z-stack images showing microglia process densities and spatial coverage area were adopted from (Baron, Babcock et al. 2014). Bars represent 50 μm.
Structural-functional aspects of microglia
In the mature brain, microglia show remarkable diversity with respect to brain anatomy (Olah, Biber et al. 2011; Grabert, Michoel et al. 2016). Under physiological conditions, their cellular density can vary considerably between different brain regions by as much as five-fold in mice (Lawson, Perry et al. 1990) and ten-fold in humans (Mittelbronn, Dietz et al. 2001). In both mice and humans, microglia are most numerous in the telencephalon, followed by the diencephalon, mesencephalon, and rhombencephalon (Lawson, Perry et al. 1990; Savchenko, McKanna et al. 2000). Myelinated regions are known to contain higher density of microglia than non-myelinated tissues of the same anatomical region (Lawson, Perry et al. 1990; Mittelbronn, Dietz et al. 2001). While white-matter microglia show elongated somata and processes that are preferentially oriented along fiber tracts, microglia in the circumventricular organs- regions characterized by a leaky blood-brain barrier, exhibit a compact morphology with a few short processes, which may resemble a primed morphology. In contrast, gray-matter microglia exhibit many elaborated, radially oriented arbors (Lawson, Perry et al. 1990).
The basic morphology of quiescent cortical microglia, as described in mice, is generally represented in a complexed branch order as follows: (i) the 1st branch order, usually between 7–9 main processes, which extend directly from the cell soma; (ii) the 2nd branch order are medium processes, which branch from the main processes to several shorter and thinner processes; and (iii) the 3rd branch order-fine processes, which are mostly devoid of Iba-1 expression (Baron, Babcock et al. 2014). Similar to the typical morphological properties of resting microglia in the mouse cortex, resting microglia in zebrafish are also highly branched with dynamic processes, which end with stick-like or bulbous tips (Peri and Nusslein-Volhard 2008; Sieger and Peri 2013).
The bulbous tip, a unique morphological feature of microglia, is formed rapidly through expansion from a stick-like ending, and stalls on the contacted neuronal soma for several minutes before gradually shrinking back (Li, Du et al. 2012). Functionally, the bulbous ending mediates the preferential contact of the microglia with neurons that show high levels of activity to ultimately regulate the spontaneous and evoked activity of the contacted neurons (Li, Du et al. 2012). Another unique morphological specialization of microglia, described based on electron microscopy studies, is finger-like protrusions that wrap around dendritic spines, followed by phagocytic inclusions of synaptic elements into the microglia (Tremblay, Lowery et al. 2010). These elements are found in the postnatal developing brain, wherein microglia play a fundamental role in “synaptic pruning”, namely, in eliminating excess synaptic connections and mediating proper maturation of excitatory synaptic transmission (Paolicelli, Bolasco et al. 2011; Zhan, Paolicelli et al. 2014). The process of synaptic pruning is dependent on neuronal activity and is achieved by microglial engulfment of both pre- and postsynaptic elements (Tremblay, Lowery et al. 2010; Paolicelli, Bolasco et al. 2011; Kettenmann, Kirchhoff et al. 2013; Schafer, Lehrman et al. 2013). Most microglial processes can contact more than one synaptic element simultaneously (Wake, Moorhouse et al. 2009; Tremblay, Lowery et al. 2010). As microglia play a role in regulating activity-triggered synaptic plasticity (Rogers, Morganti et al. 2011; Koeglsperger, Li et al. 2013; Schafer, Lehrman et al. 2013), engulfing cells undergoing apoptosis (Peri and Nusslein-Volhard 2008; Sierra, Abiega et al. 2013) and remodeling neural circuits (Schafer, Lehrman et al. 2012; Squarzoni, Thion et al. 2015), it is plausible and further shown that they play an important role in learning and memory (Rogers, Morganti et al. 2011; Tremblay and Majewska 2011; Parkhurst, Yang et al. 2013).
Although microglia in the healthy brain are typically considered to be resting and quiescent, they possess a ramified morphology with highly dynamic fine processes, continuously undergoing cycles of de novo formation and withdrawal on a time scale of minutes (Davalos, Grutzendler et al. 2005; Nimmerjahn, Kirchhoff et al. 2005; Wake, Moorhouse et al. 2009; Tremblay, Lowery et al. 2010). The dynamics of microglial process motility in the healthy brain is not a seemingly random process; rather, it was shown that the motility of the processes of resting microglia is affected by glutamatergic and GABAergic neurotransmission (Fontainhas, Wang et al. 2011), and that the frequency and duration of microglia–neuronal interactions can be influenced by local neuronal activity (Wake, Moorhouse et al. 2009; Tremblay, Lowery et al. 2010; Li, Du et al. 2012). Upon traumatic brain injury, ATP is released from damaged neural tissue and surrounding astrocytes inducing rapid microglial response towards the injury site, including chemotaxis of microglial cell bodies and processes (Davalos, Grutzendler et al. 2005; Nimmerjahn, Kirchhoff et al. 2005). ATP together with glutamate mediates rapidly propagating Ca2+ waves that provide positional information via ATP to P2YR12-expressing microglia. These Ca2+ waves define which microglia are targeted to neuronal injuries (Sieger, Moritz et al. 2012). Microglia activated simply by nerve injury involving release of ATP may then be involved in regenerative responses and phagocytotic clearance of cell debris (Streit 2002; Streit 2005).
Equipped with a rich repertoire of sensing receptors (Kettenmann, Hanisch et al. 2011), ramified surveilling microglia respond to a variety of triggers, ultimately facilitating neuronal remodeling and homeostasis. It can be hypothesized that the local environment shapes the molecular signature of these microglia, which is then translated into spatial and temporal mechanisms of surveillance morphology, contact with the target and function.
From the adult functioning brain to aging and disease
Although they maintain a quiescent phenotype in the intact brain, microglia can be activated by pattern recognition receptors (PRRs) that can sense and respond to damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). Such PRRs include CD36, CD14, Toll like receptors (TLRs), scavenger receptors (SRs), purinergic receptors, triggering receptor expressed by myeloid cells 2 (TREM2) and CD33 (Ransohoff and Perry 2009). The activation process is characterized by morphological changes and upregulation of a spectrum of proinflammatory and anti-inflammatory cytokines (Heneka and O’Banion 2007; Ransohoff and Perry 2009; Perry and Holmes 2014; Gomez-Nicola and Perry 2015). Inflammatory reactions result in thicker, less branched and motile microglial processes, which, overall, exhibit a reduced coverage area (Cunningham 2013). While aging and/or neurodegeneration may cause microglia priming characterized by morphological changes and a very mild inflammatory response, peripheral inflammation can shift this priming into a more robust microglial inflammatory response with neurotoxic characteristics (Cunningham 2013; Perry and Holmes 2014; Gomez-Nicola and Perry 2015).
Numerous studies in mice and humans have shown that, during aging, microglia acquire an activated or primed phenotype characterized by process shortening and thickening (DiPatre and Gelman 1997; Sierra, Gottfried-Blackmore et al. 2007). These aging-related morphological changes, from a typical resting state to an activated phenotype, are associated with increased production of proinflammatory mediators, such as TNF-α, IL-1β, and IL-6 (Perry, Newman et al. 2003; Lucin and Wyss-Coray 2009; Baron, Babcock et al. 2014).
Microglia in the aging CNS have been described also as dystrophic with aberrant morphological features, including de-ramification, shortening, and process twisting and fragmentation (Streit 2004; Streit, Miller et al. 2008; Baron, Babcock et al. 2014; Streit, Xue et al. 2014). It should be noted, however, that microglial senescence (dystrophy) as it appears in aging human brain differs from the aged rodent brain and is much more robust (Smith and Dragunow 2014; Streit, Xue et al. 2014). It was suggested that in humans who live much longer than rodents and are exposed to diverse environmental effects, the rates at which microglial dystrophy occurs are much higher, causing a decline in microglial neuroprotection and cognitive function (Streit, Xue et al. 2014).
Morphometric analyses of individual microglia, based on three-dimensional reconstructions of the cells or on live-imaging analyses, quantitatively defined the alterations in the fine structure of microglia processes during aging (Damani, Zhao et al. 2011; Baron, Babcock et al. 2014; Hefendehl, Neher et al. 2014). Quantitative analysis of microglial processes revealed fewer bifurcations and branches, and reduced total branch length, in old mice as compared with younger ones. Moreover, microglial coverage volume was found to be reduced by more than 50% in old mice, as compared with young mice (Baron, Babcock et al. 2014).
Similar to the changes they undergo during aging, the complexity and motility of microglia processes also appear to be compromised under neurodegenerative conditions, including Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS), presumably resulting in reduced surveillance capacity (Damani, Zhao et al. 2011; Dibaj, Steffens et al. 2011; Krabbe, Halle et al. 2013; Baron, Babcock et al. 2014; Hefendehl, Neher et al. 2014).
In AD mouse models, microglia have been shown to be more abundant at sites of Aβ plaques, with an altered morphology reminiscent of cell activation (Mucke, Masliah et al. 2000; Hickman, Allison et al. 2008; Eisenberg and Jucker 2012). In such models, microglia are clustered around Aβ plaques, leaving the surrounding tissue covered by fewer processes than usually occurs in age-matched wild-type (WT) mice (Baron, Babcock et al. 2014). Detailed analyses showed that microglia from adult amyloid precursor protein (APP)-transgenic (Tg) mice exhibit curved and twisted processes and a significant reduction in the number of bifurcations and branches in the total microglial process length. Furthermore, coverage volume by individual microglia is significantly reduced in adult APP-Tg mice by almost 50%, as compared with age-matched WT mice (Baron, Babcock et al. 2014). Co-expression of activation markers and CD39 (also known as ectonucleoside triphosphate diphosphohydrolase 1), which plays a key role in microglial migration, process formation toward ATP and phagocytosis (Davalos, Grutzendler et al. 2005; Farber, Markworth et al. 2008; Sieger, Moritz et al. 2012; Bulavina, Szulzewsky et al. 2013) was evaluated in APP/PS1 Tg mice. Overall, three layers of cells surround the plaque: first, a layer of Iba-1+/CD39high amoeboid cells, which are found in close proximity to the plaque and may resemble the activated form of microglia and/or some infiltrating monocytes; second, a layer of Iba-1+/CD39high cells that have an enlarged cytoplasm and a decreased morphological complexity (as compared with cells found in plaque-free areas), which may resemble primed cells; and third, a layer of Iba-1+/CD39+ cells, with a marked population of cells expressing low levels of CD39 and exhibiting decreased morphological complexity, as compared with cells found in plaque-free areas (Baron, Babcock et al. 2014). Microglia stratified in all three layers around Aβ plaques have a morphology and process characteristics that are distinct from those of ramified resting microglia in the healthy CNS. Interestingly, in humans, microglia appear as either ramified cells in diffuse (early) plaques or as dystrophic cells in neuritic (late) plaques, suggesting that aggregated Aβ in human amyloid plaques may promote microglial degeneration (Streit, Xue et al. 2014).
Overall, accumulating data show that, whereas microglia are tightly packed and cover the tissue uniformly in younger mice, their coverage is impaired in older mice and, even more severely so, in APP-Tg mice, in which tissue space is left devoid of microglia processes. Considering the dynamic nature of the microglial processes, such a robust loss of branches during aging and disease may significantly impair the overall sensing capacity of microglia.
It is well known that the levels of proinflammatory cytokines of the innate immune system increase in the CNS during aging (Perry, Newman et al. 2003; Lucin and Wyss-Coray 2009; Baron, Babcock et al. 2014). This suggests that, during the process of aging, microglia exhibit a proinflammatory profile that, plausibly, underlies their reduced morphological complexity.
It has been shown that the number of fine microglia processes is significantly reduced in older mice, and that overexpression of the human mutated APP and the deposition of plaques in the brain significantly accelerate this reduction (Baron, Babcock et al. 2014). In addition, in an AD mouse model, a significant reduction in the number of microglial processes surrounding Aβ plaques was observed (Baron, Babcock et al. 2014). Microglial accumulation near Aβ plaques may thus not only shift the molecular and cellular milieu to one that can enhance neurotoxicity (Varvel, Grathwohl et al. 2012; Heneka, Kummer et al. 2013), but it also causes a progressive decrease in microglial process complexity, which may impair the clearance of Aβ oligomers and modulate the synaptic network or neuronal repair processes.
A key emerging question is whether the primed or dystrophic microglia play a role in facilitating aging or age-related neurodegenerative diseases. In aging, it has been suggested that numerous microglia processes protrude into the synaptic cleft, inducing synaptic stripping (Tremblay, Zettel et al. 2012). In addition, thickening of the microglia cell body, increased granulation, impairment of remodeling, and retraction of microglia processes have been described in various brain areas (Sierra, Gottfried-Blackmore et al. 2007; Damani, Zhao et al. 2011; Tremblay, Zettel et al. 2012; Kettenmann, Kirchhoff et al. 2013). Do such impaired morphologies and functions of microglia compromise their reaction to neuronal abnormalities? Does the impairment in synaptic plasticity, caused by microglia, accelerate the cognitive decline associated with aging? Further high-resolution morphological analyses of microglia in the adult brain, and of the changes that they undergo during aging and disease, may reveal key aspects of neurodegeneration and cognitive impairment.
Conclusion
What can we learn from microglia morphology? Activation of human macrophages by various signals had led to the acquisition of 299 macrophage transcriptomes. Analysis of this dataset revealed a spectrum of macrophage activation states, which extends the current M1 versus M2-polarization model. Network analyses have identified central transcriptional regulators associated with many types of macrophage activation, complemented by regulators related to stimulus-specific programs (Xue, Schmidt et al. 2014). Microglia differ decisively from peripheral macrophages, and their activation likely does not follow these precise pathways; nevertheless, it is instructive to consider how the underlying concept of macrophage heterogeneity might apply to microglial responses. Stimulated microglia, like macrophages in other tissues, are not simply beneficial or deleterious; rather, they demonstrate a highly plastic response, modified by the nature of the stimulus and by the molecular repertoire that it affects (Gordon 2003; Ransohoff and Perry 2009). The combination of morphological criteria and molecular profiles can be useful to describe the activity of microglia in response to diverse conditions and stimuli. The functional implications of the different morphological states and dynamic changes are still not fully understood; our challenge will be to utilize state-of-the-art morphological analysis tools, in both zebrafish and mouse models, to define phenotypically heterogeneous microglia responding to distinct challenges. Such investigations may lead to further insights into neurodegeneration, aging, and cognitive decline.
Microglial ontogeny and induced pluripotent stem cell-derived microglia
Donovan Low, Kazuyuki Takata and Florent Ginhoux
Introduction
Microglia, the resident macrophages of the central nervous system (CNS), play important roles in both CNS homeostasis and inflammation. Dysregulated microglial activity is involved in the pathogenesis of neurodegenerative disorders and neuroinflammatory diseases, rendering modulation of microglial responses an attractive therapeutic target. However, rational design of novel interventions focusing on the microglial compartment must be underpinned by a thorough understanding of the origin and developmental pathways of this unique and intriguing cell type, which until now has been hindered by the difficulties in isolating microglia or generating them in vitro for study. Here, we describe the current knowledge of microglial ontogeny and its implication for the generation of microglia in vitro, with a focus on the potential advances of utilizing induced pluripotent stem cells.
The term “microglia” was first introduced by del Rio-Hortega based on a set of morphological and functional differences (such as “migratory activity” and “phagocytosis”) which delineated a distinct population of non-neuronal, non-astrocytic cells of mesodermal origin within the central nervous system (CNS). While the existence of the microglial population was undeniable, del Rio-Hortega’s claim of their mesodermal origin was highly debated as it had long been believed that microglia shared the neuro-ectodermal origin of neurons and other glial cells (Ginhoux, Lim et al. 2013; Ginhoux and Prinz 2015). The subsequent discovery that microglia express monocyte/macrophage antigens led to widespread acceptance of their myeloid nature and mesodermal origin, but for many years accurate definition of their precise ontogeny remained elusive. Initial studies proposed an embryonic origin of microglia, suggesting they were derived from yolk sac (YS) progenitors that seeded the brain rudiment during early fetal development. However, these reports could not exclude the possibility that other progenitors might supersede the YS contribution with time or under certain physiological conditions, leaving the questions on the composition and maintenance of the adult microglial compartment incompletely answered.
In fact, data continued to emerge that suggested a requirement for the contribution of blood-borne cells, such as monocytes, to both generate the post-natal microglial compartment, and to maintain it into adulthood (Ginhoux, Lim et al. 2013; Ginhoux and Prinz 2015). Microglia are essential for the immune protection of the brain against foreign pathogens (Perry and Gordon 1988) as well as for brain development and homeostasis. Microglia are involved in the proper development of the brain such as synaptic pruning (Paolicelli, Bolasco et al. 2011), interneuron positioning (Squarzoni, Oller et al. 2014), and in the regulation of neuronal activity (Li, Du et al. 2012). By clearing debris from dying neurons in steady-state and upon brain injury, microglia may also be essential to avoid plaque formation and likely play a critical role in Alzheimer’s disease (McGeer, Schulzer et al. 1996) and Parkinson’s disease (Doorn, Lucassen et al. 2012). Altogether, it is now clear that microglia serve essential functions required for tissue homeostasis far beyond their sole role of immune sentinels. Given the central role of microglia in maintaining optimal CNS function and in mediating the pathology of neurodegeneration and neuroinflammation, recent years have seen intense interest in how the microglial population might be targeted therapeutically. With advances in stem cell research, for the first time it has become feasible to consider how we might use our knowledge of microglial ontogeny and function to generate microglia-like cells in vitro for therapeutic use and study (Figure 2). Here we review the latest advances in our knowledge of the origins of the microglial population and how this might be harnessed for clinical use.
Figure 2. Microglial ontogeny and implications for the use of pluripotent stem cell-derived macrophages for therapy.
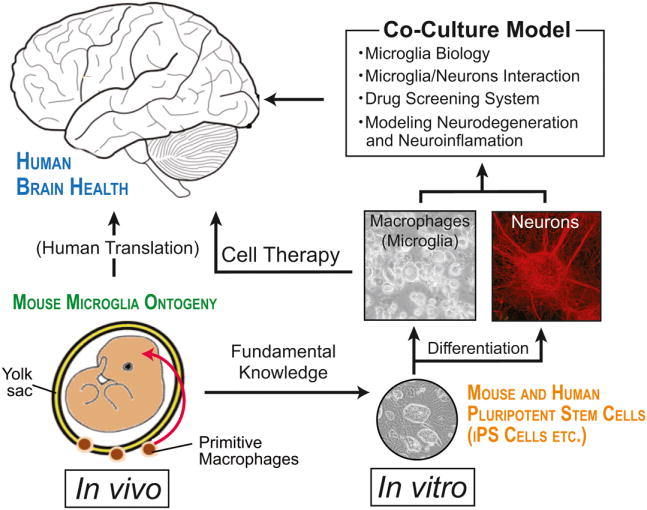
A precise understanding of microglial ontogeny in vivo may provide adequate methodologies for in vitro microglial differentiation from induced pluripotent stem cells. Findings in such in vitro models using stem cells will complement the knowledge gained in vivo. The choice of an adequate cell source for microglia will further contribute to a better understanding of microglial biology in health and disease, and will facilitate the refinement of accurate developmental and pathophysiological study models as well as drug screening systems for new microglia-targeted therapies.
Ontogeny of microglia: A primitive ancestry
Initial studies broadly described the emergence of microglia early in the developing brain at a point preceding the initiation of fetal liver and bone marrow (BM) hematopoiesis, and concomitant with the appearance of YS primitive hematopoiesis (Takahashi and Naito 1993). Primitive hematopoiesis occurs in the blood islands of the extra-embryonic YS at embryonic day 7 (E7) giving rise to ‘early’ erythro-myeloid precursors (Parkhurst, Yang et al. 2013) mainly forming primitive nucleated erythrocytes and primitive macrophages (Orkin and Zon 2008; Hoeffel, Chen et al. 2015). However the truly unique ontogeny of microglia has only recently been established following the development of new fate-mapping mouse models that allow accurate tracing of YS progenitors (Ginhoux, Greter et al. 2010; Schulz, Gomez Perdiguero et al. 2012; Sheng, Ruedl et al. 2015). Ginhoux et al. used a lineage-tracing model to label YS progenitors, including YS macrophages, based on their expression of runt-related transcription factor 1 (Runx1), which is critical for the emergence of hematopoietic stem cells. During early embryogenesis, Runx1 expression is restricted to cells of the extra-embryonic YS, allowing tracing of YS progenitors after activation of a reporter gene by tamoxifen treatment of the pregnant female within a specific timeframe (Samokhvalov, Samokhvalova et al. 2007). Using this model, the authors showed that the majority of both embryonic and adult microglia arose from YS Runx1+ progenitors (Ginhoux, Greter et al. 2010); furthermore there was minimal contribution from either fetal or adult monocytes to microglia in the brain (Ginhoux, Greter et al. 2010; Hoeffel, Chen et al. 2015), while other tissue macrophages did arise from fetal monocytes (Ginhoux, Greter et al. 2010; Hoeffel, Wang et al. 2012; Hoeffel, Chen et al. 2015; Sheng, Ruedl et al. 2015). The absence of fetal monocyte contribution to the microglial pool could result from a lack of intrinsic potential or a lack of access to the developing brain (Hoeffel, Chen et al. 2015) due to the nascent blood brain barrier as early as E13.5 (Daneman, Zhou et al. 2010).
The YS progenitor population has since been dissected further, with Kierdorf et al. first identifying the precursors of the primitive YS macrophages/microglia: at E8, c-kit+ EMPs in the YS differentiate into CD45+c-kitloCX3CR1- early precursors that mature and migrate into the developing brain as CD45+c-kit-CX3CR1+ cells (Kierdorf, Erny et al. 2013). More recently, Hoeffel et al. extended the characterization of YS EMPs using an in utero macrophage depletion system combined with genetic tagging of YS and fetal liver hematopoietic cells. Using the same Runx1 fate mapping model, the authors compared the cell populations that arose from YS progenitors labeled by tamoxifen treatment at either E7.5 or E8.5, revealing that two waves of EMPs arise sequentially in the YS: an early wave at E7.5 which differentiates locally into the YS macrophages that are the primary source of microglia, and a late wave of E8.5 EMPs that can either differentiate locally into YS macrophages, or migrate to seed the fetal liver following the establishment of blood circulation from E9.0 (Hoeffel, Chen et al. 2015). However, this study clearly showed that most of microglia arise from YS macrophages deriving from the early wave of EMPs.
