

Abstract
Gene duplication generates new genetic material, which has been shown to lead to major innovations in unicellular and multicellular organisms. A whole-genome duplication occurred in the ancestor of Saccharomyces yeast species but 92% of duplicates returned to single-copy genes shortly after duplication. The persisting duplicated genes in Saccharomyces led to the origin of major metabolic innovations, which have been the source of the unique biotechnological capabilities in the Baker’s yeast Saccharomyces cerevisiae. What factors have determined the fate of duplicated genes remains unknown. Here, we report the first demonstration that the local genome mutation and transcription rates determine the fate of duplicates. We show, for the first time, a preferential location of duplicated genes in the mutational and transcriptional hotspots of S. cerevisiae genome. The mechanism of duplication matters, with whole-genome duplicates exhibiting different preservation trends compared to small-scale duplicates. Genome mutational and transcriptional hotspots are rich in duplicates with large repetitive promoter elements. Saccharomyces cerevisiae shows more tolerance to deleterious mutations in duplicates with repetitive promoter elements, which in turn exhibit higher transcriptional plasticity against environmental perturbations. Our data demonstrate that the genome traps duplicates through the accelerated regulatory and functional divergence of their gene copies providing a source of novel adaptations in yeast.
Keywords: gene duplication, mutational genome hotspots, expression genome hotspots, environmental stress, phenotypic plasticity, adaptations, genetic redundancy
Introduction
Gene duplication is considered the most important source of novel functions (Ohno 1970, 1999). Relaxed selective constraints after gene duplication allows duplicated genes to explore novel genotypes and find new functions (Haldane 1932; Ohno 1999; Payne and Wagner 2014; Taylor and Raes 2004). However, since most emerging mutations are degenerative (Kimura 1983; Kimura and Takahata 1983), the common fate of duplicated genes is the nonfunctionalization of one of the gene copies and its subsequent erosion from the genome (Ohno 1970). An example of this is the return of 92% of the yeast Saccharomyces duplicates to single-copy genes “shortly” after the duplication of Saccharomyces ancestor genome > 100 MYA (Wolfe and Shields 1997; although see [Marcet-Houben 2015]). The remaining duplicates (around 8% of the genome) led to major metabolic innovations. Because duplication impacts genome size and can alter the genetic map of organisms, revealing the factors that determine the persistence of duplicates is an important question in evolutionary genomics.
A number of scenarios have been proposed to explain why some genes and not others persist in the genome as duplicates. Firstly, natural selection may favor individuals with an increase in gene dosage through duplication (Conant and Wolfe 2008). Secondly, purifying selection will prevent the loss of one of the gene copies after duplication if a balance between dosage-sensitive genes is required (Birchler et al. 2001, 2005; Freeling and Thomas 2006). Thirdly, highly expressed genes have been shown to be more duplicable after whole-genome duplication than lowly expressed genes because of absolute dosage constraints and constraints on dosage balance (Gout et al. 2009, 2010; Gout and Lynch 2015; Papp et al. 2003; Qian et al. 2010; Seoighe and Wolfe 1999). Fourthly, the functional backup provided by gene copies can mask the effects of degenerative mutations and be selectively advantageous (Fares et al. 2013; Keane et al. 2014), although the selective value of this masking effect remains controversial (Fares 2015). In the absence of selective advantage for the genetic redundancy provided by gene duplication, the nonfunctionalization of a gene copy and its erosion from the genome remains the most likely outcome. However, a rapid relief of genetic redundancy through the divergence between gene copies may prevent the return of duplicates to the single-copy gene status.
One way of resolving genetic redundancy is through a quick divergence between the gene copies of the duplicate. Under this scenario, the preservation of duplicated genes should be more likely in genome regions with high mutation rates than in genome regions with low mutation rates. Mutation rates vary considerably across the genome (Chuang and Li 2004), with the heterochromatic late replicating regions exhibiting remarkable differences in the mutation rates when compared with the early replicating euchromatin (Schuster-Bockler and Lehner 2012; Supek and Lehner 2015). Transcription has also been shown to be mutagenic, with highly expressed genes revealing higher net mutation rates than lowly expressed genes (Park et al. 2012). Notwithstanding the fact that most mutations would lead to the nonfunctionalization of one copy of the duplicated gene, the likelihood for the functional divergence between gene copies of duplicates is higher in genomic regions with higher mutation rates. Once a gene copy has found novel functions, purifying selection would preclude the loss of this gene copy.
In addition to functional divergence, divergence between gene copies can also take place at the expression level, such that each gene copy can be expressed under specific environmental conditions. This would allow the organism to adapt to different environments without a need to optimize the encoded function of the gene to each environment. A way of achieving a divergence in expression between gene copies is through the presence of sequence repeats in the promoters of duplicated genes. Interestingly, Sequences composed of tandem repeats, which are repeated DNA sequences adjacent to one another in a head-to-tail orientation, evolve at a higher rate than the surrounding genome (Rando and Verstrepen 2007). There is evidence that such repeats influence the expression of certain genes (Martin et al. 2005; Rockman and Wray 2002; Streelman and Kocher 2002). Moreover, genes driven by repeat-containing promoters show higher rate of transcriptional divergence (Vinces et al. 2009). Therefore, genome mutational and transcriptional hotspots can be traps for duplicated genes because duplicates at such genome regions can diverge functionally and in their expression quicker than in other regions and thus be subsequently maintained by purifying selection. We also hypothesize that such genome hotspots are sources of novel functions and adaptations.
In this study, we compare the mutational and transcriptional rates of genome regions containing duplicated genes in S. cerevisiae with the rates of genome regions containing only singletons. Duplicated genes fall preferentially within genome regions with high rates of mutation, high rates of evolution, and high transcription levels. There are important differences in terms of mutation rates between genome regions containing duplicates emerging from whole-genome duplication events (WGDs) and those with duplicates generated through small-scale duplications (SSDs). Experimentally evolved S. cerevisiae tolerates more mutations in mutational and transcriptional genome hotspots. Remarkably, the promoters of duplicates that accumulate mutations are rich in repetitive motifs, known to influence the expression of certain genes (Gemayel et al. 2010; Martin et al. 2005; Rockman and Wray 2002; Streelman and Kocher 2002; Tirosh et al. 2009; Vinces et al. 2009). Duplicates containing repetitive motifs exhibit larger regulatory plasticity under environmental perturbations. Collectively, we demonstrate that genome mutational and expression hotspots retain genes in duplicate and are the source of adaptations to environmental stress.
Materials and Methods
Identification of Duplicated Genes
Paralogs pairs of duplicated genes were identified as the resulting best reciprocal hits from all-against-all BLAST searches using BLASTP with an E-value cutoff of 1E-5 and a 50 bit score (Altschul et al. 1997). Paralogs were then divided into two groups according to the mechanism of their origin: WGDs and SSDs. WGDs are those extracted from the reconciled list provided by the YGOB (Yeast Gene Order Browser, http://wolfe.gen.tcd.ie//ygob; last accessed May 4, 2017 [Byrne and Wolfe 2005]) (555 pairs of genes), and these were not subjected to subsequent SSD. All other paralogs were considered to belong to the category of SSDs (560 pairs of genes).
Sequence Alignments and Analysis of Divergence
For each protein-coding gene of S. cerevisiae, we searched for its ortholog in the closely related species S. paradoxus using the program blastP. Pairwise sequence alignments were built using the program ClustalW. To calculate the distance between S. cerevisiae and S. paradoxus for each of the genes, we estimated the number of nonsynonymous nucleotide substitutions per nonsynonymous site (dN), synonymous substitutions per synonymous site (dS), and the nonsynonymous-to-synonymous rates ratio (ω = dN/dS) using the maximum-likelihood approach under the Goldman and Yang model (Goldman and Yang 1994) as implemented in the PAML package version 4.7 (Yang 2007).
