

Graphical abstract
Keywords: Coronavirus, 3C-like protease, MERS, HKU4, HKU5, SARS, Peptidomimetic compounds, Broad-spectrum inhibitors, Protease inhibitors, Zoonotic reservoir
Abstract
The bat coronavirus HKU4 belongs to the same 2c lineage as that of the deadly Middle East Respiratory Syndrome coronavirus (MERS-CoV) and shows high sequence similarity, therefore potentiating a threat to the human population through a zoonotic shift or ‘spill over’ event. To date, there are no effective vaccines or antiviral treatments available that are capable of limiting the pathogenesis of any human coronaviral infection. An attractive target for the development of anti-coronaviral therapeutics is the 3C-like protease (3CLpro), which is essential for the progression of the coronaviral life cycle. Herein, we report the screening results of a small, 230-member peptidomimetic library against HKU4-CoV 3CLpro and the identification of 43 peptidomimetic compounds showing good to excellent inhibitory potency of HKU4-CoV 3CLpro with IC50 values ranging from low micromolar to sub-micromolar. We established structure–activity relationships (SARs) describing the important ligand-based features required for potent HKU4-CoV 3CLpro inhibition and identified a seemingly favored peptidic backbone for HKU4-CoV 3CLpro inhibition. To investigate this, a molecular sub-structural analysis of the most potent HKU4-CoV 3CLpro inhibitor was accomplished by the synthesis and testing of the lead peptidomimetic inhibitor’s sub-structural components, confirming the activity of the favored backbone (22A) identified via SAR analysis. In order to elucidate the structural reasons for such potent HKU4-CoV 3CLpro inhibition by the peptidomimetics having the 22A backbone, we determined the X-ray structures of HKU4-CoV 3CLpro in complex with three peptidomimetic inhibitors. Sequence alignment of HKU4-CoV 3CLpro, and two other lineage C Betacoronaviruses 3CLpro’s, HKU5-CoV and MERS-CoV 3CLpro, show that the active site residues of HKU4-CoV 3CLpro that participate in inhibitor binding are conserved in HKU5-CoV and MERS-CoV 3CLpro. Furthermore, we assayed our most potent HKU4-CoV 3CLpro inhibitor for inhibition of HKU5-CoV 3CLpro and found it to have sub-micromolar inhibitory activity (IC50 = 0.54 ± 0.03 μM). The X-ray structures and SAR analysis reveal critical insights into the structure and inhibition of HKU4-CoV 3CLpro, providing fundamental knowledge that may be exploited in the development of anti-coronaviral therapeutics for coronaviruses emerging from zoonotic reservoirs.
1. Introduction
Zoonotic viruses, including coronaviruses, Ebola virus, HIV, influenza A viruses, hantaviruses and henipaviruses, are ubiquitous and can emerge as significant human pathogens capable of global epidemics and pandemics. Most often, we spend our time and resources in targeting only the human pathogens using small molecule inhibitors with the ultimate goal of developing therapeutic compounds to treat the associated diseases. A missed opportunity, however, is targeting the zoonotic host using small molecule inhibitors with the goal of utilizing the acquired chemical and structural knowledge to inform us of the evolutionary path of the virus on a biological basis. So, we believe that by using small molecule compounds as probes of the structural evolution of viral enzyme drug targets, from the zoonotic reservoirs to the human pathogens, we can gain new insights and predict a priori the structural scaffolds of small molecule compounds that can serve as lead templates for therapeutic development against emerging human pathogenic viruses such as coronaviruses.
Coronaviruses (CoVs) are enveloped, single-stranded, positive-sense RNA viruses that infect and cause disease in a variety of species including bats, birds, cats, dogs, pigs, mice, horses, whales, and humans.1, 2, 3 Coronaviral infections may range from mild to severe and can result in respiratory, enteric, hepatic, or neurological diseases in their carriers. The first two human CoV strains (HCoV-229E and HCoV-OC43) were identified in the mid-1960s, and it was not until the 21st century that a new human coronavirus (Severe Acute Respiratory Syndrome or SARS-CoV) was identified.4, 5 Currently, there are at least six known human CoVs including: HCoV-229E, HCoV-NL63, HCoV-OC43, HCoV-HKU1, SARS-CoV, and most recently, the Middle East Respiratory Syndrome coronavirus (MERS-CoV).6 MERS-CoV, formerly known as HCoV-EMC, was identified in November 2012, when it was isolated from the sputum a 60-year-old Saudi Arabian man presenting acute pneumonia and renal failure.7 Since the time of its identification, the virus has grown to be a threat to public health worldwide having a case-fatality rate of about 30%.8 To date, there are no vaccines or antiviral agents capable of preventing or treating any human coronaviral infection.
MERS-CoV belongs to lineage C in the genus Betacoronavirus of the Coronaviridae family in the Nidovirales order.9 Also of this lineage are the species Tylonycteris bat coronavirus HKU4 (HKU4-CoV) and Pipistrellus bat coronavirus HKU5 (HKU5-CoV), where the overall amino acid sequence identities of MERS-CoV to HKU4-CoV and HKU5-CoV across the conserved domains are approximately 75% and 76.7%, respectively.6, 10, 11 Though the exact origin of MERS-CoV is currently debated, a bat origin is strongly suspected as MERS-CoV is so closely related to HKU4- and HKU5-CoV and because MERS-CoV genomic RNA has been found in bats and dromedary camels in Qatar.12, 13 Though HKU4-CoV and HKU5-CoV have been found only in bats, studies have shown that their accessory proteins are capable of inhibiting human antiviral signaling pathways in vitro.14, 15 This, and the close similarity of MERS-CoV to HKU4-CoV and HKU5-CoV, suggests that a zoonotic shift from bats or camels to humans may have occurred.15 A recent investigation into the interactions between the human CD26 receptor and the receptor binding domains (RBDs) in the MERS-CoV, HKU4-CoV and HKU5-CoV envelope-embedded spike protein revealed that MERS-CoV and HKU4-CoV both engage this receptor for viral entry whereas HKU5-CoV does not.16 These observations suggest that an evolutionary pathway from bat HKU4-CoV to human MERS-CoV exists and that investigating the molecular basis of this zoonotic shift from a structural and chemical-biology perspective may allow us to predict and target the these viruses with small molecule therapeutics.
Coronaviral genomes are polycistronic, encoding for two large polyproteins, pp1a and pp1ab.17, 18, 19, 20 Initiation of coronavirus replication in cells occurs by the translation of two overlapping, open reading frames (ORF1a and ORF1b) to produce pp1a and, following a −1 ribosomal frameshift mechanism, pp1ab. These polyproteins are then proteolytically processed at 14 cleavage sites by two essential viral cysteine proteases, the papain-like protease (PLpro, or nsp3) and the 3C-like protease (3CLpro, also known as the main protease, Mpro, or nsp5). Cleavage by both proteases results in the production of 16 nonstructural proteins (nsps), where PLpro is responsible for cleavage at 3 sites and 3CLpro is responsible for cleavage at 11 sites (Fig. 1 ). The function of 3CLpro is vital for the coronaviral life cycle, making it an attractive target for the development of antiviral drugs.21, 22
Figure 1.
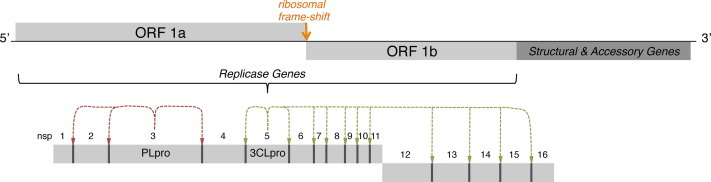
Genome and proteome organization of HKU4-, HKU5-, and MERS-CoV non-structural proteins highlighting the PLpro and 3CL-pro cleavage sites.