Further experiments in both newborn transplant models (where host CD45.2 mice were sub-lethally irradiated and subsequently reconstituted with hematopoietic cells from congenic CD45.1 mice) and parabiotic mice (where two mice are surgically attached to allow shared blood circulation while retaining separate organs) indicated that postnatal circulating hematopoietic progenitors contribute only negligibly to the adult microglial population (Ginhoux, Greter et al. 2010; Ajami, Bennett et al. 2011; Hoeffel, Chen et al. 2015). Furthermore, recent ablation experiments in adult mice showed that the cells replacing microglia after depletion are derived from CNS-intrinsic precursors (Waisman, Ginhoux et al. 2015). Of note, following depletion of microglia either in the embryo or in the adult, the microglial compartment is replenished rapidly, within 7 days (Squarzoni, Oller et al. 2014; Waisman, Ginhoux et al. 2015). The precursors of these new repopulating microglia are not yet fully characterized and it remains unknown whether they possess the same characteristics as their YS embryonic counterparts in terms of their roles in CNS development, homeostasis and neuropathologies. Altogether these data show that adult microglia unequivocally arise from YS macrophages that seed the brain at E9.5, where these cells establish the nascent microglial population, proliferate in situ and are maintained throughout adulthood. Thus the majority of the postnatal microglial compartment exists independent of circulating hematopoietic cells and can be self-renewed by local radio-resistant prenatally-seeded progenitors.
The unique ontogeny of microglia in the steady state has been established, but the situation is not as clear during certain situations, such as inflammation, where hematopoietic cells from the BM can infiltrate the brain. Several studies have reported BM-derived cells migrating into the CNS following BM transplantation and differentiating into parenchymal microglia-like cells (Ginhoux and Jung 2014; Greter, Lelios et al. 2015). Circulating myeloid cells such as monocytes can only enter the CNS if the blood-brain barrier is damaged, such as when the CNS is inflamed or upon lethal irradiation (Ajami, Bennett et al. 2007; Mildner, Mack et al. 2009). Under such conditions these cells recapitulate several of the phenotypic and functional characteristics of the pre-existing adult microglial population, giving rise to ‘microglia-like cells’ (Ajami, Bennett et al. 2011; Takahashi, Kakuda et al. 2015), which may or may not fulfill the same role as their embryonic counterparts in terms of CNS homeostasis and neuroinflammation. Interestingly, these microglia-like cells did not persist once the inflammation was resolved in a model of experimental autoimmune encephalomyelitis (Ajami, Bennett et al. 2011). Furthermore, it was also shown that monocyte-derived microglia-like cells accumulate to a much lesser extend compared to resident microglia close to amyloid plaques in the brains of Alzheimer’s disease mouse models (Prokop, Miller et al. 2015; Varvel, Grathwohl et al. 2015), suggesting that they play a different role.
Pluripotent stem cell-derived macrophages as an adequate source of microglia
Efforts to elucidate the biology of microglia have been limited by the lack of access to sufficient numbers of cells for comprehensive in vitro studies. Isolation of primary rodent microglia is generally achieved either by cell sorting or stepwise cell culture from newborn rodent brains (Ju, Zeng et al. 2015), both of which are time-consuming and often yield few cells. While of some limited use in rodents, this approach is entirely unfeasible for obtaining human microglia. Although there has been some success producing microglia-like cells from BM stem cells and circulating monocytes, as these cell populations do not share the embryonic origin of the vast majority of microglia in the homeostatic brain, they are likely to be poor models of adult microglia.
This inability to access or generate sufficient numbers of cells of true microglial origin has prevented comprehensive or large scale in vitro studies. However, a potential source of microglia might be embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs), which are reprogrammed terminally-differentiated somatic cells that resemble ESCs (Takahashi and Yamanaka 2006; Takahashi, Tanabe et al. 2007). ESCs and iPSCs have unlimited expansion potential and the ability to differentiate toward any cell type of the three germ layers (De Los Angeles, Ferrari et al. 2015). In addition, iPSCs do not suffer the limitation of having a fertilized egg as their source; instead they can be derived from a wide range of different cell types even from cancer cells and could be generated in a patient-specific manner for future therapies, thereby circumventing two of the major ethical and barrier/immune issues associated with the use of ESCs. For these reasons the use of iPSCs has opened-up new avenues for stem cell research, developmental biology, cancer research, various types of disease modeling, and the development of patient-specific disease interventions (Ohnuki and Takahashi 2015); ESCs and iPSCs are already used for research into embryonic development, as well as for cellular transplantation therapies for neurodegenerative diseases (Park, Arora et al. 2008; Kiskinis and Eggan 2010; Ben-David, Kopper et al. 2012). Moreover, the application of autologous iPSCs in the field of regenerative medicine is anticipated to provide much-needed improvements in our ability to treat/prevent organ rejection in recipient patients after engraftment, and furthermore the concept of haplo-banking with human leukocyte antigen homozygous of iPSCs has been established (Nakajima, Tokunaga et al. 2007; Nakatsuji, Nakajima et al. 2008; Zimmermann, Preynat-Seauve et al. 2012; de Rham and Villard 2014).
The differentiation pathways of hematopoietic lineages have also been investigated using iPSCs (Ackermann, Liebhaber et al. 2015). Recently, selective induction of primitive or definitive hematopoiesis has been shown to rely on Wnt signaling, providing simple selective differentiation strategies for the generation of primitive or definitive hematopoietic progenitors by Wnt-β-catenin manipulation (Kennedy, Awong et al. 2012; Sturgeon, Ditadi et al. 2014). In addition, large-scale differentiation of macrophages from human iPSCs has been reported (Lachmann, Ackermann et al. 2015). We have also developed our own “in house” xeno/serum-free differentiation protocol for the generation of immature macrophages, similar to those expected to arise from primitive hematopoiesis, as a microglial source. In this protocol, mouse iPSCs start to differentiate into primitive macrophages, which express markers including F4/80, CD11b, and CX3CR1, and exhibit phagocytic function after 8-12 days of culture. Crucially, as these cells share the same embryonic origin as microglia, they may well represent the first adequate and appropriate in vitro source of microglia for study and therapeutic investigation. We have also been establishing methods for the co-culture of iPSC-derived primitive macrophages with neurons derived from the same mouse iPSC batch, which we hope will provide insights into the mechanism of primitive macrophage maturation into microglia, and the interactions of microglia and neurons in both physiological and pathological neuroinflammatory conditions (Figure 2). Furthermore, the co-culture model has the potential to serve as an in vitro drug screening system for the development of novel therapies against brain diseases. Taken together, iPSC-derived primitive macrophages could be a useful microglial source for biological, pathophysiological and therapeutic studies.
Conclusion
Our composite knowledge of microglial origin at this time supports the idea that immature macrophages arise from primitive hematopoiesis in the YS before birth. This knowledge may be further supplemented in the near future with the advent of iPSC-based techniques that might enable us to establish an in vitro system able to generate microglia of appropriate ontogeny and in sufficient quantities for meaningful study. This will allow us to gain a better understanding of microglial biology in health and disease, and will facilitate the refinement of accurate developmental and pathophysiological study models as well as drug screening systems for new microglia-targeted therapies. Extending this to the human setting, we may soon have the opportunity to generate patient specific iPSC-derived microglia and use them for disease modeling, drug screening and even cell therapy. More focused on brain macrophages, plenty of questions remain: for example, how do resident microglia and meningeal and perivascular macrophages in the brain differ in function and cooperate to maintain a healthy CNS? This gap in knowledge also extends to macrophages arising from primitive and definitive hematopoiesis. To address these issues we will need to employ an integrated approach, incorporating in vivo studies of ontogeny as well as in vitro iPSC-based culture models; only then can the potential be realized for the development of novel drugs and/or cell replacement therapies capable of targeting microglia implicated in the pathology of brain diseases.
The Cytokine Paradox in Alzheimer’s Disease Pathogenesis
Terrence Town and M. Kerry O’Banion
Introduction
The recognition of activated glia together with inflammatory cytokines and other mediators in the brains of individuals with Alzheimer’s disease (AD) and evidence that non-steroidal anti-inflammatory drugs might protect against AD led to an early view that neuroinflammation contributed to AD pathogenesis (Akiyama, Barger et al. 2000). Over time this view has been modified to encompass a growing body of experimental evidence that neuroinflammation has both beneficial as well as detrimental roles in AD that vary with context. For example, experiments using pro-inflammatory stimuli in AD mouse models have demonstrated reduction of amyloid deposits, raising the notion that, at least under certain conditions, neuroinflammation can play a protective role (Shaftel, Griffin et al. 2008). Such stimuli include lipopolysaccharide (DiCarlo, Wilcock et al. 2001), interleukin (IL)-1β (Shaftel, Kyrkanides et al. 2007), interferon γ (Chakrabarty, Ceballos-Diaz et al. 2010), IL-6 (Chakrabarty, Jansen-West et al. 2010), and tumor necrosis factor α (Chakrabarty, Herring et al. 2011). In all cases, plaque reduction was associated with increased gliosis, suggesting the possibility that these pro-inflammatory stimuli provoked increased phagocytosis of Aβ. Indeed, in a distinct model of microglial activation caused by deletion of CX3CR1, plaque reduction was associated with increased Aβ phagocytosis and evidence of increased IL-1β expression (Lee, Varvel et al. 2010).
Interleukin-1
Using models of virally delivered IL-1β dependent reduction in local Aβ deposition, O’Banion and coworkers have investigated possible mechanisms underlying the apparent benefits of pro-inflammatory cytokine expression on AD pathology. In initial studies, overexpression of human IL-1β for one month using a viral cre-activated transgene in APP/PS-1 mice led to local reduction (e.g. one hippocampus vs. the contralateral side) in Aβ plaque load, an effect that did not appear to involve changes in APP production or Aβ processing (Shaftel, Kyrkanides et al. 2007). Further work demonstrated similar changes regardless of animal age or duration of IL-1β overexpression (1-3 months) in both the APP/PS1 and triple transgenic models (Matousek, Ghosh et al. 2012; Ghosh, Wu et al. 2013). In all cases, IL-1β overexpression was associated with increased density of Iba-1 positive cells surrounding amyloid plaques (Shaftel, Kyrkanides et al. 2007; Ghosh, Wu et al. 2013) as well as dramatic evidence of peripheral cell infiltration and a compromised blood-brain barrier (Shaftel, Carlson et al. 2007). To test the possibility that infiltrating peripheral macrophages might be responsible for the observed effects, Rivera-Escalera et al. carried out a series of studies in APP/PS-1 mice using AAV2-hIL-1β as an alternate means to drive sustained IL-1β expression (Rivera-Escalera, Matousek et al. 2014). These mice also showed reduced plaque deposition, and while bone marrow chimera experiments clearly demonstrated infiltrating Iba-1 positive cells associated with plaques, experiments with CCR2 deficient mice still showed IL-1-dependent plaque clearance despite almost complete inhibition of monocyte/macrophage infiltration (Rivera-Escalera, Matousek et al. 2014). More recently, Cherry et al. reported that sustained IL-1β overexpression induced a population of arginase-1 positive cells, derived from microglia, that appeared to have increased capacity for Aβ phagocytosis (Cherry, Olschowka et al. 2015). Sustained IL-1β overexpression also led to increased IL-4 levels in the brain, some of which appeared to derive from infiltrating CD3 positive T cells.
Indeed, when APP/PS-1 mice overexpressing IL-1β were treated with a neutralizing antibody to IL-4Rα, there was a reduction in arginase-1 positive cells that correlated with abrogation of the IL-1β dependent benefits on plaque clearance (Cherry, Olschowka et al. 2015). Moreover, IL-4 injection alone led to rapid induction of arginase-1 positive cells associated with plaque clearance in APP/PS-1 mice, an observation consistent with previous observations from some (Kiyota, Okuyama et al. 2010; Kawahara, Suenobu et al. 2012), but not all (Chakrabarty, Tianbai et al. 2012) investigators. These observations suggest that homeostatic mechanisms reacting to increased neuroinflammation in the setting of focal IL-1β overexpression result in alternative microglial activation, a process that might be harnessed for benefit in AD therapy. Interestingly, evidence of alternate microglial activation was also observed in a different model where deletion of the NLRP3 inflammasome, and subsequent failure to generate and release mature IL-1β, reduced amyloid plaque deposition in APP/PS1 mice (Heneka, Kummer et al. 2013). These findings highlight the paradoxical effects of cytokines on AD pathology: overexpression of IL-1β and absence of IL-1β both generate a similar phenotype, at least with regard to amyloid deposition. Whether the paradox can be explained by modulation of a single mechanism (e.g. microglial phenotype) or multiple mechanisms requires further investigation.
Understanding mechanisms that underlie the benefits of pro-inflammatory cytokine expression on Aβ deposition is an especially important undertaking in consideration of effects observed on tau pathology. For example, while IL-1β overexpression in triple-transgenic mice reduced Aβ plaque density, it exacerbated tau phosphorylation (Ghosh, Wu et al. 2013). Similar findings have been reported for lipopolysaccharide (Kitazawa, Oddo et al. 2005; Lee, Rizer et al. 2010) and for loss of CX3CR1 (Bhaskar, Konerth et al. 2010; Maphis, Xu et al. 2015), processes which both appear to depend on IL-1 signaling. In addition, bilateral overexpression of IL-1β in the murine hippocampus led to behavioral deficits in two hippocampal-dependent learning tasks (contextual fear conditioning and Morris water maze) (Moore, Wu et al. 2009; Hein, Stasko et al. 2010). Taken together, these results raise clear concerns about the possible application of enhancing inflammation to intervene with AD, an issue illustrated by the negative outcomes reported in initial trials of active Aβ immunization (Robinson, Bishop et al. 2004).
Rebalancing neuroinflammation to homeostasis by blocking immunosuppressive cytokines
Transforming growth factor-β
While much attention has been directed toward understanding the role(s) of pro-inflammatory cytokines in AD pathogenesis, considerably less effort has been placed on factors that limit the extent and duration of inflammatory processes. As a key immunoregulatory cytokine, transforming growth factor-β (TGF-β) ensures that inflammatory responses are carried out in an acute, controlled fashion without becoming chronic and inducing excessive tissue damage (Li and Flavell 2008). Prolonged, low-level activation of brain inflammatory processes occurs in AD and is likely pathogenic (Wyss-Coray, Masliah et al. 1997; Selkoe 2001); raising the paradoxical question of whether immunosuppressive pathways are overly activated and contribute to AD evolution. The first supporting evidence came when a transgenic mouse line overexpressing TGF-β1 (Wyss-Coray, Feng et al. 1995) was crossed with the PDAPP mouse model of cerebral amyloidosis (Games, Adams et al. 1995). In this setting, vascular Aβ deposits were markedly accelerated; suggesting an amyloidogenic role for TGF-β1 in vivo (Wyss-Coray, Masliah et al. 1997). Despite increases in vascular Aβ deposits, these bigenic animals had less parenchymal Aβ burden–suggesting opposing effects of TGF-β1 on vascular vs. parenchymal Aβ deposition (Wyss-Coray, Lin et al. 2001). These results were supported by findings in AD patients’ brains from the laboratories of Tony Wyss-Coray and Lennart Mücke. Those investigators found that TGF-β1 mRNA levels were ~3-fold increased in AD patient frontal cortex vs. non-demented controls. Further, positive correlation was noted between TGF-β1 mRNA levels and vascular Aβ deposits (Wyss-Coray, Masliah et al. 1997) while an inverse association was found between TGF-β1 mRNA and parenchymal Aβ deposits (Wyss-Coray, Lin et al. 2001). From an innate immune perspective, increased TGF-β1 abundance in AD frontal cortex may pathologically inhibit ‘beneficial neuroinflammation’ in the AD brain. Taken together, these findings raise the interesting and important question of the cellular mechanistic underpinnings of the TGF-β/AD pathology relationship.
TGF-β signaling often functions to suppress inflammation, and the initial report from the Town laboratory showed that inhibiting TGF-β signaling in peripheral macrophages led to brain infiltration of these cells and resolution of cerebral amyloidosis in concert with increased brain interleukin-10 (IL-10) levels (Town, Laouar et al. 2008). Specifically, TGF-β-Smad 2/3 signaling was genetically interrupted in peripheral macrophages (as opposed to brain-resident microglia) by engineering a CD11c promoter-driven dominant-negative TGF-β type II receptor transgene in C57BL/6 mice (CD11c-DNR mice) (Laouar, Town et al. 2008). CD11c-DNR mice were subsequently crossed with the Tg2576 AD mouse model; behavioral impairment and AD-like pathology were evaluated (Town, Laouar et al. 2008). Notably, bigenic animals exhibited partial amelioration of cognitive impairment and presented with reduced astrocytosis and concomitant ~90% attenuation of brain parenchymal Aβ and reduced cerebral amyloid angiopathy (Town, Laouar et al. 2008). These therapeutic effects were associated with increased accumulation of Aβ-containing peripheral mononuclear phagocytes in and around cerebral vessels and Aβ plaques. Further, Aβ could be localized within the cytoplasm of these cells, suggesting a productive Aβ phagocytosis/clearance response. Based on these findings, it appears that releasing TGF-β immune suppression on peripheral monocytes returns these cells to homeostasis; allowing them access into the brain while simultaneously maximizing their Aβ phagocytic potential. As ex vivo validation of the latter, there was ~3-fold increased Aβ phagocytosis by CD11c-DNR vs. wild-type macrophages (Town, Laouar et al. 2008). These results suggest that inhibition of TGF-β-Smad 2/3 signaling promotes peripheral mononuclear phagocyte recruitment to brains of cerebral amyloid-depositing transgenic mice and reduces Aβ burden via phagocytic clearance. At face value, our data and those of Wyss-Coray and Mücke might seem at odds with each other – blocking TGF-β signaling and overexpressing brain TGF-β1 both reduce parenchymal amyloid plaques. However, it is important to note that the act of genetically or pharmacologically inhibiting the TGF-β-Smad 2/3 pathway shifts downstream signaling to activate TGF-β-Smad 1/5/8 signaling in mononuclear phagocytes; thereby mimicking effects of TGF-β itself (Town, Laouar et al. 2008). Specifically, it seems that the act of blocking Smad2/3 signaling results in promotion of Smad1/5/8 signaling, which is associated with increased macrophage Aβ phagocytosis.
Interleukin-10
Often in concert with TGF-β, IL-10 suppresses overly exuberant inflammatory responses and inhibits effector function of myeloid cells by blocking pro-inflammatory cytokine pathways (Banchereau, Pascual et al. 2012). Several lines of evidence implicate aberrant IL-10 signaling in AD. Notably, elevated IL-10 signaling was observed in reactive glia neighboring β-amyloid plaques in aged Tg2576 mice (Apelt and Schliebs 2001). Additionally, a functional polymorphism within the IL10 gene (-1082 A>G) has been linked to increased risk for AD in certain populations (Lio, Licastro et al. 2003; Arosio, Trabattoni et al. 2004; Vural, Degirmencioglu et al. 2009), and two recent meta-analyses of pooled studies examining the IL10-AD risk relationship reported modest statistical significance; likely due to heterogeneity between studies (Zhang 2011; Di Bona, Rizzo et al. 2012). In the Di Bona and coworkers meta-analysis, it is interesting that the -1082 A risk relationship with AD was more evident in the oldest AD patients. While the IL10 -1082 A allele reportedly lowers IL-10 protein abundance in healthy control plasma (Ma 2005), due to unavailability of data from AD patient plasma it is not possible to draw a definitive conclusion. Nonetheless, these results could be interepreted as a ‘double edged sword,’ where blocking inflammation is beneficial early on and prior to conversion from cognitively healthy to AD-type dementia; whereas older individuals ‘on the cusp’ of dementia convert more quickly if immunity/inflammation is suppressed. This emerging concept has gained support from the first and only AD primary prevention trial for non-steroidal anti-inflammatory drugs (Breitner, Baker et al. 2011).
Interestingly, when CD11c-DNR mice were crossed with Tg2576 animals, we noted that brain IL-10 levels were elevated. There are several possible explanations for this result; cerebral IL-10 could function as 1) a compensatory response to abrogated peripheral innate immune TGF-β signaling, 2) an effector cytokine responsible for beneficial effects of TGF-β pathway blockade, or 3) simply a marker for altered innate immune phenotype.
To further explore how anti-inflammatory IL-10 signaling affects Aβ pathology, we and others have investigated the effects of IL-10 modulation in brains of APP transgenic mouse models. After finding that the IL-10 signaling pathway was elevated in AD patient brains, we interrogated the role of this cytokine in the context of AD. Specifically, the APP/PS1 mouse model of cerebral amyloidosis was crossed with a mouse deficient for Il10 (APP/PS1+Il10-/-) (Guillot-Sestier, Doty et al. 2015). Using a novel method to quantify activated Aβ phagocytic microglia by in silico 3D modeling technique (Guillot-Sestier, Doty et al. 2015), activated Aβ phagocytic microglia were observed to restrict cerebral amyloidosis in APP/PS1+Il10-/- mice (Guillot-Sestier, Doty et al. 2015). Notably, genome-wide RNA sequencing of APP/PS1+ brains showed modulation of innate immune genes that are known to promote neuroinflammation. Moreover, Il10 deficiency preserved synaptic integrity and mitigated cognitive disturbance in APP/PS1 mice, suggesting a detrimental role of IL-10 in Alzheimer pathophysiology. To probe the mechanism underlying these findings, in vitro knock-down of microglial Il10-Stat3 signaling was performed–and led to augmented Aβ phagocytosis–while addition of exogenous IL-10 had the converse effect.
In a complementary approach, Todd Golde’s group investigated the effects of adeno-associated virus (AAV2/1) delivered over-expression of IL-10 in brains of two APP transgenic mouse models. In this model, IL-10 expression resulted in increased Aβ accumulation and impaired learning and memory (Chakrabarty, Li et al. 2015). Interestingly, Il10 deficient animals had reduced apolipoprotein E (ApoE) mRNA levels by RNA sequencing (RNAseq) and quantitative real-time PCR (qPCR) (Guillot-Sestier, Doty et al. 2015), while transcriptome analyses performed by Chakrabarty et al. demonstrated enhanced IL-10 signaling with concomitantly increased ApoE expression in IL-10 expressing APP mice (Chakrabarty, Li et al. 2015). Importantly, in the Chakrabarty and colleagues report, ApoE protein was selectively increased in the plaque-associated insoluble cellular fraction, which they linked to direct interaction of ApoE with aggregated Aβ in IL-10 expressing APP transgenic mice (Chakrabarty, Li et al. 2015). In both studies, ex vivo analyses showed that IL-10 and ApoE can separately impair microglial Aβ phagocytosis. Interestingly, Il10 deficiency also partially overcame isoform-specific inhibition of human ApoE on microglial Aβ uptake (E4 > E3 > E2) (Guillot-Sestier, Doty et al. 2015). Together, these results suggest two mechanisms by which Il10 blockade enables cerebral Aβ clearance: 1) by decreasing microglial expression of ApoE, and 2) by opposing ApoE Aβ binding and therefore endorsing Aβ phagocytosis (Guillot-Sestier, Doty et al. 2015). It is important to note that our study showed that human ApoE isoforms inhibit microglial Aβ phagocytosis (E4 > E3 > E2); whereas others have recently noted beneficial effects of increasing mouse ApoE expression (Fitz, Tapias et al. 2015; Skerrett, Pellegrino et al. 2015). However, due to salient differences between human ApoE isoforms and mouse ApoE, it is not possible to compare results across species. Additionally, while both our study and that of Chakrabarty and coworkers specifically focus on ApoE in monocyte Aβ phagocytosis, this in no way precludes other function(s) of ApoE, such as its action as an Aβ molecular chaperone involved in plaque ‘seeding’ (Nilsson, Arendash et al. 2004). Collectively, these studies demonstrate a negative effect of IL-10 on Aβ proteostasis and cognition in APP mouse models, and suggest that ‘rebalancing’ innate immunity to homeostasis by blocking the IL-10 anti-inflammatory response may be therapeutically relevant for AD.
