

Abstract
Background
Approximately 20% of multiple sclerosis patients have a family history of multiple sclerosis. Studies of multiple sclerosis aggregation in families are inconclusive.
Objective
To investigate the genetic burden based on currently discovered genetic variants for multiple sclerosis risk in patients from Dutch multiple sclerosis multiplex families versus sporadic multiple sclerosis cases, and to study its influence on clinical phenotype and disease prediction.
Methods
Our study population consisted of 283 sporadic multiple sclerosis cases, 169 probands from multiplex families and 2028 controls. A weighted genetic risk score based on 102 non-human leukocyte antigen loci and HLA-DRB1*1501 was calculated.
Results
The weighted genetic risk score based on all loci was significantly higher in familial than in sporadic cases. The HLA-DRB1*1501 contributed significantly to the difference in genetic burden between the groups. A high weighted genetic risk score was significantly associated with a low age of disease onset in all multiple sclerosis patients, but not in the familial cases separately. The genetic risk score was significantly but modestly better in discriminating familial versus sporadic multiple sclerosis from controls.
Conclusion
Familial multiple sclerosis patients are more loaded with the common genetic variants than sporadic cases. The difference is mainly driven by HLA-DRB1*1501. The predictive capacity of genetic loci is poor and unlikely to be useful in clinical settings.
Keywords: Multiple sclerosis, genetic burden, genetic risk score, multiplex families, familial aggregation
Introduction
Multiple sclerosis (MS) is an inflammatory and neurodegenerative disease of the central nervous system. The aetiology of MS is complex with both genetic and environmental factors contributing to its aetiology.1 Evidence of genetic contribution is found in family studies in which approximately 20% of MS patients have an affected family member.2 In addition, the recurrence risk in siblings of MS patients is higher than in the general population.3 The main genetic locus of MS risk is the human leukocyte antigen (HLA) class II region (the classic HLA-DRB1*1501 allele). This major genetic locus has been studied and replicated extensively.4,5 In the most recent collaborative genome-wide association study (GWAS) 110 non-HLA MS risk loci were established6 and the list is still expanding.
Despite the success of the GWAS, a large part of MS heritability remains unexplained. Understanding the familial aggregation of MS will help to unravel the mechanisms of MS development. Because environmental factors probably exert their effect at the population level, although this has not been well determined for MS families, it seems likely that these families are more loaded with the genetic variants. This could result in a higher disease susceptibility in families. Several studies have investigated the genetic burden in families, but the results are inconclusive.7–12
A robust way to measure genetic loading is by a combination of the genetic variants into a cumulative genetic risk score.13,14 In this study we investigated the accumulated genetic risk in MS multiplex families and compared it to the sporadic cases in the Netherlands. We also explored the influence of genetic burden on clinical characteristics and assessed the prediction of the disease status in MS.
Materials and methods
Subjects
All patients suspected of MS who visited the neurological outpatient clinic of the Erasmus Medical Centre (EMC) between 2004 and 2009 were asked to participate in the study. During the visit to the hospital every patient was interviewed about their family history of MS. MS patients with a positive family history of MS were defined as multiplex MS cases, whereas patients with no family history of MS were defined as sporadic MS cases. A large proportion of multiplex cases are derived from the still ongoing study on gene–environment interaction in MS (GEMS) in the Netherlands. In this study multiplex MS families are included in which at least one first-degree or second-degree relative of an affected proband also has clinically definite MS. The participants of the GEMS study have filled in the questionnaire about their family structure and their family history of MS.
The diagnosis of MS in all patients was evaluated according to the standard diagnostic criteria.15,16 The following clinical information was collected: gender, age at disease onset, disease duration, clinical course, Expanded Disability Status Scale score (EDSS) and MS severity score (MSSS). A control group consisted of unrelated healthy adults from the general population enrolled in the longitudinal Rotterdam Study III (RSIII).17 Written informed consent was obtained from all participants with approval from the medical ethics committee of the EMC.