The present work was undertaken to investigate the kinetic and structural properties of HKU4-CoV 3CLpro and to utilize this knowledge to discover and develop potent inhibitors of HKU4-CoV 3CLpro. Targeting the immediate zoonotic reservoirs of coronaviruses with small molecule inhibitors can help inform structure-based design strategies aimed at creating molecular scaffolds that may also target the emerging human CoVs and ultimately aid in the development of therapeutics against coronaviral infection. Toward this goal, we first expressed, purified, and characterized the kinetic properties of HKU4-CoV 3CLpro. We then determined the inhibition of HKU4-CoV 3CLpro by a small library of 230 peptidomimetic compounds, which resulted in the identification of 43 HKU4-CoV 3CLpro inhibitors, two of which showed sub-micromolar potency. With a potent lead inhibitor compound in hand, we investigated the contributions of its individual sub-structural components to inhibitory potency and identified a favored peptidic backbone for HKU4-CoV 3CLpro inhibition. Finally, we determined the X-ray crystal structures of the three most potent inhibitors containing this favored backbone in complex with HKU4-CoV 3CLpro and elucidated the structural reasons behind such potent HKU4-CoV 3CLpro inhibition.
2. Results and discussion
2.1. Screen of peptidomimetic library for HKU4-CoV 3CLpro inhibition
A library of 230 previously reported, peptidomimetic-type compounds23, 24 was screened for inhibition of HKU4-CoV 3CLpro at a single concentration of 100 μM, and IC50 values were determined for compounds that showed greater than 50% inhibition of HKU4-CoV 3CLpro at that concentration. This screen resulted in the identification of 43 peptidomimetic compounds with inhibitory concentrations ranging from micromolar to sub-micromolar (vide infra). The peptidomimetic inhibitors can be grouped into two classes, inhibitors with a single amide bond in the inhibitor backbone (Class A) and inhibitors with a dipeptide-like backbone (Class B). The results of this screen are displayed in Table 1a (Class A) and Table 1b (Class B), where only the compounds that showed greater than 50% inhibition of HKU4-CoV 3CLpro at 100 μM are shown (see Supporting information S1 and S2 for comprehensive list of compounds tested). The compounds are listed in the tables in rank order by IC50 values starting with the lowest IC50 value. Of the 25 Class A inhibitors that were tested for inhibition of HKU4-CoV 3CLpro, 19 were found to inhibit HKU4-CoV 3CLpro above 50% at a concentration of 100 μM, and subsequently, IC50 values were determined. Remarkably, the two most potent inhibitors, 1A and 2A, proved to be sub-micromolar inhibitors of HKU4-CoV 3CLpro, having IC50’s of 0.33 ± 0.02 and 0.41 ± 0.04 μM, respectively. Upon examination of the 19 Class A inhibitors, an immediate trend is apparent where there is an overwhelming preference for a 3-thiophene at the R1 position and a 1-methylbenzotriazole at the R3 position of the peptidomimetic backbone. This is observed in every Class A inhibitor found to produce inhibition of HKU4-CoV 3CLpro over 50% at 100 μM, with the exception of the least active analog in this series, 19A. 19A only differs from the more active compound 6A in that 19A has a triazole in place of a benzotriazole; however, this difference is significant enough to decrease inhibitory potency ten-fold between the analogs, from 1.6 ± 0.1 μM for 6A to 16.0 ± 4.0 μM for 19A. Interestingly, 19A maintains a triazole ring in the R3 position, indicating that the position of the nitrogen atoms in this heterocycle is essential for good HKU4-CoV 3CLpro inhibition. Other R3 heterocycles incorporated in the less active members of Class A include 4-substituted methyltriazoles, 4-imidazoles, and 2-furans, underscoring the importance of the methylbenzotriazole at the R3 position and pointing to the potential significance of a precisely oriented hydrogen bond acceptor (see Supporting information S1 for a comprehensive list).
Table 1a.
| Compd | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|
| 1A | 0.33 ± 0.02 | |||
| 2A | 0.41 ± 0.04 | |||
| 3A | 1.2 ± 0.2 | |||
| 4A | 1.2 ± 0.06 | |||
| 5A | 1.5 ± 0.1 | |||
| 6A | 1.6 ± 0.09 | |||
| 7A | 1.7 ± 0.2 | |||
| 8A | 1.9 ± 0.09 | |||
| 9A | 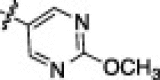 |
2.0 ± 0.2 | ||
| 10A | 2.2 ± 0.2 | |||
| 11A | 2.4 ± 0.3 | |||
| 12A | 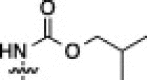 |
2.8 ± 0.4 | ||
| 13A | 3.1 ± 0.3 | |||
| 14A | 3.1 ± 0.3 | |||
| 15A | 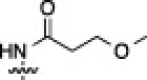 |
3.7 ± 0.4 | ||
| 16A | 4.8 ± 0.4 | |||
| 17A | 5.3 ± 0.6 | |||
| 18A | 8.8 ± 0.8 | |||
| 19A | 16.0 ± 4.0 |
Table 1b.
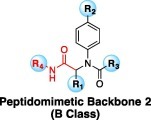
| Compd | R1 | R2 | R3 | R4 | IC50 (μM) |
|---|---|---|---|---|---|
| 1B | 1.3 ± 0.3 | ||||
| 2B | 1.5 ± 0.3 | ||||
| 3B | 1.8 ± 0.5 | ||||
| 4B | 2.2 ± 0.6 | ||||
| 5B | 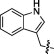 |
2.2 ± 0.4 | |||
| 6B | 2.7 ± 0.4 | ||||
| 7B | 3.4 ± 0.4 | ||||
| 8B | 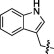 |
3.9 ± 0.7 | |||
| 9B | 4.2 ± 0.2 | ||||
| 10B | 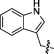 |
6.9 ± 0.7 | |||
| 11B | 7.0 ± 1.2 | ||||
| 12B | 8.6 ± 0.9 | ||||
| 13B | 9.3 ± 0.5 | ||||
| 14B | 9.5 ± 2.0 | ||||
| 15B | 11.1 ± 0.6 | ||||
| 16B | 14.7 ± 1.1 | ||||
| 17B | 15.4 ± 2.1 | ||||
| 18B | 17.2 ± 2.8 | ||||
| 19B | 18.3 ± 4.7 | ||||
| 20B | 18.7 ± 2.6 | ||||
| 21B | 22.0 ± 7.4 | ||||
| 22B | 35.6 ± 5.2 | ||||
| 23B | 52.3 ± 21.2 | ||||
| 24B | 55.6 ± 14.3 |
Of the 19 Class A peptidomimetics found to be good inhibitors of HKU4-CoV 3CLpro, 13 have amide substituents at R2, four have aromatics (5A, 7A, 9A, and 11A), and two have amines (16A and 17A). Among the inhibitors with amide substituents at R2, a trend can be observed where cyclic carboxamides are preferred (1A, 2A, 3A, 4A, and 8A) to alkyl carboxamides, with the aromatic carboxamides being the most active (1A and 2A). A preference for larger cyclic carboxamides is also observed, where 4, 5 or 6 membered cycles (1A, 2A, 3A, and 4A) are preferred over the smaller cyclopropylcarboxamide (8A).
Among the six Class A inhibitors with aromatic groups substituted at the R2 position (5A, 7A, 9A, 11A, 20A, 21A), only the four compounds 5A, 7A, 9A and 11A were found to inhibit HKU4-CoV 3CLpro above 50% at 100 μM and they each contain aromatic nitrogen heterocycles. The position of the nitrogen is of moderate significance, as a p-substituted pyridine at R2 is 2-fold more active than the m-substituted pyridine (5A vs 11A, Table 1a). Bulky substituents on R2 aromatic heterocycles, such as in 20A, decrease inhibition of HKU4-CoV 3CLpro, indicating a steric requirement within the R2 binding pocket (Table 1a and S1). Interestingly, 20A is one of only two Class A compound having a 3-thiophene at the R1 position and a 1-methylbenzotriazole at the R3 position that is not a good inhibitor of HKU4-CoV 3CLpro at 100 μM. Addition of a methoxy substituent to the R2 heterocycle is tolerated (7A and 9A, Table 1a), but does not increase or decrease inhibition significantly, indicating that the addition of another hydrogen-bond acceptor does not further engage the R2 binding pocket and that smaller aromatic substituents can be tolerated.