Conclusion
In contrast to prevailing views about the negative consequences of neuroinflammation, a growing body of literature illustrates potential benefits of activating innate immunity to mitigate AD pathology (Figure 3). Although it needs to be more fully explored whether these benefits extend to synaptic and cognitive changes seen in AD, the independent and complementary results reported here, which utilized divergent approaches, demonstrate the complex interplay between innate immunity and proteostasis in AD. Rather than shutting neuroinflammation off completely, rebalancing it toward a beneficial, homeostatic innate immune response may allow us to harness innate immunity in the fight against AD.
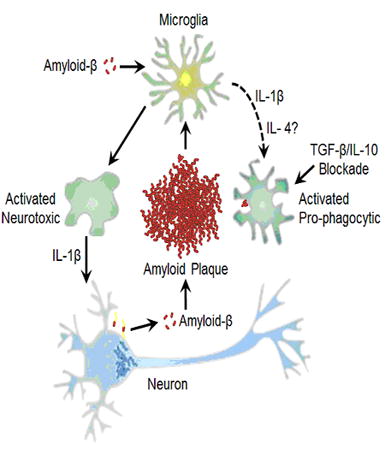
Figure 3. Paradoxical effects of cytokine signaling in amyloid-β clearance.
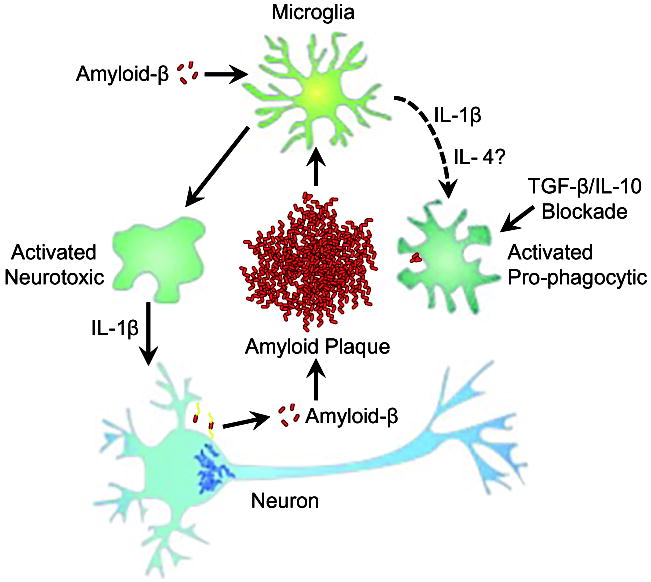
Amyloid-β induced microglial activation can be modified in the presence of anti-inflammatory cytokines (e.g. IL-4) or by genetic/pharmacologic blockade of TGFβ and/or IL-10 signaling to shift microglia from pro-inflammatory neurotoxic cells to activated pro-phagocytic cells with the capacity to reduce amyloid-β plaque load. In experimental systems, increased IL-1β expression can drive both a neurotoxic response (e.g. increased tau phosphorylation, indicated by interneuronal fibrils) as well as create an environment that supports pro-phagocytic microglial activation.
Trem2 and the role of innate immunity in neurodegenerative disease
Bruce Lamb, Marco Colonna, and Gary Landreth
Introduction
Recent studies have linked genetic variants of the triggering receptor expressed on myeloid cells 2 (TREM2) with increased disease risk, most prominently of Alzheimer’s disease. TREM2 is a critical regulator of microglia and macrophage phenotype. In the AD brain and in mouse models of AD, plaque-associated cells express high levels of TREM2. TREM2 expression increases with age and disease progression. Genetic inactivation of TREM2 results in the near absence of plaque-associated macrophages which is accompanied by alterations in plaque burden. A central question is whether these plaque-associated macrophages represent activated microglia or derive from blood borne monocytes. The expression of TREM2 and other phagocytic receptors can be induced by nuclear receptor agonists and stimulate phagocytosis. These studies have demonstrated a critical role for TREM2 in neurodegenerative disease pathogenesis.
TREM2 deficiency impacts amyloid pathology - Discrepant results on origin of myeloid cells surrounding amyloid plaques in AD models
Lamb’s work emphasized how the cells of the innate immune system respond to disease or injury-related perturbations of neuronal function. There is a substantial body of evidence demonstrating that robust and continuous communication between neurons and microglia is required for normal brain homeostasis. The Lamb lab, and others, have shown that neuronal microglial communication is mediated by cell surface ligand-receptor pairs (Kierdorf and Prinz 2013). For example, fractalkine (CX3CL1) is expressed by neurons and directly interacts with its receptor, CX3CR1 on microglia (Wolf, Yona et al. 2013). This neuron-microglia interaction results in bi-directional communication between these two cell types.
When expression of CX3CR1 is abolished in amyloidogenic murine models of AD, a reduction in the number of activated microglia and dysregulated proinflammatory cytokine expression was observed (Lee, Varvel et al. 2010). This was accompanied by diminished plaque burden, likely owing to enhanced microglial phagocytosis. CX3CR1 loss in a mouse model of tau pathology resulted in enhanced phosphorylation and aggregation of tau in neurons which was associated with behavioral impairment (Bhaskar, Konerth et al. 2010). The tau pathology was postulated to arise from elevated levels of microglial-derived IL-1β, consistent with the view that neuroinflammation is mechanistically linked to tau-related neurodegeneration (Maphis, Xu et al. 2015). These data provided direct support for the idea that the neuron-microglia interactions in the normal brain act to suppress microglial ‘activation’ and promote neuronal homeostasis. The ability of microglial dysfunction to cause neurodegeneration by perturbation of signaling through a different microglial-neuronal interaction was demonstrated in individuals with Nasu-Hakola disease (Golde, Streit et al. 2013). Nasu-Hakola disease is a progressive neurodegenerative disorder that arises from loss of function mutations in the microglial cell surface receptor TREM2 or its intracellular signaling adapter, DAP12 (aka TYROBP). Importantly, genetic variants of TREM2 have recently been shown to confer a dramatically increased risk for Alzheimer’s disease (Guerreiro, Wojtas et al. 2013; Johnson, Stewart et al. 2013; Jonsson, Stefansson et al. 2013) and other neurodegenerative diseases (reviewed in Painter, Atagi et al. 2015). The discovery of this linkage has focused attention on the critical roles of microglia /macrophages in neurodegenerative disease.
A recent study from the Colonna laboratory analyzed the impact of TREM2 deficiency and haplo-insufficiency on the microglial response to Aβ deposition in the 5XFAD mouse model of AD, which contains 5 mutations in human transgenes encoding the amyloid precursor protein (APP) and presenilin 1 (PS1) (Wang, Cella et al. 2015). Aβ deposition induced activation and accumulation of microglia around Aβ plaques in TREM2-sufficient mice; in contrast, microglia did not cluster around Aβ plaques in TREM2-deficient or haplo-insufficient mice, indicating a defective response to Aβ deposition. Moreover, transcriptional profiling of microglia demonstrated that TREM2 deficiency reduced the expression of genes associated with microglial activation, such as phagocytic receptors, costimulatory molecules, inflammatory cytokines and trophic factors. The lack of trophic factors correlated with apoptosis of TREM2 deficient microglia in vitro. These results indicate that TREM2 promotes a broad array of microglial functions in response to Aβ deposition. TREM2 deficiency resulted in increased Aβ accumulation in the hippocampus of 5XFAD mice at 8 months of age. TREM2 haploinsufficient 5XFAD mice also displayed a trend toward an increase in Aβ immunoreactivity and a significant increase in insoluble Aβ by ELISA compared to TREM2 sufficient 5XFAD mice. This finding contrasts with what was observed in 3-7 month old APP/PS1 mice hemizygous for TREM2 which exhibited no significant change in plaque burden (Ulrich, Finn et al. 2014). It will be important to determine whether these different results are due to the different AD models used or whether TREM2 might have different roles at different stages in pathology. Nonetheless, the data suggest that the TREM2-dependent activation of microglia is required to limit Aβ pathology.Wang et al. also investigated potential ligands of TREM2, which may be relevant to AD. We found that TREM2 binds anionic and zwitterionic phospholipids that may become exposed during Aβ accumulation due to neuronal and glial apoptosis, myelin degradation and formation of aggregates between Aβ and phospholipids (Wang, Cella et al. 2015). In contrast, microglia expressing the R47H variant of TREM2 associated with AD had a reduced capacity to bind phospholipids. The model emerging from this study is that TREM2 senses changes in the lipid microenvironment that result from Aβ accumulation and neuronal degeneration, which triggers signals that activate microglial capacity to limit further Aβ accumulation.
Finally, the Colonna lab addressed the origin of myeloid cells around the amyloid plaques using parabiosis experiments using TREM2-sufficient and TREM2-deficient 5XFAD mice developing Aβ accumulation. Preliminary results suggest that, in these settings, myeloid cells clustered around Aβ plaques derive from brain resident microglia rather than peripheral blood monocytes.
Recent studies reported by Jay et al. (Jay, Miller et al. 2015) found that in two mouse models of AD (APPPS1 and 5XFAD) and in humans with AD, TREM2 was highly expressed by macrophages that were associated with amyloid plaques. TREM2 expression increased progressively with age and Aβ pathology in the mouse models. Closer examination using immunohistochemistry and flow cytometry revealed that the TREM2+ cells expressed markers, which identify a subset of blood-borne monocytes. Indeed, they found an age-dependent increase in the number of cells co-expressing TREM2 and high levels of the peripheral monocyte marker CD45. These cells also expressed the canonical monocyte marker Ly6C, consistent with their systemic origin. Immunohistochemical analysis revealed that the resident microglia, identified by expression of P2YR12, did not express detectable levels of TREM2 and were not associated with plaques, whereas the TREM2+, CD45 brightly staining cells were invariably plaque-associated. These data suggest that circulating monocytes infiltrate the AD brain and migrate to plaques in the above mentioned mouse models and at the respective ages. Remarkably, plaque-associated macrophages were virtually absent in the Trem2-/- mice. Moreover, mRNA levels for CD11b, CD68, F4/80 and CD45 were dramatically reduced in Trem2-/- mice, while the microglial marker P2YR12 was unchanged. These findings provide direct evidence that TREM2 expression is required for the infiltration and/or survival of these myeloid cells and their association with deposited Aβ.
The plaque-associated macrophages in the AD brain exhibit an ‘activated’, proinflammatory phenotype, secreting cytokines and other inflammatory molecules. The Trem2-/- mice had lower transcript levels of IL-6 and IL-1β, but had elevated expression of anti-inflammatory markers Ym1 and Fizz1, reflective of the critical influence of TREM2 expressing cells on the inflammatory status in the AD brain.
The effect of TREM2 deficiency on neuroinflammation and macrophage number and distribution led to the examination of its effect on plaque burden. APP/PS1-21 (herein termed APP/PS1) mice exhibit robust plaque deposition at 4 months of age in both the cortex and the hippocampus (Radde et al., 2006). In APP/PS1 mice lacking TREM2 expression, there was a 70% reduction in plaque burden, as evaluated by 6E10 or Thioflavin S staining in the hippocampus. Analysis of Aβ40/42 levels revealed a similar reduction in both soluble and insoluble Aβ peptide concentrations. This effect was restricted to the hippocampus, as there were no significant changes observed in the cortex. It should be noted that Ulrich et al. found no change in plaque levels in APP/PS1 mice heterozygous for TREM2 deficiency (Ulrich, Finn et al. 2014).
Finally, as presented by Dr. Richard Ransohoff, the Lamb lab tried to address the origin of myeloid cells around the amyloid plaques by bone marrow chimera experiments using head sparring radiation of APP/PS1 mice, whose bone marrow was replaced with that from Trem2LacZ/+ mice. Preliminary results suggest that, following five months of reconstitution, myeloid cells clustered around Aβ plaques stained positive for β-galactosidase expression, indicating the origin of some the TREM2+ cells from peripheral blood monocytes rather than brain resident microglia.
Thus, these data suggest an unexpected role for TREM2 in AD pathology, enhancing inflammation through allowing the accumulation of peripherally derived myeloid cells around plaques and promoting amyloid accumulation. This is of particular interest since an unexplained facet of the AD is the inability of the macrophages to mount a phagocytic response and clear the deposited amyloid, despite the presence of cell surface receptors which can stimulate this response, such as TREM2, MerTK and Axl. Heppner and colleagues have demonstrated that there is an amyloid-dependent inactivation of phagocytosis, but the basis of this effect is unknown (Krabbe, Halle et al. 2013).
The phenotype of myeloid cells is regulated through the actions of ligand activated, heterodimeric type II nuclear receptors which act broadly to suppress inflammatory gene expression and promote the expression of genes associated with tissue repair and phagocytosis. In the brain, the principal type II nuclear receptors are PPARγ, PPARδ and LXR, that heterodimerize with retinoid X receptors (RXR) to form a functional transcription factor. The dimeric nuclear receptors bind to response elements positioned in the enhancers and promoters of their target genes, and directly regulate gene expression (Gosselin, Link et al. 2014). It has recently been appreciated that TREM2 expression is directly regulated by agonists of RXR (Daniel, Nagy et al. 2014; Lefterov, Schug et al. 2015). Importantly, Savage et al. (Savage, Jay et al. 2015) found that agonists of PPARγ, PPARδ and RXR stimulate the expression of the phagocytic receptors MerTK and Axl by microglia and macrophages in the brain of two different murine models of AD. Importantly, previous work documented that treatment with nuclear receptor agonists stimulated phagocytosis by these cells in vitro and in vivo, in the latter case this resulted in a reduction in plaque burden (Skerrett, Malm et al. 2014).
Immunohistochemical staining of the brains of AD mice revealed that MerTK and Axl expression was found almost exclusively on plaque-associated macrophages, and was not detected on parenchymal microglia. This finding led to experiments testing if these cells also expressed TREM2. We found that MerTK expressing macrophages were TREM2+ when evaluated by immunohistochemical staining or by flow cytometry. Moreover, the MerTK/Axl expressing cells were also CD45hi, suggesting that these cells were derived from infiltrating peripheral monocytes. Treatment of mice with the RXR agonist bexarotene resulted in no change in the number of MerTK/Axl or TREM2 expressing cells, but elevated the levels of receptor expression. Thus, these data clearly demonstrate that plaque-associated macrophages express three different receptors that function to regulate phagocytosis and these cells exhibit markers consistent with their origin as blood-borne monocytes which infiltrate the AD brain.
Subsequent studies tested if nuclear receptor activation could restore phagocytic competence to plaque-associated macrophages in the AD brain. Using an ex vivo phagocytosis assay (Krabbe, Halle et al. 2013), Krabbe and colleagues reported that phagocytosis by plaque-associated macrophages was suppressed in the AD brain. These findings are consistent with the age-dependent accumulation of plaque burden in AD patients and mouse models. We replicated these findings using 12-month-old APP/PS1 mice. We found that treatment of AD mice with bexarotene for 5 days before preparing brain slices restored phagocytic competence to the plaque-associated macrophages, coincident with the induction of MerTK/Axl expression. Moreover, activation of MerTK/Axl in APP/PS1 brain sections with a specific ligand, Gas6, resulted in phagocytic activity that was similar to that of non-transgenic mice. Importantly, an antibody blocking MerTK function inhibited phagocytosis in both wild-type and mouse models of AD. These studies directly demonstrated that nuclear receptor-stimulated phagocytosis is reliant upon MerTK expression and function.
These studies support the hypothesis that the plaque-associated macrophages are derived from monocytes in mouse models of AD. These cells express high levels of TREM2 and the phagocytic receptors MerTK and Axl, and their expression is coordinately regulated by nuclear receptors. The presence of plaque-associated macrophages is reliant upon TREM2 expression. Significantly, the TREM2+ macrophages exhibit a robust inflammatory phenotype, a finding that contravenes the postulated anti-inflammatory actions of TREM2. The phagocytic activity of these cells can be stimulated by treatment of AD mice with nuclear receptor agonists.
Conclusions and controversies
One important and consistent conclusion arising from studies in the three laboratories is that TREM2 deficient mouse models of AD exhibit reduced inflammation and myeloid cell accumulation around plaques. However, there are also a number of inconsistencies across the three studies, including the plaque burden in TREM2 deficient mouse models of AD and the findings concerning the origin of the respective myeloid cell population. Reduced plaque burden was observed in the hippocampus of 4-month-old APP/PS1 mice, whereas significantly increased plaque levels were found in the hippocampus of 8-month-old 5XFAD mice. These findings raise the possibility of age or disease progression-dependent difference in the TREM2 deficient macrophages. These studies were performed in two different mouse models of AD and TREM2 KO mice which could also influence the experimental outcomes. Efforts are currently underway to reconcile these disparate findings.
Another point of controversy is whether the plaque-associated macrophages represent the activation and accumulation of resident microglia or alternatively that these cells derived from blood borne monocytes that infiltrate the AD brain. Parabiosis experiments conducted in the Colonna lab argue that these cells are microglia, whereas flow cytometry and bone marrow chimera experiments from the Lamb and Landreth labs suggest they arise from blood borne monocytes. Clearly, additional work using genetic models is needed to resolve this important issue.
Inflammatory PGE2 receptors in models of Alzheimer’s disease
Katrin I. Andreasson
Introduction
Pathological changes in AD consist of amyloid ß accumulation, tau phosphorylation, and synaptic and neuronal loss, which develop steadily over years to decades in the context of a chronic and non-resolving pro-inflammatory response. Recent genome-wide association studies (GWAS) and systems biology approaches have confirmed a central role for the microglial inflammatory response in AD development (Lambert, Heath et al. 2009; Augustin, Lichtenthaler et al. 2011; Hollingworth, Harold et al. 2011; Naj, Jun et al. 2011; Cruchaga, Kauwe et al. 2013; Guerreiro, Wojtas et al. 2013; Lambert, Ibrahim-Verbaas et al. 2013; Zhang, Gaiteri et al. 2013; Jiang, Yu et al. 2014; Luo, Li et al. 2014; Gjoneska, Pfenning et al. 2015), and support the hypothesis that maladaptive inflammatory responses play a central role in the pre-clinical development of AD (Heneka, Golenbock et al. 2015; Malm, Jay et al. 2015; Mhatre, Tsai et al. 2015).
NSAIDs in Alzheimer disease
These recent findings complement conclusions from epidemiologic studies demonstrating reduced risk of developing AD in cognitively healthy aging populations taking non-steroidal anti-inflammatory drugs (NSAIDs) (McGeer, Schulzer et al. 1996; Stewart 1997; in t’ Veld, Ruitenberg et al. 2001; Szekely, Thorne et al. 2004; Vlad, Miller et al. 2008; Breitner, Baker et al. 2011). A primary action of NSAIDs is the enzymatic inhibition of the inflammatory cyclooxygenases COX-1 and COX-2, cytosolic enzymes that generate PGH2 from membrane stores of arachidonic acid, the precursor of the prostaglandins PGE2, PGD2, PGI2, and PGF2a, and the thromboxane TXA2. Structurally distinct NSAIDs reduce the risk of developing AD in large epidemiologic studies (Stewart 1997; in t’ Veld, Ruitenberg et al. 2001; Vlad, Miller et al. 2008) suggesting that inflammatory prostaglandin signaling may play an important role in pre-clinical development of AD. NSAIDs, however, cannot be effective for large scale AD prevention because both toxic as well as beneficial downstream prostaglandin signaling pathways are inhibited, leading to adverse renal and gastric toxicities and an increased risk for vascular and cerebrovascular disease (Funk and FitzGerald 2007). Recent genetic studies of prostaglandin receptors in mouse preclinical models have identified beneficial prostaglandin signaling pathways downstream of NSAID action, notably the vasodilatory prostacyclin (PGI2) receptor and the neuroprotective, anti-inflammatory, and vasodilatory PGE2 EP4 receptor (Egan, Lawson et al. 2004; Funk and FitzGerald 2007; Shi, Johansson et al. 2010; Liang 2011; Woodling, Wang et al. 2014). In addition, findings point to a beneficial function of PGJ2, a cyclopentenone metabolite that does not act via classical prostanoid G-protein coupled receptors, but exerts anti-inflammatory effects via the NFkB and PPARg transcription factors (reviewed in Straus and Glass 2001).
Prostacyclin signaling
Levels of prostacyclin (PGI2), considered to be anti-inflammatory and vasodilatory, are decreased in CSF of probable AD subjects (Montine, Sidell et al. 1999). Conversely levels of PGE2, widely considered an immune modulatory signaling molecule, are increased 5-fold in the cerebrospinal fluid (CSF) of patients with early probable AD (Montine, Sidell et al. 1999; Combrinck, Williams et al. 2006), suggesting that PGE2 signaling may play a role in promoting pre-clinical development of AD. PGE2 binds four G-protein coupled receptors (GPCRs) termed E-prostanoid receptors (EP1-4) that have distinct downstream signaling cascades and cellular distributions in the brain. Figure 4 depicts mechanisms of PGE2 action. In vivo, all four EP receptors are expressed in neurons, with EP2, EP3, and EP4 receptors, but not the EP1 receptor, expressed in microglia (Slawik, Volk et al. 2004; Shi, Johansson et al. 2010; Johansson 2013; Woodling, Wang et al. 2014; Johansson, Woodling et al. 2015). Activation of EP receptors leads to changes in the production of cAMP and/or phosphoinositol turnover and Ca2+ mobilization. EP2 and EP4 receptors couple positively to Gs to increase cAMP formation whereas EP3 couples negatively to cAMP through Gi.
Figure 4. Mechanisms of action of inflammatory PGE2 EP receptors in preclinical models of AD.
