

Abstract
Fibulin-3 (F3) is an important, disulfide-rich, extracellular matrix glycoprotein that has been associated with a number of diseases ranging from cancer to retinal degeneration. An Arg345Trp (R345W) mutation in F3 causes the rare, autosomal dominant macular dystrophy, Malattia Leventinese. The purpose of this study was to identify and validate novel intracellular interacting partners of wild-type (WT) and R345W F3 in retinal pigment epithelium cells. We used stable isotope labeling by amino acids in cell culture (SILAC) to generate ‘heavy’ and ‘light’ isotopically labeled ARPE-19 cell populations which were subsequently infected with adenovirus encoding for FLAG-tagged (FT) WT or R345W F3. After immunoprecipitation, interacting proteins were identified by multidimensional protein identification technology (MudPIT). We identified sixteen new intracellular F3 interacting partners, the vast majority of which are involved in protein folding and/or degradation in the endoplasmic reticulum (ER). Eight of these interactions (ANXA5, ERdj5, PDIA4, P4HB, PDIA6, RCN1, SDF2L1, and TXNDC5) were verified at the western blotting level. These F3 interactome results can serve as the basis for pursuing targeted genetic or pharmacologic approaches in an effort to alter the fate of either WT or mutant F3.
INTRODUCTION
Fibulin-3 (F3) is a 55 kDa, secreted, extracelluar matrix glycoprotein that is produced in a number of tissues throughout the body, including the lung, skin, adipose tissue, and various cell layers within the eye such as the ciliary body (Mackay et al., 2015) and the retinal pigment epithelium (RPE) (Marmorstein et al., 2002). While the exact function of F3 is still not clear, removal of F3 in mice leads to a general, aging-associated phenotype including reduced lifespan, decreased body mass/fat and organ support failure (McLaughlin et al., 2007). Surprisingly, F3 knockout mice show no indication of any ocular disorder/disease (McLaughlin et al., 2007), indicating that removal of F3 has no apparent affect on normal eye development or physiology.
Interestingly, an autosomal dominant, gain-of-toxic-function mutation (Arg345Trp [R345W]) was identified in F3 as the single cause of two nearly identical, rare retinal diseases, Malattia Leventinese (ML) and Doyne honeycomb retinal dystrophy (Stone et al., 1999). Subsequent cell culture studies found that the R345W mutation causes reductions to F3 secretion and higher intracellular steady state levels, indicating that the protein is likely misfolded (Hulleman et al., 2012; Hulleman et al., 2011; Hulleman and Kelly, 2015; Marmorstein et al., 2002; Roybal et al., 2005). R345W transgenic mouse-based work has demonstrated that this mutation increases complement activation and inflammation (Fernandez-Godino et al., 2015; Fu et al., 2007; Garland et al., 2014; Marmorstein et al., 2007). More recently, two independent studies have identified unique mutations in F3 associated with distinct eye diseases, reaffirming the notion that autosomal dominant genetic alterations in F3 have the ability to disrupt homeostasis in the eye. For example, in 2015, exome sequencing of a family with open-angle glaucoma identified an Arg140Trp (R140W) mutation in F3 (Mackay et al., 2015). Moreover, in 2016, additional sequencing efforts identified a Asp49Ala (D49A) mutation in F3 which was associated with cuticular drusen in an age-related macular degeneration (AMD) patient (Duvvari et al., 2016). Furthermore, copy number increases near the F3 locus have also been correlated with an increased likelihood of developing AMD (Meyer et al., 2011).
Given the apparent importance of F3 in a number of eye diseases, it is surprising that there is an absence of known interacting partners that bind to, and potentially influence F3 synthesis, folding and secretion. To date, there are only nine identified interacting partners of wild-type (WT) F3: tissue inhibitor of metalloproteinases-3 (TIMP3) (Klenotic et al., 2004), extracellular matrix protein 1 (ECM-1) (Sercu et al., 2009), complement factor H (CFH) (Wyatt et al., 2013), protein disulfide isomerase A3 (PDIA3) (Jessop et al., 2007), ERdj5 (Oka et al., 2013), calnexin (CNX), calreticulin (CALR), 78 kDa glucose regulated protein (GRP78), and 94 kDa glucose regulated protein (GRP94) (Hulleman and Kelly, 2015). Furthermore, only four of the listed binding partners (CNX, CALR, GRP78, GRP94), were tested for binding to the pathogenic R345W F3 variant (Hulleman and Kelly, 2015).
