

Introduction
The central nervous system (CNS) enacts a series of responses to promote homeostasis, both in pathological (e.g., neurotropic infections, neurodegenerative and autoimmune disorders1–3), and physiological (e.g. aging4) conditions. This series of responses, comprehensively referred to as “neuroinflammation”, encompasses the concerted actions of numerous cellular and molecular mechanisms designed to identify the potentially harmful event, limit its impact, and repair any consequent damage. Neuroinflammatory responses do not occur solely in the presence of a local insult (e.g., a stroke), but also when normal functioning of the CNS is challenged by distally occurring pathological events. For instance, immunocompetent CNS cells can detect pathologically enhanced afferent neuronal activity occurring after peripheral injury. In this case, neuroinflammation is termed ‘neurogenic’5, 6. Just like classical neuroinflammation, neurogenic neuroinflammation can be adaptive. For example, the CNS neuroinflammation induced by peripheral injury often results in increased pain sensitivity (hyperalgesia and allodynia), which motivates the organism to rest, thereby promoting recovery from the original insult7. However, when exaggerated, both types of neuroinflammation can become pathological, and are in fact implicated in the pathophysiology of a growing number of human disorders3, 8–10.
Because of the recent understanding that neuroinflammation is involved in various neuropathologies, interest in the development of novel methods to investigate neuroinflammatory processes, for the purpose of diagnosis and therapy monitoring, as well as to guide development of novel treatments, has surged over the past 15 years. Neuroimaging offers a wide array of non- or minimally-invasive techniques to characterize neuroinflammatory processes in vivo (Figure 1). The intent of this review is to provide a brief overview of some of the currently available methods to image neuroinflammation in the human CNS. Nuclear imaging (positron emission tomography [PET]; single photon emission computed tomography [SPECT]) and magnetic resonance imaging (MRI) technologies afford broad capability to image many different biological processes, and imaging equipment is widely accessible. For these reasons, we will focus on strategies utilizing nuclear and MRI technologies.
Figure 1.
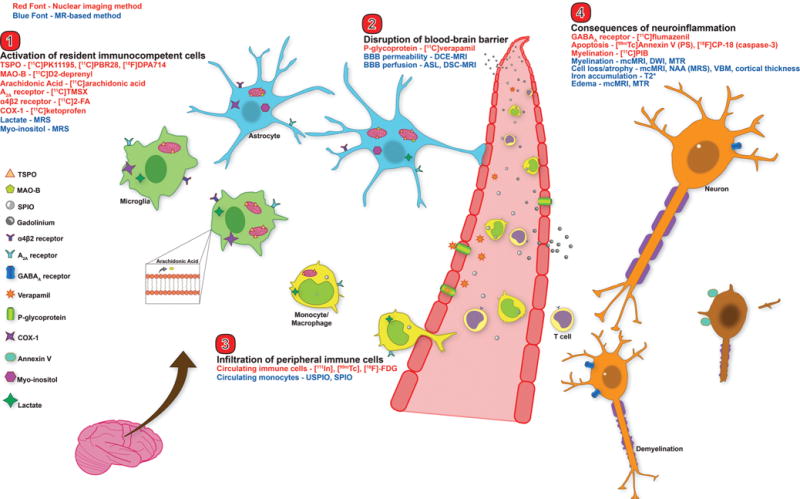
Neuroimaging targets for the major players in neuroinflammation. This figure depicts an overview of the major nuclear imaging and MRI tools to study human neuroinflammation. Nuclear imaging methods are displayed in red font, MRI methods in blue font. Categories are broken into: 1) activation of CNS immunocompetent cells (microglia and astrocytes), 2) disruption of BBB, 3) infiltration of peripheral immune cells, and 4) consequences of neuroinflammation (e.g. demyelination and cell death). TSPO – translocator protein 18kDa; MAO-B – monoamine oxidase B; SPIO – superparamagnetic iron oxide particle; USPIO – ultrasmall SPIO; COX-1 – cyclooxygenase-1; MRS – magnetic resonance spectroscopy; DCE – dynamic contrast enhanced; DSC – dynamic susceptibility contrast; ASL – arterial spin labeling; BBB – blood brain barrier; PS – phosphatidylserine; mcMRI – multiple contrast MRI; DWI – diffusion weighted imaging; MTR – magnetization transfer ratio; NAA – N-acetyl aspartate; VBM – voxel based morphometry.
Both nuclear and MR-based methods offer their own strengths and weaknesses. In general, nuclear imaging possesses superior specificity, as it images actual molecules that have been radiolableled. Meticulously developed and validated radioligands, paired with sophisticated data modeling, give nuclear imaging the ability to detect and quantify many molecular targets, often at nanomolar concentrations. Unlike nuclear methods, MRI involves no ionizing radiation, and therefore is particularly amenable to certain study designs, e.g., longitudinal studies with repeated imaging visits. Moreover, MRI possesses unique flexibility, as the same basic physical principle can be employed to measure different properties of imaged tissues (concentration of specific metabolites, perfusion, anatomical and functional properties, etc). Of course, an ideal approach is to combine imaging modalities, in order to capitalize on their complementary strengths.
Importantly, neuroinflammation is characterized by a large repertoire of local and distant responses, which can differ as a function of the type, intensity, and duration of a given proinflammatory event. For instance, increased blood-brain barrier (BBB) permeability, often considered one of the cardinal neuroinflammatory responses, is a hallmark of actively inflammatory focal lesions in multiple sclerosis (MS)11 and of ongoing inflammation in Alzheimer’s Disease (AD)12. Yet, it is unclear whether BBB compromise contributes to other conditions also thought to be characterized by neuroinflammation, such as schizophrenia8. Regardless of the specific underlying cause of the neuroinflammatory response, many of the processes are overlapping (e.g. glial activation is a factor in all three neuropathologies). The aim of this review is not to provide an exhaustive description of every neuroinflammatory element, which has been discussed extensively2, 6, 13. Rather, we will focus on methods to image in vivo biological mechanisms that are generally recognized as major players of neuroinflammation in human CNS disorders. Specifically, this review will focus on neuroimaging methods that target 1) activation of CNS immunocompetent cells, 2) compromised BBB, 3) CNS-infiltration of circulating immune cells, and 4) pathological consequences of neuroinflammation (e.g. demyelination and cell death). The relative breadth of the literature on each of these four neuroinflammatory topics will be reflected in the length of the section.
Previous reviews have discussed multitudes of potential neuroimaging targets to study neuroinflammation14, 15. However, the vast majority of these tools are currently available only for preclinical imaging, and a large percentage will never become available for human use. Additionally, observations from animal models of disease often do not translate into efficacious treatments for patients, despite very promising results at the preclinical level16. This discrepancy likely results from a combination of factors, including experimental bias, lack of negative result reports, and interspecies differences in neuroimmune targets17, 18. Therefore, this review will focus only on neuroimaging techniques that are currently available for use in human clinical research.
Imaging resident immunocompetent cells
The first line of response to an event that challenges the CNS is the activation of resident immunocompetent cells, i.e. microglia and astrocytes. Typically, although not in all circumstances19, microglia are activated first, initiating morphological changes and expression and release of pro-inflammatory mediators and other signaling molecules13. These molecules initiate astrocyte activation, leading to further release of inflammatory molecules, and influence the excitability of neurons. Homeostatic glial activation can beneficial in dealing with minor tissue dysfunction, but it can also become pathogenic if the activation is persistent20. Though oligodendrocytes have recently been suggested to participate in immune responses21, imaging methods are currently only available for microglia and astrocyte imaging in humans, and thus we will focus on techniques targeting these cells. Future efforts will hopefully lead to cell-specific imaging strategies for all CNS immunocompetent cells.
Nuclear imaging methods
TSPO
The translocator protein 18kDa (TSPO) is a five transmembrane domain protein, mainly situated in the outer mitochondrial membrane. TSPO is thought to be involved in a wide array of vital cellular functions, including steroidogenesis, mitochondrial respiration, and cellular proliferation, among others22. However, some of these functions have been the focus of several recent studies employing TSPO knockout models. Results from these studies have been equivocal, as two models suggested generally normal physiological functioning and development in knockout animals23, 24, while a third indicated that TSPO is important for both embryo viability and steroidogenesis25. Though the specific physiological function of TSPO is still a controversial subject, its features made it the molecule of choice for most PET imaging studies aimed at imaging glial activation and neuroinflammation. In the healthy CNS, TSPO is constitutively expressed by multiple cell types, including glia and neurons, but only at low levels26. During inflammatory responses, however, TSPO is substantially upregulated predominantly, if not exclusively, in glial cells27. For instance, in two animal models of neuropathic pain28, 29, ~5% of neurons and microglia, and ~30% of astrocytes expressed TSPO in the spinal cord dorsal horn prior to surgery. One week after surgery, the number of TSPO-positive neurons did not increase, whereas TSPO-positive microglia and astrocytes increased ~7-fold and ~3-fold, respectively. The colocalization of TSPO upregulation and activated glia has been observed in many animal models and human disorders30, but the specific cell type imaged by TPSO remains a large point of contention in the literature. Many studies describe TSPO tracers as markers of microglial activation, despite evidence for upregulation in astrocytes following many CNS challenges, as described above. The relative contribution of specific glial cells to the TSPO signal is likely a function of several factors, including the pathology and timecourse investigated. In some rat models of MS, TSPO upregulation is localized to microglia31, 32, whereas both astrocytes and microglia are involved in other models and human post-mortem MS data33–35, as well as in human and rodent models of ischemia35, 36 and AD33, 35, 37. As mentioned above, initial microglial activation is often followed by a delayed and prolonged astrocytic activation, although the opposite pattern has been observed in some cases29. Altogether, these observations highlight that TSPO is a marker for “glial” activation, but its ability to specifically image microglia or astrocytes may vary across studies.
Since its first use in the 1980’s, [11C]PK11195 (PK) has been the most widely used TSPO imaging agent (for a detailed review of PK, see38). Although PK imaging yielded a great deal of information on glial activation in CNS disorders, its use is suboptimal. PK has low brain penetrance and high nonspecific binding. These features limit signal to background ratio (SBR) and ability to detect subtle PET signals related to glial activation. For this reason, and given the great interest in imaging TSPO as a glial marker, a great deal of effort has recently gone into developing improved radioligands for this protein. Several of these ligands are approved for human imaging, including [11C]PBR28, [18F]DPA-714, [18F]FEPPA, [11C]DAA1106, [18F]PBR06, and [18F]PBR111 (reviewed in 22). Second-generation radioligands possess much higher SBR than PK39, and have been used to image glial activation in a number of pathologies, including brain tumors, trauma, psychiatric disorders, and chronic pain10, 40–45 (Figure 2). While second-generation TSPO ligands exhibit several advantages over PK, quantitative interpretation of their signal is confounded by the existence of two separate binding sites, one with high affinity and one with low affinity46. Differential expression of binding sites between subjects results into a tri-modal distribution of binding affinity. For instance, among people of European ancestry, ~50% express only high affinity sites (high-affinity binders; HABs), ~10% express only low affinity sites (low-affinity binders LABs), and ~40% express approximately equal numbers of high- and low-affinity sites (mixed-affinity binders; MAB)39, 47. In principle, these binding affinity differences pose potential interpretative issues, because signal differences across groups may not reflect genuine differences in glial activation, but rather binding affinity differences. Fortunately, it was recently demonstrated that a single nucleotide polymorphism (SNP) in the TSPO gene (Ala147Thr)48 fully predicts the binding affinity. Therefore, scientists can now account for global differences in binding affinity through a variety of methods, including restricting recruitment to only subjects exhibiting one genotype, using matched-pairs design, including TSPO status in the statistical model, etc39.