The two Class A peptidomimetics that have R2 amines, 16A and 17A, have similar IC50 values (4.8 ± 0.4 and 5.3 ± 0.6 μM, respectively); however, they have vastly different steric requirements (methyl vs benzyl). As opposed to the R2 amides and aromatics, this may indicate that when an amine substituent is present at the R2 position, it dictates an alternative binding orientation within the R2 binding space that does not lead to such strict steric requirements. Alternatively, the relatively hydrophobic benzyl group of 17A may decrease inhibitor solubility and consequent inhibitor availability, therefore resulting in lower% maximum inhibition and IC50 value than would otherwise be observed. This may also be the case for 21A, where the R2 aromatic ring lacks the nitrogen found in the more active inhibitors 5A and 11A (Table 1a and S1).
A comparison of 17A to the second most active inhibitor in Class A, 2A, which is only different from 17A by the presence of a carbonyl, shows that the carbonyl of 2A is crucial for sub-micromolar inhibitory potency (0.41 ± 0.04 μM for 2A vs. 5.3 ± 0.6 μM for 17A). This observed 10-fold increase in IC50 may be a consequence of the loss of a stabilizing hydrogen-bond interaction between the amide –NH group or carbonyl oxygen of the inhibitor and the HKU4-CoV 3CLpro binding site. The change in hybridization between an amine and an amide also results in a change in the molecular geometry of the R2 substituent, which may alter hydrogen-bonding properties and introduce additional effects that may be important factors in dictating good enzymatic inhibition. The X-ray structure of HKU4-CoV 3CLpro in complex with 2A, discussed below, shows that the amide –NH group of the carboxamide group of 2A forms an important interaction with the backbone carbonyl of His41.
Of the 205 Class B inhibitors that were tested for inhibition of HKU4-CoV 3CLpro, 24 were found to inhibit HKU4-CoV 3CLpro above 50% at a concentration of 100 μM, and subsequently, IC50 values were determined (Table 1b). Interestingly, we found fewer good inhibitors and no sub-micromolar inhibitors of HKU4-CoV 3CLpro among this larger, more substituted dipeptide-like class of compounds (∼76% of compounds in Class A (19 of 25 compounds tested) were good inhibitors of HKU4-CoV 3CLpro while only ∼12% of compounds in Class B (24 of 205 compounds tested) were found to be good inhibitors). This result could be a consequence of the increased steric bulk perturbing the preferred binding orientation of the Class B compounds in the HKU4-CoV 3CLpro binding site. A comparison of similar Class A and Class B inhibitors more clearly elucidates this trend, where the addition of the sterically bulky R4 t-Bu amide in the Class B compounds decreases HKU4-CoV 3CLpro inhibition in each instance (S3) (see Table S3 in the Supporting information). For example, the most active Class A inhibitor, 1A, having and IC50 of 0.33 ± 0.02 μM and 93% inhibition at 100 μM, is almost completely inactivated by the addition of the t-Bu amide as seen in the Class B compound 25B, with only 26% inhibition at 100 μM. Within the Class B inhibitors, there is a preference for the 3-thiophene at the R1 position and a 1-methylbenzotriazole at the R3 position of the peptidomimetic backbone, though it is not as strong as that observed in within Class A (25% of compounds in Class B vs. 95% of compounds in Class A). Among the series where R1 = 3-thiophene and R3 = 1-methylbenzotriazole (compounds 3B, 6B, 9B, 16B, 18B, and 21B) small carboxamide groups at the R2 position are favored as observed by the comparison of 3B to 28B and 6B to 27B (see Table S2 in the Supporting information). Possibly, Class B inhibitors with small R2 amides allow for a shift in the position of the inhibitor in the HKU4-CoV 3CLpro binding site to accommodate the bulky t-Bu amide at the R4 position. Again, an R2 amide is preferred to an amine for good inhibition of HKU4-CoV 3CLpro (IC50 = 1.8 ± 0.5 μM for 3B versus 22.0 ± 7.4 μM for 21B). Replacement of the benzotriazole within this series with a benzimidazole has dissimilar effects; for example, comparing 19B (Table 1b) to 28B (S2 and S3) where the benzimidazole of 19B has been replaced with a benzotriazole, inhibition decreases from 68% to 34% at 100 μM, respectively. In contrast, comparison of 23B to 18B shows that substitution of benzimidazole for benzotriazole increases inhibition from 56% to 76% at 100 μM (Table 1b). Interestingly, only Class B compounds having a para-substituted phenyl ring with an R2 substituent were found to be good inhibitors of HKU4-CoV 3CLpro; compounds with other, non-phenyl rings including cyclopropyl, cyclohexyl, adamantane, and bicycloheptane were not found to be active inhibitors.
2.2. 1A-RFL Sub-structural activity analysis via component synthesis
In an attempt to delineate the structural relationships that contribute to the enhanced activity of our most active inhibitor, 1A, we synthesized the sub-structural components of 1A and determined their individual inhibitory activities against HKU4-CoV 3CLpro (Scheme 1 ). The straightforward synthesis of these sub-structural 1A components, 22A, 23A, and 24A, was accomplished from the commercially available starting materials, benzotriazole and p-phenylenediamine. The synthesis of 22A was achieved in two steps. First, alkylation of benzotriazole by reaction with chloroacetic acid and sodium hydroxide yielded the benzotriazoleacetic acid 22 in 50% yield.25 Subsequent coupling of 22 with the commercially available thiophenylmethanamine using EDC produced the desired 1A component 22A in 25% yield (Scheme 1, top). The synthesis of 23A and 24A was accomplished linearly by first coupling p-phenylenediamine with the commercially available thiophenecarboxylic acid using HATU, which resulted in the desired 1A component 23A in 58% yield. Reductive amination of 23A and commercially available thiophenecarbaldehyde using sodium triacetoxyborohydride produced the desired 1A sub-structural component 24A in 81% yield. Finally, 1A was resynthesized according to the literature procedure by coupling 24A with 22 using EDC as a coupling reagent.24
Scheme 1.
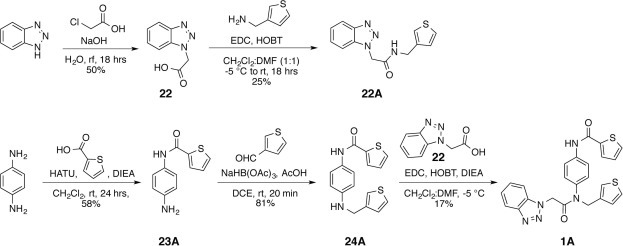
Synthesis of sub-structural components of 1A.
The sub-structural components 22A, 23A, and 24A were then individually tested for inhibition of HKU4-CoV 3CLpro at a concentration of 100 μM. The 22A component, representing the R1 and R3 substituents of the Class A peptidomimetic backbone, showed 28% inhibition of HKU4-CoV 3CLpro at 100 μM and had an IC50 of 68 μM, supporting our hypothesis that this backbone has some inhibitory capacity even in the absence of the R2 component. The 23A sub-structural component, representing the R2 substituent of the Class A peptidomimetic backbone, was found to have no inhibitory activity against HKU4-CoV 3CLpro at 100 μM, supporting R1 and R3 as essential substituents. Additionally, the 24A sub-structural component, representing the R1 and R2 substituents of the Class A peptidomimetic backbone, showed no inhibition of HKU4-CoV 3CLpro at 100 μM, indicating the crucial nature of the 1-methylbenzotriazole at the R3 position of the Class A backbone and supporting our previous observation of the need for a precisely oriented hydrogen bond acceptor at this position within the Class A peptidomimetics (vide supra). This finding establishes 22A as a minimum component necessary for inhibition of HKU4-CoV 3CLpro and suggests it may be used as a scaffold for future fragment based design of HKU4-CoV 3CLpro inhibitors. Interestingly, the work of Wong and coworkers showed that a series of benzotriazole esters, which act as potent, covalent inhibitors of SARS-3CLpro, are also able to inhibit SARS-3CLpro upon replacement of the ester oxygen with carbon, albeit weakly.26 Taken together, these findings may indicate an inhibitory preference for benzotriazoles among coronaviral 3C-like proteases.