Quality control and genetic risk score
Whole blood samples were collected from all participants and DNA was isolated by a standardised method.18 Samples were genotyped on the Illumina 610-Quad Bead array (n = 2466) and the Immuno-Chip (n = 302). Both arrays were subjected to the standard quality control (QC).19 Only subjects of European descent were included in the analysis. Study participants with unexpected relatedness (PI_HAT > 0.35) were excluded. When an individual’s genotype for a single nucleotide polymorphism (SNP) used for the calculation of the genetic risk score (see below) was missing, the sample was excluded from further analysis (see online Supplementary Appendix 1 for QC steps). After QC, 569 MS patients and 2028 controls were eligible for the analysis. Of all cases, 286 patients reported a positive family history of MS and 283 MS patients were identified as sporadic cases. For all comparative analyses we used 169 probands from the multiplex families.
After QC, 102 out of 110 MS risk SNPs6 were available for extraction from the arrays. When a SNP was not present on the Illumina 610-Quad Bead array, we used tagging SNPs with r2 > 0.6 (see online Supplementary Appendix 2). The tagging SNP rs9271366 was used for the HLA-DRB1*1501 locus (r2 = 0.957).20,21 All tested SNPs were in Hardy–Weinberg equilibrium.
The weighted genetic risk score (wGRS) was calculated as previously described13 implementing 102 available SNPs. The analyses were also conducted using only the original SNPs. Because the results (data not shown) were in the same direction, we used all available SNPs in the calculation of the wGRS. To assess the additional effect of the HLA-DRB1*1501 risk allele, we optionally included it into the model. To determine how well the wGRSHLA, wGRS102 or wGRS102+HLA models discriminate between the studied groups receiver operating characteristic (ROC) curves were constructed and the area under the curve (AUC) was calculated.
Statistical analysis
QC was completed using PLINK. SPSS statistical software (IBM Company, version 21) and GraphPad software v5 were used to analyse clinical variables (gender, age of onset, MS course, EDSS and MSSS) using the chi-square test, non-parametric Mann–Whitney test and Spearman correlation. The wGRSs and ROC curves were computed in SPSS and in R by usage of the PredictABEL package.22 Differences in predictive performance of ROC curves were tested with the De Long test using the R package ‘pROC’.23 We applied correction for multiple testing using the Benjamini–Hochberg procedure for controlling the false-positive rate.
Results
The case and control characteristics are presented in Table 1. There were no significant differences between probands from MS multiplex families (fMS) and sporadic MS cases (sMS) in terms of age of disease onset, gender ratio, disease course, EDSS or MSSS (all P > 0.05).
Table 1.
Demographic and clinical characteristics of patients with sporadic MS, multiplex MS and healthy controls.
| Controls (n = 2028) | Sporadic MS (n = 283) | Multiplex MS (all) (n = 286) | Multiplex MS (probands) (n = 169) | P valuec | |
|---|---|---|---|---|---|
| Female:male ratio (n:n) | 1.27:1 | 2.88:1 | 2.18:1 | 2.60:1 | NS |
| (1136:892) | (210:73) | (196:90) | (122:47) | ||
| Disease course (n) | N/A | RR 181 | RR 193 | RR 117 | NS |
| SP 53 | SP 41 | SP 25 | |||
| PP 49 | PP 52 | PP 27 | |||
| Relapsing-remitting onset (%) | 82.7 | 81.8 | 84 | NS | |
| Disease duration (years)a | N/A | 12.94 ± 8.80 | 15.50 ± 10.59 | 14.59 ± 9.77 | NS |
| Age at onset (years)a | N/A | 34.59 ± 10.43 | 32.82 ± 9.76 | 33.15 ± 9.21 | NS |
| EDSSb | N/A | 3.0 | 3.5 | 3.5 | NS |
| MSSSa | N/A | 4.75 ± 3.01 | 4.93 ± 2.86 | 4.81 ± 2.65 | NS |
MS: multiple sclerosis; EDSS: Expanded Disability Status Scale; MSSS: multiple sclerosis severity score; NS: not significant; N/A: not applicable; RR/SP/PP: relapsing–remitting, secondary progressive, primary progressive MS; SD: standard deviation.
For disease duration, age of onset and MSSS the mean and standard deviation are indicated.
For EDSS the median is reported.
For comparisons of sporadic MS with multiplex MS probands.