Mapping SNPs in Experimentally Evolved S. cerevisiae Genomes
The evolution experiment was performed in our previous study (Keane et al. 2014). Briefly, the evolution experiment started with a single-colony-founded population, from which we derived five evolving lineages of the S. cerevisiae strain Y06240. This clonal population was serially passaged onto YPD plates for roughly 2,200 generations of the yeast by repeated streaking, each passage resulting from restreaking a single colony. Since only one colony was passaged into the next generation, this experiment simulated a Muller ratchet dynamic, in which genome mutations in generation t, included those from generation t − 1 in addition to the new emerging mutations. The low effective population size (Ne) of our experiment implies that Ne multiplied by the mutation rate (μ) is μNe ⋘ 1, and thus the population evolved under strong genetic drift effects. Under these conditions, most of the mutations fixed in the population are likely deleterious and the fixation rate of mutations is approximated to the mutation rate—the fixation rate is 80% the mutation rate, as 20% of all mutations were estimated to be lethal (Keane, et al. 2014). Whole genome sequencing of the ancestor and each of the five evolved lineages was carried out at 2,200 generations using Illumina technology, as previously described (Keane, et al. 2014). Mapping of mutations was possible using the program breseqv0.24rc (Deatherage and Barrick 2014). Sequence reads are available at the Sequence Read Archive with accession numbers (SRP012321). Mutations were then separated into two groups: those affecting protein-coding genes and those localized within the first 600 nucleotides upstream of protein-coding genes. The second group of mutations was further divided into those mutations affecting upstream regions of duplicated genes and those affecting upstream regions of singleton genes.
Analysis of Gene Expression Under Stress in S. cerevisiae
We tested the transcriptional plasticity of S. cerevisiae genes by comparing the expression of genes in YPD to that obtained from other studies after growing S. cerevisiae in eight different stress conditions, including acidic stress (Casamayor et al. 2012), alkaline stress (Casamayor et al. 2012), wine fermentation at 12 h, heat stress (Berry and Gasch 2008), lithium stress (Bro et al. 2003), impairment of manganese (Garcia-Rodriguez et al. 2012), osmotic stress with NaCl (Berry and Gasch 2008), and glucose limitation (Jansen et al. 2005). We also performed new growth experiments in which we subjected S. cerevisiae to an additional five stress conditions (ethanol, lactic acid, glycerol, oxygen, and oxygen supplemented with dextrose) (supplementary data 1–5, Supplementary Material online). We therefore performed analyses for 13 stress conditions altogether. We considered a duplicated gene to increment significantly its expression levels under stress conditions if the proportional normalized expression of this gene increased or decreased (i.e., incremented) significantly, corresponding this to an expression increment under stress of more than 20% of the gene expression.
The transcriptomic profiling in our study was performed in the S. cerevisiae Y06240 haploid strain, with three technical replicates for each biological stress condition (3% lactic acid [YPL], 3% Ethanol [YPE], 3% glycerol [YPG], 0.25mM H2O2 [YPOx], and 0.25mM H2O2 + 1.5% glucose [YPOxD]) in comparison with the normal growth condition (YPD media). Therefore, in total S. cerevisiae was grown in YPD and five other stress conditions for 24 h. Total RNA extractions were performed with RNeasy kit (Qiagen) following manufacturer instructions. Ribosomal RNA was removed by using Ribo-Zero Gold rRNA removal yeast (Illumina) depletion kit. Stranded RNA libraries were constructed using TruSeq stranded mRNA (Illumina) from oligo-dT captured mRNAs from depleted samples. Libraries were run in NextSeq 500 (Illumina) at 75 nt single read by using High Output 75 cycles kit v2.0 (Illumina).
RNA libraries were sequenced at Genomic core facility at Servicio Central de Soporte a la Investigación Experimental (SCSIE) from University of Valencia, Spain. Raw reads were analyzed using FastQC report and cleaned with CutAdapt as implemented in RobiNA software package v 1.2.4 (Lohse et al. 2012). Low quality reads were filtered and trimmed (Pred score inferior to 20 and size <40 nt were discarded). The reads were then aligned with Bowtie (up to two mismatches accepted) to the reference transcriptome (PRJNA290217) from the reference S288c strain. Statistical assessment of differential gene expression was done either with edge R (Robinson et al. 2010) or with DeSeq (Anders and Huber 2010) as implemented in RobiNA. All newly sequenced RNA sequences are available from the Sequence Read Archive with the following accession number (SRP074821).
Genetic Interaction Data
We used the latest update of the genetic functional chart of S. cerevisiae (Costanzo et al. 2010; supplementary files S4 and S5, Supplementary Material online from http://drygin.ccbr.utoronto.ca/∼costanzo/; last accessed May 4, 2017). The genetic map is based on the synthetic genetic array methodology (Tong et al. 2001). In this methodology, synthetic lethal genetic interactions are systematically mapped to single and double mutants. In this study, two genes are considered to interact genetically if the double knock out mutant of the two genes has significantly larger or smaller effect than the multiplicative effects of simple knockouts.
Software
Calculations and statistics were performed using MS Excel and R 3.2.1. Data management was possible using in-house built PERL scripts.
Results
Duplicated Genes Fall within Evolutionary Genome Hotspots
To determine whether genes with paralogs in the genome fall preferentially within evolutionary genome hotspots, we followed two approximations: a wider genome window analysis of evolutionary rates and a more local genome region analysis of these rates. In both approximations, we compared the divergence levels across the genome between S. cerevisiae and its close phylogenetic relative S. paradoxus. Duplicates generated through WGD and SSD predate the divergence between S. cerevisiae and S. paradoxus, and thus duplicates should be all represented in these two species. Divergence levels were estimated using two measures: the nonsynonymous nucleotide distance per nonsynonymous site (i.e., nucleotide replacements that lead to amino acid changes: dN) and the synonymous changes per synonymous site (dS).
In the first approximation, we examined dN for a genome window of 40 Kilobases (kb), which we moved 40 kb across the genome in each step. This window size (40 kb) was enough to ensure that at least one duplicated gene was present in the genome region defined by the window. For each of the steps, we counted the number of duplicates—that is, genes with paralogs elsewhere in the genome (supplementary data 6, Supplementary Material online). We then compared the number of duplicates genome wide to dN for that window once duplicated genes were excluded. We found a positive significant but moderate correlation between the number of duplicates and the mean dN across the genome (Pearson correlation: r = 0.12, P = 0.034). To determine whether such a correlation is homogeneous across chromosomes, we repeated this analysis for each of the 16 chromosomes in S. cerevisiae. We found two types of chromosomes (supplementary data 7, Supplementary Material online): (a) Group 1 included those chromosome (12 out of the 16 chromosomes) in which the relationship was positive between the number of duplicates and dN and (b) Group 2 included those chromosomes (four out of the 16: I, VI, XI, and XVI) in which this relationship was negative. We took all the genome windows for the 12 chromsomes in which the relationship between the number of duplicates and dN was positive and tested the correlation between these two numbers. These chromosomes exhibited a positive correlation between dN and the number of duplicates they contained (Pearson correlation: r = 0.19, P = 0.002). Most duplicates (82.4%) belonged to chromosomes of Group 1. These results were not reproduced in the case of dS, which showed no significant relationship with the number of duplicates (Pearson correlation: r = 0.01, P = 0.892). Therefore, this first approximation showed that duplicates fell within genome regions with high rates of evolution.
