

Abstract
Several large population studies have demonstrated a negative correlation between serum bilirubin levels and the development of obesity, hepatic steatosis, and cardiovascular disease. Despite the strong correlative data demonstrating the protective role of bilirubin, the mechanism by which bilirubin can protect against these pathologies remains unknown. Bilirubin has long been known as a powerful antioxidant and also has anti-inflammatory actions, each of which may contribute to the protection afforded by increased levels. We have recently described a novel function of bilirubin as a ligand for the peroxisome proliferator-activated receptor-alpha (PPARα), which we show specifically binds to the nuclear receptor. Bilirubin may function as a selective PPAR modulator (SPPARM) to control lipid accumulation and blood glucose. However, it is not known to what degree bilirubin activation of PPARα is responsible for the protection afforded to reduce hepatic steatosis. We hypothesize that bilirubin, acting as a novel SPPARM, increases hepatic fatty acid metabolism through a PPARα-dependent mechanism which reduces hepatic lipid accumulation and protects against hepatic steatosis and non-alcoholic fatty liver disease (NAFLD).
Keywords: heme oxygenase, bilirubin, biliverdin, NAFLD, NASH, non-alcoholic fatty liver disease, PPAR, PPARα, SPPARM, nuclear receptors
Introduction
Hepatic steatosis is a serious pathologic condition in which the liver accumulates fatty acids to an excessive degree resulting in the development of non-alcoholic fatty liver disease (NAFLD) (Figure 1). Hepatic steatosis is initiated by several distinct injurious pathways rather than a true “hit” to the liver (1). When coupled with another ‘hit’, such as increased oxidative stress (reactive oxygen species), insulin resistance, or inflammation, NAFLD can lead to the development of non-alcoholic steatohepatitis (NASH). The initial ‘two-hit’ theory for explaining the progression from NAFLD to NASH is now being modified to a ‘multiple parallel hits’ hypothesis (2). In this model, the build-up of lipids causes a reduction in insulin clearance in the liver, which in turn, promotes peripheral tissue insulin resistance. Peripheral insulin resistance causes alterations in adipose lipolysis (3), which increases the delivery of free fatty acids from the adipose to the liver resulting in the first “hit”, thus, leading to hepatic steatosis. This “hit” increases the vulnerability of the liver to other factors that may follow such as increased oxidative stress and inflammation which then leads to hepatocyte injury and progression to NASH (4).
Figure 1. Schematic diagram of hypothesis.
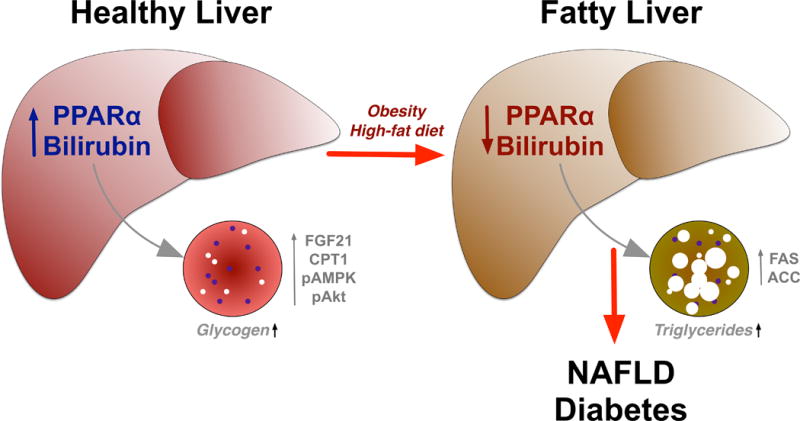
In a normal healthy liver, bilirubin and PPARα combine to reduced lipid storage through an increase of the β-oxidation pathway by an up-regulation of FGF21, CPT1, pAMPK, and pAkt. As a result of obesity or a high-fat diet, bilirubin and PPARα levels are decreased, which increases levels of FAS and ACC that enhances triglyceride accumulation. Fat accumulation in liver eventually leads to a non-alcoholic fatty liver disease (NAFLD) and type II diabetes mellitus.