The wGRS102 and wGRS102+HLA were significantly higher in sporadic and multiplex MS patients than in controls (P < 0.0001; Figure 1 and Table 2). We observed a trend towards a higher wGRS102 in fMS than in sMS (P = 0.08). The addition of HLA into the wGRS102 score resulted in a significantly higher score in fMS than in sMS (P < 0.0001). The risk allele frequency of the HLA-DRB1*1501 was significantly higher in fMS (0.35) than in sMS (0.25, P = 0.001) and healthy controls (0.13, P < 0.0001). There was no compensatory aggregation of non-HLA SNPs in affected individuals not carrying the HLA-DRB1*1501 risk allele (P = 0.314). We found no differences in genetic risk scores between men and women in multiplex MS and sporadic MS patients.
Figure 1.
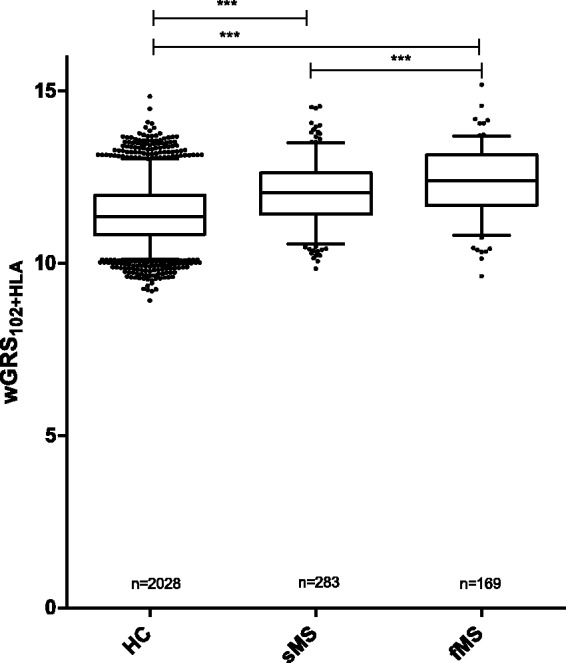
Distribution of genetic risk score in MS patients and healthy controls.
The figure shows a distribution of the weighted genetic risk score (wGRS) based on 102 non-human leukocyte antigen (HLA) loci and the HLA-DRB1*1501 allele (wGRS102+HLA) in healthy controls (HC), sporadic MS (sMS) and multiplex MS (fMS). The significance is indicated as ***P < 0.0001.
Table 2.
Mean weighted genetic risk score in healthy controls, sporadic and multiplex MS patients.
| Controls n = 2028 | Sporadic MS n = 283 | Multiplex MS n = 169 | P value controls vs sporadic MS | P value controls vs multiplex MS | P value sporadic MS vs multiplex MS | |
|---|---|---|---|---|---|---|
| wGRS102 | 11.14 ± 0.67 | 11.48 ± 0.70 | 11.60 ± 0.65 | <0.0001 | <0.0001 | 0.08 |
| wGRS102+HLA | 11.44 ± 0.86 | 12.04 ± 0.88 | 12.39 ± 0.96 | <0.0001 | <0.0001 | <0.0001 |
wGRS: weighted genetic risk score.
Means and standard deviations are indicated for wGRS.
There was a trend towards a correlation between a high wGRS102 and a low age at disease onset (AAO) in all patients with MS (P = 0.09, Figure 2(a)). Inclusion of HLA into the genetic score showed a significant correlation between a high genetic score and a low AAO in this group. After stratification, we found that a low age of onset correlated significantly with a high wGRS102 only in sporadic MS (P = 0.02) but not in familial cases (P = 0.61; Figure 2(b) and (c)). The same significant correlation was found for wGRS102+HLA and age of onset (P = 0.02) in sporadic MS but not in multiplex MS cases (P = 0.72).
Figure 2.