Because defining a genome window of 40 kb may include subregions with different mutation rates, we reanalyzed the data using a much more local genome region neighboring duplicated genes. We determined dN for genes belonging to the group of the six singletons in the immediate genome neighborhood of duplicated genes, three singletons at either side (We call these regions GRDs, fig. 1a, table 1, and supplementary data 8, Supplementary Material online). We use three singletons at either side because this number ensured a real genome neighborhood of the six genes considered in the GRDs, such that none of the six genes is located far away in the chromosome from the other five genes. We then compared the rates of evolution of GRDs with the rate of evolution of those regions that only contained single copy genes (seven singletons, one singleton neighbored by three other singletons at either side, called hereafter GRSs). In the GRDs, duplicated genes were excluded from distance estimations and only singletons were used to calculate the rates of evolution of that genomic region, thereby avoiding the bias in the results due to the contribution of dosage-sensitive genes (fig. 1a). GRDs contained genes with higher dN (Mean ± SD: 0.040 ± 0.02) than GRSs (Mean ± SD: 0.038 ± 0.03) (Wilcoxon rank test: P = 3.05 × 10−5). This indicates that the GRDs evolve faster than GRSs. Unlike dN, there was no significant difference in dS between GRDs and GRSs (supplementary data 8, Supplementary Material online; Wilcoxon rank test: P = 0.343), indicating that GRDs are hotspots of amino acid replacing mutations, that is they exhibit high rates of evolution (table 1).
Fig. 1.
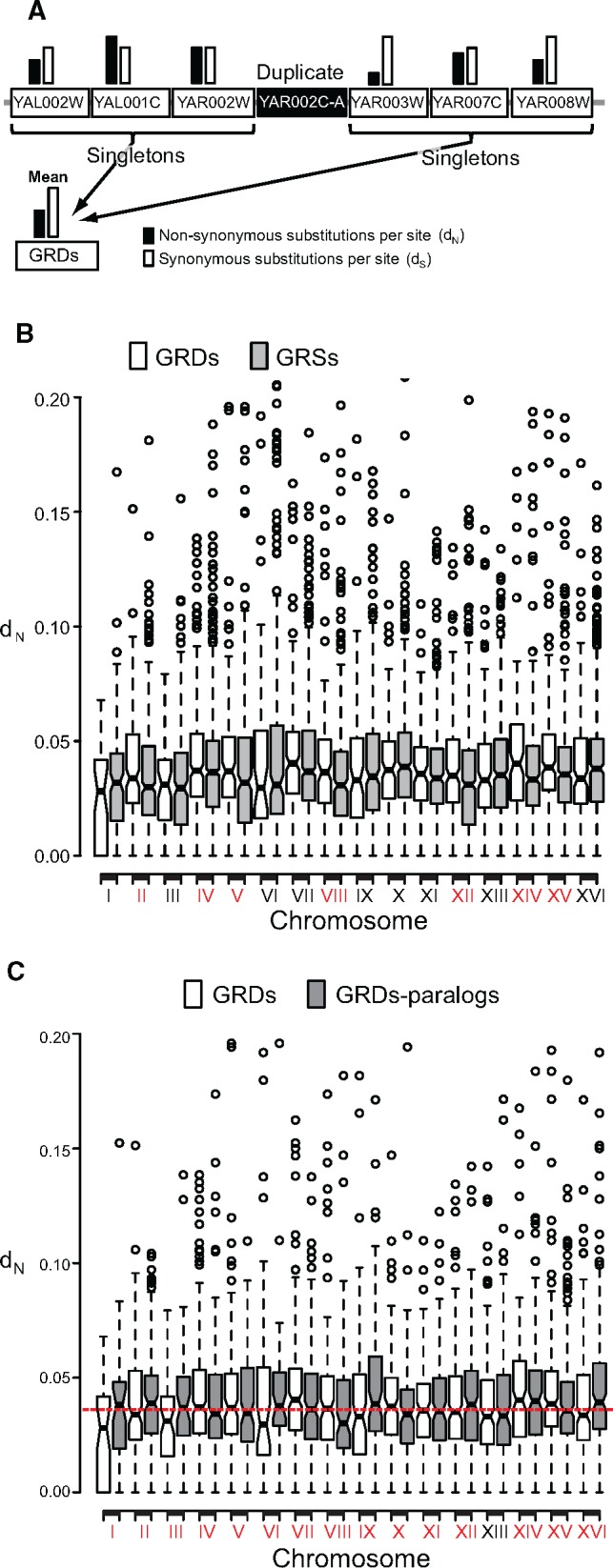
—Duplicated genes persist in genome evolutionary hotspots. (a) We estimated the mean nonsynonymous nucleotide substitutions per nonsynonymous site (dN: black column) and the synonymous substitutions per synonymous site (dS; white column), for the three singleton genes (Locus tag genes in the white rectangles) immediately flanking a duplicated gene (black rectangle) at either side. (b) The mean dN for each region containing a duplicate (GRDs) within each chromosome was calculated and compared to that of genome regions containing only singletons (GRSs). We identified seven chromosomes in which GRDs exhibited significantly higher dN than GRSs (red-labeled roman numbers in x axis). (c) For each of the duplicates contained in each GRD of each chromosome, we searched for its paralogue elsewhere in the genome. Then we compared the dN of both these groups and found that when one GRD of a chromosome exhibited a mean dN below the mean dN for GRSs (white boxes), their paralogs exhibited the inverse pattern (gray boxes), and vice versa. Red-labeled chromosomal numbers in the x axis indicate those for which evidence exist that at least one of the paralogs is in a GRD with high dN.
Table 1.
Genome Regions with Duplicates (GRD) Are Mutational, Transcriptional, and Interaction Hotspots Compared with Genome Regions with Singletons (GRS)
| GRDs | GRSs | t | d.f. | P | P (Wilcoxon) | |
|---|---|---|---|---|---|---|
| dNa | 0.04 | 0.038 | 2.24 | 4398.15 | 0.024 | 3.13 × 10−5 |
| dSb | 0.30 | 0.29 | 2.08 | 4776.90 | 0.037 | 0.343 |
| Expression | 3.33 | 3.17 | 3.58 | 4024.11 | 3.5 × 10−4 | 7.5 × 10−4 |
| GIc | 220.04 | 211.73 | 2.35 | 3927.28 | 0.018 | 3.5 × 10−3 |
Mean number of nonsynonymous nucleotide substitutions per nonsynonymous site.
Mean number of synonymous nucleotide substitutions per synonymous site.
Mean number of genetic interactions.
The preferential location of duplicates in evolutionary hotspots is not a byproduct of positive selection in duplicates: only 0.13% of all duplicates showed signature of positive selection (i.e., ω = dN/dS > 1 under the Goldman and Yang model implemented in the program PAML [Yang 2007]) against 1.9% of the singletons. GRDs were, nevertheless, more enriched for positive selection than GRSs (5.35% of the GRDs exhibited evidence of positive selection against 4.04% of the GRSs. Fisher’s exact test: F = 1.34, P = 0.008). However, removing GRDs with evidence of positive selection from the analyses yielded similar results to those before removing them: GRDs exhibited higher dN (0.038 ± 0.015) than GRSs (0.036 ± 0.026; Wilcoxon rank test: P = 1.69 × 10−4).
To determine the distribution of evolution hotspots and GRDs across the 16 chromosomes, we compared dN of GRDs and GRSs across the 16 chromosomes (supplementary data 9, Supplementary Material online and fig. 1b). Seven out of the 16 chromosomes (Chromosomes II, IV, V, VIII, XII, XIV, and XV) showed significantly higher dN for GRDs than GRSs, while in the other nine chromosomes there was no significant differences (fig. 1b). The seven chromosomes with evidence of accelerated rates of evolution for GRDs included 1,342 out of the 2,240 duplicated genes (∼60% of all duplicates), which was a higher proportion than expected given that those seven chromosomes represented 52% of all the genes in S. cerevisiae (Binomial test: P = 5.43 × 10−14). We found that for each chromosome the probability of a duplicated gene to have its paralog in the same chromosome was low (the mean proportion of duplicated genes with paralogs being located in the same chromosome was 10.27% ± 1.16), with the lowest percentage being found in chromosome XI (1.75%) and the highest in chromosome XII (17.44%). This means that the majority of duplicated genes (∼90% of all duplicates in the genome) had their paralogs in a different chromosome from the one in which they were located. Importantly, with the exception of chromosome XIII, when duplicates were in GRDs with low dN (i.e., the duplicates located in the nine chromosomes that contained 40% of duplicates) their paralogs were in GRDs with higher dN than expected (fig. 1c). In conclusion, preserved duplicated genes were those in which at least one gene copy fell within a genome region with high rates of evolution.