2.3. X-ray crystallographic analysis of HKU4-CoV 3CLpro in complex with 1A, 2A, and 3B
In order to more fully elucidate the structural factors underlying the structure–activity relationships (SARs) observed in the kinetic data, we determined the X-ray crystal structures of HKU4-CoV 3CLpro in complex with three of the most potent inhibitors, 1A, 2A, 3B and the favored backbone 22A. These complexes were chosen to determine the potential structural explanations behind our observations that Class A and B inhibitor backbones, containing a 3-thiophene and 1-methylbenzotriazole, are favored for HKU4-CoV 3CLpro inhibition. We also sought to determine the structural reasons behind the preference for inhibition by Class A compounds over Class B compounds.
Co-crystallization of 1A, 2A, 3B, and 22A with HKU4-CoV 3CLpro produced high-quality crystals suitable for X-ray data collection and structure determination. Complete X-ray data sets were collected on these four HKU4-CoV 3CLpro-inhibitor complexes to resolutions between 1.8 Å and 2.3 Å. A summary of the data collection and refinement statistics are provided in S4.
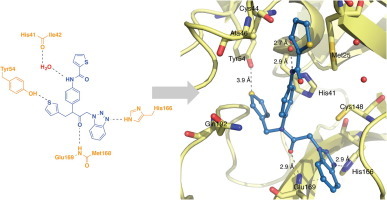
The X-ray crystal structures confirm that both the Class A (1A and 2A) and B (3B) peptidomimetics bind in the active site pocket of HKU4-Co 3CLpro, which is lined primarily with polar residues and is solvent exposed (Fig. 2 ). In each of the HKU4-CoV 3CLpro-inhibitor complexes, the carbonyl carbon of the central inhibitor backbone, that is, preceding the R3 group, is located between 4.5 and 4.8 Å from the sulfur atom of the catalytic cysteine (Cys148, Fig. 2A, C and E). Both 1A and 2A, which have a 3-thiophene at the R1 position and a 1-methylbenzotriazole at the R3 position of the peptidomimetic backbone, position these moieties in identical chemical space (Fig. 2A and C). The Class B inhibitor, 3B, which has a 3-thiophene at the R1 position and a 1-methylbenzotriazole at the R3 position, also positions the 3-thiophene and 1-methylbenzotriazole functionalities in the same chemical space utilized by the Class A inhibitors (Fig. 2E). The 3-thiophene in the R1 position of all three inhibitors occupies the S2–S4 sub-sites of the HKU4-CoV 3CLpro active site, the anilido and R2-functionality reside in the S2–S1′ sub-sites, and the R3 benzotriazole occupies the S1-subsite, acting as the P1 group of the substrate.
Figure 2.
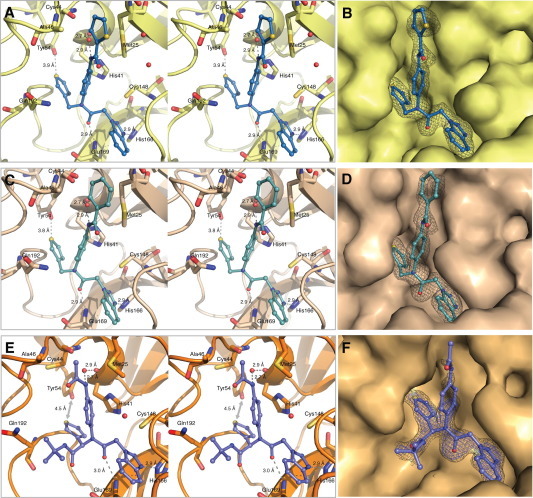
X-ray crystal structures of 1A, 2A and 3B in complex with HKU4-CoV 3CLpro. Inhibitors are colored according to atom type and shown in ball and stick representation. Wall-eye stereo-view is shown in panels A, C, and E. Water molecules are shown as red spheres. Hydrogen bonds are shown as gray dashes with distances between heteroatoms labeled (Å). Interatomic distances (Å) are displayed using a gray arrow. Electron density omit maps (Fo − Fc) are shown in grey mesh and are contoured to +3.0σ around the inhibitor only. The binding orientation of each inhibitor is the same in the active site of each monomer of the dimer in the asymmetric unit of HKU4-CoV 3CLpro, therefore only one active site is shown for clarity. (A) HKU4-CoV 3CLpro (pale yellow) in complex with 1A (sky blue), PDB 4YOI, (B) 1A electron density omit map, (C) HKU4-CoV 3CLpro (wheat) in complex with 2A (pale cyan), PDB 4YOJ, (D) 2A electron density omit map, (E) HKU4-CoV 3CLpro (light orange) in complex with 3B (slate), PDB 4YOG, (F) 3B electron density omit map contoured at +3.0σ (blue) and +2.0σ (grey).
The X-ray structures reveal the structural determinants for the inhibitory preference for Class A and B compounds with backbones containing a 3-thiophene and 1-methylbenzotriazole. This backbone allows the inhibitor to anchor into the HKU4-CoV 3CLpro active site via the formation of three key hydrogen bonds. First, there is a direct hydrogen bond that exists between the 3-nitrogen atom of benzotriazole and the tele-nitrogen of His166 in each enzyme-inhibitor complex, securing the location and position of the benzotriazole in the active site. A second hydrogen bond between the carbonyl oxygen of the inhibitor backbone and the backbone nitrogen of Glu169 in each structure further anchors the inhibitor in the HKU4-CoV 3CLpro active site. Finally, the thiophene ring is positioned within the HKU4-CoV 3CLpro active site allowing the formation of a relatively uncommon hydrogen bond to the hydroxyl hydrogen of Tyr54.27, 28, 29, 30
In each of the HKU4-CoV 3CLpro-inhibitor complexes, the R2-substituted aryl ring of both Class A and B inhibitors is positioned in the HKU4-CoV 3CLpro active site to take advantage of an edge-to-face pi-interaction with the catalytic histidine, His41 (Fig. 2A, C and E). This interaction may be important for good inhibition of HKU4-CoV 3CLpro for two reasons. First, comparing 1A and 2A to 22A, where the R2-substituted aromatic ring of 1A and 2A is absent, a greater than 200-fold increase in IC50 is observed for 22A. Second, Class B compounds having non-aromatic substituents instead of an R2-substituted aryl ring were not found to be good inhibitors of HKU4-CoV 3CLpro (vide supra). These data indicate that for low micromolar to sub-micromolar inhibition of HKU4-CoV 3CLpro, an R2-substituted aryl ring may be required. Furthermore, there is a conserved active site water molecule present in the X-ray crystal structure of each HKU4-CoV 3CLpro-inhibitor complex that mediates a hydrogen-bond between the backbone carbonyl oxygen of His41 and the amide R2 nitrogen of the inhibitor (Fig. 2A, C and E). This finding illuminates the observed preference for an amide substituent at the R2 position in both Class A and B inhibitors.
Our attempt to solve the X-ray crystal structure of 22A in complex with HKU4-CoV 3CLpro did not result in the structure of the HKU4-CoV 3CLpro-inhibitor complex, but instead resulted in the X-ray structure of the unliganded, or free, HKU4-CoV 3CLpro enzyme, which was found to crystallize in a different space group than the HKU4-CoV 3CLpro-inhibitor complexes (Fig. 3 A). The unbound, or free, HKU4-CoV 3CLpro enzyme was found to have noteworthy changes in the active site architecture in comparison to the X-ray crystal structures of the HKU4-CoV 3CLpro-inhibitor complexes. In the absence of an inhibitor, what appears to be a metal ion binds to the HKU4-CoV 3CLpro active site by coordinating to the catalytic cysteine (Cys148), which is rotated 180° from the inhibitor bound structures, resulting in a 3.2 Å positional change in the location of the sulfur atom with a distance of 2.2 Å between the sulfur of Cys148 and the metal atom. The metal is presumed to be a zinc cation, as it is present in water and likely to bind to a sulfur atom (also seen in PDB ID 2YNA), though it could also be iron. The tele-nitrogen of His41 is shifted 2.6 Å from its position in the HKU4-CoV 3CLpro-1A-RFL complex X-ray crystal structure, allowing for the coordination of the Zn2+ atom, which is located 2.4 Å away (Fig. 3A). Additionally, the positions of the Met25, Cys44, Ala46, Tyr54, and Gln192 residues all noticeably shift upon superposition of the free and inhibitor-bound forms of HKU4-CoV 3CLpro.
Figure 3.