We rationalized that identifying additional interacting partners of WT and R345W would uncover previously unknown pathways used by F3 during its maturation in the ER. Furthermore, we speculated that identification of new F3 binding partners would provide a framework of proteins which could be subsequently genetically manipulated to determine their influence on F3 protein homeostasis. To this end, we performed an unbiased identification, quantification and validation of intracellular interacting partners shared between WT and R345W F3 using stable isotope labeling by amino acids in cell culture (SILAC) in a human retinal pigment epithelial cell line.
To identify and quantify intracellular F3 binding partners, we first generated two SILAC populations of human adult retinal pigment epithelium cells (ARPE-19, American Type Culture Collection, CRL-2302, Manassas, VA verified by short tandem repeat DNA profiling) labeled with either ‘heavy’ or ‘light’ isotopes (Cambridge Isotope Laboratories, Andover, MA, 93–97% labeling efficiency, Fig. 1A). Subsequently, a confluent monolayer of the ‘light’ isotope cell line was infected with FLAG-tagged (FT) WT F3 adenovirus, while a confluent monolayer of the ‘heavy’ isotope cell line was infected with FT R345W F3 virus at an identical, low-level multiplicity of infection (MOI) of 5 for 48 h, which does not cause detectible endoplasmic reticulum (ER) stress (Hulleman and Kelly, 2015). Fresh media was incubated on cells for 24 h prior to crosslinking the proteins with dithiobis-succinimidyl propionate (DSP), which was followed by cell lysis with radioimmunoprecipitation assay (RIPA) buffer. Proteins which bound to FT F3 were immunoprecipitated (IP’d) using anti-FLAG M1 agarose beads (10 μL, 4°C, O/N, Sigma, St. Louis, MO), washed with RIPA, eluted, and combined for quantitative mass spectrometry analysis (Fig. 1A). Samples were prepared for multidimensional protein identification technology (MudPIT) mass spectrometry analysis as described previously (Tan et al., 2014) and run on a LTQ-Orbitrap Velos (Thermo Fisher, Waltham, MA). Tandem mass spectra were extracted (Raw Xtractor 1.9.1) and searched against a human database (ProLuCID, (Xu et al., 2015)). The heavy to light ratio was quantified by Census (Park et al., 2008).
Fig. 1.
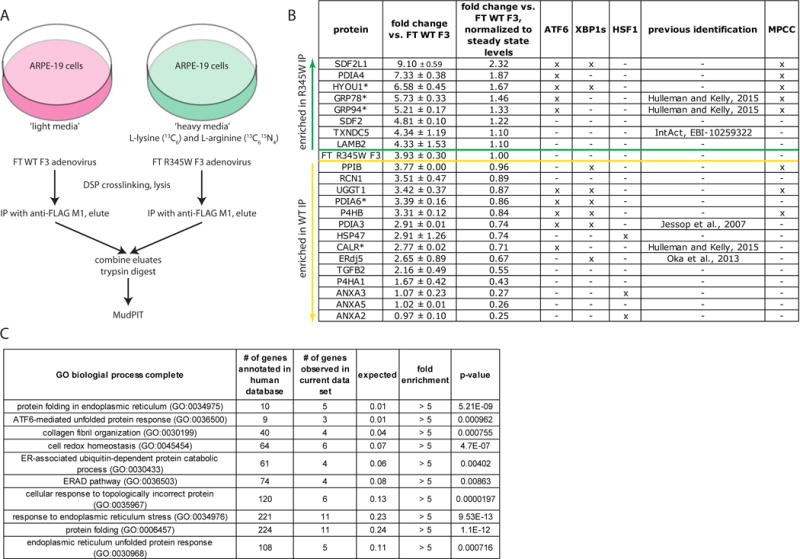
Schematic of the SILAC/MudPIT approach used to identify F3 interacting partners. (A) ARPE-19 cells were labeled with either ‘heavy’ or ‘light’ isotopes. Subsequently, the ‘light’ isotope cell line was infected with either FLAG-tagged (FT) WT F3, while the ‘heavy’ isotope cell line was infected with FT R345W F3 at an identical multiplicity of infection. Proteins which bound to FT F3 were eluted, combined, and trypsinized for quantitative mass spectrometry analysis by MudPIT. (B) List of identified F3 binding partners. An ‘x’ under ATF6, XBP1s, or HSF1 indicates that the expression of these proteins is regulated by one or more of the indicated transcription factors (Ryno et al., 2014; Shoulders et al., 2013). An ‘x’ under the MPCC column Indicates that the protein has been found in the “multi-protein chaperone complex” (MPCC) (Meunier et al., 2002). Proteins above the green line were found enriched in the R345W F3 interactome, while those below the yellow line were enriched in the WT F3 interactome. Proteins with an asterisk were among the top five enriched proteins based on spectral count. Data presented originate from two independent experiments, false discovery rate < 1%, minimum of two peptides per protein. (C) Gene ontology (GO) enrichment analysis of biological pathways of identified F3 binding partners.