Genome Regions with Different Mutation Rates Trap Differently Whole-Genome and Small-Scale Duplicates
Since the signature of high evolutionary rates for GRDs was clear when using dN but not when using dS, we investigated whether this pattern was due to the mixed signatures of evolution of GRDs containing duplicates originated through two different mechanisms: whole-genome or small-scale duplication. The mechanism of duplication is known to influence the functional fate of duplicates: duplicates originated by whole genome duplication (WGDs) are more prone to partition ancestral functions, while those generated by small-scale duplications (SSDs) are generally more prone to acquire novel functions (Carretero-Paulet and Fares 2012; Fares 2015; Fares et al. 2013; Keane et al. 2014). WGDs are known to have undergone substantial divergence in their expression levels, perhaps due to mutations in their promoter regions (Conant and Wolfe 2008; Keane et al. 2014). The gene copies of SSDs have also diverged in their expression and functional roles (Keane et al. 2014). In order to determine if GRDs exhibit different evolutionary and mutational properties when they include WGDs than SSDs, we split GRDs into those containing WGDs and SSDs and compared their rates of evolution to those of GRSs.
We analyzed the rates of evolution of genome regions containing WGDs or SSDs (supplementary data 10 and 11, Supplementary Material online). We found that GRDs of WGDs were in genome hotspots in comparison with GRSs for both dN (Mean dN ± SD for GRDs = 0.042 ± 0.02, Wilcoxon rank test: P = 5.47 × 10−9) and dS (Mean dS± SD for GRDs = 0.316 ± 0.06, mean dS for GRSs = 0.29 ± 0.1. Wilcoxon rank test: P = 2.50 × 10−4). In contrast to WGDs, dN of GRDs containing SSDs was not significantly different from that of GRSs (Mean dN for GRDs = 0.038 ± 0.02. Wilcoxon rank test: P = 0.625), while exhibited lower dS than GRSs (Mean dS for GRDs = 0.279 ± 0.13. Wilcoxon rank test: P = 0.021). Removing GRDs and GRSs that contained positively selected genes led to similar results, GRDs containing WGDs are in genome hotspots for dN (0.04 ± 0.2) (Wilcoxon rank test: P = 1.47 × 10−8) and dS (0.32 ± 0.1) (Wilcoxon rank test: P = 6.59 × 10−5), while GRDs containing SSDs showed no evidence of mutational hotpots for dN (0.035 ± 0.02) (Wilcoxon rank test: P = 0.93) and slightly significantly lower rate for dS (0.28 ± 0.13) (Wilcoxon rank test: P = 0.041). Finally, for most chromosomes, dN of GRDs containing WGDs was higher than that of GRSs (Mean of the differences: 0.003; t-paired test: t = 2.52, d.f. = 15, P = 0.023). In the case of dS, this was higher for GRDs containing WGDs than that for GRSs in most chromosomes (Mean of the differences = 0.024. Paired t-test: t = 5.05, d.f. = 15, P = 1.4 × 10−4). Unlike GRDs containing WGDs, those containing SSDs showed no evidence for higher or lower dN than GRSs (Mean of the differences = −0.002. Paired t-test: t = −1.44, d.f. = 15, P = 0.16) nor they exhibited any pattern for dS that distinguishes GRDs from GRSs (Mean of the differences = −0.013. Paired t-test: t = −1.76, d.f. = 15, P = 0.09).
Finally, using SNP data from en evolution experiment of S. cerevisiae under strong genetic drift (Keane et al. 2014), GRDs containing WGDs were significantly enriched for SNPs and indels compared to GRSs (table 2, 26.1% of GRDs with WGDs contained SNPs against the 21.95% of GRSs with SNPs; Fisher’s exact test: F = 1.26, P = 1.6 × 10−3), while GRDs containing SSDs showed no difference in terms of SNPs or indels with GRSs (table 2, 22.64% of GRDs with SSDs contained SNPs. Fisher’s exact test: F = 1.04, P = 0.60). Taken together, our results support that genome regions with high mutation rates trap differently duplicates depending on the mechanism of duplication. These data also point to higher mutation rates clearly when using WGDs but not SSDs. GRDs containing SSDs may therefore hide the difference in dS between GRDs and GRSs when the analyses do not separate these GRDs from the ones containing WGDs.
Table 2.
Distribution of SNPs among GRDs and GRSs
| # SNPs | Total # Regions | % Regions | |
|---|---|---|---|
| GRSs | 1582 | 7208 | 21.95 |
| GRDs (WGDs) | 311 | 1192 | 26.1 |
| GRDs (SSDs) | 276 | 1219 | 22.64 |
Duplicated Genes Fall within Mutational Genome Hotspots
Our previous analysis demonstrated that GRDs exhibit higher evolution rates than GRSs. We sought to investigate if GRDs also exhibit higher mutation rates than GRSs. The estimated rates of synonymous and nonsynonymous substitutions between species are largely influenced by selection, and thus is a very crude estimate of the rate of mutation. To determine whether genome mutational hotspots are traps for duplicated genes, we performed analyses on three additional data sets: (a) analysis of the correlation between the genome density of duplicates and experimentally measured mutation rates for chromosome VI of S. cerevisiae (Lang and Murray 2011); (b) analysis of yeast interstrain single nucleotide synonymous polymorphisms (SNPs) from a previous study (Agier and Fischer 2012) based on polymorphism data in yeast strains from another study (Liti et al. 2009), and (c) analysis of SNPs from an evolution experiment of S. cerevisiae under strong genetic drift effects (Keane et al. 2014).
Lang and Murray measured the mutation rate of the URA3 gene integrated at 43 different locations tiled at chromosome VI of S. cerevisiae. They found three main regions in the chromosome that are distinguished by virtue of their differential mutation rates: (a) the first 70 kb of the chromosome exhibited the highest mutation rate (we call this the fast region), (b) the 70 to 160 kb, including the centromeric and close pericentric chromosomal region, showed the lowest mutation rate (slow region), and (c) the remaining of the chromosome showed an intermediate mutation rate (intermediate region). We slid a 10-kb window across each of these three regions and determined the average number of duplicates for the slow, fast, and intermediate regions. In agreement with the distribution of mutation rates, the fast region contained the greatest number of duplicates (Mean = 2.77), which was 54% higher than that for the slow region (Mean = 1.77), while the intermediate region showed an intermediate average number of duplicates (Mean = 2.1).