Figure 2.
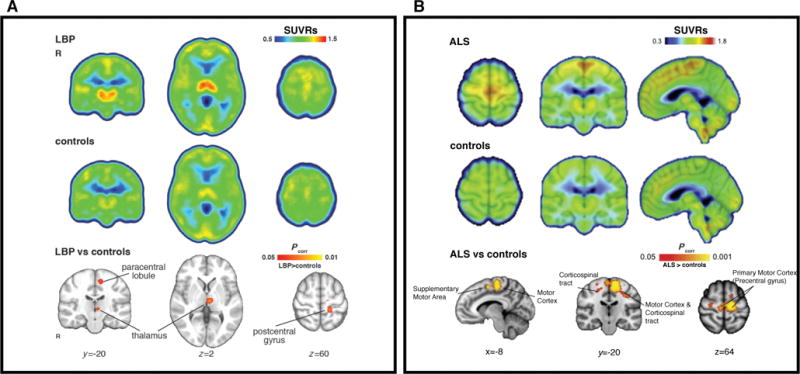
Elevated [11C]PBR28 binding in patients with chronic low back pain44 (cLBP; A) and amyotrophic lateral sclerosis42 (ALS; B) compared to healthy controls. All SUVRs represent standardized uptake value (SUV) normalized by whole-brain uptake. For both cLBP and ALS patients, regional increases in glial activation correspond to brain regions implicated in disease pathology. A) Median SUVR images for cLBP patients (top row) and controls (middle row). Bottom row – clusters are regions where SUVR in cLBP patients was significantly higher than controls. B) Mean SUVR images for ALS patients (top row) and controls (middle row), overlaid on average structural MRI. Bottom row – clusters represent voxels where ALS SUVR was significantly higher than controls. Modified from Loggia et al. “Evidence for brain glial activation in chronic pain patients”, Brain published online Jan 12. DOI: 10.1093/brain/awu37744, with permission from Oxford University Press, and Zurcher et al. “Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: Assessed with [11C]-PBR28”, NeuroImage:Clinical 7: 409–414, DOI:10.1016/j.nicl.2015.01.00942, with permission from ScienceDirect under the terms of Creative Commons Attribution-NonCommercial-No Derivatives License (CC BY NC ND; http://creativecommons.org/licenses/by-nc-nd/4.0/).
Another important issue to consider when designing a TSPO study is estimation of specific binding. The gold standard for PET quantification is kinetic modeling with an arterial plasma input function and correction for plasma free fraction (fP; fraction of tracer unbound to plasma proteins) yielding distribution volume (Vt). However, this is not as straightforward for TSPO ligands as for some other radioligands. Many studies using arterial sampling for kinetic modeling of TSPO reported substantial variability in Vt even within groups, for both first- and second-generation radioligands. For PK, this may be partially due to high binding to a plasma protein that is altered in inflammatory states49. Similar tracer binding to inflammation-susceptible plasma protein is possible, but undocumented, for second-generation ligands. Presumably, measurement of fP should account for tracer binding to plasma protein, but fP values also vary widely, likely due to methodological differences and measurement inaccuracies. In addition to blood variability, estimated Vt is highly variable within and across studies. This may be associated with inaccurate blood measurement, but high vascular TSPO binding has also been proposed to significantly affect outcome estimation50.
Because absolute quantitation is challenging, several groups have estimated TSPO binding with alternative methods, including reference tissue modeling and the use of semi-quantitative measures like standardized uptake value (SUV). Reference tissue modeling, when validated, is a suitable alternative to an arterial plasma input function51, 52. Unlike many of the neurotransmitter system tracers, there is no brain region devoid of TSPO expression, and thus a true reference region is unavailable. However, several alternatives exist. For PK, supervised clustering methods have identified reference tissue53. While these methods may be inappropriate for second-generation ligands54, a recent study with [11C]PBR28 showed that estimation of binding with a cerebellar pseudo-reference region was less variable than estimation with plasma activity, and better distinguished Alzheimer’s Disease (AD) patients from controls55. Blood-free methods like this are very attractive, due to difficulties in accurate plasma measurement described above, and patient discomfort related to arterial line placement. Normalization by within-subject PET signal has the additional benefit of partly correcting for global signal differences introduced by the TSPO genotype39, 40, 42, 44, 56. However, the use of such methods must be carefully validated both for the ligand in question and the study population.
MAO-B
Monoamine oxidase is an important enzyme that exists in two sub-types: MAO-A and MAO-B. MAO-B is more relevant for studies of neuroinflammation, due to its upregulation in reactive astrocytes in certain neuroinflammatory conditions like AD57. Selective MAO-B antagonists have been radiolabeled for PET imaging of astrocytes, including [11C]-D-deprenyl and its deuterium substituted analog, [11C]-deprenyl-D258, 59. [11C]-deprenyl-D2 is the most commonly used astrocyte tracer, because of its favorable kinetics compared to [11C]deprenyl. However, specific binding of the molecule has been questioned60. MAO-B radioligands have been used to image astrogliosis in several neuroinflammatory conditions, including AD61 and amyotrophic lateral sclerosis (ALS)62, among others. Researchers wishing to use MAO-B tracers should be aware of several limitations. MAO-B expression is not selective to astrocytes, it is also present in serotonergic and histaminergic neurons63, making interpretation of findings difficult. Additionally, the exact cellular binding of MAO-B tracers in human brain is poorly understood. Overall, MAO-B PET studies can provide useful information on astrocyte function in human neuropathology, but proper study design and cautious interpretation of results is important.
Cyclooxygenase
Cyclooxygenase-1 and -2 (COX-1 and -2) catalyze conversion of arachidonic acid to prostaglandins, which are important inflammatory signaling molecules. As for TSPO, different cell types constitutively express COX-1, and COX-2, which are upregulated following CNS insult64. There is differential upregulation depending on the type of insult and experimental model, but in general COX-1 is thought to be mostly upregulated in microglia65. COX-2 tracers are currently unavailable for human brain imaging, and thus will not be discussed here. The utility of COX-1 tracers is supported by the results of a rodent study with [11C]ketoprofen66, a COX-1 radioligand whose methyl esterified form ([11C]KTP-ME) is available for human use (but so far utilized only in a single study in healthy volunteers)67. The rodent study reported selective microglial upregulation of COX-1 following injection of inflammatory agents, but despite claims of microglial selectivity, other studies documented astrocytic changes in COX-1 expression following inflammatory challenges68, 69. These discrepancies may be a function of investigational models and techniques, as studies showing astrocytic changes in COX-1 were typically conducted in glial cell cultures, as opposed to brain tissue. Further research is needed to validate the selectivity and general utility of COX-1 tracers for human disorders, and to develop COX-2 tracers for human neuroimaging.
Arachidonic Acid
As mentioned above, arachidonic acid (AA) plays a major role in inflammatory signaling, via release from membrane glycerophospholipids and conversion to prostaglandins and leukotrienes70. Because of this, arachidonic acid is a potential imaging target for neuroinflammation studies, in spite of nonspecific cellular localization. Recently, a PET study using [11C]-labeled AA reported increased AA metabolism throughout most of the brain in AD patients71, which the authors interpreted as evidence for neuroinflammation. However, AA signaling is important for multiple aspects of healthy brain function. For instance, [11C]AA imaging has also shown cortical increases in AA metabolism as a result visual stimulation71, implicating AA involvement in neurotransmission. Therefore, AA imaging encompasses varying cellular processes, and it should be used cautiously as a marker of neuroinflammation. In addition to COX and AA, other molecules involved in the AA signaling pathway may also be useful for imaging neuroinflammation. Fatty acid amide hydrolase (FAAH) is one such molecule that has been implicated in neuroinflammation and human FAAH tracers are available72.
Adenosine Receptors
Several adenosine receptors are involved in neuroinflammatory signaling, but their specific actions during a neuroinflammatory response are not well elucidated. However, the A2A receptor (A2AR) in particular is involved in crucial neuroinflammatory processes, including LPS-induced changes in synaptic plasticity73, and microglial process retraction during activation74. Previous imaging studies assessed A2AR expression in several human neurological disorders, including PD and schizophrenia (reviewed in 75), but only one study interpreted increased A2AR expression as evidence of neuroinflammation76. Using [11C]TMSX to image secondary progressive MS patients, the authors showed increased A2AR binding in normal-appearing white matter (NAWM), which is known to contain activated glia1. They also reported a negative association between tracer binding and MR indices of white matter integrity, implicating the involvement of A2ARs in white matter pathology. However, all CNS immune cell types express A2ARs, limiting the specificity of information obtained from studies using A2A ligands, similarly to other ligands described here. Future post-mortem studies of A2AR expression with cell-specific markers, or multimodal studies as mentioned above, will aid in identifying the precise location and function of A2ARs in neuroinflammation.
α4β2 Nicotinic Acetylcholine Receptors
The nicotinic acetylcholine (NACh) system regulates many important physiological functions, including stress, arousal, cognition, and pain77, but has recently been also associated with neuroinflammation. In a multitracer animal study of ischemia, the authors demonstrated that the 2[18F]-fluoro-A85380 ligand for α4β2 NAChR (2-FA) has very similar patterns of uptake to the TSPO ligand [11C]PK1119578. Histological evidence showed concomitant upregulation of both TSPO and the α4β2 NACh receptor in activated microglia and astrocytes, suggesting that α4β2 may be a glial marker with a profile of cell-type expression similar to that of TSPO. While the 2-FA tracer has an unfavorably slow kinetic profile, requiring very long image acquisitions, kinetically favorable tracers are being developed, such as [18F]flubatine79. Thus, the study of α4β2 receptors as imaging markers of inflammation may become more widespread in the future. However, it should be noted that previous NAChR studies in neurological disorders generally reported decreased receptor binding77. This indicates drastic model-dependent differences in receptor expression, and further investigations are needed to understand how α4β2 receptors contribute to neuroinflammation.