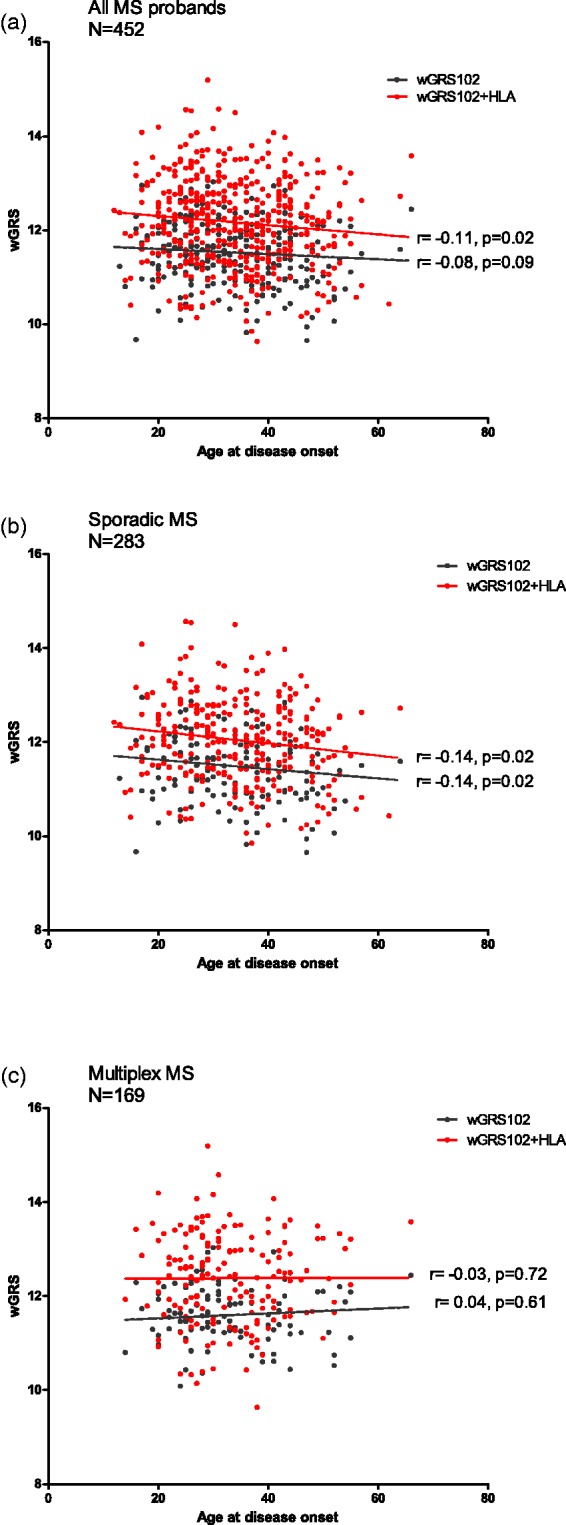
Correlation between age of disease onset and weighted genetic risk scores (wGRSs).
The figures show the correlation between wGRS and age at disease onset in (a) all multiple sclerosis (MS) probands, (b) sporadic MS and (c) multiplex MS. The linear regression lines are drawn in black for wGRS102 and red for wGRS102+HLA. The r estimates from the Spearman correlation test and P values are shown next to the regression lines.
There was no correlation between genetic risk scores and MSSS (P > 0.05). No differences were found in wGRS102 and wGRS102+HLA between primary progressive and relapsing-remitting MS patients (P > 0.05).
Both the wGRSHLA (AUC = 0.63 (0.61–0.66)), and the wGRS102 (AUC = 0.66 (0.63–0.68)) had a poor predictive capacity in discriminating between all patients and controls (Figure 3(a) and Table 3). The AUC increased significantly to 0.72 (P < 0.001) when both non-HLA SNPs and HLA-DRB1*1501 were included in the model. The predictive capacity of the genetic models was also tested in sporadic and multiplex MS patients separately (Figure 3(b) and (c)). The wGRS102+HLA had a statistically better predictive performance than the model considering only HLA-DRB1*1501 or only 102 non-HLA SNPs in both sporadic and familial MS versus controls (P < 0.001, Table 3). In addition, the wGRS102+HLA was better in discriminating familial MS from controls (AUC = 0.77 (0.73–0.81)) than the same model discriminating sporadic MS from controls (AUC = 0.69 (0.66–0.72), P =0.0033). These results parallel the Nagelkerke’s R2 results in which the proportion of genetic variability explained by different wGRS scores is represented. The maximum R2 was 0.167 in fMS and 0.0892 in sMS for wGRS102+HLA (see online Supplementary Appendix 3). We also calculated the ROC for different wGRS models to be able to discriminate between familial and sporadic MS (Figure 3(d)), but the AUC values were disappointingly low and not suitable to be used in clinical practice (Table 3).