In the second data set, we analyzed the distribution of duplicates across the genome and measured the correlation of this with the distribution of neutral SNPs identified in a previous study (Agier and Fischer 2012), which used data on polymorphism for 40 different S. cerevisiae strains (Liti et al. 2009). In this study, 144,164 SNPs were identified, of which 85,980 SNPs were synonymous. Because synonymous SNPs are more likely to be fixed neutrally than nonsynonymous SNPs. We counted the number of synonymous SNPs in GRDs and GRSs and divided this number by the number of synonymous sites for each GRD and GRS. We used this SNPs rate as a reflection of the mutation rate in each of the genome regions. SNPs rate at GRDs (rate = 0.053 synonymous SNPs per site) was 36% higher than this rate at GRSs (rate = 0.039 synonymous SNPs per site), and the difference was highly significant (Wilcoxon rank test: P < 2.2 × 10−16). Our rate estimates for GRDs can be biased by an enrichment of these regions for dosage sensitive genes. For example, it is possible that GRDs containing dosage sensitive duplicates may also contain dosage sensitive genes. We identified dosage sensitive genes in the S. cerevisiae genome as those encoding transcription factors and proteins from protein complexes (Birchler and Veitia 2012). Such genes were cataloged in a previous study, which showed that roughly 2,078 genes encoded proteins that form part of protein complexes (Pu et al. 2009). Of all the dosage sensitive genes, we identified 450 that belonged to the set of duplicated genes. We reanalyzed the mutation rates for GRDs and GRSs using the synonymous SNPs once we discarded those GRDs containing dosage-sensitive duplicates. The rate of mutation based on this data for GRDs (rate = 0.052 synonymous SNPs per site) was higher than the rate for GRSs (rate = 0.039 synonymous SNPs per site) with the difference being significant (Wilcoxon rank test: P < 2.2 × 10−16).
Agier and Fischer showed a strong correlation between replication timing and mutation rates in S. cerevisiae (Agier and Fischer 2012). Accordingly, a correlation is also expected between the density of duplicates in a genome region and the replication timing. We extracted from a previous study the data on replication timing for nearly 21,000 points in the 16 chromosomes of S. cerevisiae (Raghuraman et al. 2001), as well as the number of duplicates for these points. We generated a window with a size of 100 replication-timing points and slid it along the chromosomes, summing the minutes of replication for each window and the number of duplicates. Then, we analyzed the correlation between the replication timing in minutes and the number of duplicates. In agreement with the expectation, the number of duplicates correlated positively with replication timing, with this correlation being strong using both the parametric method (Pearson correlation: r = 0.39, P = 4.71 × 10−9) and the nonparametric method (Spearman correlation: r = 0.28, P = 2.43 × 10−5).
In the third data set, to determine if the difference in the evolutionary rates between GRDs and GRSs is the result of their different mutation rates and is not the result of differences in the selective constraints among genome regions, we used the genome SNPs data obtained in an evolution experiment of S. cerevisiae under strong genetic drift (Keane et al. 2014). In this experiment, we evolved five independent populations of a haploid S. cerevisiae strain by transferring one single colony of S. cerevisiae every 48 h to a fresh plate for 2,200 generations of the yeast. Since one single colony was transferred, and thus the effective population size is low, the efficacy of natural selection in filtering deleterious mutations out is very low, and thus the fixation rate of mutations approximates the mutation rate. We divided genes in the evolving populations into two categories, those that present a SNP in the gene or the intergenic region neighboring it and those that do not present any SNP or indel (i.e., insertion or deletion). For each of these regions we first estimated dN between S. cerevisiae and S. paradoxus for the six genes neighboring it in the genome. Regions with SNPs or indels exhibited higher dN than those without SNPs in all the five experimental populations analyzed (fig. 2a), pointing to a positive correlation between the mutation rate and the evolution rate in the genome of S. cerevisiae. Moreover, in all five mutation accumulation lines, GRDs were significantly more enriched for intergenic SNPs than GRSs: an average of 587 out of the 2,410 GRDs contained at least one SNP or indel (24.36%) against 1,582 of the 7,208 GRSs (21.95%) (Fisher’s exact test: F = 1.14, P = 0.01) (fig. 2b). When we analyzed intergenic and synonymous SNPs, we found that 45% of the GRDs contained at least one SNP against 41% of the GRSs (Fisher’s exact test: F = 1.18, P = 1.73 × 10−3). Although the number of data in the evolution experiment is very reduced compared to that of Liti et al. (2009), we found that the rate of mutation calculated as the number of synonymous SNPs per synonymous site for GRDs (Mean = 0.018 SNPs per site) was significantly greater than that for GRSs (Mean = 0.015 SNPs per site; t-test: t = 6.1, P = 1.23 × 10−9). In summary, all three data sets pinpoint the higher mutation rate in genome regions containing duplicates than in those containing only singletons.
Fig. 2.

—Experimental evolution of S. cerevisiae reveals that mutational genome hotspots are traps for duplicates. We identified mutations in the coding region and 600-nucleotide genome regions upstream genes in S. cerevisiae and calculated the nonsynonymous nucleotide substitutions (dN) between S. cerevisiae and its close phylogenetic relative S. paradoxus for the six genes surrounding that gene with a SNP in its promoter. (a) Genes that have fixed a SNP in the promoter regions (black columns) in each of the five experimentally evolving line (MA1–MA5) exhibit significantly higher (* indicates P < 0.01; ** indicates P < 0.001) dN than those without SNPs (white boxes). (b) Genome regions that contain duplicates (GRDs) exhibit more SNPs than in the promoter (black columns) fixed during the evolution experiment of each line (MA1 to MA5) (* indicates P < 0.01; ** indicates P < 0.001) than genome regions that do not contain duplicates (GRSs) (white columns).
Duplicated Genes Fall within Transcriptional Genome Hotspots
There are a number of mechanisms that ensure low error rates during transcription and translation in the cell, including tRNA aminoacetylation (Ibba and Soll 1999), Watson-Crick base pairing of codon:anticodon at the ribosome (Reynolds et al. 2010), discrimination of the elongation factor-Tu against misacylated aminoacyl tRNAs (LaRiviere et al. 2001), and ribosomal proofreading (Zaher and Green 2009). Despite the numerous quality control mechanisms for translation, the error of translation has been estimated to be in the order of 10% per codon in Escherichia coli (Ruan et al. 2008). Highly expressed regions of the genome are more prone to accumulate errors than lowly expressed genome regions (Park et al. 2012). Higher error rates of translation would ensure a reduced functional redundancy between the gene copies of duplicates, which would be followed by strong purifying selection to retain both of the functionally differentiated gene copies. We examined the distribution of gene expression hotspots in the genome of S. cerevisiae and compared the mean expression of GRDs to that of GRSs (supplementary data 12, Supplementary Material online), using the RNA sequencing data available in Supplementary table S4, Supplementary Material online from a previous study (Nagalakshmi et al. 2008), and RNA sequence data obtained for this study (see Materials and Methods) as a proxy to the rate of translation. Expression of GRDs was measured as the mean expression of neighboring singletons (supplementary data 12, Supplementary Material online). GRDs were more expressed (Mean: 3.33 ± 0.04) on average than GRSs (Mean: 3.17 ± 0.02) (Wilcoxon rank test: P = 7.35 × 10−4). WGDs are dosage sensitive (Makino et al. 2013), perhaps because their encoded functions are required at specific levels in the cell. We extracted the list of WGDs in S. cerevisiae (Fares et al. 2013; Keane et al. 2014) and reanalyzed the transcriptional features of the genome regions containing them (supplementary data 13, Supplementary Material online). GRDs formed by WGDs are more highly expressed (Mean: 3.66 ± 0.04) than GRSs (Wilcoxon test: P < 2.2 × 10−16). In contrast to GRDs formed by WGDs, those formed by SSDs showed lower mean expression levels (Mean: 3.04 ± 0.05) than GRSs (Wilcoxon rank test: P = 0.048). The question that remains is whether the promoter architecture of duplicates is different from that of singletons and allow duplicates their expression divergence in regions of the genome with high transcriptional rate.