MR-based methods
Magnetic resonance spectroscopy (MRS)
MRS uses MRI technology to extract quantitative information about brain metabolites. Several MRS measures have been used as putative markers of resident immunocompetent cells. Myo-inositol (mI) is an osmolyte, or volume-regulating molecule, proposed to be a marker of astrocytes, which become hypertrophic during an inflammatory response. In fact, mI is elevated in a wide array of pathologies thought to involve neuroinflammation (for a review, see 80). However, few studies have histologically validated the initial evidence for increased mI in reactive astrocytes81, and there is evidence of pathological mI changes in disorders without a known astrocytic component, such as Down’s syndrome82. These combined results indicate mI is a poor standalone marker for reactive astrocytosis, and neuroinflammation in general. A complementary neuroinflammatory marker should be added to MRS in order to better frame interpretations of mI in neuroinflammation. For example, a recent study in hepatitis C patients reported increases in both mI and TSPO binding in patients, strengthening evidence of neuroinflammation in this cohort83. However, the two measures were not correlated, suggesting that these target co-occurring, but distinct physiological processes.
Unlike many MRS metabolites, lactate is thought to be present only under pathological states, because of its use as an energy substrate under ischemic and hypoxic conditions. In line with this, increased lactate has been reported in acute ischemia and MS lesions84, 85. However, while average lactate concentration is relatively low in healthy brain tissue, it is a critical substrate in oxidative metabolism. Lactate concentrations increase during normal energy-consuming processes, like neural activity during visual stimulation, in quantities detectable by MRS86. Furthermore, its cell specificity is equivocal. Early research suggested a specificity for activated macrophages87, but other reports indicate that astrocytic glycolysis is the major brain source of lactate, arguing for astrocyte specificity88. Similar to mI, validation studies with animal models and immunohistochemistry are needed to better understand how lactate is involved in neuroinflammation. In addition, elevated choline has been detected in inflammatory and gliotic processes89. The choline resonance arises from several soluble components of brain myelin and fluid-cell membranes including glycerophosphocholine (GPC), phosphocholine (PCho), and choline (Cho). Elevated Cho levels have been interpreted to reflect glial activation, energy failure, macrophage infiltration, and demyelination, showing that, like mI and lactate, Cho is a nonspecific neuroinflammatory marker90.
MRS techniques are not without limitations. MRS measures of creatine are often used as an internal reference, because creatine is assumed to be relatively stable across brain parenchyma91. However, creatine levels may be altered in neuroinflammatory disorders80, suggesting that only absolute measurements should be used when comparing pathological populations. Methods used for absolute quantification include: a) phantom replacement techniques92 b) using the unsuppressed water signal as a reference93, c) the use of an external reference94 and d) reciprocity95. The spatial resolution of MRS is also much lower than structural MRI scans, making metabolites in small regions difficult to assess. Additionally, large MRS voxels contain a mixture of different tissue types, such that intracellular cannot be separated from extracellular compartments. Despite these limitations, MRS has provided and continues to provide important insights into neurochemical alterations observed during inflammation. The development of advanced MRS methods capable of increasing the number of metabolites detected (e.g., 2D-COSY)96, or fast 3D spiral encoding of spatial information97 will increase the applicability of MRS to the study of neuroinflammatory and other conditions.
Blood-brain barrier Imaging
The blood-brain barrier (BBB) is a crucial barrier that limits exposure of sensitive brain tissue to outside factors. Depending on the nature of the stimulus, BBB permeability may also be increased during a neuroinflammatory response. Astrocytes play an important role in maintaining BBB integrity, and their activation during neuroinflammation may regulate, in part, permeability of the BBB98. In the healthy CNS, the BBB limits brain entry of peripheral cells and potentially harmful molecules. However, if the CNS insult is severe enough, peripheral cells such as lymphocytes and monocytes may be recruited across the compromised BBB to help remove the debris and repair damaged tissue. There are several different strategies available to image BBB function.
Nuclear imaging methods
MRI is the predominantly used methodology for BBB imaging; however, nuclear imaging is better equipped to image efflux proteins, such as P-glycoprotein (P-gp). These proteins regulate brain molecular concentrations by actively pumping them from the brain across the BBB. Importantly, P-gp activity is known to be altered by inflammatory conditions99, which has indications for CNS delivery of therapeutic drugs. The PET tracer [11C]verapamil has been used to investigate P-gp function in healthy humans100, as well as animal models of neuroinflammation101.
MRI-based methods
Contrast-enhanced MRI (CE-MRI), generally with a gadolinium (Gd) chelate, is the workhorse for clinical BBB imaging. The simplest CE-MRI-based assessment of BBB integrity involves bolus infusion of contrast agent (e.g. Gd-DPTA), typically in conjunction with a T1-weighted MR sequence. Because Gd compounds do not readily cross a healthy BBB, changes in MR signal post-infusion versus pre-infusion indicate that contrast agent has entered the CNS via some BBB disturbance. This type of assessment provides only a semi-quantitative measurement of BBB changes, but recently more advanced kinetic models have been described to isolate individual BBB components, such as the volume transfer constant Ktrans (which is weighted by both perfusion and blood flow; reviewed in 102). These kinetic methods require dynamic data acquisition and a prolonged infusion of contrast agent, and are thus referred to as dynamic CE-MRI (DCE-MRI). T1-weighted DCE-MRI is most commonly used to assess BBB permeability, while dynamic susceptibility-enhanced MRI (DSC-MRI) with T2 or T2*-weighted sequences and contrast agent is often used to detect CNS hyper- or hypoperfusion103. Alternatively to DSC-MRI, MRI with arterial spin labeling (ASL) can be used to estimate perfusion104. As a method to image perfusion, ASL has certain advantages over DSC-MRI, as it does not require contrast agent, which causes allergic reactions in a small percentage of people and is somewhat invasive. Additionally, recent work improved BBB permeability estimates by combining Ktrans measured by DCE-MRI with ASL estimates of cerebral blood flow105.
One of the pathologies where BBB imaging is most prevalent is MS, and a great amount of information on the development and persistence of lesions has been obtained with these methods in vivo in MS patients11. While much of the literature describes imaging of MS white matter lesions with T1-weighted DCE imaging, more recently developed techniques (e.g. post-contrast T2- weighted Fluid-Attenuated Inversion Recovery [FLAIR]) enable imaging of BBB disruption in areas previously not studied with neuroimaging, such as the leptomeninges106 (Figure 3). While several studies suggest that these techniques are sensitive to BBB compromise, it should be noted that they do not provide causal information, as different mechanisms of BBB compromise may lead to similar outcomes as detected by MRI.
Figure 3.
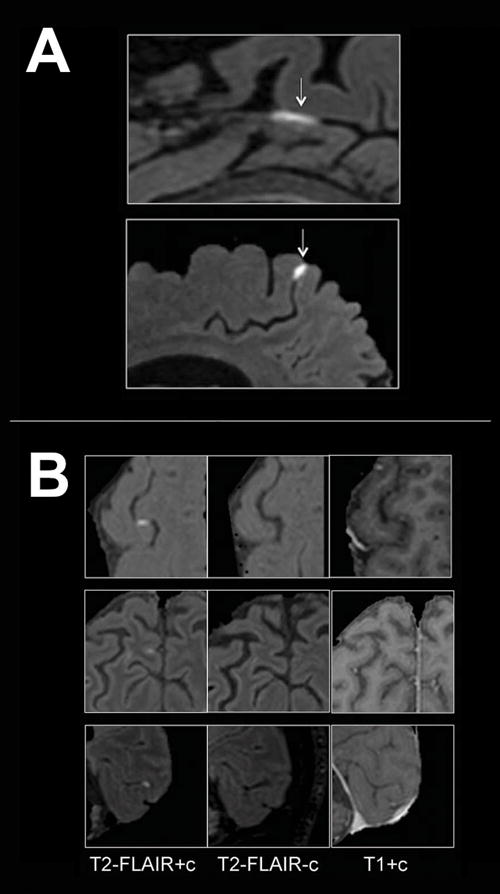
Examples of BBB imaging with contrast in MS patients. (A) Examples of leptomeningeal contrast enhancement in two representative relapsing-remitting MS cases. Foci of high signal (arrows) on 3T post-contrast T2-FLAIR images indicate leptomeningeal enhancement. Extracerebral tissues have been masked for clarity. (B) Signal intensity on different MRI sequences. Three foci of leptomeningeal enhancement are visible on post-contrast T2-FLAIR scans (left column), but not on the corresponding pre-contrast T2-FLAIR (middle column). In the right column, post-contrast T1-weighted images show minimal abnormal signal that would not routinely be classified as enhancement. T2-FLAIR = T2-weighted, fluid-attenuated inversion recovery. Reproduced from Absinta et al., “Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis”, Neurology vol. 85 no. 1, 18–28106 with permission from Wolters Kluwer Health.
Imaging infiltration of immune cells
During many neuroinflammatory events (e.g. during BBB disruption) circulating immune cells are recruited from the periphery and traffic to the CNS. Similar to the other neuroinflammatory players covered here, the contribution of peripheral immune cells to a neuroinflammatory response varies largely depending on the nature of the inciting event. Generally, the more severe the event is, the greater the likelihood of peripheral cell infiltration. For example, leukocyte and macrophage migration to CNS tissue is a hallmark of both MS and cerebral ischemia, and plays a large part in pathological progression107. However, recent evidence suggests that monocyte trafficking to the brain may also play an important role in the development of disorders not characterized by the presence of lesions, such as chronic stress108. Several neuroimaging techniques are suitable for the study of immune cell trafficking and infiltration in human neuroinflammation.
Nuclear imaging methods
For many years, human leukocyte trafficking was imaged by isolating leukocytes from blood samples, incubating with radiotracer (e.g. 111 In, 99mTc, 18F-FDG) and re-injecting the subject with autologous cells before PET or SPECT imaging (reviewed in 15). These techniques have largely fallen out of use in clinical studies, due to several disadvantages, including: a) cell preparation is time and labor intensive; b) labeled cells are also prone to loss of label in vivo via apoptosis or proliferation, as well as through non-physiological mechanisms (e.g. passive diffusion); c) if the BBB is compromised, activity leaked from labeled cells may contribute to non-specific brain signal; d) discrimination of intraparechymal from intravascular cellular location is difficult to achieve109; e) processing required to isolate and label individual cell types may be complex110, or alter cell tracking, (e.g non-metabolically active cells must be treated to increase 18F-FDG uptake)111. For all these reasons, the use of nuclear imaging to study immune cell tracking in neuroinflammation has been so far limited.