Figure 3.

Predictive performance of weighted genetic risk scores (wGRSs).
The receiver operating characteristic (ROC) curves are plotted considering three models: wGRS based on only HLA-DRB1*1501 (solid black line), 102 non-human leukocyte antigen (HLA) loci (red dashed line) and 102 non-HLA loci + HLA-DRB1*1501 allele (green dotted line). The area under the curve (AUC) was calculated to assess the predictive capacity in (a) all multiple sclerosis (MS) probands, (b) sporadic MS and (c) multiplex MS compared to controls. (d) The prediction of multiplex MS patients from sporadic cases.
Table 3.
AUC values for wGRS risk models in sporadic and multiplex MS patients.
| All MS probands vs controls |
Sporadic MS vs controls |
Multiplex MS vs controls |
Sporadic MS vs multiplex MS |
|||||
|---|---|---|---|---|---|---|---|---|
| wGRS based on | AUC | 95% CI | AUC | 95% CI | AUC | 95% CI | AUC | 95% CI |
| HLA-DRB1*1501 | 0.63 | 0.61–0.66 | 0.60 | 0.57–0.63 | 0.68 | 0.65–0.72 | 0.58 | 0.55–0.63 |
| 102 non-HLA SNPs | 0.66 | 0.63–0.68 | 0.64 | 0.60–0.67 | 0.69 | 0.65–0.73 | 0.55 | 0.49–0.60 |
| 102 non-HLA SNPs | ||||||||
| +HLA-DRB1*1501 | 0.72 | 0.69–0.75 | 0.69 | 66–0.72 | 0.77 | 0.73–0.81 | 0.61 | 0.55–0.66 |
MS: multiple sclerosis; wGRS: weighted genetic risk score; AUC: area under the receiver operating curve; CI: confidence interval; HLA: human leukocyte antigen; SNP: single nucleotide polymorphism.
Discussion
In this study we showed that the cumulative genetic risk score based on all currently known genetic risk factors including the HLA-DRB1*1501, was significantly higher in MS multiplex patients than in sporadic cases and healthy controls in the Dutch population. These results are in agreement with previous reports.7–9,12 Two studies found no difference in genetic load between multiplex and sporadic MS.10,11 Those studies were less powered than other cohorts because of a lower number of multiplex cases. Moreover, population-related differences in genetic background could result in different allele frequencies and thus a distinct genetic load in those studies.
The difference in genetic burden between Dutch multiplex and sporadic MS patients was primarily driven by a higher HLA-DRB1*1501 allele frequency in familial cases. A strong association between MS risk and HLA-DRB1*1501 has been shown previously in some MS families.24,25 Gourraud et al.8 also found a higher genetic risk score in multiplex families based solely on non-HLA variants. Our study showed a similar trend. We have conducted a post-hoc power analysis, which showed that our study was underpowered (power of 0.43) to detect a significant difference between sporadic and multiplex MS for wGRSs based on only non-HLA SNPs. The study numbers for both groups would need to increase to the study numbers used by Gourraud et al.8 to have the power of at least 0.8 at the α-level of 0.05.