Genome Regions with Duplicates Are Rich in Repeats-Containing Promoters
Is the promoter architecture of GRDs different from that of GRSs? We used the map of repeat-containing gene promoters in S. cerevisiae S288C (supplementary table S2, Supplementary Material online [Vinces et al. 2009]). This map contained 1,974 different motifs that were repeated a variable number of times in each of the 1,359 genes containing them, with the total number of repeated regions summing up to 5,699 repeats. A total of 1,341 GRDs (55.6% of all GRDs in the genome) presented at least one gene with a repeat motif in its promoter. In contrast to this, 52.3% of GRSs contained at least one gene with promoter repeats. The percentage of GRDs with repeat motifs in the gene promoters was higher than that of GRSs (Fisher’s exact test: F = 1.15, P = 5 × 10−3). Most of the genes with repeat regions in GRDs were duplicates. Indeed, 536 duplicates (i.e., 23.9% of all duplicated genes) contained repeats in their promoters against 823 singletons (i.e., 18.7% of all singletons), with duplicates being significantly more enriched for promoter repeats than singletons (Fisher’s exact test: F = 1.37, P = 8.35 × 10−7).
To determine whether such repeat regions followed gene duplication in Saccharomyces, we searched for repeat regions in Zygosaccharomyces rouxii, a yeast species predating the whole genome duplication and most small-scale duplications of Saccharomyces. To this end, we used the Yeast Genome Order Browser (Byrne and Wolfe 2005) to extract the intergenic regions up-stream of the singleton genes that were orthologs to S. cerevisiae duplicates. Only 5.3% of the repeat regions identified in S. cerevisiae were present in the orthologous intergenic regions of Z. rouxii. In most cases, however, the number and length of the repeats were lower and shorter, respectively, in Z. rouxii than in S. cerevisiae. Moreover, duplicates containing repeat regions predating the WGD, also contained other repeat regions that were absent in their Z. rouxii orthologs.
WGDs were enriched for repeat regions compared to singletons (269 WGDs, corresponding to 24.23% of all WGDs. Fisher’s exact test: F = 1.46, P = 3.49 × 10−6). Likewise, SSDs were more enriched for repeat regions than singletons (267 SSDs, corresponding to 22.25% of all SSDs. Fisher’s exact test: F = 1.31, P = 9 × 10−3). Summing the number of repeats per promoter, we found 2,333 repeats in duplicates, which was a higher number than expected (taking into account that duplicates are 33.7% of all S. cerevisiae genes, we expected 0.337 × 5,699 = 1,921) by chance (Chi-square test: χ2 = 40.01, P = 2.52 × 10−10). In contrast to duplicates, singletons represented a total of 3,366 repeat motifs, which was a lower number than expected (expected = 3,778) by chance (Chi-square test: χ2 = 23.79, P = 1.072 × 10−6).
GRDs with duplicated genes containing repeats in their promoters may enable the expression divergence between the gene copies of the duplicates under alternative environmental conditions. The enrichment of duplicates for tandem repeats may point to their higher transcriptional plasticity, perhaps to guarantee the performance of their functions in environments different to the normal ones (i.e., under stress). That is, the presence of repeat regions in the promoter of one of the gene copies may have allowed preserving the same functions as the ones performed in the sister copy but in alternative environments. If this were the case, one would expect that gene copies of duplicates in which at least one gene copy bears tandem repeat regions should exhibit more similar functions than those of duplicates without tandem repeat regions. We used the genetic interaction map of S. cerevisiae as a proxy to the functions of each of the genes (Costanzo et al. 2010). This map contains roughly 6.5 million genetic interactions and the functional chart for 75% of the S. cerevisiae genes. The number of genetic interactions for a particular gene is a proxy to the number of functions it performs (Costanzo et al. 2010). Therefore, genes sharing high number of interactions are likely to be involved in the same functions. Likewise, duplicate gene copies sharing a high proportion of their interactions are likely to perform more similar functions than those sharing a low proportion of their genetic interactions (Costanzo et al. 2010). We tested whether the proportion of shared interactions between gene copies (fig. 3a) correlates with the number or length of the repeats they contain. Duplicates with at least one gene copy containing longer repeat motifs are also those in which both gene copies performed more similar functions (Spearman correlation: r = 0.54, P = 2.80 × 10−4, fig. 3b).
Fig. 3.
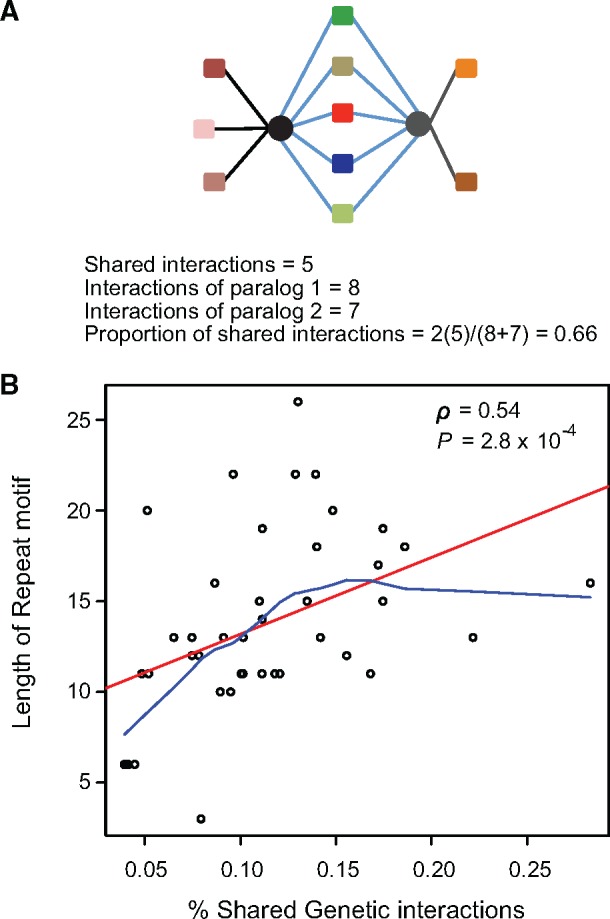
—Duplicates in which one gene copy bears tandem repeat regions (trr) in its promoter share more functions than those without trr. (a) To determine the number and kind of functions of each gene copy of a duplicate (gene copies are presented as black and gray circles) we found the significant genetic interactions of that gene (symbolized as color squares) based on the functional chart of S. cerevisiae from a previous study (Costanzo et al. 2010). The number of shared functions between the two gene copies of a duplicate was calculated as twice the number of shared interactions divided by the sum of total genetic interactions of both of the gene copies. (b) We plotted the length of the repeat units in the promoter of duplicated genes against the percentage of shared interactions (GI) of the gene copies of a duplicate. Linear and curvilinear trend adjustments are shown as red and clue lines, respectively. Pearson correlation (ρ) and its probability are shown.
Under our hypothesis, gene copies with promoter repeat motifs should be transcriptionally plastic such that variations in the number or frequency of their repeat motif should influence their expression. If the gene copy without promoter repeats is the one that is functional in constant environments, we should expect that gene copies with promoter repeat motifs should accumulate more mutations than their paralogs in such environments where they are not required. These mutations can allow the emergence of gene copy variants preadaptive to other environments. We examined the distribution of SNPs that emerged during the experimental evolution under strong genetic drift and constant rich environment of the five S. cerevisiae populations. We found that 175 out of the 536 duplicates with repeat regions (32.6% of the duplicates with tandem repeats) contained SNPs in their promoter regions (600 nucleotides upstream the coding gene), while 423 of the 1,679 genes (25.1%) with no repeat motifs accumulated SNPs. Duplicates with repeats accumulated more promoter SNPs than genes without repeats (Fisher’s exact test: F = 1.44, P = 9.61 × 10−4).