MR-based methods
MRI of human immune cell trafficking can be accomplished via cell labeling with superparamagnetic particles of iron oxide (SPIO). Typically, SPIOs are administered as a bolus to the bloodstream, where circulating monocytes internalize the particles, due to their high phagocytic activity. The iron oxide core shortens T1 and T2/T2* relaxation times, leading to hyperintense T1-weighted signal, and hypointense T2/T2*-weighted signal. SPIOs are classified by iron core diameter, ranging from 10–50 nm (ultra-small SPIO; USPIO) to 50–100 nm (SPIO) and up to >1 μm (micrometer-sized particles of iron oxide; MPIOs)112. USPIOs are the particles most commonly used for human imaging of monocyte/macrophage tracking, and have been used successfully in a number of studies of human disorders characterized by neuroinflammation, including MS and stroke (reviewed in 113). Though monocyte tracking yields important pathological information, combination with additional imaging modalities can uncover results that neither in isolation can. For example, a recent multimodal study employed longitudinal [11C]PK11195 PET and USPIO MR scanning in an animal stroke model114. The authors found both overlapping and separate stroke-induced changes in PK and USPIO signal. This observation suggests that the brain infiltration of peripheral macrophages and the activation of resident immunocompetent cells (e.g. microglia, astrocytes) in the neuroinflammatory response is partially dissociable, and further showed differences in temporal upregulation of both types of molecules.
MR studies with SPIOs have great potential for imaging human neuroinflammation, but they are also limited. In vitro labeling of autologous cells (e.g., monocytes) in humans is theoretically possible, but has not been reported. One of the potential pitfalls with in vitro labeling is that achieving enough labeling to allow for good signal detection may be challenging within the limits of a safe and tolerable dose. In vivo labeling likely results in both a smaller proportion of cells labeled and a higher concentration of cell-free iron oxide label than in vitro labeling, increasing false negative and false positive rates. Similar to radioligand labeling, in vivo-labeled cells can lose their label, resulting in signal inaccuracies as cell-free SPIOs enter the brain, particularly in disorders with increased BBB permeability. However, using MRI with both Gd and USPIO, it was shown that regions with BBB disruption were completely distinct from those showing USPIO uptake in stroke patients, suggesting that USPIOs may not cross the compromised BBB115. Finally, many SPIO-MRI studies use only qualitative or semi-quantitative signal differences as an outcome measure. A radiolabeled USPIO has been developed that would allow for absolute quantitation, but only preclinical applications have been used thus far116.
Imaging pathological consequences of neuroinflammation
Although distinct and downstream from the neuroinflammatory players discussed above, the resulting products of neuroinflammation are also important targets to investigate neuroinflammation. Like the events that cause them, the consequences of neuroinflammation are varied, and dependent on the nature of the neuroinflammatory event. Commonly studied pathological outcomes are edema, demyelination, cellular and axonal damage, and several different imaging techniques have been developed for this purpose. This type of research has important implications for diagnosis of acute insult and characterization of therapies targeting neuroinflammation.
Nuclear imaging methods
Several radiotracers have been developed for the purpose of myelin imaging, but currently only 11C-labeled Pittsburgh Compound B (PIB) has been used in human subjects15. Originally developed for β-amyloid imaging, studies have shown that [11C]PIB is able to assess demyelination117. In vitro and ex vivo experiments, however, have questioned the mechanism of PIB-white matter binding, as it seems nonsaturable and nonspecific118, 119. A recent study found strong correlations between [11C]PIB binding and expression of myelin-associated mRNA from the Allen Human Brain Atlas, suggesting that [11C]PIB does specifically bind with myelin-associated proteins120.
A great deal of interest has also gone into radiotracers for imaging cell death. The PET/SPECT ligands [11C]flumazenil (FMZ) and [123I]iomazenil, which target GABA-A receptors, have shown utility for imaging neuronal integrity in several disorders, including stroke and AD121, 122. While regional decreases in GABA-A binding seem to colocalize with regional neuronal death in human post-mortem data, some animal experiments reported poor correlation of FMZ signal with histological evidence of neuronal death123, 124. The dissociation between FMZ and neuronal death in these studies could suggest that nuclear imaging estimates of GABA-A receptor density are more specific for subtle processes, such as dendritic loss, which has been shown to be a consequence of ischemia125.
Current nuclear imaging technologies may be imperfect for detecting frank neuron loss, but there are several tracers specific for apoptotic cells (reviewed in 126). The majority of these ligands target the apoptotic marker phosphatidylserine (PS) and molecules associated with it, such as Annexin V. SPECT imaging with 99mTc-conjugated Annexin V has been used to study apoptosis for many years15, though the relatively poor resolution of SPECT incentivized PET tracer developments efforts. [18F]-labeled Annexin V synthesis has been described, but only in preclinical models126. Annexin V imaging is also limited because PS appears on necrotic as well as apoptotic cells, but several recent alternatives exist. 18F-ML-10 is a derivative of the Aposense family of apoptotic markers127. 18F-ICMT-11 and 18F-CP-18 are both specific to caspase-3, a crucial enzyme for initiation of apoptosis128, 129. Each of these tracers has received safety validation in healthy controls, but none have yet been used to study neuroinflammatory disorders.
MR-based methods
Various MR sequences can be used to study the consequences of neuroinflammation. Though many MR-methods are limited by a lack of specificity compared to nuclear methods, advances in sequence development and data analysis continue to improve, and combination with additional measures of inflammation strengthens results from individual techniques.
Voxel-based morphometry (VBM) and cortical thickness, measured with structural T1 MRI, are two of the most commonly used MR techniques to assess CNS pathology in experimental studies. The structural metrics obtained with these techniques are not direct measures of cell number or density and are likely to reflect a combination of many cellular processes130. Nonetheless, these methods are sensitive to anatomical changes that can occur in the context of pathology such as atrophy8, 130, 131, that have been linked to inflammation132. For instance, a recent multimodal study in AD demonstrated a negative association between gray matter volume and [11C]PBR28-measured TSPO levels133, thus supporting a relationship between gray matter loss and neuroinflammation. While most of the MRI structural studies focus on gray and white matter, a recent investigation used VBM to demonstrate enlarged choroid plexus volume in complex regional pain syndrome (CRPS)134. As the choroid plexus sits at an important entry point for circulating immune cells into the CNS, these results suggest yet one more link between neuroinflammation and chronic pain44, and further highlight the flexibility of MR-based methods.
N-acetyl aspartate (NAA) is a MRS-measured metabolite thought to be a marker of neuronal integrity. Therefore, studies often interpret reduced NAA (or NAA normalized by creatine levels, NAA/Cr) as evidence for neuronal loss80. Reductions in NAA signal have been reported for a wide range of neuroinflammatory disorders, but there is evidence that NAA levels can return to baseline after a period of recovery135. Therefore, NAA is likely not a specific marker of neuronal death, and some evidence suggests that NAA levels reflect more closely the functional capacity of neuronal mitochondria135.
Pathological products of neuroinflammation such as edema, demyelination, tissue loss, and iron accumulation lead to variable changes in quantitative measures of proton relaxation times (T1, T2, and T2*) as well as in semiquantitative parameters such as magnetization transfer ratio (MTR)136.
As explained in detail in 136–138, T1 relaxation times (rt) in the CNS are essentially influenced by free water protons and by the relative amount of CNS tissue components (e.g. cells, myelin, lipids, proteins). In this context, increased T1-rt indicates increased water content (i.e. due to inflammatory edema) or decreased CNS tissue “structure” (i.e. loss of cells, axons, myelin, etc.) Similarly, greater density of CNS “structure” and reduced water content as well as iron accumulation tend to reduce T1-rt.
T2 relaxometry measures the loss of spin coherence of excited water molecules, therefore reflecting the dynamic state of water protons and of their interaction with macromolecules in a tissue. An increase in T2-rt in the CNS may be due to a loss of macromolecules (e.g. myelin in white matter) and/or increased water content, which may be intracellular (e.g. neuroinflammatory gliosis) or extracellular (e.g. neuroinflammatory edema).
Iron accumulation is also considered to be a consequence of mitochondrial dysfunction, which may be provoked by CNS inflammation. Among existing relaxometry techniques, T2* relaxometry best reflects iron accumulation. The T2*-rt is a measure of the loss of transverse magnetization due to T2 relaxation and magnetic field inhomogeneities, which depend on the presence of paramagnetic or ferromagnetic ions like iron. Because of this, an increase in T2* most often indicates a loss of macromolecules or an increase in water (T2 component), while a decrease suggests an increase of macromolecular compounds (T2 component) or iron (susceptibility).
MT images are based on the interaction between macromolecule-bound immobilized protons and free water protons. A lower MTR therefore indicates a reduced spin exchange between free and bound protons, suggesting neuroaxonal damage or myelin breakdown and/or water increase. Another commonly used MR technique to assess neuroinflammation is diffusion-weighted imaging (DWI)139.
DWI measures the diffusion of water molecules, and has been widely used to study white matter changes in demyelinating and hypomyelinating disorders. Myelin-sheathed axons restrict the free diffusion of water molecules (anisotropy), and thus a reduction in directional diffusion is often assumed to indicate white matter disruption. DWI, like the methods described above, suffers from nonspecificity, and can be influenced by axonal pathology as well as changes in myelin content140. However, advanced DWI models appear to improve correlations with histology141.
The combination of multiple MR contrasts (mcMRI) provides high specificity and sensitivity to detect CNS inflammatory processes. Thanks to recent hardware and software development, it is nowadays feasible to acquire multiple quantitative and semi-quantitative MRI techniques in a clinically compatible protocol137, 138. Combining T1, T2 and T2* relaxometry with MTI, Bonnier et al.137 showed subtle increases in T2- and T2*-rt in NAWM of early stage and minimally impaired MS patients. T2/T2* increases, in the absence of significant changes in T1-rt, suggested extracellular water accumulation in patients suffering from chronic autoimmune neuroinflammation. In addition, by combining the four contrasts above, the authors classified MS lesion pathology and identified groups of lesions with predominant inflammatory or degenerative components138.
The multimodal approach above shows once again that results from somewhat nonspecific MR methods may also be strengthened with a multimodal approach. Similarly, a recent TSPO PET imaging study in MS also acquired MTR, and found differential glial activation in nonlesional regions dependent on high or low MTR, which was assumed to be myelin-related in the context of the study142.
In sum, MRI metrics are important for characterization of neuroinflammatory disorders, and some have already proven their usefulness for diagnosis, and follow-up of patients affected by neuroinflammation. Future efforts should focus on exploring the clinical feasibility of combined mcMRI and DWI acquisitions with techniques like MRI fingerprinting143 and on the reproducibility and standardization of MRI measurements of neuroinflammation.
Conclusions
As new findings increasingly suggest the involvement in neuroinflammation in a host of different human disorders, sensitive and specific in vivo imaging methods will continue to be important for accurate depiction of complex neuroinflammatory processes. Advances in our understanding of neuroinflammation occurring independently of neuroimaging are crucial for this field of research to mature. Ideally, translational collaborations between basic and clinical scientists will lead to more specific and relevant neuroimaging targets to study neuroinflammation. In addition to development of novel techniques, neuroimaging researchers need to design studies using existing methods in innovative ways to overcome the inherent limitations described here. This could encompass refining existing data analysis techniques, or the clever use of multimodal studies, where results from one imaging technique can inform results obtained with the other. Several such studies have been highlighted in this review, showing that relatively unspecific methods often can yield more specific information about pathology when paired with a complementary technique. We expect that neuroimaging, complemented by basic science discoveries, will make fundamental contributions to research and clinical investigations of neuroinflammation in CNS pathology.