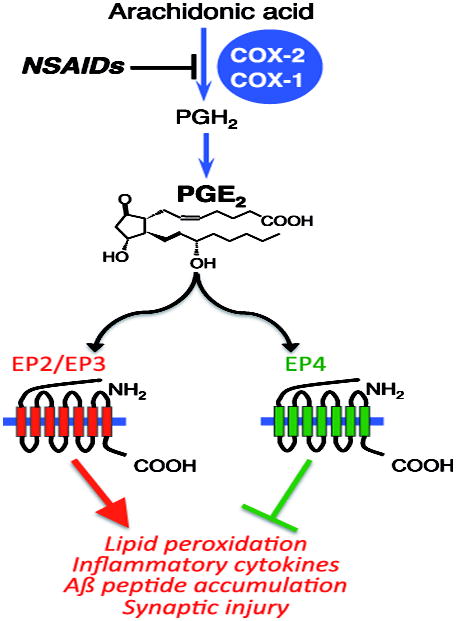
The primary action of NSAIDs is inhibition of COX-1/COX-2, with one consequence being reduction of downstream PGE2 production and signaling. The PGE2 EP2 and EP3 receptors enhance inflammatory oxidative stress, pro-inflammatory gene expression and are pro-amyloidogenic. In contrast, EP4 signaling is anti-inflammatory and enhances Aβ peptide phagocytosis. Reduction of PGE2 generation in response to NSAIDs may reduce deleterious EP2/EP3 signaling but also reduce beneficial EP4 signaling, which plays a beneficial role early in development of pathology in the APPSwe-PS1∆E9 model of familial AD.
APP transgenic models of AD display loss of synapses associated with development of spatial memory deficits along with inflammation. Synaptic injury and behavioral deficits in murine AD models are associated with the presence of Aß oligomers, which are directly toxic to synapses (Mucke and Selkoe 2012), and neuroactive inflammatory mediators, for example IL1-β (Hein, Stasko et al. 2010) or TNF-α (Medeiros, Prediger et al. 2007).
Early studies examining the in vivo function of the PGE2 EP2 receptor in models of innate immunity demonstrated a dramatic reduction in lipid peroxidation following intracerebroventricular (ICV) administration of lipopolysaccharide (LPS) in EP2 null mice (Montine, Milatovic et al. 2002). Additionally, in vitro microglial EP2 signaling elicited paracrine neurotoxicity in co-cultures of neurons and LPS-primed microglia, an effect dependent on increased inducible nitric oxide synthase (iNOS) and COX-2 activities (Shie, Montine et al. 2005). In vivo, APP/PS1, global deletion of EP2 led not only to significant decreases in lipid peroxidation and amyloid accumulation (Liang 2007), but also to reduced expression of inflammatory enzymes iNOS and NADPH (Liang, Wang et al. 2008).
A cell-specific role for microglial PGE2 EP2 signaling in suppressing phagocytosis of Aβ fibrils was initially demonstrated in an ex vivo preparation using AD brain sections coated with EP2-/- microglia (Shie, Breyer et al. 2005). This observation was subsequently confirmed in vivo, where deletion of EP2 in the APP/PS1 model reduced amyloid plaque load, an outcome reflecting both a more benign inflammatory milieu and an enhanced clearance of Aβ peptides (Liang, Wang et al. 2005; Keene, Chang et al. 2010). Recent conditional knockout strategies of microglial EP2 receptor using the Cd11bCre recombinase line, which excises loxP-flanked sequences in myeloid cells (Johansson 2013), restored healthy microglial responses to Aβ peptides (Johansson, Woodling et al. 2015), increasing microglial clearance of Aβ peptides and suppressing toxic inflammatory gene expression. Transcriptomics of microglia isolated from adult mice lacking microglial EP2 receptor additionally revealed that ablation of microglial EP2 receptor increased expression of insulin-like growth factor-1 (IGF-1) in response to Aβ peptide stimulation (Johansson, Woodling et al. 2015) and enhanced both IGF-1 and PPAR signaling; in this screen expression of RXRγ was enhanced with ablation of microglial EP2, and RXRγ along with its binding partner PPARγ reduce proinflammatory gene expression (Heneka, Landreth et al. 2007) and enhance clearance of Aβ peptides (Yamanaka, Ishikawa et al. 2012). The upregulation of these genes in microglial EP2 knockout mice suggests that EP2-deficient microglia respond to Aβ42 peptides in vivo by inducing anti-inflammatory and Aβ-clearing nuclear hormone receptor signaling genes. In support of a beneficial effect of reduced EP2 signaling, conditional deletion of microglial EP2 in the APP/PS1 model prevented synaptic injury and spatial memory deficits (Johansson, Woodling et al. 2015).
Toxic effects of microglial E-prostanoid receptors
The toxic effects of microglial EP2 contrast significantly with the beneficial anti-inflammatory effects of microglial EP4 in vivo. Initial in vivo studies of innate immune responses to LPS identified a pronounced anti-inflammatory effect of microglial EP4 signaling associated with reduced nuclear translocation of NF-κB subunits p65 and p50 (Shi, Johansson et al. 2010) in myeloid cells. Subsequent studies confirmed the anti-inflammatory function of microglial EP4 signaling, where in primary microglial cells, EP4 stimulation attenuated levels of Aβ42-induced inflammatory factors and potentiated phagocytosis of Aβ42 (Woodling, Wang et al. 2014). Transcriptomic studies demonstrated that microglial EP4 signaling broadly opposed Aβ42-driven gene expression changes, with a significant suppression of IRF1, IRF7, and NF-κB transcription factor regulated genes. In vivo, APP/PS1 mice deficient for microglial EP4 demonstrated increased inflammatory gene expression, oxidative protein modification, and Aβ deposition in brain at early but not late stages of pathology, suggesting an anti-inflammatory function of microglial EP4 signaling early in the APP/PS1 model. Interestingly, both EP2 and EP4 receptors are positively coupled to cAMP, yet have opposing inflammatory functions in models of Aβ peptide mediated neuroinflammation. This may be explained in part by the carboxy terminal cytoplasmic tail of the EP4 receptor, which is significantly longer than that of the EP2 receptor and may recruit distinct signaling molecules (Takayama, Sukhova et al. 2006; Minami, Shimizu et al. 2008).
The EP3 receptor is a central component in the regulation of the febrile response (Ushikubi, Segi et al. 1998; Lazarus, Yoshida et al. 2007). In the brain, EP3 is mainly expressed in neurons, although it can be induced in striatal microglia following injection of the excitotoxin quinolinic acid (Slawik, Volk et al. 2004). The function of EP3 signaling has been examined in models of AD, including the intracerebroventricular (ICV) Aβ injection model, which elicits a potent and long lasting inflammatory response to Aβ peptides (Letiembre, Liu et al. 2007; Walter, Letiembre et al. 2007) and in the APP/PS1 model. In a recent study (Shi, Wang et al. 2012), the induction of pro-inflammatory gene expression, cytokine generation, and lipid peroxidation following ICV Aβ42 was markedly reduced in EP3 null mice, suggesting that inflammatory EP3 signaling triggers a deleterious response to Aβ peptides. In the APP/PS1 model, deletion of EP3 reduced expression of proteins capable of increasing oxidative injury, including iNOS, components of the NADPH oxidase complex, and COX-2. Moreover, suggestive of a role in Aβ clearance, APP/PS1 mice lacking either one or both EP3 alleles exhibited lower amyloid accumulation. However, EP3 deletion also affected the generation of Aβ peptide from APP, as prior studies have correlated increased oxidative stress and inflammation with increased expression and activity of β-secretase (BACE) (Tamagno, Bardini et al. 2002; Apelt, Bigl et al. 2004). Indeed, deletion of the EP3 receptor in the APP/PS1 background decreased BACE expression and activity. This would suggest that in the APP/PS1 model, EP3 signaling both suppresses Aβ clearance and enhances Aβ peptide generation through increased BACE expression and activity. Interestingly, loss of just one allele of EP3 in the APP/PS1 background had beneficial effects in lowering Aß peptide levels.
Conclusion
In conclusion, studies examining mechanisms underlying the preventive effects of NSAIDs in AD development have identified maladaptive inflammatory responses mediated by the EP2 and EP3 receptors, but at the same time beneficial immune modulatory responses mediated by the EP4 receptor. This dichotomy of beneficial vs toxic prostaglandin signaling argues that NSAIDs are not an optimal mechanism to halt preclinical development of AD. The opposing actions of the EP2/EP3 and EP4 receptors highlight the importance of targeting selected EP receptors downstream of COX-1/COX-2. This is a potential future indication, as we await the identification and validation of biomarkers that can reliably predict subjects at risk for AD.
Arginine Metabolism: Neuroinflammation and Neurodegenerative Disorders
Leslie A. Sandusky, Maj-Linda B. Selenica, Daniel C. Lee
Introduction
Arginine metabolism represents a critical branch-point that impacts several systems, notably the production of nitric oxide (NO) signaling and polyamines. Arginases (Arg) (including arginase 1 and 2; Arg1 and Arg2, respectively) or nitric oxide synthases (NOSes), comprised of inducible, neuronal, and epithelial nitric oxide synthase, metabolize arginine to generate either ornithine and subsequent polyamines (putrescine, spermidine, and spermine) or nitric oxide, respectively (Morris 2004) (Figure 5). Polyamines remain critical for growth, as they are known to interact with macromolecules such as DNA and RNA, both electrostatically and covalently, promoting different cellular effects (Minois, Carmona-Gutierrez et al. 2011; Lightfoot and Hall 2014). Polyamines carry positive charges on nitrogen at physiological pH and become “supercations”; yet they distribute the charge along the entire length of the carbon chain making them structurally unique and distinct from point-charges of other cellular bivalent cations. Polyamines represent phylogenetically old molecules; and they continue to provide new purpose for biology and diseases. For example, an alternative arginine metabolism pathway also leads to polyamine production through the putative neurotransmitter agmatine plus carbon dioxide via arginine decarboxylase (ADC) and subsequently to putrescine via agmatinase (Halaris and Plietz 2007; Bernstein, Derst et al. 2011). Agmatine binds several receptors notably imidazoline receptors (I1R, I2R) with high affinity in the CNS, alpha 2 adrenoceptors (α-2), NMDA receptors and 5-HT receptors (Molderings and Haenisch 2012). It also produces a variety of physiological and pharmacological effects. So, while this endogenous molecule has been known for 100 years, its biosynthesis and therapeutic potential remain relatively unexplored. It was only recently that decarboxylation of arginine was found in the brain. The controversy and neglect in agmatine research arose in relation to the difficulty of demonstrating ADC activity in mammalian systems compared to bacteria, plants and fish. It wasn’t until 1994 that Ries and colleges demonstrated ADC activity and identified agmatine in the mammalian brain (Li, Regunathan et al. 1994).
Figure 5. L-Arginine pathways.
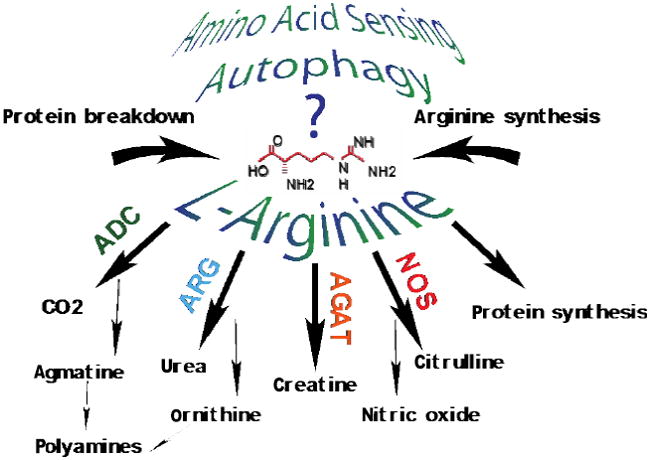
L-arginine represents a branch point of metabolic pathways including arginine decarboxylase (ADC), arginases (ARG), glycine amidotransferase (AGAT), and nitric oxide synthases (NOS). Arginine is essential for protein synthesis and amino acid turnover and may serve as a sensor for amino acid deprivation and autophagy activation.
Arginine metabolism and its by-products become essential in regards to the inflammatory challenge presented or the cytokines induced (proinflammatory versus anti-inflammatory). Two enzymes capable of regenerating the pool of L-arginine comprised of argininosuccinate synthetase 1 (ASS1) which converts citrulline (a NOS product) to argininosuccinate and argininosuccinate lyase (ASL), which converts argininosuccinate to L-arginine (Wiesinger 2001. Interestingly, proinflammatory stimuli can also induce both ASS1 and ASL concomitantly with NOS, suggesting a feedforward or recycling of citrulline and L-arginine in response to inflammation (Nagasaki, Gotoh et al. 1996). Furthermore, coinduction of NOS and arginase 1 with LPS has been observed (Sonoki, Nagasaki et al. 1997). Many reports show increased arginase 1 expression through the conical IL-4/ IL-13 JAK/ STAT pathway (Welch, Escoubet-Lozach et al. 2002; Mantovani, Sica et al. 2004; Nolan, Maher et al. 2005; Martinez, Helming et al. 2009; Varin and Gordon 2009) but it is also upregulated by proinflammatory stimuli and other insults (Rodriguez, Hernandez et al. 2005; El Kasmi, Qualls et al. 2008; Lee, Rizer et al. 2010; Lee, Ruiz et al. 2013; Fenn, Hall et al. 2014; Cherry, Olschowka et al. 2015). It remains clear that arginine metabolism will be governed by the inflammatory milieu, cell type, and combination of stimuli elicited. While arginine metabolism is historically considered to increase primarily in microglia, arginase is also expressed in neurons at lower levels and may compete with neuronal NOS (Yu, Iyer et al. 2001; Yu, Iyer et al. 2002; Yu, Yoo et al. 2003; Peters, Berger et al. 2013; Quirie, Demougeot et al. 2013) which may independently impact neural transmission and subsequent cognitive processing. Furthermore, by-products of the arginine metabolism that produce polyamines and nitric oxide can provide opportunities for post-translational modifications of proteins including polyamination and S-nitrosylation, which can modify aggregation-prone/disordered proteins including tau, (Tucholski, Kuret et al. 1999; Pevalova, Filipcik et al. 2006; Reynolds, Reyes et al. 2006; Reyes, Reynolds et al. 2008; Reyes, Fu et al. 2011; Reyes, Geula et al. 2012; Fontaine, Sabbagh et al. 2015), alpha synuclein (Kotzbauer, Giasson et al. 2004; Yu, Xu et al. 2010; Liu, Qiang et al. 2011) and Aβ (Kummer, Hermes et al. 2011). This suggests an interaction of polyamine metabolism, neuroinflammation and neuropathological markers associated with neurodegnerative disease states.
Altered Arginine and Polyamine Metabolism in Alzheimer’s disease
Two independent studies show that modulating arginine metabolism and/ or the polyamine system in animal models of tau or amyloid deposition alter disease pathology (Hunt, Nash et al. 2015; Kan, Lee et al. 2015). Additionally, in the previous two studies, amyloid and tau pathology changed L-arginine levels. More specifically, viral-mediated tau overexpression reduced arginase 1 mRNA levels and rTg4510 tau transgenic mice showed increased levels of L-arginine. Several studies showed altered polyamines, polyamine metabolites and L-arginine metabolism in CSF, plasma, or brain tissue of patients with mild cognitive impairment (MCI) and AD (Morrison and Kish 1995; Inoue, Tsutsui et al. 2013; Trushina, Dutta et al. 2013; Liu, Fleete et al. 2014; Graham, Chevallier et al. 2015). A recent report using untargeted blood-based metabolic profiling revealed that L-arginine and polyamine metabolism was differentially disrupted between control patients, patients with MCI and AD converters, which could be used to predict converters for up to two years (Graham, Chevallier et al. 2015). As more evidence surfaces regarding altered arginine and polyamine metabolism in AD, it is tempting to speculate activation of the urea cycle in the brain under conditions of chronic inflammation or neurodegeneration. Although this idea has been previously suggested in AD (Hansmannel, Sillaire et al. 2010), the activation of the urea cycle and changes in arginine metabolism may be disease-specific (Patassini, Begley et al. 2015).
Impact of L-arginine and Polyamine on Autophagy and Inflammation
The impact on cell biology versus pathology likely depends on the interplay of arginase, NOS, and their downstream products, polyamines or nitric oxide, and the availability of L-arginine for neurons versus microglia. Recently, increasing autophagy has been suggested as a therapeutic strategy for aggregation-prone/disordered proteins. Mammalian target of rapamycin (mTOR) is a negative regulator of autophagy and sensitive to amino acid availability and inhibition of mTOR signaling has been shown to impact neuropathology in certain models of AD (Wullschleger, Loewith et al. 2006; Sancak, Peterson et al. 2008; Caccamo, De Pinto et al. 2014). Furthermore, recent evidence has shown that mTOR and components of the mTOR complex are reduced during continual arginase 1 overexpression (Hunt, Nash et al. 2015). Additionally, arginase 1 overexpression significantly reduced multiple aspects of the tau phenotype associated with inflammation and microglial/macrophage activation including Iba-1 and CD45 along with various cytokines (i.e. IL-1β, TNFα, IL-12, IL-10, KC-Gro) (Hunt, Nash et al. 2015), while conditional deletion of arginase 1 in myeloid cells showed increased phospho-tau pathology following adeno-associated viral tau truncated at D421 (a caspase cleaved site which is preferentially cleared through autophagy (Dolan and Johnson 2010) than mice with sufficient arginase 1 (Hunt, Nash et al. 2015). However, it remains uncertain as to which mediated the autophagy response, arginine deprivation or polyamine production. Yet, this is consistent with other reports demonstrating that L-arginine and L-arginine deprivation act as a sensor for autophagy arguably through mTOR signaling (Ban, Shigemitsu et al. 2004; Yao, Yin et al. 2008; Savaraj, You et al. 2010; Wu, Liang et al. 2011; Hsueh, Knebel et al. 2012; Banik, Renner Viveros et al. 2013; Angcajas, Hirai et al. 2014). However, making the association less clear, the longer chain polyamine spermidine also acts as an inducer of autophagy but may not occur through mTOR signaling (Eisenberg, Knauer et al. 2009; Morselli, Galluzzi et al. 2009; Morselli, Marino et al. 2011), yet it has been demonstrated to increase longevity, restore memory in various models through autophagy, and is elevated in centenarians (Eisenberg, Knauer et al. 2009; Morselli, Galluzzi et al. 2009; Madeo, Eisenberg et al. 2010; Marino, Morselli et al. 2011; Minois, Carmona-Gutierrez et al. 2011; Morselli, Marino et al. 2011; Gupta, Scheunemann et al. 2013; Minarini, Zini et al. 2013).
Conclusion
Overall, increased metabolism of arginine through inflammation, aging, and/or injury may modulate various biological pathways that impact neurodegenerative diseases. Recent work in animal studies would suggest a role for arginase and NOS in amyloid and tau neuropathology (Hunt, Nash et al. 2015; Kan, Lee et al. 2015); however, lack of arginine metabolism or accumulation may also influence pathways including autophagy.
Systemic Inflammation and Alzheimer’s Disease
Clive Holmes, Jessica Teeling
Introduction
In AD, inflammation is thought to be largely mediated by the CNS resident cells with a dominant role being played by cytokines. However, there is also a developing interest in the potential role for peripherally derived cells and other mediators of inflammation. Furthermore, there is increasing recognition that neuroinflammatory processes in the AD brain are markedly influenced by inflammation that occurs outside the CNS (Perry and Holmes 2014; Takeda, Sato et al. 2014; Goldeck, Witkowski et al. 2016)
Pre-clinical studies
In the past the parenchyma of the CNS was considered an ‘immunologically privileged site’ because the blood brain barrier (BBB) was thought to prevent the entry, or exit, of many molecules including antibodies from the periphery and was largely devoid of macrophages, neutrophils and lymphocytes. This historical concept has led to research on CNS inflammation being viewed in isolation and independent of systemic inflammation occurring outside the CNS. However, communication of the presence of systemic inflammation to the brain is clearly supported by the constellation of centrally derived symptoms that occur in animals following administration of TLR agonist to mimic an acute systemic bacterial or viral infection. This syndrome is known as ‘sickness behavior’ and includes anorexia; depression; somnolence; decreased social interaction; decreased concentration and fever, and is an evolutionary conserved, homeostatic mechanism that allows the body to adapt to an insult (Hart 1988). The state of the BBB in AD is still uncertain and, although controversial, it is possible that there may be, in limited circumstances, some direct entry of immune cells (monocytes, macrophages) from the periphery into the brain. Thus, it is known that sepsis (Lee and Slutsky 2010) and persistent peripheral inflammatory stimuli may compromise the BBB because of a breakdown of the intercellular tight junctions caused by lipopolysaccharide (LPS) and peptidoglycans (Nau, Sorgel et al. 2010). However, there are a number of other communication routes from the periphery to the brain that occur despite the presence of an intact BBB. These include:
Stimulation of peripheral nerves (e.g. the vagus) by cytokines and prostaglandin which signal to the medulla oblongata and are relayed to the hypothalamus (Ek, Kurosawa et al. 1998; Dantzer, Konsman et al. 2000).
Direct actions of LPS or pro-inflammatory cytokines at brain areas lacking a BBB e.g. the circumventricular organs (Blatteis, Bealer et al. 1983).
Direct action of LPS or pro-inflammatory cytokines of perivascular macrophages in the neurovascular unit of the BBB (Matsumura and Kobayashi 2004).
In animals, unless the peripheral inflammatory event is extreme, sickness behavior induced by LPS is a relatively benign and transient phenomenon (Perry 2010). Damage limitation in the brain is in part due to the wide number of regulatory mechanisms that reduce the pro-inflammatory response to an acute challenge within the brain. Thus, following a peripheral signal the pro-inflammatory response in the brain appears to be suppressed by the increased production of a large variety of proteins from microglial cells including anti-inflammatory cytokines IL-10; TGFβ and other cytokine signaling protein suppressors (Rivest 2009).
Systemic inflammation induced by real life bacteria, for example S. typhimurium, leads to robust systemic inflammation and a delayed innate immune activation in the brain, with cerebral endothelial and microglial cells showing prolonged phenotype and functional changes, including enhanced sensitivity to a secondary challenge (Puntener, Booth et al. 2012).
With aging and in AD, microglial cells also show enhanced sensitivity to inflammatory stimuli with an upregulation of a range of cell surface receptors and an exaggerated microglial response to a second inflammatory stimulus. This phenomenon is called priming and its exact origins are unknown. It might be caused by microglial senescence, by prolonged exposure to the aged neuronal environment including the presence of Aβ protein, or by sustained systemic inflammation. The result is that in aged animal or AD mouse models, priming results in microglial cells that show a markedly increased production of IL-1β, and an increased production of reactive oxygen species by microglia to even a very modest peripheral inflammatory event. Thus, priming results in a microglial state that once activated cuts across the simple classification of M1 or M2 states by having elements of both (Perry, Nicoll et al. 2010; Perry and Holmes 2014).