Because in our cohort there was a large number of patients without the HLA-DRB1*1501 risk allele, it was interesting to look at the aggregation of non-HLA SNPs in these patients. We did not observe a compensatory increase in non-HLA SNPs in the HLA non-carriers, as earlier reported.8
The factors contributing to the difference between men and women in MS susceptibility are unknown. Our study showed that the genetic risk score is not different between men and women with MS, and is thus probably not responsible for the gender differences in the disease susceptibility. Our results are in line with previous studies.10,14
The accumulation of a higher number of susceptibility alleles, such as the case in multiplex MS, might predispose to an earlier disease onset. However, we did not find a younger age at onset in familial MS than in sporadic MS, supported by several other studies.8,26,27 Furthermore, the genetic risk score was not associated with the age at onset in Dutch multiplex cases. According to a post-hoc power analysis the participant number in the multiplex MS group was too low to detect a statistically significant result. When all sporadic and multiplex cases were taken together a significant inverse correlation was found between genetic risk score and age at onset. This finding is consistent with some10,11 but not all studies.8,14 The result of our study probably reflects the known association of the HLA-DRB1*1501 with a younger age at onset.28 We did not find any association between genetic risk score and other clinical parameters such as MSSS and the disease course, as previously reported.8,11,14
Finally, the genetic risk model based on all risk SNPs was modest in discriminating MS patients from controls and this result was comparable with previous findings.13,29 Our study showed that the model based on all risk loci was better in discriminating multiplex MS than sporadic MS from healthy controls. In the treatable and preventable diseases such as coronary heart disease an AUC of about 0.77 is used (Framingham risk score).30 Although the AUC for MS found in our study is approaching this value, a value with a higher specificity is required for a relatively rare condition such as MS. In order to obtain higher AUCs a considerable number of additional common variants or stronger associated variants with higher odds ratios (ORs) are needed.31 The addition of environmental factors into the model could also be beneficial in disease prediction.13
We have conducted correction for multiple testing using the Benjamini–Hochberg procedure for controlling the false-positive rate, and can confirm that all results with significant P values remained significant after the application of this correction.
There were a few limitations to our study. First, our current findings are based on a simplistic modeling of only one tagging SNP of the major HLA class II locus. As well as HLA-DRB1*1501, other alleles are also associated with MS risk.5 Although these alleles are independently associated with MS, their contribution to MS risk is smaller than that of the HLA-DRB1*1501. Second, the sample size of our study was moderate and lacked power for some comparisons, but despite this the results were in the same direction as in the largest study.9 Third, the effect sizes used for the calculation of the genetic risk score originate from a GWAS performed in a general MS population. Effect sizes of the SNPs might be different for specifically multiplex cases. However, it cannot be ruled out that some of the GWAS participants have a family history of MS and already account for the effect size of the found associations in the GWAS. In our own data (data not shown) we observed slightly higher ORs for the majority of the original SNPs in fMS versus controls than in sMS versus controls. Because of the low numbers in both case groups we could not prove whether the differences in ORs were significant. D’Netto et al.7 also suggest that the effect sizes of genetic variants in multiplex families are probably increased compared to sporadic cases. Unfortunately, that study was also underpowered to be able to make reliable conclusions about the mentioned differences. Fourth, the accuracy of the wGRS calculation might be influenced by the use of the proxy SNPs, which might blur the differences between the groups. In our study the impact of the use of proxy SNPs is limited for two reasons: (a) even with the use of the proxy SNPs we were able to observe differences between cases and controls; (b) we also conducted calculations of wGRS based on only original SNPs and observed that the effect was in the same direction as with the wGRS based on original and proxy SNPs.
In conclusion, our results indicate that patients from multiplex MS families are more genetically loaded with common variants, especially with the HLA-DRB1*1501, than the sporadic MS patients. A greater genetic burden leads to an increased risk of MS in families. Because of the elevated frequencies of the genetic variants, patients from multiplex families are valuable and may increase the power of case–control studies. For further understanding of familial MS aggregation the research should also focus on rare variants and environmental influences on MS risk in families. Combining this information with the genetic risk score might help us to explain familial MS aggregation and also to achieve a better disease prediction.
Acknowledgements
The authors would like to thank all MS patients and healthy controls who participated in this study.
Conflicts of interest.
RQ Hintzen has participated in trials with Biogen Idec, Merck-Serono, Roche and Novartis, and serves on the editorial board of Multiple Sclerosis and Related Disorders. The remaining authors report no conflict of interest.