Table 1.
Summary of neuroimaging agents targeting neuroinflammatory processes
| Neuroinflammatory Target | Modality | Imaging Agent/Technique |
|---|---|---|
| CNS Immunocompetent cells | ||
| TSPO | PET | [11C]PK11195, [11C]Ro5-4864, [11C]vinpocetine |
| [11C]PBR28, [11C]DAA1106, [11C]DPA-713 | ||
| [18F]DPA-714, [18F]FEDAA1106, [18F]PBR111 | ||
| [18F]PBR06, [18F]FEPPAA | ||
| SPECT | [123I]PK11195, [123I]CLINDE | |
| MAO-B | PET | [11C]-D-deprenyl, [11C]-deprenyl-D2 |
| COX-1 | PET | [11C]ketoprofen, [11C]KTP-Me |
| Arachidonic Acid | PET | [11C]AA |
| Adenosine A2A | PET | [11C]TMSX, [11C]SCH442416 |
| α4β2 NAChR | PET | [18F]2-FA, [18F]flubatine |
| Myo-inositol | MRI | MR Spectroscopy |
| Lactate | MRI | MR Spectroscopy |
| Choline | MRI | MR Spectroscopy |
| Blood-brain barrier | ||
| P-glycoprotein | PET | [11C]verapamil, [11C]loperamide |
| [11C]desmethyl-loperamide | ||
| BBB permeability | MRI | DCE-MRI with gadolinium chelates |
| BBB perfusion | MRI | DSC-MRI with gadolinium chelates, ASL |
| Infiltration of immune cells | ||
| Leukoctyes | PET | [18F]FDG |
| SPECT | 111 In, 99mTc | |
| Monocytes | MRI | Superparamagnetic iron oxide particles |
| (SPIO, USPIO, MPIO) | ||
| Pathological consequences of neuroinflammation | ||
| Demyelination | PET | [11C]PIB |
| MRI | mcMRI, DWI, MTR | |
| Apoptosis | ||
| Phosphatidylserine | SPECT | [99m]Tc-Annexin V |
| Caspase-3 | PET | [11C]CP-18, [18F]ICMT-11 |
| Aposense | [18F]FM-10 | |
| Neuronal loss | ||
| GABAA | PET | [11C]flumazenil |
| N-Acetyl Aspartate | MRI | MR Spectroscopy |
| Non-specific | MRI | mcMRI, VBM, cortical thickness |
| Iron Accumulation | MRI | T2* |
| Edema | MRI | mcMRI, MTR |
Only imaging agents that have been approved for human use are included in the table. CNS – central nervous system; TSPO – translocator protein; PET – positron emission tomography; SPECT – single photon emission computed tomography; MAO-B – monoamine oxidase B; COX-1 – cyclooxygenase 1; NAChR – Nicotinic acetylcholine receptor; MRI – magnetic resonance imaging; DCE-MRI – dynamic contrast enhanced-MRI; DSC-MRI – dynamic susceptibility enhanced-MRI; ASL – arterial spin labeling; SPIO – superparamagnetic particle of iron oxide; USPIO – ultrasmall SPIO; MPIO – micrometer-sized particle of iron oxide; mcMRI – multiple contrast MRI; DWI – diffusion weighted imaging; MTR – magnetization transfer ratio.
Acknowledgments
We would like to acknowledge the following funding mechanisms for support of this project: 5T32EB13180 (T32 supporting Dr. Albrecht), 1R21NS087472-01A1 (MLL), and DoD W81XWH-14-1-0543 (MLL). We would also like to thank Dr. Eva Ratai for helpful feedback on the manuscript.
References
- 1.Airas L, Rissanen E, Rinne JO. Imaging neuroinflammation in multiple sclerosis using TSPO-PET. Clinical and Translational Imaging. 2015;3:461–473. doi: 10.1007/s40336-015-0147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- 3.Littlejohn G. Neurogenic neuroinflammation in fibromyalgia and complex regional pain syndrome. Nature reviews Rheumatology. 2015;11:639–648. doi: 10.1038/nrrheum.2015.100. [DOI] [PubMed] [Google Scholar]
- 4.Gabuzda D, Yankner BA. Physiology: Inflammation links ageing to the brain. Nature. 2013;497:197–198. doi: 10.1038/nature12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren K, Dubner R. Activity-triggered tetrapartite neuron-glial interactions following peripheral injury. Current opinion in pharmacology. 2016;26:16–25. doi: 10.1016/j.coph.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 7.Walters ET. Nociceptors as chronic drivers of pain and hyperreflexia after spinal cord injury: an adaptive-maladaptive hyperfunctional state hypothesis. Frontiers in physiology. 2012;3:309. doi: 10.3389/fphys.2012.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasternak O, Kubicki M, Shenton ME. In vivo imaging of neuroinflammation in schizophrenia. Schizophrenia research. 2015 doi: 10.1016/j.schres.2015.05.034. published online Jun 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathbone AT, Tharmaradinam S, Jiang S, Rathbone MP, Kumbhare DA. A review of the neuro- and systemic inflammatory responses in post concussion symptoms: Introduction of the “post-inflammatory brain syndrome” PIBS. Brain Behav Immun. 2015;46:1–16. doi: 10.1016/j.bbi.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 10.van der Doef TF, Doorduin J, van Berckel BN, Cervenka S. Assessing brain immune activation in psychiatric disorders: clinical and preclinical PET imaging studies of the 18-kDa translocator protein. Clinical and Translational Imaging. 2015;3:449–460. doi: 10.1007/s40336-015-0140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaitan MI, Shea CD, Evangelou IE, Stone RD, Fenton KM, Bielekova B, Massacesi L, Reich DS. Evolution of the blood-brain barrier in newly forming multiple sclerosis lesions. Annals of neurology. 2011;70:22–29. doi: 10.1002/ana.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowman GL, Quinn JF. Alzheimer’s disease and the Blood-Brain Barrier: Past, Present and Future. Aging health. 2008;4:47–55. doi: 10.2217/1745509X.4.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russo MV, McGavern DB. Immune Surveillance of the CNS following Infection and Injury. Trends in immunology. 2015;36:637–650. doi: 10.1016/j.it.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobs AH, Tavitian B, consortium, I. N. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012;32:1393–1415. doi: 10.1038/jcbfm.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pulli B, Chen JW. Imaging Neuroinflammation - from Bench to Bedside. Journal of clinical & cellular immunology. 2014;5 doi: 10.4172/2155-9899.1000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chesselet MF, Carmichael ST. Animal models of neurological disorders. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2012;9:241–244. doi: 10.1007/s13311-012-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edmondson DE, Binda C, Wang J, Upadhyay AK, Mattevi A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry. 2009;48:4220–4230. doi: 10.1021/bi900413g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsilidis KK, Panagiotou OA, Sena ES, Aretouli E, Evangelou E, Howells DW, Al-Shahi Salman R, Macleod MR, Ioannidis JP. Evaluation of excess significance bias in animal studies of neurological diseases. PLoS biology. 2013;11:e1001609. doi: 10.1371/journal.pbio.1001609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loth MK, Choi J, McGlothan JL, Pletnikov MV, Pomper MG, Guilarte TR. TSPO in a murine model of Sandhoff disease: presymptomatic marker of neurodegeneration and disease pathophysiology. Neurobiology of disease. 2016;85:174–186. doi: 10.1016/j.nbd.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94:1077–1098. doi: 10.1152/physrev.00041.2013. [DOI] [PubMed] [Google Scholar]
- 21.Zeis T, Enz L, Schaeren-Wiemers N. The immunomodulatory oligodendrocyte. Brain Res. 2015 doi: 10.1016/j.brainres.2015.09.021. published online Sep 28. [DOI] [PubMed] [Google Scholar]
- 22.Herrera-Rivero M, Heneka MT, Papadopoulos V. Translocator protein and new targets for neuroinflammation. Clinical and Translational Imaging. 2015;3:391–402. [Google Scholar]
- 23.Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, Selvaraj V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. The Journal of biological chemistry. 2014;289:27444–27454. doi: 10.1074/jbc.M114.578286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banati RB, Middleton RJ, Chan R, Hatty CR, Kam WW, Quin C, Graeber MB, Parmar A, Zahra D, Callaghan P, Fok S, Howell NR, Gregoire M, Szabo A, Pham T, Davis E, Liu GJ. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nature communications. 2014;5:5452. doi: 10.1038/ncomms6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan J, Campioli E, Midzak A, Culty M, Papadopoulos V. Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:7261–7266. doi: 10.1073/pnas.1502670112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, Lee SC. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306–328. doi: 10.1111/j.1365-2990.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacology & therapeutics. 2008;118:1–17. doi: 10.1016/j.pharmthera.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Liu H, Xu S, Tang Z, Xia W, Cheng Z, Li W, Jin Y. Spinal translocator protein alleviates chronic neuropathic pain behavior and modulates spinal astrocyte-neuronal function in rats with L5 spinal nerve ligation model. Pain. 2016;157:103–116. doi: 10.1097/j.pain.0000000000000339. [DOI] [PubMed] [Google Scholar]
- 29.Wei XH, Wei X, Chen FY, Zang Y, Xin WJ, Pang RP, Chen Y, Wang J, Li YY, Shen KF, Zhou LJ, Liu XG. The upregulation of translocator protein (18 kDa) promotes recovery from neuropathic pain in rats. J Neurosci. 2013;33:1540–1551. doi: 10.1523/JNEUROSCI.0324-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nature reviews Drug discovery. 2010;9:971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 31.Abourbeh G, Theze B, Maroy R, Dubois A, Brulon V, Fontyn Y, Dolle F, Tavitian B, Boisgard R. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J Neurosci. 2012;32:5728–5736. doi: 10.1523/JNEUROSCI.2900-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, Price G, Wegner F, Giovannoni G, Miller DH, Perkin GD, Smith T, Hewson AK, Bydder G, Kreutzberg GW, Jones T, Cuzner ML, Myers R. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123(Pt 11):2321–2337. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- 33.Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, Zhang MR, Suzuki K, Ando K, Staufenbiel M, Trojanowski JQ, Lee VM, Higuchi M, Suhara T. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer’s and other CNS pathologies. J Neurosci. 2008;28:12255–12267. doi: 10.1523/JNEUROSCI.2312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen MK, Guilarte TR. Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol Sci. 2006;91:532–539. doi: 10.1093/toxsci/kfj172. [DOI] [PubMed] [Google Scholar]