ACC, Acetyl-CoA carboxylase, pAMPK, phosphorylated AMP-activated protein kinase, pAkt, phosphorylated Akt, CPT1, Carnitine palmitoyltransferase I, FAS, fatty acid synthase, FGF21, fibroblast growth factor 21, NAFLD, non-alcoholic fatty liver disease.
We and others have previously shown that the lower expression and activity of PPARα in the obese is directly linked to hepatic lipid accumulation and glucose intolerance (5–12). PPARα is a transcription factor that upon activation promotes uptake, utilization and catabolism of fatty acids by the upregulation of genes involved in fatty acid transport and peroxisomal and mitochondrial fatty acid β-oxidation. We have demonstrated that increasing plasma bilirubin levels attenuated adiposity in obese mice by significantly increasing expression of PPARα (6). Interestingly, bilirubin levels are also decreased in human obese patients as well as several rodent models of obesity (Figure 1) (6, 13–15). Bilirubin is primarily known as a scavenger of free radicals and acts as an antioxidant (16–18). Bilirubin is a product derived from the breakdown of heme released from red blood cells by heme oxygenase enzymes in the spleen. Heme oxygenase is found in two forms, the constitutively expressed form, HO-2, and the inducible form HO-1. Heme oxygenase enzymes metabolize heme to biliverdin which is rapidly converted to bilirubin by the ubiquitous enzyme biliverdin reductase. Bilirubin can also be produced intracellularly as a result of recycling of heme contained in cytochrome P450 proteins. Bilirubin also protects against other forms of cellular stress including endoplasmic reticulum (ER) stress in a model of type II diabetes (19). In addition to being a potent antioxidant, bilirubin also has anti-inflammatory actions through regulation of cytokine production as well as disruption of immune cell adhesion molecule-mediated migration (20, 21). These are well-known and established functions of bilirubin. The antioxidant properties of bilirubin protect against increases in ROS, which contributes to the increased oxidative stress that has been linked to the development of hepatocyte injury and progression to NASH.
Serum levels of bilirubin are regulated by the hepatic UDP-glucuronosyltransferase 1A1 (UGT1A1) which conjugates bilirubin for elimination in the bile (22, 23). Mutations in the UGT system results in elevated plasma levels of unconjugated bilirubin. Gilbert’s syndrome (GS) is the most common hereditary cause of hyperbilirubinemia affecting approximately 5% to 10% of the population. GS is the result of reduced activity of the UGT enzyme, UGT1A1, resulting in higher plasma bilirubin levels. GS patients exhibiting mildly elevated levels of bilirubin were found to have reduced the risk of cardiovascular events and a lower risk for future heart disease (24). Several large-scale population studies have demonstrated a negative relationship between serum bilirubin levels and the development of cardiovascular disease as well as obesity and diabetes (25–29). Studies in several different patient populations have also demonstrated a negative relationship between serum bilirubin levels and hepatic steatosis (30–33). Despite these correlative studies, the mechanism by which increases in serum bilirubin levels affords protection against hepatic steatosis is unknown. However, we have recently discovered a novel function for bilirubin as a selective modulator of the PPAR family of nuclear receptors (SPPARM), or more specifically its’ direct binding to PPARα (34). The interaction of bilirubin with other PPARs is unknown. We hypothesize that bilirubin may protect against hepatic steatosis through activation of PPARα and it’s associated pathways that promote β-oxidation of fatty acids and decrease fatty acid synthesis.
The hypothesis
Serum bilirubin and non-alcoholic fatty liver disease
NAFLD is an emerging liver pathology which is closely linked to the growing obesity epidemic (35). It is estimated that the global incidence of NAFLD is 25% (36). This percentage is likely to increase to parallel the global obesity epidemic which is estimated to affect well over a third of the world’s population. The negative correlation between serum bilirubin levels and NAFLD has been observed in several different patient populations including children (30–33, 37). Both total, as well as unconjugated serum bilirubin levels, have been reported to be negatively correlated with the development of NAFLD (31, 32). Serum bilirubin levels have also been demonstrated to be negatively correlated with NASH, which is a pathological inflammatory condition of the liver that often results in the development of cirrhosis (37). It is interesting that only a 1.5–2 fold difference in serum bilirubin levels affords protection against NAFLD and NASH in most patient populations observed. Moderate increases in serum bilirubin levels obtain the protective effects of bilirubin against NAFLD.