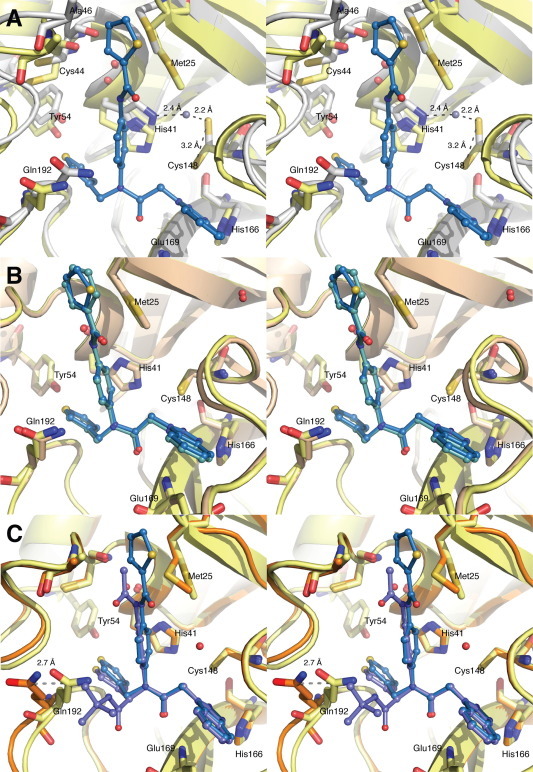
(A) Superposition of unbound HKU4-CoV 3CLpro (gray, PDB 4YO9) and HKU4-CoV 3CLpro co-crystallized with inhibitor 1A (where HKU4-CoV 3CLpro is shown in ribbon representation in light yellow and 1A is shown in sky blue and colored by element, PDB 4YOI). The Zn atom in the unbound HKU4-CoV 3CLpro is represented as a gray sphere. (B) Superposition of HKU4-CoV 3CLpro co-crystallized with 1A (where HKU4-CoV 3CLpro is shown in ribbon representation in light yellow and 1A is shown in sky blue and colored by element, PDB 4YOI) and HKU4-CoV 3CLpro co-crystallized with 2A (where HKU4-CoV 3CLpro is shown as cartoon in wheat and 2A is shown in pale cyan and colored by element, PDB 4YOJ). (C) Superposition of HKU4-CoV 3CLpro co-crystallized with 1A (where HKU4-CoV 3CLpro is shown in ribbon representation in light yellow and 1A is shown in sky blue and colored by element, PDB 4YOI) and HKU4-CoV 3CLpro co-crystallized with 3B (where HKU4-CoV 3CLpro is shown as cartoon in light orange and 3B is shown in slate and colored by element, PDB 4YOG). Wall-eye stereo-view is shown in each panel.
Comparing the most potent Class B inhibitor having the 1-methylbenzotriazole and 3-thiophene backbone, 3B, to the top HKU4-CoV 3CLpro inhibitor identified, 1A, elucidates the observed preference for Class A inhibitors over Class B inhibitors (Fig. 3C). The (R)-enantiomer of the Class B compound, 3B, was found in the HKU4-CoV 3CLpro active site, with the backbone of the inhibitor oriented similarly to the backbones of the Class A inhibitors 1A and 2A, utilizing the same hydrogen bonds; however, the (R)-R4 t-Bu amide substituent of 3B, which is absent in the Class A inhibitors, marginally rotates the thiophene away from Tyr54, increasing the distance to the hydrogen-bond donor Tyr54 from 3.9 to 4.5 Å compared to 1A, and moving the thiophene out of hydrogen-bonding distance to Tyr54 (distance measured to heteroatoms). The R4 t-Bu amide of 3B extends out toward the solvent exposed side of the active site, inducing a structural rearrangement of HKU4-CoV 3CLpro where Gln192, which encloses 1A and 2A in the active site, is moved 2.7 Å to accommodate the steric bulk of the R4 substituent of 3B. The combination of these factors may be attributed for the observed 5-fold increase in IC50 when comparing 1A and 2A to 3B and likely account for the reduced inhibitory activity of Class B compounds relative to Class A compounds.
The Class B peptidomimetic compounds were synthesized and tested for HKU4-CoV 3CLpro inhibition as racemates, which may lead to significant differences in IC50 values. An example of this can be seen by the comparison of (R)-13B to racemic- and (S)-13B (Fig. 4 ). The enantiopure (R)-13B has an IC50 of 9.3 ± 0.5 μM while the 13B racemate has an IC50 of 11.1 ± 0.6 μM and (S)-13B has a 50% reduction in inhibition relative to the racemate, with only 44% inhibition of HKU4-CLpro at 100 μM. Interestingly, (R)-3B was found in the active site of HKU4-CoV 3CLpro in the X-ray crystal structure (Fig. 2E and F), suggesting a preference for (R)-stereochemistry in the inhibitor for good inhibition of HKU4-CoV 3CLpro, despite the nature of the R1 and R3 heterocyclic groups.
Figure 4.
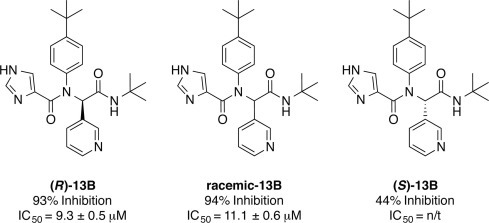
Effect of 13B stereocenter configuration on HKU4-CoV 3CLpro inhibition. Where% inhibition is determined at 100 μM inhibition concentration, n/t = not tested.
The X-ray structures of HKU4-CoV 3CLpro in complex with inhibitors provide vital insights into the types of compounds that can be developed as therapeutics against lineage C Betacoronaviruses. A sequence alignment of HKU4-CoV, HKU5-CoV, and MERS-CoV 3CLpro, all of which are lineage C Betacoronaviruses, show that HKU4-CoV and HKU5-CoV are 81.0% and 82.7% identical to MERS-CoV 3CLpro, and HKU4-CoV is 83.7% identical to HKU5-CoV 3CLpro (Fig. 5 ).31, 32, 33 Moreover, the pertinent HKU4-CoV 3CLpro active site residues (Met25, His41, Cys44, Ala46, Tyr54, Cys148, His166, Glu169, and Gln192, Figure 3, Figure 4) identified in the X-ray crystal structures that line the active site and interact with the peptidomimetic inhibitors are all conserved in HKU5-CoV and MERS-CoV 3CLpro. These data indicate that the active site architectures among HKU4-CoV, HKU5-CoV, and MERS-CoV 3CLpro may be similar enough to design broad-spectrum antiviral therapeutics against lineage C Betacoronaviruses and have implications for the structure and inhibition of lineage C Betacoronaviruses yet to emerge. To test this hypothesis, we tested whether our lead HKU4-CoV 3CLpro inhibitor, compound 1A, would also have inhibition of HKU5-CoV 3CLpro. We found that compound 1A is a good inhibitor of HKU5-CoV 3CLpro, having 91% inhibition at 100 μM and a sub-micromolar IC50 value of 0.54 ± 0.03 μM which is comparable to the IC50 value of 0.33 ± 0.02 μM against HKU4-3CLpro. Based on these data, we predict compound 1A will show inhibition of MERS-CoV 3CLpro as well.
Figure 5.
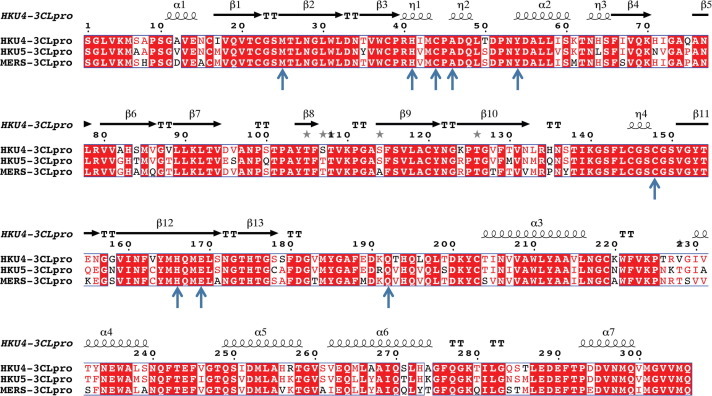
Sequence alignment of HKU4-CoV, HKU5-CoV, and MERS-CoV 3CLpro where the top row indicates the secondary structure of HKU4-3CLpro at a particular residue position, conserved residues among all three 3CLpro’s are shaded in red, and residues that have similar steric or electronic properties are shown in red text with no shading. Non-conserved residues are displayed in black text. The pertinent active site residues that border and interact with the peptidomimetic inhibitors (Met25, His41, Cys44, Ala46, Tyr54, Cys148, His166, Glu169, and Gln192) are indicated by blue arrows.