- 35.Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, Lee SC. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306–328. doi: 10.1111/j.1365-2990.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rojas S, Martin A, Arranz MJ, Pareto D, Purroy J, Verdaguer E, Llop J, Gomez V, Gispert JD, Millan O, Chamorro A, Planas AM. Imaging brain inflammation with [(11)C]PK11195 by PET and induction of the peripheral-type benzodiazepine receptor after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2007;27:1975–1986. doi: 10.1038/sj.jcbfm.9600500. [DOI] [PubMed] [Google Scholar]
- 37.Gulyas B, Makkai B, Kasa P, Gulya K, Bakota L, Varszegi S, Beliczai Z, Andersson J, Csiba L, Thiele A, Dyrks T, Suhara T, Suzuki K, Higuchi M, Halldin C. A comparative autoradiography study in post mortem whole hemisphere human brain slices taken from Alzheimer patients and age-matched controls using two radiolabelled DAA1106 analogues with high affinity to the peripheral benzodiazepine receptor (PBR) system. Neurochem Int. 2009;54:28–36. doi: 10.1016/j.neuint.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Progress in neurobiology. 2006;80:308–322. doi: 10.1016/j.pneurobio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owen DR, Guo Q, Rabiner EA, Gunn RN. The impact of the rs6971 polymorphism in TSPO for quantification and study design. Clinical and Translational Imaging. 2015:1–6. doi: 10.1007/s40336-015-0141-z. published online 2015/10/15. [DOI] [Google Scholar]
- 40.Coughlin JM, Wang Y, Munro CA, Ma S, Yue C, Chen S, Airan R, Kim PK, Adams AV, Garcia C, Higgs C, Sair HI, Sawa A, Smith G, Lyketsos CG, Caffo B, Kassiou M, Guilarte TR, Pomper MG. Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiology of disease. 2015;74:58–65. doi: 10.1016/j.nbd.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banati RB, Cagnin A, Brooks DJ, Gunn RN, Myers R, Jones T, Birch R, Anand P. Long-term trans-synaptic glial responses in the human thalamus after peripheral nerve injury. Neuroreport. 2001;12:3439–3442. doi: 10.1097/00001756-200111160-00012. [DOI] [PubMed] [Google Scholar]
- 42.Zurcher NR, Loggia ML, Lawson R, Chonde DB, Izquierdo-Garcia D, Yasek JE, Akeju O, Catana C, Rosen BR, Cudkowicz ME, Hooker JM, Atassi N. Increased glial activation in patients with amyotrophic lateral sclerosis: Assessed with [C]-PBR28. NeuroImage Clinical. 2015;7:409–414. doi: 10.1016/j.nicl.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janczar K, Su Z, Raccagni I, Anfosso A, Kelly C, Durrenberger PF, Gerhard A, Roncaroli F. The 18-kDa mitochondrial translocator protein in gliomas: from the bench to bedside. Biochemical Society transactions. 2015;43:579–585. doi: 10.1042/BST20150064. [DOI] [PubMed] [Google Scholar]
- 44.Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, Hill E, Hsu S, Izquierdo-Garcia D, Ji RR, Riley M, Wasan AD, Zurcher NR, Albrecht DS, Vangel MG, Rosen BR, Napadow V, Hooker JM. Evidence for brain glial activation in chronic pain patients. Brain. 2015 doi: 10.1093/brain/awu377. published online Jan 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niccolini F, Su P, Politis M. PET in multiple sclerosis. Clinical nuclear medicine. 2015;40:e46–52. doi: 10.1097/RLU.0000000000000359. [DOI] [PubMed] [Google Scholar]
- 46.Owen DR, Howell OW, Tang SP, Wells LA, Bennacef I, Bergstrom M, Gunn RN, Rabiner EA, Wilkins MR, Reynolds R, Matthews PM, Parker CA. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab. 2010;30:1608–1618. doi: 10.1038/jcbfm.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL, Zoghbi SS, Hyde T, Kleinman JE, Pike VW, McMahon FJ, Innis RB, Biomarkers Consortium P. E. T. R. P. T A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab. 2013;33:53–58. doi: 10.1038/jcbfm.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, Rhodes C, Pulford DJ, Bennacef I, Parker CA, StJean PL, Cardon LR, Mooser VE, Matthews PM, Rabiner EA, Rubio JP. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5. doi: 10.1038/jcbfm.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lockhart A, Davis B, Matthews JC, Rahmoune H, Hong G, Gee A, Earnshaw D, Brown J. The peripheral benzodiazepine receptor ligand PK11195 binds with high affinity to the acute phase reactant alpha1-acid glycoprotein: implications for the use of the ligand as a CNS inflammatory marker. Nuclear medicine and biology. 2003;30:199–206. doi: 10.1016/s0969-8051(02)00410-9. [DOI] [PubMed] [Google Scholar]
- 50.Turkheimer Federico E, Rizzo G, Bloomfield Peter S, Howes O, Zanotti-Fregonara P, Bertoldo A, Veronese M. The methodology of TSPO imaging with positron emission tomography. Biochemical Society transactions. 2015;43:586–592. doi: 10.1042/BST20150058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 52.Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- 53.Turkheimer FE, Edison P, Pavese N, Roncaroli F, Anderson AN, Hammers A, Gerhard A, Hinz R, Tai YF, Brooks DJ. Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med. 2007;48:158–167. [PubMed] [Google Scholar]
- 54.Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, Bloomfield MA, Bonoldi I, Kalk N, Turkheimer F, McGuire P, de Paola V, Howes OD. Microglial Activity in People at Ultra High Risk of Psychosis and in Schizophrenia: An [C]PBR28 PET Brain Imaging Study. The American journal of psychiatry. 2015 doi: 10.1176/appi.ajp.2015.14101358. published online Oct 16. appiajp201514101358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyoo CH, Ikawa M, Liow JS, Zoghbi SS, Morse CL, Pike VW, Fujita M, Innis RB, Kreisl WC. Cerebellum Can Serve As a Pseudo-Reference Region in Alzheimer Disease to Detect Neuroinflammation Measured with PET Radioligand Binding to Translocator Protein. J Nucl Med. 2015;56:701–706. doi: 10.2967/jnumed.114.146027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coughlin JM, Wang Y, Ma S, Yue C, Kim PK, Adams AV, Roosa HV, Gage KL, Stathis M, Rais R, Rojas C, McGlothan JL, Watkins CC, Sacktor N, Guilarte TR, Zhou Y, Sawa A, Slusher BS, Caffo B, Kassiou M, Endres CJ, Pomper MG. Regional brain distribution of translocator protein using [(11)C]DPA-713 PET in individuals infected with HIV. Journal of neurovirology. 2014;20:219–232. doi: 10.1007/s13365-014-0239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ekblom J, Jossan SS, Oreland L, Walum E, Aquilonius SM. Reactive gliosis and monoamine oxidase B. Journal of neural transmission Supplementum. 1994;41:253–258. doi: 10.1007/978-3-7091-9324-2_33. [DOI] [PubMed] [Google Scholar]
- 58.Fowler JS, Wang GJ, Logan J, Xie S, Volkow ND, MacGregor RR, Schlyer DJ, Pappas N, Alexoff DL, Patlak C, et al. Selective reduction of radiotracer trapping by deuterium substitution: comparison of carbon-11-L-deprenyl and carbon-11-deprenyl-D2 for MAO B mapping. J Nucl Med. 1995;36:1255–1262. [PubMed] [Google Scholar]
- 59.Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, Christman D, Logan J, Smith M, Sachs H, et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science. 1987;235:481–485. doi: 10.1126/science.3099392. [DOI] [PubMed] [Google Scholar]
- 60.Linnman C, Appel L, Fredrikson M, Gordh T, Soderlund A, Langstrom B, Engler H. Elevated [11C]-D-deprenyl uptake in chronic Whiplash Associated Disorder suggests persistent musculoskeletal inflammation. PLoS One. 2011;6:e19182. doi: 10.1371/journal.pone.0019182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scholl M, Carter SF, Westman E, Rodriguez-Vieitez E, Almkvist O, Thordardottir S, Wall A, Graff C, Langstrom B, Nordberg A. Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Scientific reports. 2015;5:16404. doi: 10.1038/srep16404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johansson A, Engler H, Blomquist G, Scott B, Wall A, Aquilonius SM, Langstrom B, Askmark H. Evidence for astrocytosis in ALS demonstrated by [11C](L)-deprenyl-D2 PET. Journal of the neurological sciences. 2007;255:17–22. doi: 10.1016/j.jns.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 63.Saura J, Bleuel Z, Ulrich J, Mendelowitsch A, Chen K, Shih JC, Malherbe P, Da Prada M, Richards JG. Molecular neuroanatomy of human monoamine oxidases A and B revealed by quantitative enzyme radioautography and in situ hybridization histochemistry. Neuroscience. 1996;70:755–774. doi: 10.1016/s0306-4522(96)83013-2. [DOI] [PubMed] [Google Scholar]
- 64.Aid S, Bosetti F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie. 2011;93:46–51. doi: 10.1016/j.biochi.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yagami T, Koma H, Yamamoto Y. Pathophysiological Roles of Cyclooxygenases and Prostaglandins in the Central Nervous System. Molecular neurobiology. 2015 doi: 10.1007/s12035-015-9355-3. published online Sep 2. [DOI] [PubMed] [Google Scholar]
- 66.Shukuri M, Takashima-Hirano M, Tokuda K, Takashima T, Matsumura K, Inoue O, Doi H, Suzuki M, Watanabe Y, Onoe H. In vivo expression of cyclooxygenase-1 in activated microglia and macrophages during neuroinflammation visualized by PET with 11C-ketoprofen methyl ester. J Nucl Med. 2011;52:1094–1101. doi: 10.2967/jnumed.110.084046. [DOI] [PubMed] [Google Scholar]
- 67.Ohnishi A, Senda M, Yamane T, Sasaki M, Mikami T, Nishio T, Ikari Y, Nishida H, Shukuri M, Takashima T, Mawatari A, Doi H, Watanabe Y, Onoe H. Human whole-body biodistribution and dosimetry of a new PET tracer, [(11)C]ketoprofen methyl ester, for imagings of neuroinflammation. Nuclear medicine and biology. 2014;41:594–599. doi: 10.1016/j.nucmedbio.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 68.Soliman ML, Ohm JE, Rosenberger TA. Acetate reduces PGE2 release and modulates phospholipase and cyclooxygenase levels in neuroglia stimulated with lipopolysaccharide. Lipids. 2013;48:651–662. doi: 10.1007/s11745-013-3799-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Perdomo A, Marquez-Kisinousky L, Santalucia T, Planas AM. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. The Journal of biological chemistry. 2012;287:6454–6468. doi: 10.1074/jbc.M111.327874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. J Neurochem. 2007;101:577–599. doi: 10.1111/j.1471-4159.2006.04371.x. [DOI] [PubMed] [Google Scholar]