MudPIT mass spectrometry analysis identified 22 common interacting partners of both WT and R345W F3 in ARPE-19 cells (Fig. 1B), all of which were either ER resident proteins, secreted proteins, or typically membrane-bound. Of the 22 identified interacting partners, six were identified previously in prior studies; CALR, GRP78, and GRP94 (Hulleman and Kelly, 2015), PDIA3 (Jessop et al., 2007), ERdj5 (Oka et al., 2013), and thioredoxin domain-containing protein 5 (TXNDC5, IntAct (Orchard et al., 2014), [http://www.ebi.ac.uk/intact/], EBI-10259322). Thus, of the 22 identified proteins, 16 were considered novel binding partners of both WT and R345W F3. The five most prominent F3 interacting proteins based on total spectral count were: hypoxia up-regulated-1 (HYOU1), GRP78, GRP94, protein disulfide isomerase 6 (PDIA6), and CALR (Fig. 1B, asterisk).
Eight of the 22 total identified proteins were enriched in the R345W F3 IP between 1.10 and 2.32 fold (Fig. 1B, above green line) when compared to their levels in the WT F3 IP, and after accounting for higher R345W F3 intracellular steady-state levels (i.e., 3.93 ± 0.30 fold vs WT F3, Fig. 1B). Of these proteins, the majority (5/8) were members of a multi-protein chaperone complex (MPCC) identified previously to associate with unassembled, incompletely folded immunoglobulin heavy chain or other unfolded substrates (Meunier et al., 2002). In contrast, 14/22 of the remaining proteins were enriched in the WT F3 IP (Fig. 1B, below yellow line), again after accounting for higher R345W F3 intracellular steady-state levels. Fifteen of the interacting proteins have been demonstrated to be regulated by unfolded protein-triggered stress-responsive signaling pathways; the majority of identified proteins (12/22) are downstream targets of either the inositol-requiring enzyme 1 (IRE1, via spliced X-box protein 1, XBP1s) and/or activating transcription factor 6 (ATF6) arms of the unfolded protein response (UPR) (Shoulders et al., 2013), whereas three additional proteins (annexin A2 [ANXA2], annexin A3 [ANXA3], and heat shock protein 47 [HSP47]) are downstream targets of activated heat shock factor 1 (HSF1) (Ryno et al., 2014) (Fig. 1B). Furthermore, gene ontology (GO, http://geneontology.org/) pathway analysis (Ashburner et al., 2000; Gene Ontology, 2015) of the 22 identified proteins demonstrated >5 fold enrichment in proteins involved in protein folding, unfolded protein response, collagen fibril organization and ER-associated degradation, amongst other folding-related pathways (Fig. 1C).
To confirm the hits identified by mass spectrometry, we validated a subset of the F3 interacting partners by western blotting using an elevated MOI of 25. This level of expression was necessary to visualize a number of F3 interactions by the less sensitive western blotting method. However, this extent of F3 over-expression also leads to noticeable UPR activation, especially in cells expressing the R345W variant (Hulleman and Kelly, 2015; Roybal et al., 2005).