This hypothesis is supported by animal studies showing an exaggerated inflammatory and oxidative stress response to peripheral stimuli in aged mice (Godbout and Johnson 2009), increased concentrations of IL-1β in the CNS and neuronal apoptosis in the ME7 prion mouse after peripheral challenge with the bacterial mimic LPS or the viral mimic polyinosinic-polycytidylic acid (Cunningham, Wilcockson et al. 2005; Cunningham, Campion et al. 2009; Field, Campion et al. 2010).
Clinical studies
Blood markers of peripheral inflammation
A number of studies have tried to establish differences in serum or plasma markers of inflammation between AD populations and aged matched control groups in cross sectional studies. A meta-analysis of these studies (Swardfager, Lanctot et al. 2010) suggests that overall there are increases in pro-inflammatory cytokines in AD compared with control groups, although clearly cross-sectional studies cannot establish whether this is cause or effect. A proteomics study with no a priori candidates reported the presence of a number of plasma inflammatory proteins including complement factors and clusterin to be associated with hippocampal atrophy and clinical progression in MCI and AD (Thambisetty and Lovestone 2010). In another AD cohort, evidence of acute and chronic systemic inflammatory diseases was associated with raised serum TNFα levels and an exacerbation of sickness behaviour like symptoms and increased cognitive decline (Holmes, Cunningham et al. 2009; Holmes, Cunningham et al. 2011).
Imaging studies
Functional magnetic resonance imaging studies of healthy human volunteers have shown that low dose Salmonella typhi LPS are associated with significant increases in systemic IL-6 with higher cytokine levels being associated with increased neuronal activity in the substantia nigra (Brydon, Harrison et al. 2008). PET imaging studies with direct examination of microglial activation using the Translocator Protein (TSPO) ligand have also shown preliminary evidence of increased 11C-PK11195 binding in the brains of subjects with chronic aseptic systemic inflammation (Drake, Boutin et al. 2011). AD cross sectional PET studies support evidence of an up to 50% increased binding of 11C-PK11195 in the Frontal and Temporal Cortex (Diorio, Welner et al. 1991; Cagnin, Brooks et al. 2001; Edison, Archer et al. 2008; Okello, Edison et al. 2009; Venneti, Lopresti et al. 2009; Wiley, Lopresti et al. 2009). Interestingly, AD studies that have examined both microglial and Aβ markers have found significant correlations between cognitive scores and both TSPO ligands 11C-PK11195 and 11C-PBR28 (Kreisl, Lyoo et al. 2013) but not with the amyloid ligand Pittsburgh compound B (11C-PIB) binding (Edison, Archer et al. 2008; Yokokura, Mori et al. 2011) supporting the view that microglial activation rather than Aβ alone may be a key change leading to neurodegeneration.
Therapeutic strategies
Small trials in early AD with indomethacin suggested some evidence of reduced cognitive decline (Rogers, Kirby et al. 1993). However, this study was not replicated in a later follow up study (de Jong, Jansen et al. 2008). Large scale studies of other NSAIDs including naproxen (Aisen, Schafer et al. 2003) and rofecoxib (Reines, Block et al. 2004) in AD have also been unsuccessful. Randomised trials with a range of other anti-inflammatory drugs, including prednisone (Aisen, Davis et al. 2000), hydroxychloroquine (Van Gool, Weinstein et al. 2001), simvastatin (Simons, Schwarzler et al. 2002) atorvastatin (Sparks, Sabbagh et al. 2005; Feldman, Doody et al. 2010), aspirin (Bentham, Gray et al. 2008) and rosiglitazone (Harrington, Sawchak et al. 2011) have also shown no clinically significant changes in primary cognitive outcomes in patients with AD. More recently, we have conducted a small study of AD subjects using the TNF-α inhibiting agent Etanercept in AD subjects on the basis that a more targeted approach to completely block peripheral TNF-α may have a more beneficial effect. The study showed that the drug was well tolerated in the AD population and preliminary evidence of a reduction of decline on a number of clinical outcomes (Butchart, Brook et al. 2015). This study design is now being replicated in patients with early AD (amyloid positive PET scans in amnestic Mild Cognitive Impairment) as part of the INMIND consortium with an examination of the effects of Etanercept on neuroimaging markers of inflammation as the primary outcome.
Conclusions
Sepsis and persistent peripheral inflammatory stimuli may compromise intercellular tight junctions and subsequently lead to BBB breakdown. However, even with intact BBB, periphery-to-brain signaling may take place, e.g. through stimulation of peripheral nerves by cytokines and prostaglandin which signal to the medulla oblongata and are relayed to the hypothalamus; furthermore, LPS or pro-inflammatory cytokines may act directly at brain areas lacking a BBB such as the circumventricular organs; or in the neurovascular unit of the BBB. Microglial cells may fence the pro-inflammatory response in the brain by increasing production of anti-inflammatory cytokines IL-10; TGFβ and other cytokine signaling protein. Despite promising results in previous studies using NSAIDs, thus far no clinically relevant benefit in terms of cognitive improvement could be replicated on a large scale. However, further trials are underway using more targeted study designs, e.g. on the basis of TNF-α inhibition.
Long-term potentiation and neuroinflammation
Marina A. Lynch
Introduction
It is well documented that neuroinflammatory changes, that are characteristic of the aged brain and evident in neurodegenerative diseases, are associated with compromised cognitive function. This is also reflected in animal models where the deficit in cognitive function is accompanied by impaired synaptic plasticity. Specifically it has been consistently shown that a decrease in hippocampal long-term potentiation (LTP), which is perhaps the most studied form of synaptic plasticity, is coupled with a decrease in performance in hippocampal-dependent tasks. Neuroinflammatory changes result from loss of homeostatic control and, although several cells play contributory roles in driving neuroinflammation, activation of microglia, and also astrocytes, is crucial in the sense that they are the primary producers of inflammatory mediators, which impact negatively on synaptic plasticity (Figure 6).
Figure 6. Microglia in LTP.
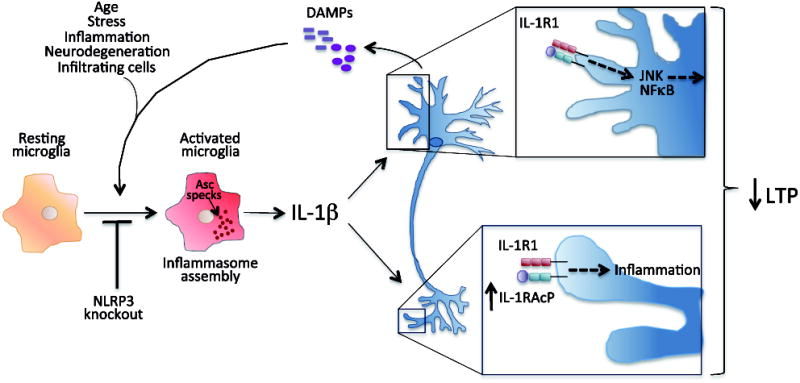
Several factors stimulate microglia to adopt an inflammatory phenotype and, in most cases, inflammasome activation and the consequent production and release of IL-1β. IL-1β interacts with IL-1R1 activating signalling molecules like JNK and NFκB that exert an inhibitory effect on LTP. In addition, the age-related increase in IL-1RAcP at synapses results in an exaggerated effect locally that can also depress LTP. In age, neurons have been shown to release DAMPs that can feed back to enhance microglial activation, contributing to a damaging cycle of events.
Inhibitors of synaptic plasticity
It has been known for 2 decades or more that inflammatory cytokines, including IL-1β, IL-6, and both type I and type II interferons (IFN) inhibit synaptic plasticity, in particular LTP (Lynch 2015). Since learning and consolidation of memory are dependent on changes at the level of the synapse, it is not surprising that these inflammatory cytokines also inhibit various forms of memory (Donzis and Tronson 2014). Perhaps the cytokine that has elicited most interest is IL-1β and it has been shown by many groups that application of IL-1β to hippocampal slices or central administration of IL-1β inhibits LTP in the major afferent pathways in the hippocampus (Lynch 2015). The inhibitory effect of IL-1β has been shown to be blocked by the endogenous receptor antagonist, IL-1ra (Loscher, Mills et al. 2003). However, the effect of IL-1β on LTP is complex and probably dependent on concentration, the specific afferent pathway stimulated, the method of delivery of IL-1β and the timing (Yirmiya and Goshen 2011). Thus a deficit in LTP in IL-1R1-/- mice has been described (Avital, Goshen et al. 2003) while it has also been shown that IL-1ra can inhibit LTP (Schneider, Pitossi et al. 1998; Loscher, Mills et al. 2003).
Given that age-related neuroinflammatory changes are undisputed and a negative correlation between inflammation and LTP exists, it is not surprising that LTP is impaired in aged animals. It is similarly reduced in lipopolysaccharide (LPS)-treated animals and in Aβ-treated transgenic mouse models of AD, which trigger microglial activation and increased production of inflammatory cytokines (Lynch 2015). Inflammatory stimuli, such as Aβ, LPS and IL-1β exert a greater effect in aged, compared with young, animals (Minogue, Lynch et al. 2007; Patterson 2015) perhaps because the microglial activation state has shifted and the cells are primed. However, a recent study revealed that IL-1β acts to inhibit LTP because of a direct effect at the synapse and that there was an age-related increase in sensitivity to IL-1β that could be explained by an increase in the IL-1 receptor accessory protein, IL-1RAcP, which promotes inflammatory changes (Prieto, Snigdha et al. 2015). This study indicates that microglial priming is not the only factor in the age-related sensitivity to IL-1β, at least as far as LTP in synaptosomes is concerned. The authors argue that the increased expression of IL-1RAcP is driven by the age-related increase in IL-1β, so the question of the source of the IL-1β and the reason for its increased concentration remain to be addressed. One possibility is an increase in inflammasome assembly with the consequent increases in caspase 1 activation and IL-1β processing (Youm, Grant et al. 2013). Significantly, microglial activation, caspase 1 activation and hippocampal and cortical IL-1β concentrations were markedly decreased in aged NLRP3-deficient mice compared with wild type mice. These changes were associated with improved performance in hippocampal-dependent tasks providing further evidence of an inverse relationship between hippocampal IL-1β and cognitive function (Youm, Grant et al. 2013). Knocking out NLRP3 in a mouse model of AD (APP/PS1 mice) was shown to exert similar effects; the increases in caspase 1 activation and IL-1β concentration, and the deficits in hippocampal-dependent tasks and LTP that are characteristic of APP/PS1 mice, were all attenuated in NLRP3-deficient APP/PS1 mice (Heneka, Kummer et al. 2013). Interestingly, microglia in NLRP3-deficient APP/PS1 mice had characteristics of the M2-like phenotype and were more phagocytic, and consequently Aβ accumulation was reduced in these animals. Overall, these findings point to a detrimental effect of IL-1β on hippocampal function. Several factors can stimulate NLRP3 activation but the key factors that trigger this in age remain to be determined. Among the multiple possibilities are factors like HMGB1 and ATP that are released from damaged neurons and can act on TLR2/4 and purinergic receptors respectively, providing signals 1 and 2. HMGB1 has been shown to be increased in the hippocampus with age (Barrett, Costello et al. 2015) and its inhibitory effect on LTP has been described (Costello, Watson et al. 2011).
Microglia in the aged brain
The broader question of the factors that stimulate microglia to adopt a persistent inflammatory phenotype in the aged brain remains to be answered. These cells are maintained in a relatively quiescent state because of the suppressive microenvironment in the brain where there is a relatively high concentration of anti-inflammatory factors, where neurotransmitters like noradrenaline exert a calming influence on the cells and where microglia interact with other cells, for example neurons, through ligand-receptor interactions that also reduce cell activation. The interaction between neuronal CD200, CX3CL1, CD45 and their respective receptors on microglia prevents microglial activation. However, ageing is associated with a disruption in ligand-receptor interactions and age-related decreases in CD200 and CX3CL1, associated with microglial activation and inflammatory changes, have been described (Lyons, Downer et al. 2007; Lyons, Lynch et al. 2009; Lynch 2010; Fenn, Smith et al. 2013).
The impact of the immune system on the brain is profound (Ransohoff, Schafer et al. 2015) and the findings that peripheral immune cells access the brain under certain circumstances bring into focus the importance of understanding the impact of these infiltrating cells on resident cells in the brain. We have found that the age-related increase in blood brain barrier permeability is associated with increased infiltration of macrophages, especially in APP/PS1 mice (Minogue, Jones et al. 2014). Although macrophages that infiltrate the brain are efficient phagocytes, particularly of Aβ, our evidence suggests that macrophages from aged animals, and also APP/PS1 mice, are more responsive to inflammatory stimuli (Barrett, Costello et al. 2015; Barrett, Minogue et al. 2015). Therefore, when these macrophages encounter the inflammatory environment that exists in these mice, they have the potential to exacerbate the already-existing neuroinflammation (Costello et al. unpublished). Macrophages may also be the source of the increase in IFNγ observed with age and in APP/PS1 mice (Minogue, Jones et al. 2014). Significantly, IFNγ inhibits LTP (Kelly, Minogue et al. 2013) and synergizes with Aβ to increase microglial activation (Jones, Minogue et al. 2015). Infiltration of T cells also occurs with age, particularly in APP/PS1 mice, and our evidence indicates that Th1 and also Th17 cells activate microglia in vitro and in vivo while their presence in the brain of APP/PS1 mice negatively impacts hippocampal-dependent cognitive function (Browne, McQuillan et al. 2013; McManus, Higgins et al. 2014; McManus, Mills et al. 2015).
Conclusion
While there are multiple questions that remain to be answered with respect to the microglial activation states, the evidence overwhelmingly suggests that when the cells adopt an inflammatory phenotype, synaptic function and cognition are impaired. To make significant advances, clarification is needed with respect to the beneficial/damaging effects of microglial activation in age and in AD and this relies on a greater understanding of multiple activated cell subtypes and their functional characteristics. The likelihood that cells in different activation states co-exist needs to be addressed and an appreciation of how these cells interact is likely to provide answers to what currently presents as intractable issues.
TBI and Alzheimer’s risk: Targeting dysregulated glial activation and innate immunity
Linda J. Van Eldik, PhD and Adam D. Bachstetter, PhD
Introduction
Traumatic brain injury (TBI) is exceedingly common, with nearly 2 million people in the United States seen annually in emergency departments for head injuries (Faul, Xu et al. 2010), and many more who do not seek medical treatment. For those with a single mild TBI, the likelihood of a favorable recovery is high, and there may be no residual cognitive or neurological symptoms immediately evident. However, subclinical pathophysiological changes to the CNS caused by TBI persist, which increase the risk for later life neurologic impairments, including dementia (for review see Johnson, Stewart et al. 2010; Shively, Scher et al. 2012). The multifaceted mechanisms through which a TBI increases the risk for developing a neurodegenerative disease is not currently well defined. One potential mechanism is dysregulation of glial activation and a priming of the innate immune system.
Glial dysregulation and innate immune system priming as risk factors for dementia
The evidence suggesting involvement of aberrant glial activation and a priming of the innate immune system on increasing the risk of developing dementia largely comes from animal models. However, a few notable studies in humans have shown persistent signs of inflammation months-to-years after a person suffered a moderate-to-severe TBI. For example, a PET imaging study using a ligand that binds to activated microglia showed increased signal present up to 17 years after TBI, demonstrating an unresolved chronic inflammatory response (Ramlackhansingh, Brooks et al. 2011). Consistent with this, persistent elevated microglial activation has been documented in human autopsy brain by increased CD68+ and MHCII+ macrophage / microglia staining in long term (up to 18 years) survivors of head injury (Johnson, Stewart et al. 2013).
While the clinical literature is limited, extensive work has been done in preclinical models of TBI, describing the temporal onset of glial activation and innate immune response in a number of disease models and injury severities. Even with diverse models of TBI, differences in severities of injury, and differences in animal species, the acute inflammatory response to the injury is remarkably stereotyped (for review see Morganti-Kossmann, Satgunaseelan et al. 2007; Donnelly and Popovich 2008; Loane and Byrnes 2010). Figure 7, largely adapted from two recent studies in mouse models of diffuse brain injury (Bachstetter, Rowe et al. 2013; Webster, Van Eldik et al. 2015), summarizes the temporal pattern of the neuroinflammatory response to TBI. Within the first minutes to hours after the injury, cytokine / chemokine levels begin to rise, and subsequently peak 6-12 hours later. The cytokine / chemokine levels may return to baseline or remain chronically elevated over the sham levels (Bachstetter, Rowe et al. 2013; Webster, Van Eldik et al. 2015). Morphological changes in microglia begin within minutes and can clearly be seen by at least 6 hours after the injury (Bachstetter, Rowe et al. 2013; Roth, Nayak et al. 2014). The microglia response largely peaks by 1 week after the injury. Similar to human TBI, the microglia response in mouse models of TBI may stay elevated or worsen with time compared to sham-injured mice (Webster, Van Eldik et al. 2015). Interestingly, the recruitment of peripheral immune cells to the brain appears to occur in a narrow window between 1-7 days after the injury, and corresponds to increased levels of the chemokine CCL2. The CCL2-CCR2 pathway has been shown to be important for recruitment of peripheral immune cells to the brain after TBI (Hsieh, Niemi et al. 2014; Morganti, Jopson et al. 2015). Morphological changes to astrocytes follow a similar temporal pattern of activation post-injury, albeit slightly more delayed in onset (Bachstetter, Rowe et al. 2013; Webster, Van Eldik et al. 2015).
Figure 7. Targeting dysregulated glial activation and innate immunity in TBI and AD.
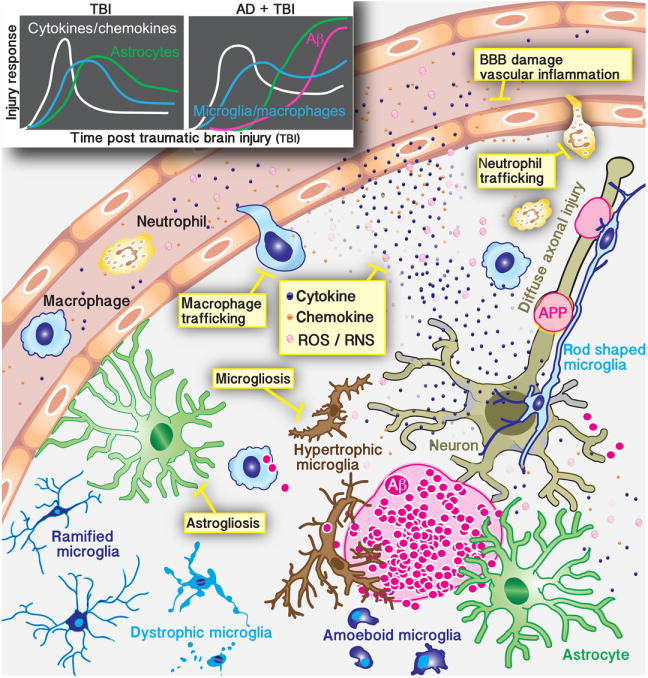
Top left panels show a conceptualized profile of the temporal changes in cytokines / chemokines, astrocytes, and microglia / macrophages following TBI, and how the neuroinflammatory responses are heightened and prolonged in the presence of beta-amyloid (Aβ) pathology (Webster, Van Eldik et al. 2015). In the figure, potential areas for therapeutic intervention targeting glial activation and innate immunity are highlighted in the yellow boxes. APP=amyloid precursor protein, BBB=blood-brain barrier, ROS/RNS=reactive oxygen species/reactive nitrogen species.
Microglial priming
Repetitive CNS stressors can alter this pattern of acute post-injury neuroinflammatory responses and resolution of the inflammation, leading to an exaggerated and unresolved neuroinflammatory response. Microglial priming, a process whereby a primary insult can sensitize, or “prime,” microglia to exhibit an exaggerated response to a subsequent injury, is increasingly being recognized as a potential mechanism by which a state of heightened microglial reactivity elicited by a prior insult can dramatically influence the responses to subsequent insults (for a review on microglia priming see Norden, Muccigrosso et al. 2015). Stressors which have been described to prime the neuroinflammatory response include a prior TBI, advanced age and neurodegenerative pathology. For example, a role for amplified and prolonged microglia activation has been postulated to contribute to the development of chronic traumatic encephalopathy, a progressive neurodegenerative disease seen in individuals with a history of repetitive brain trauma (Norden, Muccigrosso et al. 2015). Another example of how the neuroinflammatory response can be primed by exposure to multiple CNS stressors is a recent study combining a TBI with an amyloidosis mouse model of Alzheimer’s disease (AD) pathology (Webster, Van Eldik et al. 2015). The combination of AD pathology and brain injury resulted in a heightened and prolonged neuroinflammatory response to the injury (see top left panels in Figure 7), and worsened cognitive performance on a behavioral task compared to mice that had only a TBI or only AD pathology. A small molecule experimental therapeutic that selectively restores injury- or disease- induced overproduction of proinflammatory cytokines towards homeostasis was shown to rescue the cognitive deficits in the AD + TBI mice, providing evidence of a link between the dysregulated neuroinflammatory response and cognitive deficits in the AD + TBI mice (Webster, Van Eldik et al. 2015). These data suggest that selective targeting of abnormal neuroinflammatory responses may be a viable therapeutic approach to reduce the risk of neurologic impairment after TBI. As is indicated by the yellow boxes in Figure 7, there are multiple secondary injury mechanisms associated with dysregulated glial activation and innate immunity that could be points for therapeutic intervention following a TBI. Even within broad classes of secondary injury mechanisms, such as blood-brain-barrier (BBB) damage or immune cell trafficking, there are many specific molecular targets regulating these biological responses that could be explored for development of selective therapeutic interventions. An important concept for intervening in dysregulated glial activation and innate immunity following a TBI is that of therapeutic window: targeting a mechanism at the appropriate time after injury. For example, the acute CNS proinflammatory cytokine surge occurs in a treatable time frame (hours after injury), and has been shown to be amenable to therapeutic intervention with small molecule experimental therapeutics (Lloyd, Somera-Molina et al. 2008; Bachstetter, Webster et al. 2015). Suppression of the acute neuroinflammatory responses to TBI also provides an example of the concept of pharmacodynamics, i.e. the biochemical and physiological effects of a drug on the body. When an upstream process, such as proinflammatory cytokine overproduction, drives downstream detrimental events, then inhibition of the upstream process suppresses the downstream process whether the drug is still present or not (if the injury/activating stimulus is no longer present).