PPARα and non-alcoholic fatty liver disease
Given the profound effects that PPARα has on lipid metabolism, agonists have been developed as potential therapeutics for the treatment of NAFLD (38, 39). PPARα levels are decreased in animal models of NAFLD and patients with NASH (Figure 1) (40, 41). Treatment with PPARα agonists has been demonstrated to protect against dietary-induced NAFLD in several different animal models (42–44). PPARα agonists are believed to protect against hepatic steatosis through up-regulation of genes responsible for increasing β-oxidation of fatty acids. Despite the relative efficacy of PPARα agonists to protect against NAFLD they are not without their limitations. For example, the PPARα agonist, fenofibrate, was demonstrated to increase hepatomegaly despite decreasing steatosis, necro-inflammation, and collagen deposition in a dietary model of NAFLD in the rat (45). The use of fibrates is also associated with gastrointestinal side effects such as nausea, stomach cramps, or diarrhea. Fibrates can also interact with other drugs such as blood thinners and statin medications to alter their effectiveness. These side effects observed with fibrate-based PPARα agonists have not been reported in bilirubin treated animals or patients with moderately increased levels.
Bilirubin and PPARα
We hypothesize that bilirubin acting through PPARα will attenuate and reverse NAFLD (Figure 1). This hypothesis is based upon several lines of evidence. The first being the pyrrole-ring like structure of bilirubin which is very similar to known PPARα ligands such as WY-14643 and fenofibrate. Secondly, in silco modeling clearly demonstrates that bilirubin binds to the activation site of PPARα in an identical fashion as the known agonist, GW735 (34). Thirdly, physiological concentrations of bilirubin and biliverdin activate a PPARα reporter construct in cultured cells (34). Lastly, our hypothesis is based on in vivo data demonstrating that bilirubin is not able to activate PPARα target genes in the liver of PPARα KO mice (34). Taken together, these data demonstrate for the first time that bilirubin is a SPPARM that activates PPARα. This is a novel function of bilirubin which has long been thought to only function as an antioxidant or the cause of jaundice which can be a pathological complication of liver disease.
In addition to its potential role to prevent and reverse NAFLD though activation of PPARα, bilirubin has several beneficial cardiovascular actions. We have demonstrated that moderate hyperbilirubinemia is able to prevent angiotensin-II induced hypertension in mice (46). The blood pressure lowering actions of moderate hyperbilirubinemia were associated with the preservation of renal blood flow and improvement in glomerular filtration rate in angiotensin II induced hypertension (47). Interestingly, we recently demonstrated that the antioxidant actions of bilirubin only account for a small fraction of the blood pressure lowering effects of moderate hyperbilirubinemia in angiotensin II induced hypertension (48). These findings open up the possibility that bilirubin activation of PPARα may also be responsible for the beneficial cardiovascular effects of moderate hyperbilirubinemia.
Evaluation of the hypothesis
We have demonstrated that bilirubin is a SPPARM for PPARα. However, specific studies examining the relative contribution of bilirubin activation of PPARα for the anti-steatotic actions have yet to be performed. We hypothesize that bilirubin functioning as a SPPARM and binding to PPARα prevents hepatic steatosis by increasing genes involved in the β-oxidation pathway, as well as suppression of fatty acid synthesis (Figure 1).
We will test this hypothesis in bilirubin treated wild-type and PPARα knockout mice fed a high-fat diet for several weeks to promote hepatic steatosis. The level of hepatic steatosis will be determined by EchoMRI measurement of liver fat, biochemical measurement of hepatic triglyceride levels, and Oil Red O staining of liver sections. If our hypothesis is true, we predict that bilirubin treatment will decrease hepatic steatosis and increase levels of proteins involved in the β-oxidation pathway such as carnitine palmitoyltransferase I (CPT1) and fibroblast growth factor 21 (FGF21), as well as fatty acid synthase (FAS) and phosphorylated Acetyl-CoA carboxylase (ACC) that regulate de novo fatty acid production. These changes in hepatic steatosis and proteins regulating β-oxidation and synthesis of fatty acids will be markedly attenuated in PPARα knockout mice.