3. Conclusions
In conclusion, 43 of the 230 peptidomimetics were found to inhibit HKU4-CoV 3CLpro, 19 of which belonged to the Class A peptidomimetic series and 24 of which belonged to the Class B peptidomimetic series. Overall, the Class A peptidomimetics were found to be better inhibitors relative to the Class B compounds, and two of the Class A compounds were determined to have sub-micromolar inhibitory potency against HKU4-CoV 3CLpro, 1A and 2A. Analysis of the inhibition data from the 230 peptidomimetics resulted in the identification of a favored inhibitor backbone consisting of 3-thiophene and 1-methylbenzotriazole functionalities. To evaluate the relative contribution of the favored inhibitor backbone to the total HKU4-CoV 3CLpro inhibition observed for the molecule, we synthesized the sub-structural components of the lead inhibitor, 1A, and tested them for inhibition of HKU4-CoV 3CLpro. The simple compound, 22A, which represents the R1 and R3 inhibitor functionalities and consists of the 3-thiophene and 1-methylbenzotriazole of the favored backbone, was found to inhibit HKU4-CoV 3CLpro even in the absence of the anilido and R2 components of the inhibitor. We then determined the X-ray crystal structures of the three most potent inhibitors containing this favored backbone, 1A, 2A, 3B, in complex with HKU4-CoV 3CLpro. These X-ray crystal structures prove that the three Class A and B inhibitors bind in the HKU4-CoV 3CLpro active site, utilizing the same set of three hydrogen-bonding interactions between the favored backbone and the HKU4-CoV 3CLpro active site residues, serving to anchor compounds with this backbone into the active site of the enzyme. Our attempt to solve the X-ray structure of the privileged backbone compound, 22A, in complex with HKU4-CoV 3CLpro resulted in the structure of unbound HKU4-CoV 3CLpro, which is presumably as a consequence of weak inhibition by 22A (28% at 100 μM). Superposition of the unbound and inhibitor-bound forms of HKU4-CoV 3CLpro revealed several subtle, but important changes in the active site architecture upon inhibitor binding. Furthermore, sequence alignment of HKU4-CoV, HKU5-CoV, and MERS-CoV 3CLpro show that the residues involved in inhibitor binding are conserved, indicating that identification of broad spectrum 3CLpro inhibitors of lineage C Betacoronaviruses is possible. In support of this, we found our most active HKU4-CoV 3CLpro inhibitor to have inhibition of HKU5-CoV 3CLpro in the sub-micromolar range (IC50 = 0.54 ± 0.03 μM). These findings, and the structural details underlying the potent inhibition of HKU4-CoV 3CLpro by compounds containing 3-thiophene and 1-methylbenzotriazole backbones, can be used in the future to design compounds with the ability to more fully exploit the HKU4-CoV 3CLpro active site, leading ultimately to the development of more highly potent and selective inhibitors against human pathogens such as SARS and MERS.
4. Experimental
4.1. Expression and purification of HKU4-CoV 3CLpro
The gene encoding the 3CLpro of HKU4-CoV (residues 3292-3597 in the Tylonycteris bat-CoV polyprotein)9 was codon optimized for expression in Escherichia coli and cloned into pET-11a expression vector with an N-terminal (His)6-tag followed by nsp4-/5 auto-cleavage site by BioBasic Inc. This construct results in the expression of HKU4-CoV 3CLpro without an N-terminal or C-terminal extension. E. coli BL21(DE3) cells, transformed with pET11a-HKU4 3CLpro plasmid were grown in Super LB media (3 g potassium phosphate monobasic, 6 g sodium phosphate dibasic, 20 g tryptone, 5 g yeast extracts, 5 g sodium in 1 L water, pH 7.2 adjusted with 1 M NaOH) in the presence of 1 mL 100 mg/mL carbenicillin, 25 mL 8% lactose, 10 mL 60% glycerol, and 5 mL of 10% glucose per 1 L of expression culture for 24 h at 25 °C. The cells were harvested by centrifugation (8400g for 20 min) to yield 15.5 g/L of cells. The cell pellet was then re-suspended in 5 mL of Buffer A/1 g cell pellet (50 mM Tris pH 7.5, 0.2 M ammonium sulfate, 0.05 mM EDTA, 5 mM BME) containing 1 mg/mL lysozyme. After the cells were homogenized, they were lysed via sonication for 10 min with 10 s pulses at 50% amplitude using a Branson Digital Sonifier. The cell lysate was clarified by pelleting the cell debris via centrifugation (28,960g, 4 °C, 20 min) and loaded onto a 30 mL Phenyl Sepharose 6 Fast Flow HiSub column equilibrated with Buffer A. Protein was eluted with a gradient to 100% Buffer B (20 mM Tris pH 7.5, 0.05 mM EDTA, 5 mM BME) over five column volumes (150 mL) collecting 5 mL fractions. Fractions containing HKU4 3CLpro were pooled and loaded onto a 60 mL DEAE Sepharose Fast Flow column equilibrated with Buffer B. Protein was eluted with a gradient to 50% Buffer C (50 mM Tris pH 7.5, 1 M sodium chloride, 0.05 mM EDTA, 5 mM BME, 10% glycerol) over five column volumes (300 mL) collecting 5 mL fractions. Fractions containing pure HKU4-CoV 3CLpro were pooled, dialyzed into storage buffer (25 mM HEPES, pH 7.5, 2.5 mM DTT, 10% glycerol), and concentrated. The protein was then aliquoted into 1.5 mL push-cap Eppendorf tubes, flash frozen in liquid nitrogen, and stored at -80 °C in a freezer until further use.
4.2. IC50 Determination of inhibitors against HKU4-CoV 3CLpro at 25 °C and 37 °C
Each of the 230 dipeptide-like inhibitors were first screened for inhibition of HKU4 3CLpro at a concentration of 100 μM in duplicate assays containing the following assay buffer (50 mM HEPES, 0.1 mg/mL BSA, 0.01% TritonX-100, 1 mM DTT). The assays were carried out in Costar 3694 EIA/RIA 96-Well Half Area, Flat Bottom, Black Polystyrene plates from Corning Incorporated. 1 μL of 100× inhibitor stock in DMSO was added to 79 μL of enzyme in assay buffer and the enzyme-inhibitor mixture was incubated for 10 min. The reaction was initiated by the addition of 20 μL of 10 μM UIVT3 substrate, a custom synthesized Förster resonance energy transfer substrate peptide with the following sequence: HilyteFluorTM488-ESARLQSGLRKAK-QXL520TM-NH2, producing final concentrations of 100 nM and 100 μM for the 3CLpro enzyme and UIVT3 substrate, respectively. The fluorescence intensity of the reaction was then measured over time as relative fluorescence units (RFUt) for a period of 10 min, using an excitation wavelength of 485 and bandwidth of 20 nM and monitoring emission at 528 and bandwidth of 20 nM using a BioTek Synergy H1 multimode microplate reader.
The inhibition of HKU4-CoV 3CLpro by inhibitor compounds was monitored by following the change in RFUs over time, using the initial slope of the progress curve to determine the initial rate (Vi). The percent inhibition of the 3CLpro enzymes was determined using the following equation:
Full IC50 data were acquired for the compounds that showed greater than 50% inhibition of HKU4-CoV 3CLpro at 100 μM of inhibitor compound. The IC50 values were determined at ambient temperature from 100 μL assays performed in triplicate in the following buffer: 50 mM HEPES, 0.1 mg/mL BSA, 0.01% TritonX-100, 1 mM DTT. Kinetic assays were carried out in Costar 3694 EIA/RIA 96-Well Half Area, Flat Bottom, Black Polystyrene plates from Corning Incorporated. Each inhibitor was tested at concentrations of 0.313, 0.652, 1.25, 2.5, 5.0, 10.0, 20.0, 40.0, 60.0, 80.0, 100.0, and 120.0 μM; 1 μL of 100× inhibitor stock in DMSO was added to 79 μL of enzyme in assay buffer and the enzyme-inhibitor mixture was incubated for 10 min. The reaction was initiated by the addition of 20 μL of 10 μM UIVT3 substrate, producing final concentrations of 100 nM and 2 μM for the 3CLpro enzyme and UIVT3 substrate, respectively. The fluorescence intensity of the reaction was then measured over time as RFUt for a period of 20 min, using an excitation wavelength of 485 and bandwidth of 20 nM and monitoring emission at 528 and bandwidth of 20 nM using a BioTek Synergy H1 multimode microplate reader.