Duplicates Provide Transcriptional Plasticity under Stress
To determine the transcriptional plasticity of duplicates, we compared the alteration in the transcriptional levels of duplicates and singletons in S. cerevisiae after growing it in 13 different stress conditions (see Materials and Methods). If duplicates provide plasticity to adapt to stress conditions, the number of altered genes under stress should be greater among duplicates than singletons. The number of duplicated genes with significantly increased expression levels was higher than that of singleton genes in all 13 conditions of stress (fig. 4a). Examination of the duplicates that contained one gene copy with and one without tandem repeats in their promoters revealed that the gene copy with tandem repeats systematically incremented more its expression under the 13 stress conditions examined than the gene copy without repeats (Mean of the differences: 25.74, Paired t-test: t = 8.51, d.f. = 12, P = 1.99 × 10−6; fig. 4b). In conclusion, the high transcriptional plasticity of certain genome regions allowed trapping duplicates with particular promoter architecture. This architecture involved enrichment for repetitive motifs that allowed the expression divergence of the gene copies and their increased transcriptional plasticity, increasing the adaptability of S. cerevisiae to stress.
Fig. 4.

—Duplicates exhibit high transcriptional plasticity under stress. (a) We compared the transcriptional plasticity of duplicates to singletons of S. cerevisiae growing under one of a set of 13 different stress conditions. RNA sequence data of eight of the conditions (Acidic stress, Alkaline, Wine fermentation, Heat stress, Lithium stress, Manganese stress, Osmotic stress, and Glucose limitation) were extracted from previously published data and which are available from the Saccharomyces Genome Database (SGD; http://www.yeastgenome.org/download-data/expression; last accessed May 4, 2017). RNA sequence data for five of the conditions (Ethanol stress, Lactic stress, Glycerol stress, Oxidative stress, and Oxidative stress with glucose) were obtained in the laboratory after growing populations of the yeast under these conditions. We calculated the increments in the expression levels (ΔEi−j) by comparing the normalized RNA sequence counts in normal conditions (i) versus those for stress conditions (j). Increments of expression were calculated as . We considered identified genes with significant increments in expression and with at least 20% increments from the normal to the stress conditions. The proportion of genes with significant increments for Duplicates (D) and singletons (S) were estimated (color coded in the figure) and these were compared using a Fisher’s exact test (Probability plot). (b) For each of the 23 stress conditions, we calculated the percentage of cases in which the duplicated gene copy with repeats in its promoter regions (Drep) exhibited a larger increment in expression (ΔEi−j) than its paralog with no repeats in its promoter (black portions of the columns), and viceversa (D) (gray portions of the columns).
Discussion
In this study, we present evidence that support a higher persistence of duplicates in genome regions with high mutation rates and transcription levels. This persistence seems to be the result of a quick divergence in the functions and expressions of the gene copies of duplicates after duplication, followed by purifying selection to maintain both of the gene copies. An alternative explanation to this is that duplicate provides a selective advantage because of the masking effect of deleterious mutations of the gene copies, a phenomenon that is more important in genome regions with high mutation rates. However, the number of scenarios that render the mutational masking effects of duplicates selectively advantageous is limited. Fisher realized that in an idealized population with infinite size, two genes with identical functions can only be mutually maintained by the masking of deleterious mutations if both bear identical mutation rates to defective alleles (Fisher 1935). In finite populations, the duplicate is effectively neutral and vulnerable to eventual loss by genetic drift (Clark 1994; Lynch 2007; Lynch et al. 2001; O'Hely 2006). This prediction is in agreement with the loss of 92% of all Saccharomyces cerevisiae WGDs (Wolfe and Shields 1997). Therefore, it is more likely that the higher persistence of duplicates in genome regions with high mutation and translation rates is the result of faster divergence between the gene copies, thereby increasing the strength of purifying selection on each gene copy.
An intriguing result derived from our analyses is that WGDs but not SSDs have been maintained in mutational and transcriptional genomic hotspots. WGDs are more enriched than SSDs for dosage-sensitive genes as well as for genes encoding protein complexes that require keeping a stoichiometric balance and previous reports have shown stronger evidence for functional and transcriptional subfunctionalization in WGDs than SSDs (Fares et al. 2013; Keane et al. 2014). The enrichment of genome mutational hotspots for WGDs may therefore be due to stronger selective constraints against the loss of one gene copy in order to preserve the stoichiometric balance and the set of ancestral functions in the cell.
It has been shown that the birth-death dynamics of duplicated genes depend on the functional features of the duplicates, however such dependency relaxes with regards to WGDs (Guan et al. 2007; Wapinski et al. 2007). Since WGDs were particularly preserved in genome mutational hotspots, we expect such pattern to be independent of the functional features of duplicates. Importantly, these authors also highlighted that the regulatory divergence after gene duplication may play a more important role in the origin of adaptations than the biochemical divergence, well in support of our observations.
The enrichment of genome mutational and transcriptional hotspots for duplicated genes has important implication for the origin of evolutionary innovations and adaptations. The persistence of duplicates in genome error hotspots can allow the fixation of polymorphisms in the population at both the regulatory and coding levels and the eventual emergence of exaptations, preadaptations to conditions never before encountered by the organism. In support of this scenario, GRDs are enriched for duplicates with promoter repetitive elements more so than expected by chance and they bear more SNPs in their promoters than GRSs and then expected by chance. Duplicates with repeat regions are known to allow both the transcriptional plasticity of these regions (Vinces et al. 2009) and transcriptional reprogramming of the gene copy when its sister copy is silenced (Kafri et al. 2005). This transcriptional plasticity has been proposed to be key to the origin of adaptations to stress in S. cerevisiae and certainly the source of biological innovations (Fares 2015). Accordingly, we show that duplicates with repeats in their promoters are transcriptionally more plastic than those without repeats. This plasticity may be correlated with the ability of yeast to grow under stress conditions (Mattenberger et al. 2017a, b). Taken together, our results strongly pinpoint the role of the genome context on the fate of duplicated genes and on the origin of evolutionary adaptations.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This study was supported by a grant (reference: FEDER-BFU2015-66073-P) from the Spanish Ministerio de Economía y Competitividad-FEDER and a grant (reference: ACOMP/2015/026) from the local government Consellería de Educación Investigación, Cultura y Deporte, Generalitat Valenciana to M.A.F. C.T. was supported by a grant Juan de la Cierva from the Spanish Ministerio de Economía y Competitividad (reference: JCA-2012-14056).