If our hypothesis is correct, we predict that reductions in hepatic steatosis and alterations in genes regulating β-oxidation and synthesis of fatty acids are mediated to a large degree via activation of PPARα by bilirubin treated mice. This hypothesis could explain how moderate increases in serum bilirubin levels attenuate the development of NAFLD via bilirubin functioning as a SPPARM in addition to its better-known roles as an antioxidant and anti-inflammatory molecule. Taken together, these results could provide further rationale for the therapeutic potential of moderate hyperbilirubinemia to treat NAFLD, which is a growing problem, and there are currently no available FDA-approved treatments.
Acknowledgments
Sources of funding
This work was supported by the National Institutes of Health L32MD009154 (T.D.H.), the National Heart, Lung, and Blood Institute [K01HL-125445] (T.D.H.) and (PO1HL-051971), [HL088421] (D.E.S.), and the National Institute of General Medical Sciences (P20GM-104357) (D.E.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest
The authors declare they have no conflicts of interest.
References
- 1.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 2.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–46. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 3.John K, Marino JS, Sanchez ER, Hinds TD., Jr The Glucocorticoid Receptor: Cause or Cure for Obesity? American journal of physiology Endocrinology and metabolism. 2015 doi: 10.1152/ajpendo.00478.2015. ajpendo 00478 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith BW, Adams LA. Non-alcoholic fatty liver disease. Crit Rev Clin Lab Sci. 2011;48(3):97–113. doi: 10.3109/10408363.2011.596521. [DOI] [PubMed] [Google Scholar]
- 5.Stec DE, John K, Trabbic CJ, Luniwal A, Hankins MW, Baum J, et al. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PloS one. 2016;11(4):e0153427. doi: 10.1371/journal.pone.0153427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinds TD, Jr, Sodhi K, Meadows C, Fedorova L, Puri N, Kim DH, et al. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity. 2014;22(3):705–12. doi: 10.1002/oby.20559. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Shiomi Y, Yamauchi T, Iwabu M, Okada-Iwabu M, Nakayama R, Orikawa Y, et al. A Novel Peroxisome Proliferator-activated Receptor (PPAR)alpha Agonist and PPARgamma Antagonist, Z-551, Ameliorates High-fat Diet-induced Obesity and Metabolic Disorders in Mice. The Journal of biological chemistry. 2015;290(23):14567–81. doi: 10.1074/jbc.M114.622191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, et al. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes. 2005;54(12):3358–70. doi: 10.2337/diabetes.54.12.3358. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Jia Y, Fu T, Viswakarma N, Bai L, Rao MS, et al. Sustained activation of PPARalpha by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26(2):628–38. doi: 10.1096/fj.11-194019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura R, Takahashi N, Murota K, Yamada Y, Niiya S, Kanzaki N, et al. Activation of peroxisome proliferator-activated receptor-alpha (PPARalpha) suppresses postprandial lipidemia through fatty acid oxidation in enterocytes. Biochemical and biophysical research communications. 2011;410(1):1–6. doi: 10.1016/j.bbrc.2011.05.057. [DOI] [PubMed] [Google Scholar]
- 11.Lee JY, Hashizaki H, Goto T, Sakamoto T, Takahashi N, Kawada T. Activation of peroxisome proliferator-activated receptor-alpha enhances fatty acid oxidation in human adipocytes. Biochemical and biophysical research communications. 2011;407(4):818–22. doi: 10.1016/j.bbrc.2011.03.106. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Liu X, Jiao Y, Xiong X, Wang E, Wang X, et al. Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARalpha. The Journal of clinical investigation. 2014;124(8):3501–13. doi: 10.1172/JCI74438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Wang L, Tian X, Liu L, Wong WT, Zhang Y, et al. Unconjugated bilirubin mediates heme oxygenase-1-induced vascular benefits in diabetic mice. Diabetes. 2014 doi: 10.2337/db14-1391. [DOI] [PubMed] [Google Scholar]