The percent inhibition of the 3CLpro enzymes was then plotted as a function of inhibitor concentration. The SigmaPlot Enzyme Kinetics Wizard was used to fit the triplicate percent inhibition data and associated standard error to a non-linear Michaelis-Menten type regression model and determine the IC50 for each enzyme using the following equation:
where %I max is the percent maximum inhibition of 3CLpro and the error in IC50 values was determined as the error in the fitted parameter.
4.3. Crystallization and X-ray structure determination of HKU4 3C-like protease in complex with inhibitors
The HKU4 3CLpro-inhibitor complexes were co-crystallized from four different crystallization solutions. Briefly, the hanging-drop, vapor-diffusion method was used for crystallization by setting up drops and adding 1 μL of purified HKU4-CoV 3CLpro (2.5 mg/mL, 75 μM), that had been incubated for three hours with a 3 molar excess of the appropriate inhibitor, and 1 μL of reservoir solution. For 1A: 15% PEG-3350, 2% tacsimate, 5% isopropanol, 0.1 M imidazole pH 6; for 3B: 20% PEG-3350, 0.2 M ammonium acetate, 0.1 M Bis-Tris pH 5.5; for 2A: 18% PEG-3350, 5% tacsimate, 0.2 M ammonium sulfate, 0.1 M Bis-Tris pH5.5; for 22A: 10% PEG-3350, 2% tacsimate, 5% isopropanol, 0.1 M imidazole pH 6.5. Protein crystals appeared between 14 h and 7 days after setting up crystallization plates. Crystals were harvested with a nylon loop, which was then swiped through the same mother-liquor solution supplemented with 15% MPD. The crystals were cooled by plunging into liquid nitrogen and stored in shipping dewars containing liquid nitrogen until X-ray data collection at an available synchrotron could be performed.
All diffraction data were collected at 100 K at the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source (APS) at Argonne National Laboratories. Crystals were transferred from shipping dewars into automated dewars and then and mounted robotically on a goniostat while under a stream of N2. X-ray data sets of HKU4 3CLpro-inhibitor complexes were collected on a Rayonix 225 HE detector at a wavelength of 0.98 Å. X-ray data were processed and scaled using the program HLK2000, crystals of HKU4-CoV 3CLpro complexes with the inhibitors 1A, 2A, and 3B belonged to the space group P212121, and HKU4-CoV 3CLpro crystallized as a dimer in the asymmetric unit. Co-crystallization of HKU4-CoV 3CLpro with 22A did not result in an HKU4-CoV 3CLpro:inhibitor co-complex, but instead yielded the apo form of HKU4-CoV 3CLpro, which crystallized in a different space group, P3121 with one dimer per asymmetric unit.
The initial phases for HKU4-CoV 3CLpro in complex with 1A, 2A, and 3B were determined by molecular replacement using Phaser-MR (simple interface) in the Phenix suite and the protein database PDB file 2YNA.34 The inhibitors were built and optimized from their SMILES strings using eLBOW and fit into the density using LigandFit in the Phenix suite and the program COOT. Structures were refined using Phenix. Water molecules were added manually to the 2Fo−Fc density peaks that were greater than 3.0σ and iterative rounds of refinement were carried out until Rwork and Rfree reached their lowest values. The electron density maps presented in the figures were calculated using Phenix and COOT and the figures generated using the program PyMol.
4.4. General chemistry methods
All reagents and chemicals were purchased from SigmaAldrich or Acros Organics and used without further purification. Anhydrous solvents were purchased or purified by passage through a solvent column composed of activated alumina and a supported copper redox catalyst. Analytical thin-layer chromatography was performed using Sorbent Technologies Glass-Backed Silica Gel HL TLC plates w/UV254. Flash chromatography was performed with Sorbent Technologies 200–400 mesh silica gel. Nuclear magnetic resonance (NMR) data was acquired using either a Varian Inova300, Bruker ARX400, or Bruker AV-III-800 spectrometers at 300, 400, or 800 MHz for 1H or 75, 100, or 200 MHz for 13C. All compounds were purified by flash chromatography to >90% purity by 1H NMR.
4.4.1. 2-(1H-Benzo[d][1,2,3]triazol-1-yl)-N-(thiophen-3-ylmethyl)acetamide (22A)
To 2-(1H-benzo[d][1,2,3]triazol-1-yl)acetic acid (1.5 equiv) in anhydrous dichloromethane (0.2 M) at 0 °C under nitrogen atmosphere was added EDC (3.0 equiv) and HOBT (3.0 equiv). The mixture was allowed to stir for five minutes before the addition of DIEA (13.0 equiv). The mixture was allowed to stir for an additional 20 min before the addition of thiophen-3-ylmethanamine (1.0 equiv). The reaction mixture was allowed to warm to ambient temperature and stirred under nitrogen atmosphere for 16 h. The reaction was quenched by addition of saturated potassium bicarbonate and the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were washed with water (1×) and brine (1×), dried over sodium sulfate, and concentrated. The crude residue was recrystallized from hot ethanol to provide the product as a beautiful white, crystalline solid in 30% yield. 1H NMR (800 MHz, CD3OD) δ (ppm): 8.03 (1H, d, J = 8.37), 7.72 (1H, d, J = 8.37), 7.59 (1H, dd, J = 8.37, 0.96), 7.48 (1H, dd, J = 8.37, 0.96), 7.40 (1H, dd, J = 4.94, 2.97), 7.29 (1H, m), 7.08 (1H, dd, J = 4.93, 1.30), 5.51 (2H, s), 4.46 (2H, s). 13C NMR (200 MHz, CD3OD) δ (ppm): 166.47, 145.30, 138.67, 133.83, 127.63, 126.92, 125.81, 124.28, 121.90, 118.60, 110.11, 49.87, 38.19. ESI-MS(+): 273 [M+1], 295 [M+23].
4.4.2. N-(4-Aminophenyl)thiophene-2-carboxamide (23A)
Thiophene-2-carboxylic acid (1.0 equiv) and HATU (1.4 equiv) were added to anhydrous dichloromethane (0.2 M) at room temperature followed by drop-wise addition of DIEA (3.0 equiv) under nitrogen atmosphere. The mixture was allowed to stir at ambient temperature for 5 min before benzene-1,4-diamine was added in one portion. The reaction mixture was allowed to stir for 14 h before it was quenched with saturated potassium bicarbonate. The aqueous layer was extracted with ethyl acetate (3×) and the combined organic extracts were washed with water (1×) and brine (1×), dried over sodium sulfate, and concentrated. The crude yellow residue was purified by flash chromatography using a gradient of 20% to 70% ethyl acetate in hexanes to give the final product in 50% yield. This reaction was performed from mg to g scale with no detrimental effect on yield. 1H NMR (800 MHz, CD3OD) δ (ppm): 7.84 (1H, d, J = 3.74), 7.67 (1H, dd, J = 4.97, 1.08), 7.37 (2H, d, J = 8.56), 7.15 (1H, dd, J = 4.97, 3.74). 13C NMR (200 MHz, CD3OD) δ (ppm): 161.26, 144.66, 139.63, 130.56, 128.71, 128.31, 127.49, 122.85. ESI-MS(+): 219 [M+1], 241 [M+23].