Input lanes prior to IP demonstrated that ERdj5 (Proteintech, Chicago, IL), protein disulfide isomerase A4 (PDIA4, Proteintech), prolyl 4-hydroxylase beta polypeptide (P4HB, Bethyl Laboratories, Montgomery, TX), protein disulfide isomerase A6 (PDIA6, Bethyl Laboratories), and stromal-derived factor 2-like 1 (SDF2L1, Santa Cruz, Santa Cruz, CA) were all stress-responsive (as expected), increasing in abundance with WT and/or R345W F3 over-expression (Fig. 2). Intriguingly, two proteins, TXNDC5 (Proteintech) and reticulocalbin 1 (RCN1, Bethyl Laboratories) were also increased in cells expressing WT F3, and even more so in cells expressing R345W F3 (Fig. 2), suggesting that the expression of these proteins is regulated by UPR activation in ARPE-19 cells. Steady-state input and IP levels of R345W F3 were increased by 2–3 fold over WT F3 (Fig. 2, anti-FLAG M2, Sigma), in general accordance with the MudPIT results (Fig. 1B) and previous observations (Hulleman et al., 2011; Hulleman and Kelly, 2015; Marmorstein et al., 2002; Roybal et al., 2005).
Fig. 2.
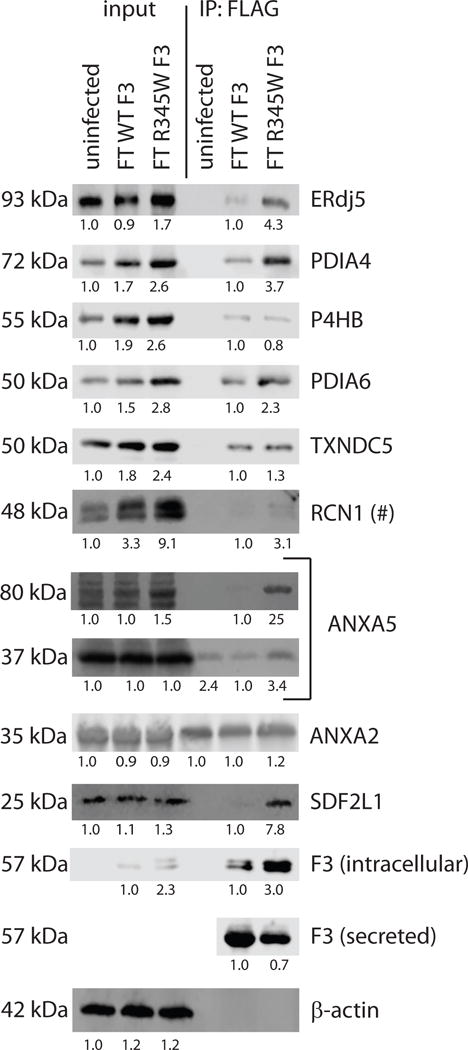
Western blot validation of identified F3 binding partners. ARPE-19 cells were infected with adenovirus encoding for FT WT F3 or FT R345W F3 at an MOI of 25 and F3 binding partners were co-IP’d with anti-FLAG M1 agarose beads. Eluted proteins were run on an SDS-PAGE followed by western blotting. The numbers below each lane are the average intensities relative to either uninfected (input lanes) or FT WT F3 (IP lanes) samples. Representative images of ≥ 3 independent experiments for all proteins but ANXA2 (n =2). (#) – RCN1 bound to both WT and R345W F3, but the relative levels in the IP lanes were highly variable. Western blots were imaged and quantified using a LI-COR Odyssey Fc (LI-COR, Lincoln, NE).
Qualitatively, the vast majority (8/9) of the selected proteins we analyzed by western blotting were verified as F3 interacting partners of both WT and R345W F3. However, in a number of instances, the stiochometry with which the proteins bound to WT or R345W F3 differed from the mass spectrometry results (Fig. 2). After taking into account higher steady state levels of R345W F3 in the IP, ERdj5, PDIA4, ANXA5 (Bethyl Laboratories), and SDF2L1 demonstrated preference for binding to R345W F3, whereas P4HB, PDIA6 and TXNDC5 demonstrated preference for binding to WT F3 according to the western blotting results (Fig. 2). While binding of RCN1 to F3 was confirmed, these results were variable across the independent triplicates (Fig. 2, hashtag). Annexin proteins (ANXA5 and ANXA2 [Santa Cruz]) demonstrated a strong preference for binding to the M1 agarose beads, independent of F3, and ANXA2 was the only mass spectrometry hit we identified as clearly non-specific (Fig. 2). Furthermore, while the canonical ~35 kDa isoform of ANXA5 also demonstrated an affinity for the IP beads (Fig. 2), a previously uncharacterized, immunoreactive 80 kDa species bound to F3, with a strong preference for R345W F3 (~25 fold, Fig. 2).