- 14.O’Brien L, Hosick PA, John K, Stec DE, Hinds TD., Jr Biliverdin reductase isozymes in metabolism. Trends Endocrinol Metab. 2015;26(4):212–20. doi: 10.1016/j.tem.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersson C, Weeke P, Fosbol EL, Brendorp B, Kober L, Coutinho W, et al. Acute effect of weight loss on levels of total bilirubin in obese, cardiovascular high-risk patients: an analysis from the lead-in period of the Sibutramine Cardiovascular Outcome trial. Metabolism. 2009;58(8):1109–15. doi: 10.1016/j.metabol.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–6. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 17.Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. 2004;6(5):841–9. doi: 10.1089/ars.2004.6.841. [DOI] [PubMed] [Google Scholar]
- 18.Lanone S, Bloc S, Foresti R, Almolki A, Taille C, Callebert J, et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J. 2005;19(13):1890–2. doi: 10.1096/fj.04-2368fje. [DOI] [PubMed] [Google Scholar]
- 19.Dong H, Huang H, Yun X, Kim DS, Yue Y, Wu H, et al. Bilirubin increases insulin sensitivity in leptin-receptor deficient and diet-induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology. 2014;155(3):818–28. doi: 10.1210/en.2013-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu H, Wang J, Jiang H, Ma Y, Pan S, Reddy S, et al. Bilirubin protects grafts against nonspecific inflammation-induced injury in syngeneic intraportal islet transplantation. Exp Mol Med. 2010;42(11):739–48. doi: 10.3858/emm.2010.42.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel ME, Zucker SD. Bilirubin acts as an endogenous regulator of inflammation by disrupting adhesion molecule-mediated leukocyte migration. Inflamm Cell Signal. 2016;3(1) doi: 10.14800/ics.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke DJ, Moghrabi N, Monaghan G, Cassidy A, Boxer M, Hume R, et al. Genetic defects of the UDP-glucuronosyltransferase-1 (UGT1) gene that cause familial non-haemolytic unconjugated hyperbilirubinaemias. Clin Chim Acta. 1997;266(1):63–74. doi: 10.1016/s0009-8981(97)00167-8. [DOI] [PubMed] [Google Scholar]
- 23.Ishihara T, Kaito M, Takeuchi K, Gabazza EC, Tanaka Y, Higuchi K, et al. Role of UGT1A1 mutation in fasting hyperbilirubinemia. J Gastroenterol Hepatol. 2001;16(6):678–82. doi: 10.1046/j.1440-1746.2001.02495.x. [DOI] [PubMed] [Google Scholar]
- 24.Vitek L, Jirsa M, Brodanova M, Kalab M, Marecek Z, Danzig V, et al. Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis. 2002;160(2):449–56. doi: 10.1016/s0021-9150(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 25.Lin JP, O’Donnell CJ, Schwaiger JP, Cupples LA, Lingenhel A, Hunt SC, et al. Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham Heart Study. Circulation. 2006;114(14):1476–81. doi: 10.1161/CIRCULATIONAHA.106.633206. [DOI] [PubMed] [Google Scholar]
- 26.Schwertner HA, Vitek L. Gilbert syndrome, UGT1A1*28 allele, and cardiovascular disease risk: possible protective effects and therapeutic applications of bilirubin. Atherosclerosis. 2008;198(1):1–11. doi: 10.1016/j.atherosclerosis.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Cheriyath P, Gorrepati VS, Peters I, Nookala V, Murphy ME, Srouji N, et al. High Total Bilirubin as a Protective Factor for Diabetes Mellitus: An Analysis of NHANES Data From 1. J Clin Med Res. 2010;2(5):201–6. doi: 10.4021/jocmr425w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi SH, Yun KE, Choi HJ. Relationships between serum total bilirubin levels and metabolic syndrome in Korean adults. Nutr Metab Cardiovasc Dis. 2013;23(1):31–7. doi: 10.1016/j.numecd.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Jung CH, Lee MJ, Kang YM, Hwang JY, Jang JE, Leem J, et al. Higher serum bilirubin level as a protective factor for the development of diabetes in healthy Korean men: A 4year retrospective longitudinal study. Metabolism. 2013 doi: 10.1016/j.metabol.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Jang BK. Elevated serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol. 2012;18(4):357–9. doi: 10.3350/cmh.2012.18.