- 71.Esposito G, Giovacchini G, Liow JS, Bhattacharjee AK, Greenstein D, Schapiro M, Hallett M, Herscovitch P, Eckelman WC, Carson RE, Rapoport SI. Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET. J Nucl Med. 2008;49:1414–1421. doi: 10.2967/jnumed.107.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henry RJ, Kerr DM, Finn DP, Roche M. FAAH-mediated modulation of TLR3-induced neuroinflammation in the rat hippocampus. J Neuroimmunol. 2014;276:126–134. doi: 10.1016/j.jneuroim.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 73.Rebola N, Simoes AP, Canas PM, Tome AR, Andrade GM, Barry CE, Agostinho PM, Lynch MA, Cunha RA. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J Neurochem. 2011;117:100–111. doi: 10.1111/j.1471-4159.2011.07178.x. [DOI] [PubMed] [Google Scholar]
- 74.Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nature neuroscience. 2009;12:872–878. doi: 10.1038/nn.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mishina M, Ishiwata K. Adenosine receptor PET imaging in human brain. International review of neurobiology. 2014;119:51–69. doi: 10.1016/B978-0-12-801022-8.00002-7. [DOI] [PubMed] [Google Scholar]
- 76.Rissanen E, Virta JR, Paavilainen T, Tuisku J, Helin S, Luoto P, Parkkola R, Rinne JO, Airas L. Adenosine A2A receptors in secondary progressive multiple sclerosis: a [(11)C]TMSX brain PET study. J Cereb Blood Flow Metab. 2013;33:1394–1401. doi: 10.1038/jcbfm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends in pharmacological sciences. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 78.Martin A, Szczupak B, Gomez-Vallejo V, Domercq M, Cano A, Padro D, Munoz C, Higuchi M, Matute C, Llop J. In vivo PET imaging of the alpha4beta2 nicotinic acetylcholine receptor as a marker for brain inflammation after cerebral ischemia. J Neurosci. 2015;35:5998–6009. doi: 10.1523/JNEUROSCI.3670-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sabri O, Becker GA, Meyer PM, Hesse S, Wilke S, Graef S, Patt M, Luthardt J, Wagenknecht G, Hoepping A, Smits R, Franke A, Sattler B, Habermann B, Neuhaus P, Fischer S, Tiepolt S, Deuther-Conrad W, Barthel H, Schonknecht P, Brust P. First-in-human PET quantification study of cerebral alpha4beta2* nicotinic acetylcholine receptors using the novel specific radioligand (−)-[(18)F]Flubatine. Neuroimage. 2015;118:199–208. doi: 10.1016/j.neuroimage.2015.05.065. [DOI] [PubMed] [Google Scholar]
- 80.Chang L, Munsaka SM, Kraft-Terry S, Ernst T. Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J Neuroimmune Pharmacol. 2013;8:576–593. doi: 10.1007/s11481-013-9460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brand A, Richter-Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Developmental neuroscience. 1993;15:289–298. doi: 10.1159/000111347. [DOI] [PubMed] [Google Scholar]
- 82.Fisher SK, Novak JE, Agranoff BW. Inositol and higher inositol phosphates in neural tissues: homeostasis, metabolism and functional significance. J Neurochem. 2002;82:736–754. doi: 10.1046/j.1471-4159.2002.01041.x. [DOI] [PubMed] [Google Scholar]
- 83.Grover VP, Pavese N, Koh SB, Wylezinska M, Saxby BK, Gerhard A, Forton DM, Brooks DJ, Thomas HC, Taylor-Robinson SD. Cerebral microglial activation in patients with hepatitis C: in vivo evidence of neuroinflammation. Journal of viral hepatitis. 2012;19:e89–96. doi: 10.1111/j.1365-2893.2011.01510.x. [DOI] [PubMed] [Google Scholar]
- 84.Butteriss DJ, Ismail A, Ellison DW, Birchall D. Use of serial proton magnetic resonance spectroscopy to differentiate low grade glioma from tumefactive plaque in a patient with multiple sclerosis. The British journal of radiology. 2003;76:662–665. doi: 10.1259/bjr/85069069. [DOI] [PubMed] [Google Scholar]
- 85.Harris JL, Yeh HW, Choi IY, Lee P, Berman NE, Swerdlow RH, Craciunas SC, Brooks WM. Altered neurochemical profile after traumatic brain injury: (1)H-MRS biomarkers of pathological mechanisms. J Cereb Blood Flow Metab. 2012;32:2122–2134. doi: 10.1038/jcbfm.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Prichard J, Rothman D, Novotny E, Petroff O, Kuwabara T, Avison M, Howseman A, Hanstock C, Shulman R. Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:5829–5831. doi: 10.1073/pnas.88.13.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lopez-Villegas D, Lenkinski RE, Wehrli SL, Ho WZ, Douglas SD. Lactate production by human monocytes/macrophages determined by proton MR spectroscopy. Magnetic resonance in medicine. 1995;34:32–38. doi: 10.1002/mrm.1910340107. [DOI] [PubMed] [Google Scholar]
- 88.Magistretti PJ. Role of glutamate in neuron-glia metabolic coupling. The American journal of clinical nutrition. 2009;90:875S–880S. doi: 10.3945/ajcn.2009.27462CC. [DOI] [PubMed] [Google Scholar]
- 89.Meyerhoff DJ, Bloomer C, Cardenas V, Norman D, Weiner MW, Fein G. Elevated subcortical choline metabolites in cognitively and clinically asymptomatic HIV+ patients. Neurology. 1999;52:995–1003. doi: 10.1212/wnl.52.5.995. [DOI] [PubMed] [Google Scholar]
- 90.Zahr NM, Mayer D, Rohlfing T, Sullivan EV, Pfefferbaum A. Imaging neuroinflammation? A perspective from MR spectroscopy. Brain Pathol. 2014;24:654–664. doi: 10.1111/bpa.12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Buonocore MH, Maddock RJ. Magnetic resonance spectroscopy of the brain: a review of physical principles and technical methods. Reviews in the neurosciences. 2015;26:609–632. doi: 10.1515/revneuro-2015-0010. [DOI] [PubMed] [Google Scholar]
- 92.Jacobs MA, Horska A, van Zijl PC, Barker PB. Quantitative proton MR spectroscopic imaging of normal human cerebellum and brain stem. Magnetic resonance in medicine. 2001;46:699–705. doi: 10.1002/mrm.1248. [DOI] [PubMed] [Google Scholar]
- 93.Barker PB, Soher BJ, Blackband SJ, Chatham JC, Mathews VP, Bryan RN. Quantitation of proton NMR spectra of the human brain using tissue water as an internal concentration reference. NMR in biomedicine. 1993;6:89–94. doi: 10.1002/nbm.1940060114. [DOI] [PubMed] [Google Scholar]
- 94.Hennig J, Pfister H, Ernst T, Ott D. Direct absolute quantification of metabolites in the human brain with in vivo localized proton spectroscopy. NMR in biomedicine. 1992;5:193–199. doi: 10.1002/nbm.1940050406. [DOI] [PubMed] [Google Scholar]
- 95.Roser W, Steinbrich W, Radue EW. Results and consequences of frequent quality control of quantitative clinical 1H-MR-spectroscopy. RoFo: Fortschritte auf dem Gebiete der Rontgenstrahlen und der Nuklearmedizin. 1997;166:554–557. doi: 10.1055/s-2007-1015476. [DOI] [PubMed] [Google Scholar]
- 96.Ramadan S, Andronesi OC, Stanwell P, Lin AP, Sorensen AG, Mountford CE. Use of in vivo two-dimensional MR spectroscopy to compare the biochemistry of the human brain to that of glioblastoma. Radiology. 2011;259:540–549. doi: 10.1148/radiol.11101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Andronesi OC, Gagoski BA, Sorensen AG. Neurologic 3D MR spectroscopic imaging with low-power adiabatic pulses and fast spiral acquisition. Radiology. 2012;262:647–661. doi: 10.1148/radiol.11110277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 99.McCaffrey G, Staatz WD, Sanchez-Covarrubias L, Finch JD, Demarco K, Laracuente ML, Ronaldson PT, Davis TP. P-glycoprotein trafficking at the blood-brain barrier altered by peripheral inflammatory hyperalgesia. J Neurochem. 2012;122:962–975. doi: 10.1111/j.1471-4159.2012.07831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu L, Collier AC, Link JM, Domino KB, Mankoff DA, Eary JF, Spiekerman CF, Hsiao P, Deo AK, Unadkat JD. Modulation of P-glycoprotein at the Human Blood-Brain Barrier by Quinidine or Rifampin Treatment: A Positron Emission Tomography Imaging Study. Drug metabolism and disposition: the biological fate of chemicals. 2015;43:1795–1804. doi: 10.1124/dmd.114.058685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Doorduin J, de Vries EF, Dierckx RA, Klein HC. P-glycoprotein activity in the blood-brain barrier is affected by virus-induced neuroinflammation and antipsychotic treatment. Neuropharmacology. 2014;85:548–553. doi: 10.1016/j.neuropharm.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 102.Heye AK, Culling RD, Valdes Hernandez Mdel C, Thrippleton MJ, Wardlaw JM. Assessment of blood-brain barrier disruption using dynamic contrast-enhanced MRI. A systematic review. NeuroImage Clinical. 2014;6:262–274. doi: 10.1016/j.nicl.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sourbron SP, Buckley DL. Classic models for dynamic contrast-enhanced MRI. NMR in biomedicine. 2013;26:1004–1027. doi: 10.1002/nbm.2940. [DOI] [PubMed] [Google Scholar]
- 104.Mirasol RV, Bokkers RP, Hernandez DA, Merino JG, Luby M, Warach S, Latour LL. Assessing reperfusion with whole-brain arterial spin labeling: a noninvasive alternative to gadolinium. Stroke; a journal of cerebral circulation. 2014;45:456–461. doi: 10.1161/STROKEAHA.113.004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu HL, Chang TT, Yan FX, Li CH, Lin YS, Wong AM. Assessment of vessel permeability by combining dynamic contrast-enhanced and arterial spin labeling MRI. NMR in biomedicine. 2015;28:642–649. doi: 10.1002/nbm.3297. [DOI] [PubMed] [Google Scholar]