4.4.3. N-(4-((Thiophen-3-ylmethyl)amino)phenyl)thiophene-2-carboxamide (24A)
23A (1.0 equiv) and thiophene-3-carbaldehyde (1.0 equiv) were dissolved in anhydrous 1,2-DCE (0.2 M) at ambient temperature under nitrogen atmosphere. Sodium triacetoxyborohydride (1.6 equiv) and acetic acid (1.0 equiv) were then added in one portion. The reaction mixture was allowed to stir for 30 min before the reaction was quenched with 1 N sodium hydroxide. The aqueous layer was extracted with ethyl acetate (3×) and the combined organic extracts were washed with water (1×) and brine (1×), dried over sodium sulfate, and concentrated. The crude yellow residue was purified by flash chromatography using a gradient of 20–70% ethyl acetate in hexanes to give the final product in 50–80% yield. 1H NMR (800 MHz, CD3OD) δ (ppm): 7.85 (1H, m), 7.69 (1H, m), 7.36 (3H, m), 7.26 (1H, m), 7.17 (1H, d, J = 5.15), 7.12 (1H, d, J = 5.15), 6.70 (2H, m), 4.34–4.35 (2H, rotomeric peaks). 13C NMR (200 MHz, CD3OD) δ (ppm): 172.76, 146.24, 141.08, 130.47, 128.20, 127.65, 127.43, 126.82, 125.27, 122.87, 120.85, 112.81, 43.08). ESI-MS(+): 315 [M+1], 337 [M+23].
Author contributions
Sarah E. St. John (SSJ) expressed, purified, and characterized HKU4-3CLpro. SSJ designed and performed all of the experiments and synthesis detailed herein and was responsible for crystallizing unbound and bound HKU4-3CLpro:inhibitor complexes and solving their X-ray structures. SSJ wrote the research article and made all figures/schemes/tables. Sakshi Tomar (ST) designed the HKU4- and HKU5-3CLpro expression plasmids, and over expressed and purified the HKU5-3CLpro enzyme and tested for inhibition by 1A. The original compound library (S1 and S2) was synthesized in the laboratory of Shaun R. Stauffer (SRS), who also edited this article. Andrew D. Mesecar (ADM) supervised this research study.
Acknowledgements
This work was supported in part by Grants (AI085089 and AI26603) to A.D.M. from the National Institutes of Health via the National Institute of Allergy and Infectious Diseases. A.D.M. also wishes to also acknowledge partial support from the Walther Cancer Foundation. Sakshi Tomar is supported by a Grant from the Purdue University Research Foundation. Crystallization and DNA sequencing were partially supported by the Purdue Center for Cancer Research Macromolecular Crystallography and DNA Sequencing Shared Resources which are partially supported by an NIH Grant (P30 CA023168). The authors would also like to acknowledge the scientists at the Vanderbilt Specialized Chemistry Center for synthesis of the initial compound library used in this work, which was supported in part by the MLPCN (1U54 MH084659 and MH084512). Finally, the authors would like to acknowledge the LS-CAT beamline staff for their help in acquiring X-ray data. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.06.039.
Supplementary data
Supporting information.
References and notes
- 1.McIntosh K., Dees J.H., Becker W.B., Kapikian A.Z., Chanock R.M. Proc. Natl. Acad. Sci. U.S.A. 1967;57:933. doi: 10.1073/pnas.57.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenblatt M.B., Vrbanac V., Tivey T., Tsang K., Tager A.M., Aliprantis A.O. PLoS ONE. 2012;7:e44664. doi: 10.1371/journal.pone.0044664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perlman S., Netland J. Nat. Rev. Microbiol. 2009;7:439. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 5.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 6.Chan J.F., Lau S.K., Woo P.C. J. Formos. Med. Assoc. 2013;112:372. doi: 10.1016/j.jfma.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. N. Engl. J. Med. 2012;367:1814. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 8.WHO In MERS-CoV Update 16; World Health Organization: 2014; Vol. 2014.
- 9.Woo P.C., Wang M., Lau S.K., Xu H., Poon R.W., Guo R., Wong B.H., Gao K., Tsoi H.W., Huang Y., Li K.S., Lam C.S., Chan K.H., Zheng B.J., Yuen K.Y. J. Virol. 2007;81:1574. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A., Galiano M., Gorbalenya A.E., Memish Z.A., Perlman S., Poon L.L., Snijder E.J., Stephens G.M., Woo P.C., Zaki A.M., Zambon M., Ziebuhr J. J. Virol. 2013;87:7790. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Boheemen S., de Graaf M., Lauber C., Bestebroer T.M., Raj V.S., Zaki A.M., Osterhaus A.D., Haagmans B.L., Gorbalenya A.E., Snijder E.J., Fouchier R.A. MBio. 2012:3. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haagmans B.L., Al Dhahiry S.H.S., Reusken C.B.E.M., Raj V.S., Galiano M.G., Myers R., Godeke G.J., Jonges M., Farag E., Diab A., Ghobashy H., Alhajri F., Al-Thani M., Al-Marri S.A., Al Romaihi H.E., Al Khal A., Bermingham A., Osterhaus A.D.M.E., Al Hajri M.M., Koopmans M.P.G. Lancet. Infect. Dis. 2014;14:140. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Memish Z.A., Mishra N., Olival K.J., Fagbo S.F., Kapoor V., Epstein J.H., Alhakeem R., Durosinloun A., Al Asmari M., Islam A., Kapoor A., Briese T., Daszak P., Al Rabeeah A.A., Lipkin W.I. Emerg. Infect. Dis. 2013;19:1819. doi: 10.3201/eid1911.131172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niemeyer D., Zillinger T., Muth D., Zielecki F., Horvath G., Suliman T., Barchet W., Weber F., Drosten C., Muller M.A. J. Virol. 2013;87:12489. doi: 10.1128/JVI.01845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews K.L., Coleman C.M., van der Meer Y., Snijder E.J., Frieman M.B. J. Gen. Virol. 2014;95:874. doi: 10.1099/vir.0.062059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q.H., Qi J.X., Yuan Y., Xuan Y.F., Han P.C., Wan Y.H., Ji W., Li Y., Wu Y., Wang J.W., Iwamoto A., Woo P.C.Y., Yuen K.Y., Yan J.H., Lu G.W., Gao G.F. Cell Host Microbe. 2014;16:328. doi: 10.1016/j.chom.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Virus Res. 2006;117:17. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziebuhr J., Snijder E.J., Gorbalenya A.E. J. Gen. Virol. 2000;81:853. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 20.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 21.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 22.Thiel V., Herold J., Schelle B., Siddell S.G. J. Virol. 2001;75:6676. doi: 10.1128/JVI.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., Mielech A.M., Baker S.C., Lindsley C.W., Hodder P., Mesecar A., Stauffer S.R. J. Med. Chem. 2013;56:534. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turlington M., Chun A., Tomar S., Eggler A., Grum-Tokars V., Jacobs J., Daniels J.S., Dawson E., Saldanha A., Chase P., Baez-Santos Y.M., Lindsley C.W., Hodder P., Mesecar A.D., Stauffer S.R. Bioorg. Med. Chem. Lett. 2013;23:6172. doi: 10.1016/j.bmcl.2013.08.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajput A.P.G., Gore R.P. J. Chem. Pharma. Res. 2010;2:52. [Google Scholar]
- 26.Wu C.Y., King K.Y., Kuo C.J., Fang J.M., Wu Y.T., Ho M.Y., Liao C.L., Shie J.J., Liang P.H., Wong C.H. Chem. Biol. 2006;13:261. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooke S.A., Corlett G.K., Legon A.C. Chem. Phys. Lett. 1998;291:269. [Google Scholar]
- 28.Cooke S.A., Corlett G.K., Legon A.C. J. Chem. Soc. Faraday Trans. 1998;94:1565. [Google Scholar]
- 29.Laurence C., Brameld K.A., Graton J., Le Questel J.Y., Renault E. J. Med. Chem. 2009;52:4073. doi: 10.1021/jm801331y. [DOI] [PubMed] [Google Scholar]
- 30.Legon A.C., Ottaviani P. Phys. Chem. Chem. Phys. 2004;6:488. [Google Scholar]
- 31.Gouet P., Robert X., Courcelle E. Nucleic Acids Res. 2003;31:3320. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gouet P., Courcelle E., Stuart D.I., Metoz F. Bioinformatics. 1999;15:305. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 33.Cockerill M. Trends Biochem. Sci. 1993;18:106. [Google Scholar]
- 34.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. Acta Crystallogr., Sect. D. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.