Funding
The MS center ErasMS is financially supported by the MS Research Foundation, Voorschoten, the Netherlands.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet 2008; 372: 1502–1517. [DOI] [PubMed] [Google Scholar]
- 2.Sadovnick AD, Baird PA, Ward RH. Multiple sclerosis: updated risks for relatives. Am J Med Genet 1988; 29: 533–541. [DOI] [PubMed] [Google Scholar]
- 3.O'Gorman C, Lin R, Stankovich J, et al. Modelling genetic susceptibility to multiple sclerosis with family data. Neuroepidemiology 2013; 40: 1–12. [DOI] [PubMed] [Google Scholar]
- 4.Jersild C, Fog T, Hansen GS, et al. Histocompatibility determinants in multiple sclerosis, with special reference to clinical course. Lancet 1973; 2: 1221–1225. [DOI] [PubMed] [Google Scholar]
- 5.Patsopoulos NA, Barcellos LF, Hintzen RQ, et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet 2013; 9: e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beecham AH, Patsopoulos NA, Xifara DK, et al. International Multiple Sclerosis Genetics Consortium. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013; 45: 1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Netto MJ, Ward H, Morrison KM, et al. Risk alleles for multiple sclerosis in multiplex families. Neurology 2009; 72: 1984–1988. [DOI] [PubMed] [Google Scholar]
- 8.Gourraud PA, McElroy JP, Caillier SJ, et al. Aggregation of multiple sclerosis genetic risk variants in multiple and single case families. Ann Neurol 2011; 69: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isobe N, Damotte V, Lo Re V, et al. Genetic burden in multiple sclerosis families. Genes Immun 2013; 14: 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harbo HF, Isobe N, Berg-Hansen P, et al. Oligoclonal bands and age at onset correlate with genetic risk score in multiple sclerosis. Mult Scler 2014; 20: 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esposito F, Guaschino C, Sorosina M, et al. Impact of MS genetic loci on familial aggregation, clinical phenotype, and disease prediction. Neurol Neuroimmunol Neuroinflamm 2015; 2: e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia Z, White CC, Owen EK, et al. Genes and Environment in Multiple Sclerosis project: a platform to investigate multiple sclerosis risk. Ann Neurol 2016; 79: 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Jager PL, Chibnik LB, Cui J, et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurol 2009; 8: 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilven K, Patsopoulos NA, Dubois B, et al. Burden of risk variants correlates with phenotype of multiple sclerosis. Mult Scler 2015; 21: 1670–1680. [DOI] [PubMed] [Google Scholar]
- 15.Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 1983; 13: 227–231. [DOI] [PubMed] [Google Scholar]
- 16.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol 2001; 50: 121–127. [DOI] [PubMed] [Google Scholar]
- 17.Hofman A, van Duijn CM, Franco OH, et al. The Rotterdam Study: 2012 objectives and design update. Eur J Epidemiol 2011; 26: 657–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 1988; 16: 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson CA, Pettersson FH, Clarke GM, et al. Data quality control in genetic case–control association studies. Nat Protoc 2010; 5: 1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Bakker PI, McVean G, Sabeti PC, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 2006; 38: 1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Field J, Browning SR, Johnson LJ, et al. A polymorphism in the HLA-DPB1 gene is associated with susceptibility to multiple sclerosis. PLoS One 2010; 5: e13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kundu S, Aulchenko YS, van Duijn CM, et al. PredictABEL: an R package for the assessment of risk prediction models. Eur J Epidemiol 2011; 26: 261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011; 12: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawcer S, Ban M, Maranian M, et al. A high-density screen for linkage in multiple sclerosis. Am J Hum Genet 2005; 77: 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barcellos LF, Sawcer S, Ramsay PP, et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet 2006; 15: 2813–2824. [DOI] [PubMed] [Google Scholar]
- 26.Ebers GC, Koopman WJ, Hader W, et al. The natural history of multiple sclerosis: a geographically based study: 8: familial multiple sclerosis. Brain 2000; 123(Pt 3): 641–649. [DOI] [PubMed] [Google Scholar]
- 27.Weinshenker BG, Bulman D, Carriere W, et al. A comparison of sporadic and familial multiple sclerosis. Neurology 1990; 40: 1354–1358. [DOI] [PubMed] [Google Scholar]
- 28.Sawcer S, Hellenthal G, Pirinen M, et al. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Disanto G, Dobson R, Pakpoor J, et al. The refinement of genetic predictors of multiple sclerosis. PLoS One 2014; 9: e96578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson PW, D'Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation 1998; 97: 1837–1847. [DOI] [PubMed] [Google Scholar]
- 31.Jafari N, Broer L, van Duijn CM, et al. Perspectives on the use of multiple sclerosis risk genes for prediction. PLoS One 2011; 6: e26493. [DOI] [PMC free article] [PubMed] [Google Scholar]