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar R, Rastogi A, Maras JS, Sarin SK. Unconjugated hyperbilirubinemia in patients with non-alcoholic fatty liver disease: a favorable endogenous response. Clin Biochem. 2012;45(3):272–4. doi: 10.1016/j.clinbiochem.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 32.Kwak MS, Kim D, Chung GE, Kang SJ, Park MJ, Kim YJ, et al. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol. 2012;18(4):383–90. doi: 10.3350/cmh.2012.18.4.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin YC, Chang PF, Hu FC, Chang MH, Ni YH. Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics. 2009;124(6):e1221–e7. doi: 10.1542/peds.2008-3087. [DOI] [PubMed] [Google Scholar]
- 34.Stec DE, John K, Trabbic CJ, Luniwal A, Hankins MW, Baum J, et al. Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PLoS One. 2016;11(4):e0153427. doi: 10.1371/journal.pone.0153427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masuoka HC, Chalasani N. Nonalcoholic fatty liver disease: an emerging threat to obese and diabetic individuals. Ann N Y Acad Sci. 2013;1281:106–22. doi: 10.1111/nyas.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global Epidemiology of Non-Alcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence and Outcomes. Hepatology. 2015 doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 37.Puri K, Nobili V, Melville K, Corte CD, Sartorelli MR, Lopez R, et al. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr. 2013;57(1):114–8. doi: 10.1097/MPG.0b013e318291fefe. [DOI] [PubMed] [Google Scholar]
- 38.Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144(6):2201–7. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 39.Harano Y, Yasui K, Toyama T, Nakajima T, Mitsuyoshi H, Mimani M, et al. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 2006;26(5):613–20. doi: 10.1111/j.1478-3231.2006.01265.x. [DOI] [PubMed] [Google Scholar]
- 40.Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol. 2015;63(1):164–73. doi: 10.1016/j.jhep.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 41.Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr. 2011;141(4):603–10. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seo YS, Kim JH, Jo NY, Choi KM, Baik SH, Park JJ, et al. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J Gastroenterol Hepatol. 2008;23(1):102–9. doi: 10.1111/j.1440-1746.2006.04819.x. [DOI] [PubMed] [Google Scholar]
- 43.Barbosa-da-Silva S, Souza-Mello V, Magliano DC, Marinho Tde S, Aguila MB, Mandarim-de-Lacerda CA. Singular effects of PPAR agonists on nonalcoholic fatty liver disease of diet-induced obese mice. Life Sci. 2015;127:73–81. doi: 10.1016/j.lfs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Yan F, Wang Q, Xu C, Cao M, Zhou X, Wang T, et al. Peroxisome proliferator-activated receptor alpha activation induces hepatic steatosis, suggesting an adverse effect. PLoS One. 2014;9(6):e99245. doi: 10.1371/journal.pone.0099245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong XZ, Li LD, Wu LM. Effects of fenofibrate and xuezhikang on high-fat diet-induced non-alcoholic fatty liver disease. Clin Exp Pharmacol Physiol. 2007;34(1–2):27–35. doi: 10.1111/j.1440-1681.2007.04547.x. [DOI] [PubMed] [Google Scholar]
- 46.Vera T, Granger JP, Stec DE. Inhibition of bilirubin metabolism induces moderate hyperbilirubinemia and attenuates ANG II-dependent hypertension in mice. Am J Physiol Regul Integr Comp Physiol. 2009;297(3):R738–R43. doi: 10.1152/ajpregu.90889.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vera T, Stec DE. Moderate hyperbilirubinemia improves renal hemodynamics in ANG II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;299(4):R1044–R9. doi: 10.1152/ajpregu.00316.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stec DE, Storm MV, Pruett BE, Gousset MU. Antihypertensive actions of moderate hyperbilirubinemia: role of superoxide inhibition. Am J Hypertens. 2013;26(7):918–23. doi: 10.1093/ajh/hpt038. [DOI] [PMC free article] [PubMed] [Google Scholar]