In the case of a neurodegenerative disease, where the activating stimulus (protein misfolding and aggregation, age, priming, etc.) remains, the situation is more complex and a more chronic administration of drug is likely to be necessary to target the chronically dysregulated glial activation and innate immunity. In animal models of AD, selective suppression of the proinflammatory cytokine overproduction has been shown to be an effective therapeutic strategy. In addition, dysregulated cytokines/neuroinflammation can be suppressed by targeting more than one intracellular signaling pathway: p38α-dependent and p38α-independent pathways (Bachstetter, Norris et al. 2012; Watterson, Grum-Tokars et al. 2013; Roy, Grum-Tokars et al. 2015). Targeting proinflammatory cytokines is only one example of a potential avenue to suppress dysregulated glial activation and innate immunity following CNS injury or in CNS disease. Selected other examples include: (1) pharmacological scavenging antioxidants to neutralize reactive oxygen and nitrogen species (Hall 2015); (2) CCR2 antagonist or CD11d blocking antibody to block the trafficking of macrophages, monocytes and neutrophils to the CNS (Bao, Shultz et al. 2012; Morganti, Jopson et al. 2015); (3) inhibition of the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway to target astrogliosis (Furman, Sama et al. 2012).
A consideration in targeting dysregulated glial activation and innate immunity is the growing evidence for a remarkable heterogeneity in microglia. For example, a recent study (Bachstetter, Van Eldik et al. 2015) demonstrated variations in human microglial morphology that showed some degree of selectivity towards different neurodegenerative diseases, including AD, hippocampal sclerosis of aging, and dementia with Lewy bodies. The microglia heterogeneity includes the well-described hypertrophic and ameboid microglia which, in comparison to the ramified microglia, are generally associated with microglia “activation”. In addition, rod shaped microglia, which have also been found in animal models of TBI associated with diffuse axonal injury (Bachstetter, Rowe et al. 2013; Webster, Van Eldik et al. 2015), are seen in the hippocampus of aged individuals (Bachstetter, Van Eldik et al. 2015). The function of rod shaped microglia is an area of microglia biology that needs further exploration. Finally, as previously described (Streit 2006), dystrophic microglia with processes that are spheroidal, beaded, de-ramified or fragmented are commonly seen in the aged brain. In the case of hippocampal sclerosis of aging, dystrophic microglia are proportionally the most prevalent microglia phenotype in the CA1 region of the hippocampus (Bachstetter, Van Eldik et al. 2015). Similar to rod shaped microglia, the influence of dystrophic microglia on CNS pathophysiology is unknown.
Conclusion
The diversity and heterogeneity of the dysregulated glial activation and innate immunity responses to CNS injury and disease provide a number of druggable pathways and opportunities for therapeutic intervention. However, the complexity of neuroinflammatory responses also highlights the critical need for continued research to better understand which responses are beneficial and which are harmful (Heneka, Kummer et al. 2014; Heppner, Ransohoff et al. 2015).
A novel 3D culture system that gives astrocytes and neurons the third dimension – a new tool for assessing (patho)physiological responses of cells in vitro
Milos Pekny and Till Puschmann
Introduction
Since the advent of cell culture systems, their advantages and shortcomings were obvious. The maintainance of live cells independent of the body to which they once belonged, involves attending to their preservation and propagation, and offers countless experimental opportunities that would often be difficult to achieve in experimental animals and impossible in human patients. However, cell cultures constitute highly artificial systems, in which the surviving cells have gone through multiple selection processes that have no in vivo counterparts. Moreover, cells in cultures have been removed from their natural context, lost the multitude of regulatory and feedback loop mechanisms, as well as cellular partners, with which they normally interact, and thus resemble a solitary player of a symphony orchestra who has lost the conductor, fellow musicians and whose musical scores got scrambled. One severe limitation of cell culture systems has been the lack of the third dimension: only a very limited number of cell types in any mammalian organ do not extend in the third dimension. Thus, the enforced planar tiling of adjacent cells in cultures is almost always highly artificial and imposes major cellular stress that changes the morphology, rheological and functional properties of such cells.
Astrocyte cell cultures
Astrocytes in the CNS fulfill a wide range of homeostatic functions, are important regulators of neuronal functions and, when made reactive by the disease process, profoundly affect both acute and later responses in brain diseases including neurodegenerative disorders (Pekny and Pekna 2014). Astrocytes also emerge as pivotal regulators of CNS inflammatory mechanisms with important implications for a whole range of neurological diseases (Sofroniew 2015). Astrocyte culture systems have long been used to maintain, propagate and experimentally challenge the brain or spinal cord astrocytes, enabling us to study interactions between astrocytes and other cell types (Booher and Sensenbrenner 1972; McCarthy and de Vellis 1980; Saura 2007). These systems advanced the understanding of the function of astrocytes in healthy brain and spinal cord, including their roles in neurotransmitter recycling, synaptogenesis, metabolism and signaling within the astrocyte network; they also allowed aspects of disease situations to be modelled, such as neurodegenerative diseases, hypoxia, or neurotrauma (Sullivan, Martinez et al. 1976; Cornell-Bell, Finkbeiner et al. 1990; Ellis, McKinney et al. 1995; Abramov, Canevari et al. 2003; Neary, Kang et al. 2003; Wyss-Coray, Loike et al. 2003; Abramov, Canevari et al. 2004; Garwood, Pooler et al. 2011; Lange, Bak et al. 2012; Schousboe 2012). However, the currently available two dimensional (2D) culture systems for astrocytes have many limitations, such as a highly reduced morphological complexity, enforced tiling of adjacent cells, and a prominent undesired baseline reactivity (Lange, Bak et al. 2012; Puschmann, Zanden et al. 2013).
Three-dimensional cell cultures
New methods for astrocyte preparation (Foo, Allen et al. 2011) and three-dimensional cell culture systems (Puschmann, Zanden et al. 2013) allow some of these limitations to be overcome (Figure 8). The undesired baseline reactivity of astrocytes in 2D cultures that exhibit prominent signs of cellular stress is overcome in the newly developed Bioactive3D culture system, a bioactively coated polyurethane nanofiber scaffold-based cell culture system which was previously developed for astrocytes (Puschmann, Zanden et al. 2013) (Figure 9). Levels of intermediate filament proteins GFAP, nestin and vimentin, and of the Hsp70 stress protein, signs of astrocyte reactivity and cellular stress, were several fold lower in astrocytes in Bioactive3D compared to standard 2D cultures (Figure 9). The filopodia of astrocytes grown in Bioactive3D were more motile, and those of adjacent astrocytes interacted much more with each other than in standard 2D culture systems (Puschmann, Zanden et al. 2013). Immunocytochemical and Western blot analyses of connexin-43 showed a large reduction in the number of gap-junction clusters per astrocyte in Bioactive3D, which by supporting their complex morphology allows cell–cell interactions only at certain areas of astrocyte processes, similar to what is seen in vivo (Puschmann, Zanden et al. 2013). This is in contrast to the enforced planar tiling of astrocytes in 2D cultures, which are extensively, but artificially, connected through gap junctions. Astrocytes cultured in Bioactive3D showed decreased proliferation (Puschmann, Zanden et al. 2013), a desirable feature for many in vitro experiments. Stimulation by heparin-binding epidermal growth factor (HB-EGF), which was previously proposed as a serum replacement for astrocyte cultures (Foo, Allen et al. 2011), was sufficient for cell survival, induced Mapk/Erk pathway activation, and supported cell proliferation in both culture systems, with a stronger HB-EGF-induced proliferative response of Bioactive3D grown astrocytes, possibly due to their lower baseline reactivity (Puschmann, Zanden et al. 2013; Puschmann, Zanden et al. 2013).
Figure 8. The Bioactive3D culture system supports the complex cell morphology of astrocytes and comes as a novel tool to study and modulate astrocyte activation in situations such as nerodegeneration, ischemia or neurotrauma.
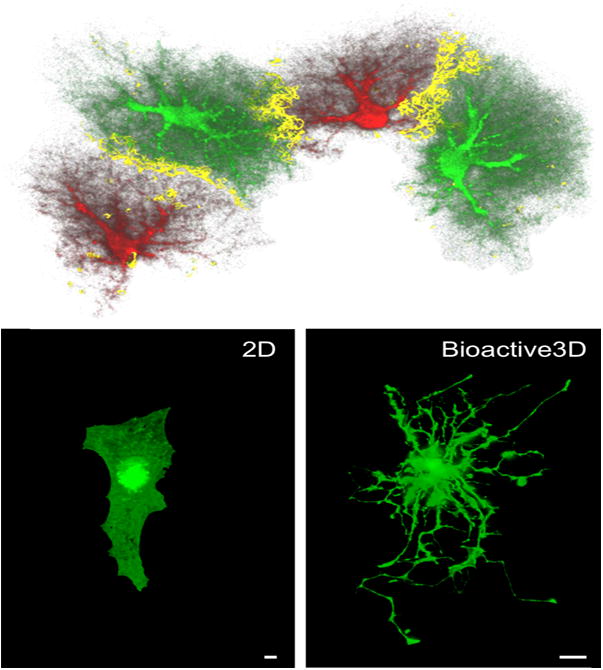
Upper panel. Astrocytes are highly complex cells with a multitude of cellular processes as demonstrated on this 3D reconstruction of adjacent astrocytes in the adult mouse hippocampus. Astrocytes were filled with either Alexa 568 or Lucifer yellow dyes. The gray matter is largely subdivided into individual astrocyte domains. Astrocytes are interconnected via gap junctions into a highly dynamic network, however in the healthy adult brain, astrocytes do enter domains of their astrocyte neighbors. Hence the limited overlap zone between adjacent astrocytes reflecting the interdigitation of fine cellular processes of adjacent astrocytes (shown in yellow). Reproduced from (Wilhelmsson, Bushong et al. 2006). Lower panel. Primary astrocytes cultured in the Bioactive3D system, which is composed of polyurethane nanofibers coated with poly-L-ornithin and laminin (Puschmann, Zanden et al. 2013), preserve some of the complex morphological and biochemical features of in vivo astrocytes that are normally lost upon 2D culture (compare the left and right panel). Space bar, 10 μm. The primary astrocytes were derived from mice expressing enhanced green fluorescein protein in their astrocytes (Nolte, Matyash et al. 2001).
Figure 9. The Bioactive3D culture system highly reduces the cellular stress seen in conventional 2D cell culture systems.
A) and B) Western blot analysis and levels of intermediate filament proteins GFAP, nestin, synemin and vimentin, which are known to be upregulated upon astrocyte activation and cellular stress, in freshly isolated astrocytes and astrocytes cultured in a standard 2D and Bioactive3D system. The undesired baseline activation of astrocytes seen upon 2D culture is absent in Bioactive3D; ***, p<0.001. C) The levels of intermediate filament proteins nestin and vimentin in astrocytes initially cultured in a standard 2D and then transferred to Bioactive3D, decrease correspondingly, indicating that astrocyte cultures can be established and expanded in 2D cultures and move to Bioactive3D just prior to experimental manipulations; days, days after transfer from 2D to Bioactive3D. D) The expression of HSP70, a cellular stress marker, is highly reduced in astrocytes cultured in Bioactive3D. E) Western blot analysis of phosphorylated MapK/Erk1/2 in astrocytes maintained in standard 2D cultures or in Bioactive3D and treated for 1 hour with 100 μM ATP shows that astrocytes grown in Bioactive3D are highly responsive to activation. Modified from (Puschmann, Zanden et al. 2013).
Transferability from 2D- to 3D culture
Astrocytes established and maintained for 7 days in a standard 2D system and then transferred to Bioactive3D behaved as those established in Bioactive3D, thus proving the usefulness of Bioactive3D in situations where the initiation or expansion of cultures in 2D culture systems are preferable or become obligatory (Figure 9). Astrocytes grown in Bioactive3D remained highly responsive to activation. Treatment with ATP (100 μM for 1h following serum deprivation for 4h) led to a comparable activation of the MapK/Erk1/2 signaling cascade in astrocytes grown in Bioactive3D and in a standard 2D system (Figure 9). Thus, Bioactive3D makes astrocytes only minimally reactive under basal conditions and allows experiments in which activation of astrocytes is to be studied - alone or in co-cultures with other cell types - either in a pathological context (e.g. neurodegeneration, trauma or ischemia), or while evaluating possible new treatment strategies aiming at modulation of astrocyte activity. The Bioactive 3D culture system has already proven its usefulness for assessing (patho)physiological responses of astrocytes (Puschmann, Zanden et al. 2013; Puschmann, Zanden et al. 2013) and can be used for identification of clinically relevant targets and leads targeting astrocytes in disease.
The Bioactive3D culture system has now also been optimized to support 3D culture of neurons (Puschmann, de Pablo et al. 2014). Similar to astrocytes, morphologically complex neurons of the brain or spinal cord spread out in all three dimensions, and this opportunity is lost in 2D cell cultures. The Bioactive 3D was modified to support the formation of complex 3D neuronal networks allowing neurons to extend neurites into the z-direction, i.e. into the nanofiber scaffold (Puschmann, de Pablo et al. 2014). The neurite outgrowth was found to be the function of the nanofiber diameter and the pore size (Puschmann, de Pablo et al. 2014). Confocal image analyses of neurons labeled for neuron-specific beta-III-tubulin showed that neurons grown in the 3D system extended their neurites through the entire thickness of the poly-D-lysine coated nanofiber scaffold. Western blot and immunocytochemical analyses showed that despite the presence of a comparable number of glutamine synthetase positive contaminating astrocytes in 2D and 3D culture systems, the astrocytes in the 3D system expressed lower levels of the intermediate filament (nanofilament) protein GFAP, a marker of reactive astrocytes.
In vitro 5-ethynyl-2-deoxyuridine (EdU) labeling and Western blot analysis of the proliferation marker MCM2 demonstrated that the fraction of proliferating astrocytes that contaminated the 3D-grown neuronal cultures was lower than in 2D cultures. This shows that the contaminating astrocytes in 3D neuronal cultures are less reactive and less proliferative than in 2D-grown neuronal cultures. Western blot analysis of the pre-synaptic marker synaptotagmin showed that synaptogenesis, a key attribute of neuronal cell cultures, was not negatively affected by less reactive astrocytes in the 3D system.
Conclusion
As outlined above, the 3D cell culture system for neurons provides a highly useful platform for the growth of and experimentation on live neurons.
Acknowledgments
Donovan Low, Kazuyuki Takata and Florent Ginhoux thank Dr. Lucy Robison for editing the manuscript.
Terrence Town acknowledges support from NIH R01 NS076794, an Alzheimer’s Association Zenith Fellows Award (ZEN-10-174633), an American Federation of Aging Research/Ellison Medical Foundation Julie Martin Mid-Career Award in Aging Research (M11472), and a Cure Alzheimer’s Fund Award, as well as startup funds from the Zilkha Neurogenetic Institute, which helped to make this work possible.
M. Kerry O’Banion acknowledges support from NIH R21 NS048522, NIH R01 AG30149, and awards from the Killian J. and Caroline F. Schmitt Foundation Program on Integrative Brain Research.
Bruce Lamb acknowledges support from NIH R01AG023012, NIH R01NS074804, Alzheimer’s Association and BrightFocus Foundation.
Marco Colonna is supported by Cure Alzheimer’s Fund (CAF) and NIH RF1 AG051485 01.
Gary Landreth acknowledges support trom the Alzheimer’s Association, and NIH R01AG043522.
Clive Holmes received funding from Pfizer pharmaceuticals for some of the clinical trial work.
Katrin I. Andreasson acknowledges support from NIH R01AG030209, NIH R21AG033914, NIH P50 AG047366, Alzheimer’s Association, American Federation for Aging Research, Bright Focus.
Clive Holmes and Jessica Teeling acknowledge funding from the Wellcome Trust and the Alzheimer’s Society UK.
Marina Lynch acknowledges support from Science Foundation Ireland (11/PI/2014) and the Higher Education Authority, Ireland.
Linda J. Van Eldik and Adam D. Bachstetter acknowledge support from NIH R01 NS093920, U01 AG050636, P30 AG028383, K99 AG044445, and grants from the Alzheimer’s Association, Alzheimer’s Drug Discovery Foundation, Kentucky Spinal Cord & Head Injury Research Trust (KSCHIRT), and Thome Memorial Foundation.
Milos Pekny acknowledges support from the Swedish Medical Research Council (project 11548), AFA Research Foundation, ALF Göteborg (project 11392), Sten A. Olsson Foundation for Research and Culture, Hjärnfonden, Hagströmer’s Foundation Millennium, the Swedish Stroke Foundation, Amlöv’s Foundation, NanoNet COST Action (BM1002), the EU FP 7 Program EduGlia (237956), and the EU FP 7 Program TargetBraIn (279017).
Abbreviations
- AD
Alzheimer disease
- BBB
blood-brain barrier
- CNS
central nervous system
- CSF-1R
colony stimulating factor receptor
- DR
delayed rectifier
- EMPs
erythro-myeloid precursors
- ESC
embryonic stem cell
- iPSC
induced pluripotent stem cell
- P2YR12
Purinergic Receptor P2Y, G-Protein Coupled, 12
- TGF-β
transforming growth factor-β
- YS
yolk sac
Footnotes
Conflict of interest disclosures
The authors have no conflict of interests to declare. All experiments were conducted with the ARRIVE guidelines.