To the best of our knowledge, this is the first comprehensive analysis of F3 intracellular binding partners. The redundancy of a portion of our hits, in addition to the observation that eight out of nine interactions were qualitatively validated by western blotting, lends credence to the data set and suggests that the identified proteins are likely bona fide binding partners of F3. The majority of these interacting partners are components of the protein folding/degradation machinery of the ER and are increased during conditions of cellular stress, typically in response to unfolded proteins either in the cytosol (Ryno et al., 2014), or in the ER (Shoulders et al., 2013). These results, combined with the GO analysis, indicate that the F3 which was IP’d may be misfolded, or in the process of being folded/refolded by ER chaperones and/or protein disulfide isomerases.
Given the number of cysteine residues in F3 (40) and the number of disulfide bonds required for F3 folding and secretion (15), it is not necessarily surprising that multiple proteins involved in disulfide bond formation or isomerization such as prolyl 4-hydroxylase subunit alpha 1 (P4HA1), P4HB, ERdj5, PDIA3, PDIA4, PDIA6, and TXNDC5 interact with F3. Furthermore, since F3 has a number of domains (6 epidermal growth factor (EGF) domains followed by a C-terminal fibulin-type domain) that need to arrange prior to secretion, it is conceivable that F3 requires interacting with chaperones or protein folding enzymes such as GRP78, GRP94, HYOU1, or peptidylprolyl isomerase B (PPIB). Moreover, as an O- and N-glycosylated protein (Djokic et al., 2013; Hulleman and Kelly, 2015), F3 was found to bind to stromal derived factor 2 (SDF2), SDF2L1 (two O-mannosyltransferase-related proteins), UDP-glucose glycoprotein glucosyltransferase 1 (UGGT1, an enzyme that reglucosylates unfolded glycoproteins (Ferris et al., 2013)), and the ER lectin chaperone, CALR. Nonetheless, the actual identity of many of the isomerases, chaperones or glycosylation-dependent proteins remained elusive until now. Collectively, these data indicate that F3 synthesis, folding, and secretion is complex and involves a number of prominent protein ER pathways.
Consistent with its potential role in modulating the extracellular matrix (ECM) environment (Fernandez-Godino et al., 2015; Fu et al., 2007; Marmorstein et al., 2007), we found that F3 bound to several proteins involved in ECM formation, including laminin subunit beta-2 (LAMB2), a major noncollagenous constituent of basement membranes (Sasaki et al., 2004), HSP47, which is normally thought to be a collagen-specific chaperone (Ishida and Nagata, 2011), RCN1, a largely uncharacterized protein that has the ability to bind to collagen I (DiChiara et al., 2016), and the known ECM modulator and cytokine, transforming growth factor beta 2 (TGFB2) (Fuchshofer et al., 2005). Furthermore, GO analysis showed an enrichment in proteins involved in collagen fibril organization, an intriguing observation given that i) the sub-RPE deposits observed in ML mice expressing F3 are composed of wide-spaced collagen (Fu et al., 2007; Garland et al., 2014), ii) collagen VI was found to be increased in the Bruch’s membrane of ML mice (Garland et al., 2014), and iii) collagen IV was found to be a significant component of drusen isolated from ML patients (Sohn et al., 2014). These data indicate that F3 may be able to directly or indirectly affect the composition or and/or amount of ECM deposited by either binding to, or sequestering proteins that influence ECM formation or degradation.
A surprising feature of our data set is that for the most part WT and R345W F3 bind to the same partners within the ER. This observation begs the question of whether R345W F3 actually causes ML pathogenesis through intracellular or extracellular means (Hulleman, 2016). Recent work using primary RPE cultures strongly suggests the latter (Fernandez-Godino et al., 2015). Future studies will be directed toward identifying extracellular F3 binding partners, which may more clearly delineate binding partner differences between WT and R345W F3. Nonetheless, our findings provide a starting point for exploring how validated interacting partners affect F3, and it is conceivable that other homologous fibulin protein family members (e.g., fibulin-4, or fibulin-5), may also use the same partners for their maturation in the ER, and thus may be important in a number of other fibulin-based physiologic or disease-related processes.
Acknowledgments
This work was funded in part by an endowment from the Roger and Dorothy Hirl Research Fund (JDH), a National Eye Institute Visual Science Core Grant (EY020799), an unrestricted grant from Research to Prevent Blindness, and a Career Development Award from Research to Prevent Blindness (JDH).