- 106.Absinta M, Vuolo L, Rao A, Nair G, Sati P, Cortese IC, Ohayon J, Fenton K, Reyes-Mantilla MI, Maric D, Calabresi PA, Butman JA, Pardo CA, Reich DS. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology. 2015;85:18–28. doi: 10.1212/WNL.0000000000001587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pinheiro MAL, Kooij G, Mizee MR, Kamermans A, Enzmann G, Lyck R, Schwaninger M, Engelhardt B, de Vries HE. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2015;1862:461–471. doi: 10.1016/j.bbadis.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 108.Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Frontiers in neuroscience. 2014;8:447. doi: 10.3389/fnins.2014.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wunder A, Klohs J, Dirnagl U. Non-invasive visualization of CNS inflammation with nuclear and optical imaging. Neuroscience. 2009;158:1161–1173. doi: 10.1016/j.neuroscience.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 110.Kelsen J, Agnholt J, Falborg L, Nielsen JT, Romer JL, Hoffmann HJ, Dahlerup JF. Indium-labelled human gut-derived T cells from healthy subjects with strong in vitro adhesion to MAdCAM-1 show no detectable homing to the gut in vivo. Clinical and experimental immunology. 2004;138:66–74. doi: 10.1111/j.1365-2249.2004.02578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Van Hemert FJ, Voermans C, Van Eck-Smit BL, Bennink RJ. Labeling monocytes for imaging chronic inflammation. The quarterly journal of nuclear medicine and molecular imaging: official publication of the Italian Association of Nuclear Medicine. 2009;53:78–88. [PubMed] [Google Scholar]
- 112.Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nature reviews Clinical oncology. 2011;8:677–688. doi: 10.1038/nrclinonc.2011.141. [DOI] [PubMed] [Google Scholar]
- 113.Neuwelt A, Sidhu N, Hu CA, Mlady G, Eberhardt SC, Sillerud LO. Iron-based superparamagnetic nanoparticle contrast agents for MRI of infection and inflammation. AJR American journal of roentgenology. 2015;204:W302–313. doi: 10.2214/AJR.14.12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Walter HL, Walberer M, Rueger MA, Backes H, Wiedermann D, Hoehn M, Neumaier B, Graf R, Fink GR, Schroeter M. In vivo analysis of neuroinflammation in the late chronic phase after experimental stroke. Neuroscience. 2015;292:71–80. doi: 10.1016/j.neuroscience.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 115.Nighoghossian N, Wiart M, Cakmak S, Berthezene Y, Derex L, Cho TH, Nemoz C, Chapuis F, Tisserand GL, Pialat JB, Trouillas P, Froment JC, Hermier M. Inflammatory response after ischemic stroke: a USPIO-enhanced MRI study in patients. Stroke; a journal of cerebral circulation. 2007;38:303–307. doi: 10.1161/01.STR.0000254548.30258.f2. [DOI] [PubMed] [Google Scholar]
- 116.Normandin MD, Yuan H, Wilks MQ, Chen HH, Kinsella JM, Cho H, Guehl NJ, Absi-Halabi N, Hosseini SM, El Fakhri G, Sosnovik DE, Josephson L. Heat-Induced Radiolabeling of Nanoparticles for Monocyte Tracking by PET. Angewandte Chemie. 2015;54:13002–13006. doi: 10.1002/anie.201505525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stankoff B, Freeman L, Aigrot MS, Chardain A, Dolle F, Williams A, Galanaud D, Armand L, Lehericy S, Lubetzki C, Zalc B, Bottlaender M. Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-(1)(1)C]-2-(4′-methylaminophenyl)- 6-hydroxybenzothiazole. Annals of neurology. 2011;69:673–680. doi: 10.1002/ana.22320. [DOI] [PubMed] [Google Scholar]
- 118.Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li QX, Masters CL, Cappai R, Villemagne VL. Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med. 2009;50:198–204. doi: 10.2967/jnumed.108.057984. [DOI] [PubMed] [Google Scholar]
- 119.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 120.Veronese M, Bodini B, Garcia-Lorenzo D, Battaglini M, Bongarzone S, Comtat C, Bottlaender M, Stankoff B, Turkheimer FE. Quantification of [(11)C]PIB PET for imaging myelin in the human brain: a test-retest reproducibility study in high-resolution research tomography. J Cereb Blood Flow Metab. 2015;35:1771–1782. doi: 10.1038/jcbfm.2015.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baron JC, Yamauchi H, Fujioka M, Endres M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J Cereb Blood Flow Metab. 2014;34:2–18. doi: 10.1038/jcbfm.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pascual B, Prieto E, Arbizu J, Marti-Climent JM, Penuelas I, Quincoces G, Zarauza R, Pappata S, Masdeu JC. Decreased carbon-11-flumazenil binding in early Alzheimer’s disease. Brain. 2012;135:2817–2825. doi: 10.1093/brain/aws210. [DOI] [PubMed] [Google Scholar]
- 123.Giffard C, Landeau B, Kerrouche N, Young AR, Barre L, Baron JC. Decreased chronic-stage cortical 11C-flumazenil binding after focal ischemia-reperfusion in baboons: a marker of selective neuronal loss? Stroke; a journal of cerebral circulation. 2008;39:991–999. doi: 10.1161/STROKEAHA.107.489419. [DOI] [PubMed] [Google Scholar]
- 124.Vivash L, Gregoire MC, Bouilleret V, Berard A, Wimberley C, Binns D, Roselt P, Katsifis A, Myers DE, Hicks RJ, O’Brien TJ, Dedeurwaerdere S. In vivo measurement of hippocampal GABAA/cBZR density with [18F]-flumazenil PET for the study of disease progression in an animal model of temporal lobe epilepsy. PLoS One. 2014;9:e86722. doi: 10.1371/journal.pone.0086722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang S, Boyd J, Delaney K, Murphy TH. Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J Neurosci. 2005;25:5333–5338. doi: 10.1523/JNEUROSCI.1085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zeng W, Wang X, Xu P, Liu G, Eden HS, Chen X. Molecular imaging of apoptosis: from micro to macro. Theranostics. 2015;5:559–582. doi: 10.7150/thno.11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hoglund J, Shirvan A, Antoni G, Gustavsson SA, Langstrom B, Ringheim A, Sorensen J, Ben-Ami M, Ziv I. 18F-ML-10, a PET tracer for apoptosis: first human study. J Nucl Med. 2011;52:720–725. doi: 10.2967/jnumed.110.081786. [DOI] [PubMed] [Google Scholar]
- 128.Challapalli A, Kenny LM, Hallett WA, Kozlowski K, Tomasi G, Gudi M, Al-Nahhas A, Coombes RC, Aboagye EO. 18F-ICMT-11, a caspase-3-specific PET tracer for apoptosis: biodistribution and radiation dosimetry. J Nucl Med. 2013;54:1551–1556. doi: 10.2967/jnumed.112.118760. [DOI] [PubMed] [Google Scholar]
- 129.Doss M, Kolb HC, Walsh JC, Mocharla V, Fan H, Chaudhary A, Zhu Z, Alpaugh RK, Lango MN, Yu JQ. Biodistribution and radiation dosimetry of 18F-CP-18, a potential apoptosis imaging agent, as determined from PET/CT scans in healthy volunteers. J Nucl Med. 2013;54:2087–2092. doi: 10.2967/jnumed.113.119800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cauda F, Palermo S, Costa T, Torta R, Duca S, Vercelli U, Geminiani G, Torta DM. Gray matter alterations in chronic pain: A network-oriented meta-analytic approach. NeuroImage Clinical. 2014;4:676–686. doi: 10.1016/j.nicl.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matsuda H. MRI morphometry in Alzheimer’s disease. Ageing research reviews. 2016 doi: 10.1016/j.arr.2016.01.003. published online Jan 23. [DOI] [PubMed] [Google Scholar]
- 132.Frodl T, Amico F. Is there an association between peripheral immune markers and structural/functional neuroimaging findings? Progress in neuro-psychopharmacology & biological psychiatry. 2014;48:295–303. doi: 10.1016/j.pnpbp.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 133.Kreisl WC, Lyoo CH, McGwier M, Snow J, Jenko KJ, Kimura N, Corona W, Morse CL, Zoghbi SS, Pike VW, McMahon FJ, Turner RS, Innis RB, Biomarkers Consortium, P. E. T. R. P. T In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain. 2013;136:2228–2238. doi: 10.1093/brain/awt145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhou G, Hotta J, Lehtinen MK, Forss N, Hari R. Enlargement of choroid plexus in complex regional pain syndrome. Scientific reports. 2015;5:14329. doi: 10.1038/srep14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Maddock RJ, Buonocore MH. MR spectroscopic studies of the brain in psychiatric disorders. Current topics in behavioral neurosciences. 2012;11:199–251. doi: 10.1007/7854_2011_197. [DOI] [PubMed] [Google Scholar]
- 136.Granziera C, Sprenger T. Brain Inflammation, Degeneration, and Plasticity in Multiple Sclerosis A2 - Toga. In: Arthur W, editor. Brain Mapping. Academic Press; Waltham: 2015. pp. 917–927. [Google Scholar]
- 137.Bonnier G, Roche A, Romascano D, Simioni S, Meskaldji D, Rotzinger D, Lin YC, Menegaz G, Schluep M, Du Pasquier R, Sumpf TJ, Frahm J, Thiran JP, Krueger G, Granziera C. Advanced MRI unravels the nature of tissue alterations in early multiple sclerosis. Annals of clinical and translational neurology. 2014;1:423–432. doi: 10.1002/acn3.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bonnier G, Roche A, Romascano D, Simioni S, Meskaldji DE, Rotzinger D, Lin YC, Menegaz G, Schluep M, Du Pasquier R, Sumpf TJ, Frahm J, Thiran JP, Krueger G, Granziera C. Multicontrast MRI Quantification of Focal Inflammation and Degeneration in Multiple Sclerosis. BioMed research international. 2015;2015:569123. doi: 10.1155/2015/569123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Enzinger C, Barkhof F, Ciccarelli O, Filippi M, Kappos L, Rocca MA, Ropele S, Rovira A, Schneider T, de Stefano N, Vrenken H, Wheeler-Kingshott C, Wuerfel J, Fazekas F, group, M. s. Nonconventional MRI and microstructural cerebral changes in multiple sclerosis. Nature reviews Neurology. 2015;11:676–686. doi: 10.1038/nrneurol.2015.194. [DOI] [PubMed] [Google Scholar]
- 140.Klawiter EC, Schmidt RE, Trinkaus K, Liang HF, Budde MD, Naismith RT, Song SK, Cross AH, Benzinger TL. Radial diffusivity predicts demyelination in ex vivo multiple sclerosis spinal cords. Neuroimage. 2011;55:1454–1460. doi: 10.1016/j.neuroimage.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sepehrband F, Clark KA, Ullmann JF, Kurniawan ND, Leanage G, Reutens DC, Yang Z. Brain tissue compartment density estimated using diffusion-weighted MRI yields tissue parameters consistent with histology. Human brain mapping. 2015;36:3687–3702. doi: 10.1002/hbm.22872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Colasanti A, Guo Q, Muhlert N, Giannetti P, Onega M, Newbould RD, Ciccarelli O, Rison S, Thomas C, Nicholas R, Muraro PA, Malik O, Owen DR, Piccini P, Gunn RN, Rabiner EA, Matthews PM. In Vivo Assessment of Brain White Matter Inflammation in Multiple Sclerosis with (18)F-PBR111 PET. J Nucl Med. 2014;55:1112–1118. doi: 10.2967/jnumed.113.135129. [DOI] [PubMed] [Google Scholar]
- 143.Ma D, Gulani V, Seiberlich N, Liu K, Sunshine JL, Duerk JL, Griswold MA. Magnetic resonance fingerprinting. Nature. 2013;495:187–192. doi: 10.1038/nature11971. [DOI] [PMC free article] [PubMed] [Google Scholar]