Literature Cited
- Agier N, Fischer G.. 2012. The mutational profile of the yeast genome is shaped by replication. Mol Biol Evol. 29:905–913. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W.. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DB, Gasch AP.. 2008. Stress-activated genomic expression changes serve a preparative role for impending stress in yeast. Mol Biol Cell. 19:4580–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Bhadra U, Bhadra MP, Auger DL.. 2001. Dosage-dependent gene regulation in multicellular eukaryotes: implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev Biol. 234:275–288 [DOI] [PubMed] [Google Scholar]
- Birchler JA, Riddle NC, Auger DL, Veitia RA.. 2005. Dosage balance in gene regulation: biological implications. Trends Genet. 21:219–226. [DOI] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA.. 2012. Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci U S A. 109:14746–14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bro C, et al. 2003. Transcriptional, proteomic, and metabolic responses to lithium in galactose-grown yeast cells. J Biol Chem. 278:32141–32149. [DOI] [PubMed] [Google Scholar]
- Byrne KP, Wolfe KH.. 2005. The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 15:1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero-Paulet L, Fares MA.. 2012. Evolutionary dynamics and functional specialization of plant paralogs formed by whole and small-scale genome duplications. Mol Biol Evol. 29:3541–3551. [DOI] [PubMed] [Google Scholar]
- Casamayor A, et al. 2012. The role of the Snf1 kinase in the adaptive response of Saccharomyces cerevisiae to alkaline pH stress. Biochem J. 444:39–49. [DOI] [PubMed] [Google Scholar]
- Chuang JH, Li H.. 2004. Functional bias and spatial organization of genes in mutational hot and cold regions in the human genome. PLoS Biol. 2:E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG. 1994. Invasion and maintenance of a gene duplication. Proc Natl Acad Sci U S A. 91:2950–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH.. 2008. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 9:938–950. [DOI] [PubMed] [Google Scholar]
- Costanzo M, et al. 2010. The genetic landscape of a cell. Science 327:425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE.. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 1151:165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares MA. 2015. The origins of mutational robustness. Trends Genet. 31:373–381. [DOI] [PubMed] [Google Scholar]
- Fares MA, Keane OM, Toft C, Carretero-Paulet L, Jones GW.. 2013. The roles of whole-genome and small-scale duplications in the functional specialization of Saccharomyces cerevisiae genes. PLoS Genet. 9:e1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1935. The sheltering of lethals. Am Nat. 69:10. [Google Scholar]
- Freeling M, Thomas BC.. 2006. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res. 16:805–814. [DOI] [PubMed] [Google Scholar]
- Garcia-Rodriguez N, et al. 2012. Impaired manganese metabolism causes mitotic misregulation. J Biol Chem. 287:18717–18729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ.. 2010. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 44:445–477. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z.. 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 11:725–736. [DOI] [PubMed] [Google Scholar]
- Gout JF, Duret L, Kahn D.. 2009. Differential retention of metabolic genes following whole-genome duplication. Mol Biol Evol. 26:1067–1072. [DOI] [PubMed] [Google Scholar]
- Gout JF, Kahn D, Duret L, Paramecium Post-Genomics C.. 2010. The relationship among gene expression, the evolution of gene dosage, and the rate of protein evolution. PLoS Genet. 6:e1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout JF, Lynch M.. 2015. Maintenance and loss of duplicated genes by dosage subfunctionalization. Mol Biol Evol. 32:2141–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Dunham MJ, Troyanskaya OG.. 2007. Functional analysis of gene duplications in Saccharomyces cerevisiae. Genetics 175:933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. 1932. The causes of evolution London: Green and Co. [Google Scholar]
- Ibba M, Soll D.. 1999. Quality control mechanisms during translation. Science 286:1893–1897. [DOI] [PubMed] [Google Scholar]
- Jansen ML, et al. 2005. Prolonged selection in aerobic, glucose-limited chemostat cultures of Saccharomyces cerevisiae causes a partial loss of glycolytic capacity. Microbiology 151:1657–1669. [DOI] [PubMed] [Google Scholar]
- Kafri R, Bar-Even A, Pilpel Y.. 2005. Transcription control reprogramming in genetic backup circuits. Nat Genet. 37:295–299. [DOI] [PubMed] [Google Scholar]
- Keane OM, Toft C, Carretero-Paulet L, Jones GW, Fares MA.. 2014. Preservation of genetic and regulatory robustness in ancient gene duplicates of Saccharomyces cerevisiae. Genome Res. 24:1830–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1983. Rare variant alleles in the light of the neutral theory. Mol Biol Evol. 1:84–93. [DOI] [PubMed] [Google Scholar]
- Kimura M, Takahata N.. 1983. Selective constraint in protein polymorphism: study of the effectively neutral mutation model by using an improved pseudosampling method. Proc Natl Acad Sci U S A. 80:1048–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Murray AW.. 2011. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol. 3:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRiviere FJ, Wolfson AD, Uhlenbeck OC.. 2001. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science 294:165–168. [DOI] [PubMed] [Google Scholar]
- Liti G, et al. 2009. Population genomics of domestic and wild yeasts. Nature 458:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, et al. 2012. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 40:W622–W627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sunderland, MA: Sinauer Associates, Inc. [Google Scholar]
- Lynch M, O'Hely M, Walsh B, Force A.. 2001. The probability of preservation of a newly arisen gene duplicate. Genetics 159:1789–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino T, McLysaght A, Kawata M.. 2013. Genome-wide deserts for copy number variation in vertebrates. Nat Commun. 4:2283. [DOI] [PubMed] [Google Scholar]
- Marcet-Houben MG, T.. 2015. Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the Baker's yeast lineage. PLoS Biol. 13:e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Makepeace K, Hill SA, Hood DW, Moxon ER.. 2005. Microsatellite instability regulates transcription factor binding and gene expression. Proc Natl Acad Sci U S A. 102:3800–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattenberger F, Sabater-Munoz B, Hallsworth JE, Fares MA.. 2017a. Glycerol stress in Saccharomyces cerevisiae: cellular responses and evolved adaptations. Environ Microbiol. 19:990–1007. [DOI] [PubMed] [Google Scholar]
- Mattenberger F, Sabater-Munoz B, Toft C, Fares MA.. 2017b. The phenotypic plasticity of duplicated genes in Saccharomyces cerevisiae and the origin of adaptations. G3 (Bethesda) 7:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, et al. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hely M. 2006. A diffusion approach to approximating preservation probabilities for gene duplicates. J Math Biol. 53:215–230. [DOI] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Berlin: Springer Verlag.
- Ohno S. 1999. Gene duplication and the uniqueness of vertebrate genomes circa 1970–1999. Semin Cell Dev Biol. 10:517–522. [DOI] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD.. 2003. Dosage sensitivity and the evolution of gene families in yeast. Nature 424:194–197. [DOI] [PubMed] [Google Scholar]
- Park C, Qian W, Zhang J.. 2012. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 13:1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JL, Wagner A.. 2014. The robustness and evolvability of transcription factor binding sites. Science 343:875–877. [DOI] [PubMed] [Google Scholar]
- Pu S, Wong J, Turner B, Cho E, Wodak SJ.. 2009. Up-to-date catalogues of yeast protein complexes. Nucleic Acids Res. 37:825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Liao BY, Chang AY, Zhang J.. 2010. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 26:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman MK, et al. 2001. Replication dynamics of the yeast genome. Science 294:115–121. [DOI] [PubMed] [Google Scholar]
- Rando OJ, Verstrepen KJ.. 2007. Timescales of genetic and epigenetic inheritance. Cell 128:655–668. [DOI] [PubMed] [Google Scholar]
- Reynolds NM, et al. 2010. Cell-specific differences in the requirements for translation quality control. Proc Natl Acad Sci U S A. 107:4063–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Wray GA.. 2002. Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol. 19:1991–2004. [DOI] [PubMed] [Google Scholar]
- Ruan B, et al. 2008. Quality control despite mistranslation caused by an ambiguous genetic code. Proc Natl Acad Sci U S A. 105:16502–16507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Bockler B, Lehner B.. 2012. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488:504–507. [DOI] [PubMed] [Google Scholar]
- Seoighe C, Wolfe KH.. 1999. Yeast genome evolution in the post-genome era. Curr Opin Microbiol. 2:548–554. [DOI] [PubMed] [Google Scholar]
- Streelman JT, Kocher TD.. 2002. Microsatellite variation associated with prolactin expression and growth of salt-challenged tilapia. Physiol Genomics 9:1–4. [DOI] [PubMed] [Google Scholar]
- Supek F, Lehner B.. 2015. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JS, Raes J.. 2004. Duplication and divergence: the evolution of new genes and old ideas. Annu Rev Genet. 38:615–643. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Barkai N, Verstrepen KJ.. 2009. Promoter architecture and the evolvability of gene expression. J Biol. 8:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294:2364–2368. [DOI] [PubMed] [Google Scholar]
- Vinces MD, Legendre M, Caldara M, Hagihara M, Verstrepen KJ.. 2009. Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324:1213–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A.. 2007. Natural history and evolutionary principles of gene duplication in fungi. Nature 449:54–61. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC.. 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387:708–713. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Zaher HS, Green R.. 2009. Quality control by the ribosome following peptide bond formation. Nature 457:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.