References
- Abramov AY, Canevari L, et al. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci. 2003;23(12):5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, et al. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24(2):565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abutbul S, Shapiro J, et al. TGF-beta signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia. 2012;60(7):1160–1171. doi: 10.1002/glia.22343. [DOI] [PubMed] [Google Scholar]
- Ackermann M, Liebhaber S, et al. Lost in translation: pluripotent stem cell-derived hematopoiesis. EMBO Mol Med. 2015 doi: 10.15252/emmm.201505301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aisen PS, Davis KL, et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology. 2000;54(3):588–593. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Schafer KA, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, et al. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14(9):1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, et al. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angcajas AB, Hirai N, et al. Diversity of amino acid signaling pathways on autophagy regulation: A novel pathway for arginine. Biochem Biophys Res Commun. 2014 doi: 10.1016/j.bbrc.2014.01.117. [DOI] [PubMed] [Google Scholar]
- Apelt J, Bigl M, et al. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int J Dev Neurosci. 2004;22(7):475–484. doi: 10.1016/j.ijdevneu.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Apelt J, Schliebs R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001;894(1):21–30. doi: 10.1016/s0006-8993(00)03176-0. [DOI] [PubMed] [Google Scholar]
- Arosio B, Trabattoni D, et al. Interleukin-10 and interleukin-6 gene polymorphisms as risk factors for Alzheimer’s disease. Neurobiol Aging. 2004;25(8):1009–1015. doi: 10.1016/j.neurobiolaging.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Augustin R, Lichtenthaler SF, et al. Bioinformatics identification of modules of transcription factor binding sites in Alzheimer’s disease-related genes by in silico promoter analysis and microarrays. Int J Alzheimers Dis. 2011;2011:154325. doi: 10.4061/2011/154325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avital A, Goshen I, et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. 2003;13(7):826–834. doi: 10.1002/hipo.10135. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Norris CM, et al. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer’s disease-related pathology. J Neurosci. 2012;32(30):10201–10210. doi: 10.1523/JNEUROSCI.1496-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Rowe RK, et al. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci. 2013;33(14):6143–6153. doi: 10.1523/JNEUROSCI.5399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Van Eldik LJ, et al. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun. 2015;3:32. doi: 10.1186/s40478-015-0209-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Webster SJ, et al. Attenuation of traumatic brain injury-induced cognitive impairment in mice by targeting increased cytokine levels with a small molecule experimental therapeutic. J Neuroinflammation. 2015;12 doi: 10.1186/s12974-015-0289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban H, Shigemitsu K, et al. Arginine and Leucine regulate p70 S6 kinase and 4E-BP1 in intestinal epithelial cells. Int J Mol Med. 2004;13(4):537–543. [PubMed] [Google Scholar]
- Banchereau J, Pascual V, et al. From IL-2 to IL-37: the expanding spectrum of anti-inflammatory cytokines. Nat Immunol. 2012;13(10):925–931. doi: 10.1038/ni.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik S, Renner Viveros P, et al. Giardia duodenalis arginine deiminase modulates the phenotype and cytokine secretion of human dendritic cells by depletion of arginine and formation of ammonia. Infect Immun. 2013;81(7):2309–2317. doi: 10.1128/IAI.00004-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Shultz SR, et al. A CD11d Monoclonal Antibody Treatment Reduces Tissue Injury and Improves Neurological Outcome after Fluid Percussion Brain Injury in Rats. J Neurotrauma. 2012;29(14):2375–2392. doi: 10.1089/neu.2012.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, Babcock AA, et al. Accelerated microglial pathology is associated with Abeta plaques in mouse models of Alzheimer’s disease. Aging Cell. 2014;13(4):584–595. doi: 10.1111/acel.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JP, Costello DA, et al. Bone marrow-derived macrophages from aged rats are more responsive to inflammatory stimuli. J Neuroinflammation. 2015;12:67. doi: 10.1186/s12974-015-0287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JP, Minogue AM, et al. Bone marrow-derived macrophages from AbetaPP/PS1 mice are sensitized to the effects of inflammatory stimuli. J Alzheimers Dis. 2015;44(3):949–962. doi: 10.3233/JAD-142076. [DOI] [PubMed] [Google Scholar]
- Ben-David U, Kopper O, et al. Expanding the boundaries of embryonic stem cells. Cell Stem Cell. 2012;10(6):666–677. doi: 10.1016/j.stem.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Bentham P, Gray R, et al. Aspirin in Alzheimer’s disease (AD2000): a randomised open-label trial. Lancet Neurol. 2008;7(1):41–49. doi: 10.1016/S1474-4422(07)70293-4. [DOI] [PubMed] [Google Scholar]
- Bernstein HG, Derst C, et al. The agmatine-degrading enzyme agmatinase: a key to agmatine signaling in rat and human brain? Amino Acids. 2011;40(2):453–465. doi: 10.1007/s00726-010-0657-5. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, et al. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatteis CM, Bealer SL, et al. Suppression of fever after lesions of the anteroventral third ventricle in guinea pigs. Brain Res Bull. 1983;11(5):519–526. doi: 10.1016/0361-9230(83)90124-7. [DOI] [PubMed] [Google Scholar]
- Booher J, Sensenbrenner M. Growth and cultivation of dissociated neurons and glial cells from embryonic chick, rat and human brain in flask cultures. Neurobiology. 1972;2(3):97–105. [PubMed] [Google Scholar]
- Breitner JC, Baker LD, et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7(4):402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne TC, McQuillan K, et al. IFN-gamma Production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J Immunol. 2013;190(5):2241–2251. doi: 10.4049/jimmunol.1200947. [DOI] [PubMed] [Google Scholar]
- Bruttger J, Karram K, et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity. 2015;43(1):92–106. doi: 10.1016/j.immuni.2015.06.012. [DOI] [PubMed] [Google Scholar]
- Brydon L, Harrison NA, et al. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry. 2008;63(11):1022–1029. doi: 10.1016/j.biopsych.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulavina L, Szulzewsky F, et al. NTPDase1 activity attenuates microglial phagocytosis. Purinergic Signal. 2013;9(2):199–205. doi: 10.1007/s11302-012-9339-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchart J, Brook L, et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. 2015;84(21):2161–2168. doi: 10.1212/WNL.0000000000001617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, De Pinto V, et al. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014;34(23):7988–7998. doi: 10.1523/JNEUROSCI.0777-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Chakrabarty P, Ceballos-Diaz C, et al. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184(9):5333–5343. doi: 10.4049/jimmunol.0903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Herring A, et al. Hippocampal expression of murine TNFalpha results in attenuation of amyloid deposition in vivo. Mol Neurodegener. 2011;6:16. doi: 10.1186/1750-1326-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Jansen-West K, et al. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24(2):548–559. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Li A, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85(3):519–533. doi: 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Tianbai L, et al. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol Neurodegener. 2012;7:36. doi: 10.1186/1750-1326-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, et al. Arginase 1+ microglia reduce Abeta plaque deposition during IL-1beta-dependent neuroinflammation. J Neuroinflammation. 2015;12(1):203. doi: 10.1186/s12974-015-0411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combrinck M, Williams J, et al. Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2006;77(1):85–88. doi: 10.1136/jnnp.2005.063131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, et al. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247(4941):470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Costello DA, Watson MB, et al. Interleukin-1alpha and HMGB1 mediate hippocampal dysfunction in SIGIRR-deficient mice. J Neurosci. 2011;31(10):3871–3879. doi: 10.1523/JNEUROSCI.6676-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78(2):256–268. doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65(4):304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, et al. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25(40):9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damani MR, Zhao L, et al. Age-related alterations in the dynamic behavior of microglia. Aging Cell. 2011;10(2):263–276. doi: 10.1111/j.1474-9726.2010.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, et al. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel B, Nagy G, et al. The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes Dev. 2014;28(14):1562–1577. doi: 10.1101/gad.242685.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Konsman JP, et al. Neural and humoral pathways of communication from the immune system to the brain: parallel or convergent? Auton Neurosci. 2000;85(1-3):60–65. doi: 10.1016/S1566-0702(00)00220-4. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- de Jong D, Jansen R, et al. No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PLoS One. 2008;3(1):e1475. doi: 10.1371/journal.pone.0001475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Los Angeles A, Ferrari F, et al. Hallmarks of pluripotency. Nature. 2015;525(7570):469–478. doi: 10.1038/nature15515. [DOI] [PubMed] [Google Scholar]
- de Rham C, Villard J. Potential and limitation of HLA-based banking of human pluripotent stem cells for cell therapy. J Immunol Res. 2014;2014:518135. doi: 10.1155/2014/518135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio-Hortega P. Microglia. In: P W, editor. Cytology and Cellular Pathology of the Nervous System. New York: Hoeber; 1932. 482–1924–1534. [Google Scholar]
- Di Bona D, Rizzo C, et al. Association between interleukin-10 polymorphisms and Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2012;29(4):751–759. doi: 10.3233/JAD-2012-111838. [DOI] [PubMed] [Google Scholar]
- Dibaj P, Steffens H, et al. In Vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. PLoS One. 2011;6(3):e17910. doi: 10.1371/journal.pone.0017910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, et al. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22(6):1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Diorio D, Welner SA, et al. Peripheral benzodiazepine binding sites in Alzheimer’s disease frontal and temporal cortex. Neurobiol Aging. 1991;12(3):255–258. doi: 10.1016/0197-4580(91)90106-t. [DOI] [PubMed] [Google Scholar]
- DiPatre PL, Gelman BB. Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol. 1997;56(2):143–149. doi: 10.1097/00005072-199702000-00004. [DOI] [PubMed] [Google Scholar]
- Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010;285(29):21978–21987. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209(2):378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzis EJ, Tronson NC. Modulation of learning and memory by cytokines: signaling mechanisms and long term consequences. Neurobiol Learn Mem. 2014;115:68–77. doi: 10.1016/j.nlm.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorn KJ, Lucassen PJ, et al. Emerging roles of microglial activation and non-motor symptoms in Parkinson’s disease. Prog Neurobiol. 2012;98(2):222–238. doi: 10.1016/j.pneurobio.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Drake C, Boutin H, et al. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav Immun. 2011;25(6):1113–1122. doi: 10.1016/j.bbi.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P, Archer HA, et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32(3):412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Egan KM, Lawson JA, et al. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306(5703):1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- Eglitis MA, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc Natl Acad Sci U S A. 1997;94(8):4080–4085. doi: 10.1073/pnas.94.8.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148(6):1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11(11):1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- Ek M, Kurosawa M, et al. Activation of vagal afferents after intravenous injection of interleukin-1beta: role of endogenous prostaglandins. J Neurosci. 1998;18(22):9471–9479. doi: 10.1523/JNEUROSCI.18-22-09471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kasmi KC, Qualls JE, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. 2008;9(12):1399–1406. doi: 10.1038/ni.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis EF, McKinney JS, et al. A new model for rapid stretch-induced injury of cells in culture: characterization of the model using astrocytes. J Neurotrauma. 1995;12(3):325–339. doi: 10.1089/neu.1995.12.325. [DOI] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber K, Markworth S, et al. The ectonucleotidase cd39/ENTPDase1 modulates purinergic-mediated microglial migration. Glia. 2008;56(3):331–341. doi: 10.1002/glia.20606. [DOI] [PubMed] [Google Scholar]
- Faul M, Xu L, et al. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control Atlanta (GA) 2010 [Google Scholar]
- Feldman HH, Doody RS, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956–964. doi: 10.1212/WNL.0b013e3181d6476a. [DOI] [PubMed] [Google Scholar]
- Fenn AM, Hall JC, et al. IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci. 2014;34(26):8904–8917. doi: 10.1523/JNEUROSCI.1146-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Smith KM, et al. Increased micro-RNA 29b in the aged brain correlates with the reduction of insulin-like growth factor-1 and fractalkine ligand. Neurobiol Aging. 2013;34(12):2748–2758. doi: 10.1016/j.neurobiolaging.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field R, Campion S, et al. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. 2010;24(6):996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Tapias V, et al. Opposing effects of Apoe/Apoa1 double deletion on amyloid-beta pathology and cognitive performance in APP mice. Brain. 2015;138(Pt 12):3699–3715. doi: 10.1093/brain/awv293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine SN, Sabbagh JJ, et al. Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol Life Sci. 2015;72(10):1863–1879. doi: 10.1007/s00018-015-1839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontainhas AM, Wang M, et al. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One. 2011;6(1):e15973. doi: 10.1371/journal.pone.0015973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, et al. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71(5):799–811. doi: 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol. 2007;50(5):470–479. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- Furman JL, Sama DM, et al. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32(46):16129–16140. doi: 10.1523/JNEUROSCI.2323-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, et al. Alzheimer-Type Neuropathology in Transgenic Mice Overexpressing V717f Beta-Amyloid Precursor Protein. Nature. 1995;373(6514):523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Garwood CJ, Pooler AM, et al. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167. doi: 10.1038/cddis.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Wu MD, et al. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33(11):5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, et al. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol. 2015;7(8):a020537. doi: 10.1101/cshperspect.a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature. 2015;518(7539):365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol Allergy Clin North Am. 2009;29(2):321–337. doi: 10.1016/j.iac.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Golde TE, Streit WJ, et al. Alzheimer’s disease risk alleles in TREM2 illuminate innate immunity in Alzheimer’s disease. Alzheimers Res Ther. 2013;5(3):24. doi: 10.1186/alzrt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldeck D, Witkowski JM, et al. Peripheral immune signatures in Alzheimer Disease. Curr Alzheimer Res. 2016 doi: 10.2174/1567205013666160222112444. [DOI] [PubMed] [Google Scholar]
- Gomez-Nicola D, Perry VH. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist. 2015;21(2):169–184. doi: 10.1177/1073858414530512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Link VM, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159(6):1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabert K, Michoel T, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016 doi: 10.1038/nn.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham SF, Chevallier OP, et al. Untargeted metabolomic analysis of human plasma indicates differentially affected polyamine and L-arginine metabolism in mild cognitive impairment subjects converting to Alzheimer’s disease. PLoS One. 2015;10(3):e0119452. doi: 10.1371/journal.pone.0119452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Lelios I, et al. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front Immunol. 2015;6:249. doi: 10.3389/fimmu.2015.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Doty KR, et al. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015;85(3):534–548. doi: 10.1016/j.neuron.2014.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Doty KR, et al. Innate Immunity Fights Alzheimer’s Disease. Trends Neurosci. 2015;38(11):674–681. doi: 10.1016/j.tins.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta VK, Scheunemann L, et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat Neurosci. 2013;16(10):1453–1460. doi: 10.1038/nn.3512. [DOI] [PubMed] [Google Scholar]
- Halaris A, Plietz J. Agmatine : metabolic pathway and spectrum of activity in brain. CNS Drugs. 2007;21(11):885–900. doi: 10.2165/00023210-200721110-00002. [DOI] [PubMed] [Google Scholar]
- Hall ED. The contributing role of lipid peroxidation and protein oxidation in the course of CNS injury neurodegeneration and neuroprotection: an overview. In: Kobeissy FHP, editor. Brain Neurotrauma: Molecular Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): 2015. [PubMed] [Google Scholar]
- Hansmannel F, Sillaire A, et al. Is the urea cycle involved in Alzheimer’s disease? J Alzheimers Dis. 2010;21(3):1013–1021. doi: 10.3233/JAD-2010-100630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington C, Sawchak S, et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: two phase 3 studies. Curr Alzheimer Res. 2011;8(5):592–606. doi: 10.2174/156720511796391935. [DOI] [PubMed] [Google Scholar]
- Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- Hefendehl JK, Neher JJ, et al. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. 2014;13(1):60–69. doi: 10.1111/acel.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein AM, Stasko MR, et al. Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24(2):243–253. doi: 10.1016/j.bbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Golenbock DT, et al. Innate immunity in Alzheimer’s disease. Nat Immunol. 2015;16(3):229–236. doi: 10.1038/ni.3102. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, et al. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–477. doi: 10.1038/nri3705. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Landreth GE, et al. Drug insight: effects mediated by peroxisome proliferator-activated receptor-gamma in CNS disorders. Nat Clin Pract Neurol. 2007;3(9):496–504. doi: 10.1038/ncpneuro0586. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease. J Neuroimmunol. 2007;184(1-2):69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Ransohoff RM, et al. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- Herbomel P, Thisse B, et al. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. 2001;238(2):274–288. doi: 10.1006/dbio.2001.0393. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, et al. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Chen J, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42(4):665–678. doi: 10.1016/j.immuni.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Wang Y, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. 2012;209(6):1167–1181. doi: 10.1084/jem.20120340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Cunningham C, et al. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77(3):212–218. doi: 10.1212/WNL.0b013e318225ae07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Cunningham C, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Witkowsky LB, et al. Nucleosomes impede Cas9 access to DNA and. Elife. 2016;5 doi: 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Niemi EC, et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma. 2014;31(20):1677–1688. doi: 10.1089/neu.2013.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh EC, Knebel SM, et al. Deprivation of arginine by recombinant human arginase in prostate cancer cells. J Hematol Oncol. 2012;5:17. doi: 10.1186/1756-8722-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt JB, Jr, Nash KR, et al. Sustained Arginase 1 Expression Modulates Pathological Tau Deposits in a Mouse Model of Tauopathy. J Neurosci. 2015;35(44):14842–14860. doi: 10.1523/JNEUROSCI.3959-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- in t’ Veld BA, Ruitenberg A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345(21):1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Inoue K, Tsutsui H, et al. Metabolic profiling of Alzheimer’s disease brains. Scientific reports. 2013;3:2364. doi: 10.1038/srep02364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212(3):287–295. doi: 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Yu JT, et al. CD33 in Alzheimer’s disease. Mol Neurobiol. 2014;49(1):529–535. doi: 10.1007/s12035-013-8536-1. [DOI] [PubMed] [Google Scholar]
- Johansson JU, Pradhan S, Lokteva LA, Woodling NS, Ko N, Brown HD, Wang Q, Loh C, Cekanaviciute E, Buckwalter M, Manning-Boğ AB, Andreasson KI. Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. J Neurosci. 2013;33:16012–16032. doi: 10.1523/JNEUROSCI.2203-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson JU, Woodling NS, et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J Clin Invest. 2015;125(1):350–364. doi: 10.1172/JCI77487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136(Pt 1):28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, et al. Traumatic brain injury and amyloid-β pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11(5):361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RS, Minogue AM, et al. Inhibition of JAK2 attenuates the increase in inflammatory markers in microglia from APP/PS1 mice. Neurobiol Aging. 2015;36(10):2716–2724. doi: 10.1016/j.neurobiolaging.2015.04.018. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju L, Zeng H, et al. Dual polarization of microglia isolated from mixed glial cell cultures. J Neurosci Res. 2015;93(9):1345–1352. doi: 10.1002/jnr.23563. [DOI] [PubMed] [Google Scholar]
- Kan MJ, Lee JE, et al. Arginine deprivation and immune suppression in a mouse model of Alzheimer’s disease. J Neurosci. 2015;35(15):5969–5982. doi: 10.1523/JNEUROSCI.4668-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara K, Suenobu M, et al. Intracerebral microinjection of interleukin-4/interleukin-13 reduces beta-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience. 2012;207:243–260. doi: 10.1016/j.neuroscience.2012.01.049. [DOI] [PubMed] [Google Scholar]
- Keene CD, Chang RC, et al. Suppressed accumulation of cerebral amyloid {beta} peptides in aged transgenic Alzheimer’s disease mice by transplantation with wild-type or prostaglandin E2 receptor subtype 2-null bone marrow. Am J Pathol. 2010;177(1):346–354. doi: 10.2353/ajpath.2010.090840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RJ, Minogue AM, et al. Glial Activation in A beta PP/PS1 Mice is Associated with Infiltration of IFN gamma-Producing Cells. Journal of Alzheimers Disease. 2013;37(1):63–75. doi: 10.3233/JAD-130539. [DOI] [PubMed] [Google Scholar]
- Kennedy M, Awong G, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2(6):1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, et al. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, et al. Microglia: new roles for the synaptic stripper. Neuron. 2013;77(1):10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, Eggan K. Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest. 2010;120(1):51–59. doi: 10.1172/JCI40553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Oddo S, et al. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25(39):8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Okuyama S, et al. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. Faseb J. 2010;24(8):3093–3102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeglsperger T, Li S, et al. Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-beta1 in the CNS. Glia. 2013;61(6):985–1002. doi: 10.1002/glia.22490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzbauer PT, Giasson BI, et al. Fibrillization of alpha-synuclein and tau in familial Parkinson’s disease caused by the A53T alpha-synuclein mutation. Experimental Neurology. 2004;187(2):279–288. doi: 10.1016/j.expneurol.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Krabbe G, Halle A, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8(4):e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl WC, Lyoo CH, et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain. 2013;136(Pt 7):2228–2238. doi: 10.1093/brain/awt145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, et al. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;71(5):833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Lachmann N, Ackermann M, et al. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Reports. 2015;4(2):282–296. doi: 10.1016/j.stemcr.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Heath S, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SC, Bak LK, et al. Primary cultures of astrocytes: their value in understanding astrocytes in health and disease. Neurochem Res. 2012;37(11):2569–2588. doi: 10.1007/s11064-012-0868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laouar Y, Town T, et al. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105(31):10865–10870. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, et al. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lazarus M, Yoshida K, et al. EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci. 2007;10(9):1131–1133. doi: 10.1038/nn1949. [DOI] [PubMed] [Google Scholar]
- Lee DC, Rizer J, et al. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. Journal of neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Rizer J, et al. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Ruiz CR, et al. Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol Aging. 2013;34(6):1610–1620. doi: 10.1016/j.neurobiolaging.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Varvel NH, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med. 2010;363(7):689–691. doi: 10.1056/NEJMcibr1007320. [DOI] [PubMed] [Google Scholar]
- Lefterov I, Schug J, et al. RNA-sequencing reveals transcriptional up-regulation of Trem2 in response to bexarotene treatment. Neurobiol Dis. 2015;82:132–140. doi: 10.1016/j.nbd.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letiembre M, Liu Y, et al. Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Li G, Regunathan S, et al. Agmatine: an endogenous clonidine-displacing substance in the brain. Science. 1994;263(5149):966–969. doi: 10.1126/science.7906055. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity. 2008;28(4):468–476. doi: 10.1016/j.immuni.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Li Y, Du XF, et al. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell. 2012;23(6):1189–1202. doi: 10.1016/j.devcel.2012.10.027. [DOI] [PubMed] [Google Scholar]
- Liang X, Lin L, Wang Q, Woodling NS, Anacker C, Pan T, Merchant M, Andreasson K. Neuronal and vascular protection by the prostaglandin E2 EP4 receptor in a mouse model of cerebral ischemia. J Clin Invest. 2011;121(11):4362–4371. doi: 10.1172/JCI46279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, Lokteva L, Nguyen T, Breyer RM, Montine TM, Andreasson K. The PGE2 PE2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Soc Neuroscience. 2007;2007(160.16) doi: 10.1002/ana.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, et al. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci. 2005;25(44):10180–10187. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang Q, et al. The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann Neurol. 2008;64(3):304–314. doi: 10.1002/ana.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfoot HL, Hall J. Endogenous polyamine function--the RNA perspective. Nucleic Acids Res. 2014;42(18):11275–11290. doi: 10.1093/nar/gku837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio D, Licastro F, et al. Interleukin-10 promoter polymorphism in sporadic Alzheimer’s disease. Genes Immun. 2003;4(3):234–238. doi: 10.1038/sj.gene.6363964. [DOI] [PubMed] [Google Scholar]
- Liu P, Fleete MS, et al. Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Liu Y, Qiang M, et al. A novel molecular mechanism for nitrated {alpha}-synuclein-induced cell death. Journal of molecular cell biology. 2011;3(4):239–249. doi: 10.1093/jmcb/mjr011. [DOI] [PubMed] [Google Scholar]
- Lloyd E, Somera-Molina K, et al. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7(4):366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher CE, Mills KH, et al. Interleukin-1 receptor antagonist exerts agonist activity in the hippocampus independent of the interleukin-1 type I receptor. J Neuroimmunol. 2003;137(1-2):117–124. doi: 10.1016/s0165-5728(03)00072-9. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64(1):110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Li S, et al. Meta-analysis of the association between CR1 polymorphisms and risk of late-onset Alzheimer’s disease. Neurosci Lett. 2014;578:165–170. doi: 10.1016/j.neulet.2014.06.055. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Age-related neuroinflammatory changes negatively impact on neuronal function. Front Aging Neurosci. 2010;1:6. doi: 10.3389/neuro.24.006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. Neuroinflammatory changes negatively impact on LTP: A focus on IL-1beta. Brain Res. 2015;1621:197–204. doi: 10.1016/j.brainres.2014.08.040. [DOI] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, et al. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27(31):8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Lynch AM, et al. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. 2009;110(5):1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- Ma SL, T NL, Lam LC, Chiu HF. The association between promoter polymorphism of the interleukin-10 gene and Alzheimer’s disease. Neurobiol Aging. 2005;26:1005–1010. doi: 10.1016/j.neurobiolaging.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Madeo F, Eisenberg T, et al. Spermidine: a novel autophagy inducer and longevity elixir. Autophagy. 2010;6(1):160–162. doi: 10.4161/auto.6.1.10600. [DOI] [PubMed] [Google Scholar]