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiChiara AS, Taylor RJ, Wong MY, Doan ND, Rosario AM, Shoulders MD. Mapping and Exploring the Collagen-I Proteostasis Network. ACS Chem Biol. 2016;11:1408–1421. doi: 10.1021/acschembio.5b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djokic J, Fagotto-Kaufmann C, Bartels R, Nelea V, Reinhardt DP. Fibulin-3, -4, and -5 are highly susceptible to proteolysis, interact with cells and heparin, and form multimers. The Journal of biological chemistry. 2013;288:22821–22835. doi: 10.1074/jbc.M112.439158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvvari MR, van de Ven JP, Geerlings MJ, Saksens NT, Bakker B, Henkes A, Neveling K, Rosario MD, Westra D, van den Heuvel LP, Schick T, Fauser S, Boon CJ, Hoyng CB, Jong EK, Hollander AI. Whole Exome Sequencing in Patients with the Cuticular Drusen Subtype of Age-Related Macular Degeneration. PLoS One. 2016;11:e0152047. doi: 10.1371/journal.pone.0152047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Godino R, Garland DL, Pierce EA. A local complement response by RPE causes early-stage macular degeneration. Human molecular genetics. 2015;24:5555–5569. doi: 10.1093/hmg/ddv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris SP, Jaber NS, Molinari M, Arvan P, Kaufman RJ. UDP-glucose:glycoprotein glucosyltransferase (UGGT1) promotes substrate solubility in the endoplasmic reticulum. Mol Biol Cell. 2013;24:2597–2608. doi: 10.1091/mbc.E13-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Garland D, Yang Z, Shukla D, Rajendran A, Pearson E, Stone EM, Zhang K, Pierce EA. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Human molecular genetics. 2007;16:2411–2422. doi: 10.1093/hmg/ddm198. [DOI] [PubMed] [Google Scholar]
- Fuchshofer R, Birke M, Welge-Lussen U, Kook D, Lutjen-Drecoll E. Transforming growth factor-beta 2 modulated extracellular matrix component expression in cultured human optic nerve head astrocytes. Investigative ophthalmology & visual science. 2005;46:568–578. doi: 10.1167/iovs.04-0649. [DOI] [PubMed] [Google Scholar]
- Garland DL, Fernandez-Godino R, Kaur I, Speicher KD, Harnly JM, Lambris JD, Speicher DW, Pierce EA. Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Human molecular genetics. 2014;23:52–68. doi: 10.1093/hmg/ddt395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulleman JD. Malattia Leventinese/Doyne Honeycomb Retinal Dystrophy: Similarities to Age-Related Macular Degeneration and Potential Therapies. Adv Exp Med Biol. 2016;854:153–158. doi: 10.1007/978-3-319-17121-0_21. [DOI] [PubMed] [Google Scholar]
- Hulleman JD, Balch WE, Kelly JW. Translational attenuation differentially alters the fate of disease-associated fibulin proteins. FASEB J. 2012;26:4548–4560. doi: 10.1096/fj.11-202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulleman JD, Kaushal S, Balch WE, Kelly JW. Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Mol Biol Cell. 2011;22:4765–4775. doi: 10.1091/mbc.E11-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulleman JD, Kelly JW. Genetic ablation of N-linked glycosylation reveals two key folding pathways for R345W fibulin-3, a secreted protein associated with retinal degeneration. FASEB J. 2015;29:565–575. doi: 10.1096/fj.14-255414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Nagata K. Hsp47 as a collagen-specific molecular chaperone. Methods Enzymol. 2011;499:167–182. doi: 10.1016/B978-0-12-386471-0.00009-2. [DOI] [PubMed] [Google Scholar]
- Jessop CE, Chakravarthi S, Garbi N, Hammerling GJ, Lovell S, Bulleid NJ. ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J. 2007;26:28–40. doi: 10.1038/sj.emboj.7601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenotic PA, Munier FL, Marmorstein LY, Anand-Apte B. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1). Implications for macular degenerations. The Journal of biological chemistry. 2004;279:30469–30473. doi: 10.1074/jbc.M403026200. [DOI] [PubMed] [Google Scholar]
- Mackay DS, Bennett TM, Shiels A. Exome Sequencing Identifies a Missense Variant in EFEMP1 Co-Segregating in a Family with Autosomal Dominant Primary Open-Angle Glaucoma. PLoS One. 2015;10:e0132529. doi: 10.1371/journal.pone.0132529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, McLaughlin PJ, Peachey NS, Sasaki T, Marmorstein AD. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Human molecular genetics. 2007;16:2423–2432. doi: 10.1093/hmg/ddm199. [DOI] [PubMed] [Google Scholar]
- Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D, Traboulsi E, Marmorstein AD. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13067–13072. doi: 10.1073/pnas.202491599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin PJ, Bakall B, Choi J, Liu Z, Sasaki T, Davis EC, Marmorstein AD, Marmorstein LY. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Human molecular genetics. 2007;16:3059–3070. doi: 10.1093/hmg/ddm264. [DOI] [PubMed] [Google Scholar]
- Meunier L, Usherwood YK, Chung KT, Hendershot LM. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell. 2002;13:4456–4469. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KJ, Davis LK, Schindler EI, Beck JS, Rudd DS, Grundstad AJ, Scheetz TE, Braun TA, Fingert JH, Alward WL, Kwon YH, Folk JC, Russell SR, Wassink TH, Stone EM, Sheffield VC. Genome-wide analysis of copy number variants in age-related macular degeneration. Hum Genet. 2011;129:91–100. doi: 10.1007/s00439-010-0904-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka OB, Pringle MA, Schopp IM, Braakman I, Bulleid NJ. ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol Cell. 2013;50:793–804. doi: 10.1016/j.molcel.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, Duesbury M, Dumousseau M, Galeota E, Hinz U, Iannuccelli M, Jagannathan S, Jimenez R, Khadake J, Lagreid A, Licata L, Lovering RC, Meldal B, Melidoni AN, Milagros M, Peluso D, Perfetto L, Porras P, Raghunath A, Ricard-Blum S, Roechert B, Stutz A, Tognolli M, van Roey K, Cesareni G, Hermjakob H. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Venable JD, Xu T, Yates JR., 3rd A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods. 2008;5:319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal CN, Marmorstein LY, Vander Jagt DL, Abcouwer SF. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Investigative ophthalmology & visual science. 2005;46:3973–3979. doi: 10.1167/iovs.05-0070. [DOI] [PubMed] [Google Scholar]
- Ryno LM, Genereux JC, Naito T, Morimoto RI, Powers ET, Shoulders MD, Wiseman RL. Characterizing the Altered Cellular Proteome Induced by the Stress-Independent Activation of Heat Shock Factor 1. ACS Chem Biol. 2014 doi: 10.1021/cb500062n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Fassler R, Hohenester E. Laminin: the crux of basement membrane assembly. The Journal of cell biology. 2004;164:959–963. doi: 10.1083/jcb.200401058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sercu S, Lambeir AM, Steenackers E, El Ghalbzouri A, Geentjens K, Sasaki T, Oyama N, Merregaert J. ECM1 interacts with fibulin-3 and the beta 3 chain of laminin 332 through its serum albumin subdomain-like 2 domain. Matrix Biol. 2009;28:160–169. doi: 10.1016/j.matbio.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell reports. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn EH, Wang K, Thompson S, Riker MJ, Hoffmann JM, Stone EM, Mullins RF. Comparison of Drusen and Modifying Genes in Autosomal Dominant Radial Drusen and Age-Related Macular Degeneration. Retina. 2014 doi: 10.1097/IAE.0000000000000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH, Vandenburgh K, Cousin P, Nishimura D, Swiderski RE, Silvestri G, Mackey DA, Hageman GS, Bird AC, Sheffield VC, Schorderet DF. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nature genetics. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- Tan YL, Genereux JC, Pankow S, Aerts JM, Yates JR, 3rd, Kelly JW. ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher’s disease. Chem Biol. 2014;21:967–976. doi: 10.1016/j.chembiol.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt MK, Tsai JY, Mishra S, Campos M, Jaworski C, Fariss RN, Bernstein SL, Wistow G. Interaction of complement factor h and fibulin3 in age-related macular degeneration. PLoS One. 2013;8:e68088. doi: 10.1371/journal.pone.0068088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, Wong CC, Fonslow B, Delahunty C, Gao Y, Shah H, Yates JR., 3rd ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics. 2015;129:16–24. doi: 10.1016/j.jprot.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]