- Malm TM, Jay TR, et al. The evolving biology of microglia in Alzheimer’s disease. Neurotherapeutics. 2015;12(1):81–93. doi: 10.1007/s13311-014-0316-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Maphis N, Xu G, et al. Loss of tau rescues inflammation-mediated neurodegeneration. Front Neurosci. 2015;9:196. doi: 10.3389/fnins.2015.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N, Xu G, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(Pt 6):1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Morselli E, et al. Longevity-relevant regulation of autophagy at the level of the acetylproteome. Autophagy. 2011;7(6):647–649. doi: 10.4161/auto.7.6.15191. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, et al. Alternative activation of macrophages: an immunologic functional perspective. Annual Review of Immunology. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Matousek SB, Ghosh S, et al. Chronic IL-1beta-mediated neuroinflammation mitigates amyloid pathology in a mouse model of Alzheimer’s disease without inducing overt neurodegeneration. J Neuroimmune Pharmacol. 2012;7(1):156–164. doi: 10.1007/s11481-011-9331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K, Kobayashi S. Signaling the brain in inflammation: the role of endothelial cells. Front Biosci. 2004;9:2819–2826. doi: 10.2741/1439. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Schulzer M, et al. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- McManus RM, Higgins SC, et al. Respiratory infection promotes T cell infiltration and amyloid-beta deposition in APP/PS1 mice. Neurobiol Aging. 2014;35(1):109–121. doi: 10.1016/j.neurobiolaging.2013.07.025. [DOI] [PubMed] [Google Scholar]
- McManus RM, Mills KH, et al. T Cells-Protective or Pathogenic in Alzheimer’s Disease? J Neuroimmune Pharmacol. 2015 doi: 10.1007/s11481-015-9612-2. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Prediger RD, et al. Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J Neurosci. 2007;27(20):5394–5404. doi: 10.1523/JNEUROSCI.5047-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre SD, Tsai CA, et al. Microglial Malfunction: The Third Rail in the Development of Alzheimer’s Disease. Trends Neurosci. 2015;38(10):621–636. doi: 10.1016/j.tins.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Mack M, et al. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132(Pt 9):2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- Minami M, Shimizu K, et al. Prostaglandin E receptor type 4-associated protein interacts directly with NF-kappaB1 and attenuates macrophage activation. J Biol Chem. 2008;283(15):9692–9703. doi: 10.1074/jbc.M709663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minarini A, Zini M, et al. Synthetic polyamines activating autophagy: effects on cancer cell death. Eur J Med Chem. 2013;67:359–366. doi: 10.1016/j.ejmech.2013.06.044. [DOI] [PubMed] [Google Scholar]
- Minogue AM, Jones RS, et al. Age-associated dysregulation of microglial activation is coupled with enhanced blood-brain barrier permeability and pathology in APP/PS1 mice. Neurobiol Aging. 2014;35(6):1442–1452. doi: 10.1016/j.neurobiolaging.2013.12.026. [DOI] [PubMed] [Google Scholar]
- Minogue AM, Lynch AM, et al. Modulation of amyloid-beta-induced and age-associated changes in rat hippocampus by eicosapentaenoic acid. J Neurochem. 2007;103(3):914–926. doi: 10.1111/j.1471-4159.2007.04848.x. [DOI] [PubMed] [Google Scholar]
- Minois N, Carmona-Gutierrez D, et al. Polyamines in aging and disease. Aging (Albany NY) 2011;3(8):716–732. doi: 10.18632/aging.100361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbronn M, Dietz K, et al. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001;101(3):249–255. doi: 10.1007/s004010000284. [DOI] [PubMed] [Google Scholar]
- Molderings GJ, Haenisch B. Agmatine (decarboxylated L-arginine): physiological role and therapeutic potential. Pharmacol Ther. 2012;133(3):351–365. doi: 10.1016/j.pharmthera.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Milatovic D, et al. Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. J Neurochem. 2002;83(2):463–470. doi: 10.1046/j.1471-4159.2002.01157.x. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Sidell KR, et al. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 1999;53(7):1495–1498. doi: 10.1212/wnl.53.7.1495. [DOI] [PubMed] [Google Scholar]
- Moore AH, Wu M, et al. Sustained expression of interleukin-1beta in mouse hippocampus impairs spatial memory. Neuroscience. 2009;164(4):1484–1495. doi: 10.1016/j.neuroscience.2009.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, et al. Modulation of immune response by head injury. Injury. 2007;38(12):1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Morganti JM, Jopson TD, et al. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. 2015;35(2):748–760. doi: 10.1523/JNEUROSCI.2405-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM., Jr Recent advances in arginine metabolism. Curr Opin Clin Nutr Metab Care. 2004;7(1):45–51. doi: 10.1097/00075197-200401000-00009. [DOI] [PubMed] [Google Scholar]
- Morrison LD, Kish SJ. Brain polyamine levels are altered in Alzheimer’s disease. Neurosci Lett. 1995;197(1):5–8. doi: 10.1016/0304-3940(95)11881-v. [DOI] [PubMed] [Google Scholar]
- Morselli E, Galluzzi L, et al. Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging (Albany NY) 2009;1(12):961–970. doi: 10.18632/aging.100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli E, Marino G, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol. 2011;192(4):615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20(11):4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ. Neurotoxicity of Amyloid beta-Protein: Synaptic and Network Dysfunction. Cold Spring Harb Perspect Med. 2012;2(7):a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaki A, Gotoh T, et al. Coinduction of nitric oxide synthase, argininosuccinate synthetase, and argininosuccinate lyase in lipopolysaccharide-treated rats. RNA blot, immunoblot, and immunohistochemical analyses. J Biol Chem. 1996;271(5):2658–2662. doi: 10.1074/jbc.271.5.2658. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima F, Tokunaga K, et al. Human leukocyte antigen matching estimations in a hypothetical bank of human embryonic stem cell lines in the Japanese population for use in cell transplantation therapy. Stem Cells. 2007;25(4):983–985. doi: 10.1634/stemcells.2006-0566. [DOI] [PubMed] [Google Scholar]
- Nakatsuji N, Nakajima F, et al. HLA-haplotype banking and iPS cells. Nat Biotech. 2008;26:739–740. doi: 10.1038/nbt0708-739. [DOI] [PubMed] [Google Scholar]
- Nau R, Sorgel F, et al. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858–883. doi: 10.1128/CMR.00007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary JT, Kang Y, et al. Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. J Neurosci. 2003;23(6):2348–2356. doi: 10.1523/JNEUROSCI.23-06-02348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson LN, Arendash GW, et al. Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol Aging. 2004;25(9):1153–1167. doi: 10.1016/j.neurobiolaging.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, et al. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nolan Y, Maher FO, et al. Role of interleukin-4 in regulation of age-related inflammatory changes in the hippocampus. J Biol Chem. 2005;280(10):9354–9362. doi: 10.1074/jbc.M412170200. [DOI] [PubMed] [Google Scholar]
- Nolte C, Matyash M, et al. GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia. 2001;33(1):72–86. [PubMed] [Google Scholar]
- Norden DM, Muccigrosso MM, et al. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96:29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuki M, Takahashi K. Present and future challenges of induced pluripotent stem cells. Philos Trans R Soc Lond B Biol Sci. 2015;370(1680) doi: 10.1098/rstb.2014.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello A, Edison P, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72(1):56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Biber K, et al. Microglia phenotype diversity. CNS Neurol Disord Drug Targets. 2011;10(1):108–118. doi: 10.2174/187152711794488575. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MM, Atagi Y, et al. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener. 2015;10:43. doi: 10.1186/s13024-015-0040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Park IH, Arora N, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134(5):877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patassini S, Begley P, et al. Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington’s disease. Biochem Biophys Res Commun. 2015 doi: 10.1016/j.bbrc.2015.10.140. [DOI] [PubMed] [Google Scholar]
- Patterson SL. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1beta, BDNF and synaptic plasticity. Neuropharmacology. 2015;96(Pt A):11–18. doi: 10.1016/j.neuropharm.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94(4):1077–1098. doi: 10.1152/physrev.00041.2013. [DOI] [PubMed] [Google Scholar]
- Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133(5):916–927. doi: 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010;120(3):277–286. doi: 10.1007/s00401-010-0722-x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988;11(6):273–277. doi: 10.1016/0166-2236(88)90110-5. [DOI] [PubMed] [Google Scholar]
- Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- Perry VH, Newman TA, et al. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4(2):103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, et al. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Peters D, Berger J, et al. Arginase and Arginine Decarboxylase - Where Do the Putative Gate Keepers of Polyamine Synthesis Reside in Rat Brain? PLoS One. 2013;8(6):e66735. doi: 10.1371/journal.pone.0066735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevalova M, Filipcik P, et al. Post-translational modifications of tau protein. Bratisl Lek Listy. 2006;107(9-10):346–353. [PubMed] [Google Scholar]
- Prieto GA, Snigdha S, et al. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1beta in the aged hippocampus. Proc Natl Acad Sci U S A. 2015;112(36):E5078–5087. doi: 10.1073/pnas.1514486112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop S, Miller KR, et al. Impact of peripheral myeloid cells on amyloid-beta pathology in Alzheimer’s disease-like mice. J Exp Med. 2015;212(11):1811–1818. doi: 10.1084/jem.20150479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntener U, Booth SG, et al. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puschmann TB, de Pablo Y, et al. A novel method for three-dimensional culture of central nervous system neurons. Tissue Eng Part C Methods. 2014;20(6):485–492. doi: 10.1089/ten.TEC.2013.0445. [DOI] [PubMed] [Google Scholar]
- Puschmann TB, Zanden C, et al. Bioactive 3D cell culture system minimizes cellular stress and maintains the in vivo-like morphological complexity of astroglial cells. Glia. 2013;61(3):432–440. doi: 10.1002/glia.22446. [DOI] [PubMed] [Google Scholar]
- Puschmann TB, Zanden C, et al. HB-EGF affects astrocyte morphology, proliferation, differentiation, and the expression of intermediate filament proteins. Journal of neurochemistry. 2013 doi: 10.1111/jnc.12519. [DOI] [PubMed] [Google Scholar]
- Quirie A, Demougeot C, et al. Effect of stroke on arginase expression and localization in the rat brain. Eur J Neurosci. 2013;37(7):1193–1202. doi: 10.1111/ejn.12111. [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, et al. Inflammation after Trauma: Microglial Activation and Traumatic Brain Injury. Ann Neurol. 2011;70(3):374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Schafer D, et al. Neuroinflammation: Ways in Which the Immune System Affects the Brain. Neurotherapeutics. 2015 doi: 10.1007/s13311-015-0385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines SA, Block GA, et al. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62(1):66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- Reyes JF, Fu Y, et al. Tyrosine nitration within the proline-rich region of Tau in Alzheimer’s disease. The American Journal of Pathology. 2011;178(5):2275–2285. doi: 10.1016/j.ajpath.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JF, Geula C, et al. Selective tau tyrosine nitration in non-AD tauopathies. Acta neuropathologica. 2012;123(1):119–132. doi: 10.1007/s00401-011-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JF, Reynolds MR, et al. A possible link between astrocyte activation and tau nitration in Alzheimer’s disease. Neurobiology of disease. 2008;31(2):198–208. doi: 10.1016/j.nbd.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Reyes JF, et al. Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer’s disease and other tauopathies. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26(42):10636–10645. doi: 10.1523/JNEUROSCI.2143-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie P, Male D. Mesoglia & microglia--a historical review of the concept of mononuclear phagocytes within the central nervous system. J Hist Neurosci. 2002;11(4):325–374. doi: 10.1076/jhin.11.4.325.8531. [DOI] [PubMed] [Google Scholar]
- Rivera-Escalera F, Matousek SB, et al. Interleukin-1beta mediated amyloid plaque clearance is independent of CCR2 signaling in the APP/PS1 mouse model of Alzheimer’s disease. Neurobiol Dis. 2014;69:124–133. doi: 10.1016/j.nbd.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- Robinson SR, Bishop GM, et al. Lessons from the AN 1792 Alzheimer vaccine: lest we forget. Neurobiol Aging. 2004;25(5):609–615. doi: 10.1016/j.neurobiolaging.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Rodriguez PC, Hernandez CP, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202(7):931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Kirby LC, et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology. 1993;43(8):1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. 2011;31(45):16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Nayak D, et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505(7482):223–+. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SM, Grum-Tokars VL, et al. Targeting human central nervous system protein kinases: An isoform selective p38alphaMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem Neurosci. 2015;6(4):666–680. doi: 10.1021/acschemneuro.5b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samokhvalov IM, Samokhvalova NI, et al. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature. 2007;446(7139):1056–1061. doi: 10.1038/nature05725. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura J. Microglial cells in astroglial cultures: a cautionary note. J Neuroinflammation. 2007;4:26. doi: 10.1186/1742-2094-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage JC, Jay T, et al. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J Neurosci. 2015;35(16):6532–6543. doi: 10.1523/JNEUROSCI.4586-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaraj N, You M, et al. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr Mol Med. 2010;10(4):405–412. doi: 10.2174/156652410791316995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savchenko VL, McKanna JA, et al. Microglia and astrocytes in the adult rat brain: comparative immunocytochemical analysis demonstrates the efficacy of lipocortin 1 immunoreactivity. Neuroscience. 2000;96(1):195–203. doi: 10.1016/s0306-4522(99)00538-2. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, et al. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia. 2013;61(1):24–36. doi: 10.1002/glia.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T, Nitsch R, et al. Astrocyte-released cytokines induce ramification and outward K+ channel expression in microglia via distinct signalling pathways. Eur J Neurosci. 2001;14(3):463–473. doi: 10.1046/j.0953-816x.2001.01661.x. [DOI] [PubMed] [Google Scholar]
- Schneider H, Pitossi F, et al. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci U S A. 1998;95(13):7778–7783. doi: 10.1073/pnas.95.13.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe A. Studies of Brain Metabolism: A Historical Perspective. Adv Neurol. 2012;4:909–920. [Google Scholar]
- Schulz C, Gomez Perdiguero E, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Carlson TJ, et al. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27(35):9301–9309. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Griffin WS, et al. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, et al. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117(6):1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J, Ruedl C, et al. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity. 2015;43(2):382–393. doi: 10.1016/j.immuni.2015.07.016. [DOI] [PubMed] [Google Scholar]
- Shi J, Johansson J, et al. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol. 2010;184(12):7207–7218. doi: 10.4049/jimmunol.0903487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang Q, et al. Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann Neurol. 2012;72(5):788–798. doi: 10.1002/ana.23677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau CE, Monk KR, et al. An anti-inflammatory NOD-like receptor is required for microglia development. Cell Rep. 2013;5(5):1342–1352. doi: 10.1016/j.celrep.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie FS, Breyer RM, et al. Microglia lacking E Prostanoid Receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am J Pathol. 2005;166(4):1163–1172. doi: 10.1016/s0002-9440(10)62336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie FS, Montine KS, et al. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia. 2005;52(1):70–77. doi: 10.1002/glia.20220. [DOI] [PubMed] [Google Scholar]
- Shively S, Scher AI, et al. Dementia resulting from traumatic brain injury: what is the pathology? Arch Neurol. 2012;69(10):1245–1251. doi: 10.1001/archneurol.2011.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieger D, Moritz C, et al. Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev Cell. 2012;22(6):1138–1148. doi: 10.1016/j.devcel.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Sieger D, Peri F. Animal models for studying microglia: the first, the popular, and the new. Glia. 2013;61(1):3–9. doi: 10.1002/glia.22385. [DOI] [PubMed] [Google Scholar]
- Sierra A, Abiega O, et al. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. doi: 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore AC, et al. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55(4):412–424. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- Simons M, Schwarzler F, et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol. 2002;52(3):346–350. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- Skerrett R, Malm T, et al. Nuclear Receptors in Neurodegenerative Diseases. Neurobiol Dis. 2014 doi: 10.1016/j.nbd.2014.05.019. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerrett R, Pellegrino MP, et al. Combined Liver X Receptor/Peroxisome Proliferator-activated Receptor gamma Agonist Treatment Reduces Amyloid beta Levels and Improves Behavior in Amyloid Precursor Protein/Presenilin 1 Mice. J Biol Chem. 2015;290(35):21591–21602. doi: 10.1074/jbc.M115.652008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawik H, Volk B, et al. Microglial expression of prostaglandin EP3 receptor in excitotoxic lesions in the rat striatum. Neurochem Int. 2004;45(5):653–660. doi: 10.1016/j.neuint.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Smith AM, Dragunow M. The human side of microglia. Trends Neurosci. 2014;37(3):125–135. doi: 10.1016/j.tins.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoki T, Nagasaki A, et al. Coinduction of nitric-oxide synthase and arginase I in cultured rat peritoneal macrophages and rat tissues in vivo by lipopolysaccharide. J Biol Chem. 1997;272(6):3689–3693. doi: 10.1074/jbc.272.6.3689. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Sabbagh MN, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol. 2005;62(5):753–757. doi: 10.1001/archneur.62.5.753. [DOI] [PubMed] [Google Scholar]
- Squarzoni P, Oller G, et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8(5):1271–1279. doi: 10.1016/j.celrep.2014.07.042. [DOI] [PubMed] [Google Scholar]
- Squarzoni P, Thion MS, et al. Neuronal and microglial regulators of cortical wiring: usual and novel guideposts. Front Neurosci. 2015;9:248. doi: 10.3389/fnins.2015.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21(3):185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40(2):133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and Alzheimer’s disease pathogenesis. J Neurosci Res. 2004;77(1):1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and neuroprotection: implications for Alzheimer’s disease. Brain Res Brain Res Rev. 2005;48(2):234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 2006;29(9):506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Miller KR, et al. Microglial degeneration in the aging brain--bad news for neurons? Front Biosci. 2008;13:3423–3438. doi: 10.2741/2937. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Xue QS, et al. Microglial pathology. Acta Neuropathol Commun. 2014;2:142. doi: 10.1186/s40478-014-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon CM, Ditadi A, et al. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat Biotechnol. 2014;32(6):554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan HG, Martinez J, et al. Fluid-percussion model of mechanical brain injury in the cat. J Neurosurg. 1976;45(5):521–534. [PubMed] [Google Scholar]
- Swardfager W, Lanctot K, et al. A meta-analysis of cytokines in Alzheimer’s disease. Biol Psychiatry. 2010;68(10):930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Thorne JE, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology. 2004;23(4):159–169. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kakuda Y, et al. Differentiation of Donor-Derived Cells Into Microglia After Umbilical Cord Blood Stem Cell Transplantation. J Neuropathol Exp Neurol. 2015;74(9):862–866. doi: 10.1097/NEN.0000000000000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Naito M. Development, differentiation, and proliferation of macrophages in the rat yolk sac. Tissue Cell. 1993;25(3):351–362. doi: 10.1016/0040-8166(93)90077-x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takayama K, Sukhova GK, et al. A novel prostaglandin E receptor 4-associated protein participates in antiinflammatory signaling. Circ Res. 2006;98(4):499–504. doi: 10.1161/01.RES.0000204451.88147.96. [DOI] [PubMed] [Google Scholar]
- Takeda S, Sato N, et al. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: relevance to pathogenesis and therapy. Front Aging Neurosci. 2014;6:171. doi: 10.3389/fnagi.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagno E, Bardini P, et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10(3):279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- Thambisetty M, Lovestone S. Blood-based biomarkers of Alzheimer’s disease: challenging but feasible. Biomark Med. 2010;4(1):65–79. doi: 10.2217/bmm.09.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL, et al. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8(11):e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Majewska AK. A role for microglia in synaptic plasticity? Commun Integr Biol. 2011;4(2):220–222. doi: 10.4161/cib.4.2.14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Zettel ML, et al. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia. 2012;60(4):541–558. doi: 10.1002/glia.22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Dutta T, et al. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS One. 2013;8(5):e63644. doi: 10.1371/journal.pone.0063644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucholski J, Kuret J, et al. Tau is modified by tissue transglutaminase in situ: possible functional and metabolic effects of polyamination. J Neurochem. 1999;73(5):1871–1880. [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20. doi: 10.1186/1750-1326-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushikubi F, Segi E, et al. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature. 1998;395(6699):281–284. doi: 10.1038/26233. [DOI] [PubMed] [Google Scholar]
- Van Gool WA, Weinstein HC, et al. Effect of hydroxychloroquine on progression of dementia in early Alzheimer’s disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet. 2001;358(9280):455–460. doi: 10.1016/s0140-6736(01)05623-9. [DOI] [PubMed] [Google Scholar]
- Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009;214(7):630–641. doi: 10.1016/j.imbio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, et al. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A. 2012;109(44):18150–18155. doi: 10.1073/pnas.1210150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, et al. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-beta deposition in mouse models of Alzheimer’s disease. J Exp Med. 2015;212(11):1803–1809. doi: 10.1084/jem.20150478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, et al. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol Aging. 2009;30(8):1217–1226. doi: 10.1016/j.neurobiolaging.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, et al. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70(19):1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vural P, Degirmencioglu S, et al. The combinations of TNFalpha-308 and IL-6 -174 or IL-10 -1082 genes polymorphisms suggest an association with susceptibility to sporadic late-onset Alzheimer’s disease. Acta Neurol Scand. 2009;120(6):396–401. doi: 10.1111/j.1600-0404.2009.01230.x. [DOI] [PubMed] [Google Scholar]
- Waisman A, Ginhoux F, et al. Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol. 2015;36(10):625–636. doi: 10.1016/j.it.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, et al. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29(13):3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S, Letiembre M, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem. 2007;20(6):947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson DM, Grum-Tokars VL, et al. Development of novel chemical probes to address CNS protein kinase involvement in synaptic dysfunction. PLoS One. 2013;8(6):e66226. doi: 10.1371/journal.pone.0066226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Van Eldik LJ, et al. Closed head injury in an age-related alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J Neurosci. 2015;35(16):6554–6569. doi: 10.1523/JNEUROSCI.0291-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Escoubet-Lozach L, et al. TH2 cytokines and allergic challenge induce Ym1 expression in macrophages by a STAT6-dependent mechanism. J Biol Chem. 2002;277(45):42821–42829. doi: 10.1074/jbc.M205873200. [DOI] [PubMed] [Google Scholar]
- Wiesinger H. Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog Neurobiol. 2001;64(4):365–391. doi: 10.1016/s0301-0082(00)00056-3. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Lopresti BJ, et al. Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009;66(1):60–67. doi: 10.1001/archneurol.2008.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Bushong EA, et al. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci U S A. 2006;103(46):17513–17518. doi: 10.1073/pnas.0602841103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y, Yona S, et al. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci. 2013;7:26. doi: 10.3389/fncel.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodling NS, Wang Q, et al. Suppression of Alzheimer-Associated Inflammation by Microglial Prostaglandin-E2 EP4 Receptor Signaling. J Neurosci. 2014;34(17):5882–5894. doi: 10.1523/JNEUROSCI.0410-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FL, Liang YF, et al. RNA interference of argininosuccinate synthetase restores sensitivity to recombinant arginine deiminase (rADI) in resistant cancer cells. J Biomed Sci. 2011;18:25. doi: 10.1186/1423-0127-18-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, et al. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Feng L, et al. Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-beta 1. Am J Pathol. 1995;147(1):53–67. [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7(5):612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, et al. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature. 1997;389(6651):603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, et al. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32(48):17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K, Yin YL, et al. Dietary arginine supplementation increases mTOR signaling activity in skeletal muscle of neonatal pigs. J Nutr. 2008;138(5):867–872. doi: 10.1093/jn/138.5.867. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25(2):181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Yokokura M, Mori N, et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2011;38(2):343–351. doi: 10.1007/s00259-010-1612-0. [DOI] [PubMed] [Google Scholar]
- Youm YH, Grant RW, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Iyer RK, et al. Expression of arginase isozymes in mouse brain. J Neurosci Res. 2001;66(3):406–422. doi: 10.1002/jnr.1233. [DOI] [PubMed] [Google Scholar]
- Yu H, Iyer RK, et al. Arginase expression in mouse embryonic development. Mech Dev. 2002;115(1-2):151–155. doi: 10.1016/s0925-4773(02)00089-8. [DOI] [PubMed] [Google Scholar]
- Yu H, Yoo PK, et al. Widespread expression of arginase I in mouse tissues. Biochemical and physiological implications. J Histochem Cytochem. 2003;51(9):1151–1160. doi: 10.1177/002215540305100905. [DOI] [PubMed] [Google Scholar]
- Yu Z, Xu X, et al. Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PloS one. 2010;5(4):e9956. doi: 10.1371/journal.pone.0009956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17(3):400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153(3):707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, Narvaez A, Honrada R, Ruvalcaba D, McGrath MS. The-1082G/A polymorphism in IL-10 gene is associated with risk of Alzheimer’s disease: a meta-analysis. J Neurol Sci. 2011;303:133–138. doi: 10.1016/j.jns.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Zimmermann A, Preynat-Seauve O, et al. Haplotype-based banking of human pluripotent stem cells for transplantation: potential and limitations. Stem Cells Dev. 2012;21(13):2364–2373. doi: 10.1089/scd.2012.0088. [DOI] [PubMed] [Google Scholar]
- Parpura V, et al. Glial cells in (patho)physiology. J Neurochem. 2012;121(1):4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim Biophys Acta. 2016;1862(3):483–491. doi: 10.1016/j.bbadis.2015.11.014. [DOI] [PubMed] [Google Scholar]
- Pekny M, et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 2016;131(3):323–345. doi: 10.1007/s00401-015-1513-1. [DOI] [PubMed] [Google Scholar]