

Abstract
Downregulation in the astroglial glutamate transporter EAAT2 in amyotrophic lateral sclerosis (ALS) patients and mutant SOD1 mouse models of ALS is believed to contribute to the death of motor neurons by excitotoxicity. We previously reported that caspase-3 cleaves EAAT2 at a unique cleavage consensus site located in its c-terminus domain, a proteolytic cleavage that also occurs in vivo in the mutant SOD1 mouse model of ALS and leads to accumulation of a sumoylated EAAT2 C-Terminus fragment (CTE-SUMO1) beginning around onset of disease. CTE-SUMO1 accumulates in PML nuclear bodies of astrocytes and causes them to alter their mature phenotypes and secrete factors toxic to motor neurons.
Here, we report that mutating the caspase-3 consensus site in the EAAT2 sequence with an aspartate to asparagine mutation (D504N), thereby inhibiting caspase-3 cleavage of EAAT2, confers protection to the SOD1-G93A mouse. EAAT2-D504N knock-in mutant mice were generated and crossed with SOD1-G93A mice to assess the in vivo pathogenic relevance for ALS symptoms of EAAT2 cleavage. The mutation did not affect normal EAAT2 function nor non-ALS mice. In agreement with the timing of CTE-SUMO1 accumulation, while onset of disease was not affected, the mutation caused an extension in progression time, a delay in the development of hindlimb and forelimb muscle weakness, and a significant increase in the lifespan of SOD1-G93A mice.
Keywords: Excitotoxicity, glutamate, astrocyte, amyotrophic lateral sclerosis, EAAT2, GLT-1, SOD1
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with no effective treatment. Upper and lower motor neurons are primarily affected and their degeneration leads to symptoms such as muscle fasciculations and wasting (Turner et al., 2013). While a small subset of patients lives longer, the vast majority die of respiratory failure within 3–5 years of symptom onset. With an incidence of 1.75 per 100,000 people per year, ALS is a fairly rare, yet devastating diagnosis (**Marin et al., 2016). The only FDA-approved disease modifying therapy is riluzole, which prolongs life by only a few months, necessitating greater understanding of the disease and the generation of more effective therapeutics (Bellingham, 2011; Bensimon et al., 1994).
Understanding the mechanisms of ALS is challenging, as it is an extremely heterogeneous disease in cause, in progression, and in symptom development (Robberecht and Philips, 2013; Turner et al., 2013). The ~10% of cases that are familial are caused by mutations in several genes, some just being discovered and some still unknown (Fifita et al., 2016; Robberecht and Philips, 2013). Sporadic cases predominate, though risk loci have begun to be uncovered (van Rheenen et al., 2016). A mutation in one of the first discovered causative genes, SOD1-G93A, has been used to develop cell and animal models of the disease (Gurney et al., 1994). New models are being developed to mimic the more recently discovered genes, in particular the C9orf72 repeat expansion, which accounts for ~40% of familial ALS and a portion (~10%) of sporadic disease (Haeusler et al., 2016; Liu et al., 2016; Wegorzewska and Baloh, 2011; Yin et al., 2012). It is also becoming possible to model ALS with unknown cause, as induced pluripotent stem cell (iPSC) are being generated from sporadic ALS patients (Burkhardt et al., 2013; Y. Li et al., 2015).
Expression of causative mutations in only some cell types has demonstrated the importance of cells other than motor neurons in causing degeneration, in a process known as non-cell autonomous toxicity (Beers et al., 2006; Boillée et al., 2006; Clement et al., 2003; Nagai et al., 2007; Robberecht and Philips, 2013). Astrocytes, for example, normally clear the vast majority of the excitatory neurotransmitter glutamate from the synapse using the excitatory amino acid transporter 2 (EAAT2, or sometimes referred to as GLT-1 in rodents) (Lauriat and McInnes, 2007). Decreases in EAAT2 expression and function have been reported in models of ALS and in patients, leading to over stimulation of post-synaptic neurons, eventually causing excitotoxicity (Bruijn et al., 1997; Lauriat and McInnes, 2007). Motor neurons are particularly susceptible to excitotoxicity, implicating the relevance of this pathway in ALS (Foran and Trotti, 2009).
While attempting to understand the mechanism of EAAT2 loss in ALS, we found that EAAT2 has a caspase-3 consensus sequence and can be cleaved by activated caspase-3 in vitro (Boston-Howes et al., 2006; Gibb et al., 2007). We also found evidence of this process in the diseased SOD1-G93A ALS mouse model, as a truncated fragment of 55 kDa and a sumoylated C-terminal end (CTE-SUMO1) accumulated with disease course. Mutation of a critical amino acid aspartate (D) to asparagine (N) of the caspase-3 cleavage consensus sequence (D505N in human EAAT2 or D504N in mouse EAAT2) successfully inhibits cleavage by caspase-3 in vitro (Boston-Howes et al., 2006). Interestingly, while this cleavage likely contributes to EAAT2 decreases in ALS, it also leads to the accumulation of CTE-SUMO1 associated with PML nuclear bodies of astrocytes and causes them to secrete neurotoxic factors (Couratier et al., 1993; Foran et al., 2011; Sen et al., 2005).
Here, we analyzed the impact of the EAAT2 cleavage pathway on the disease course in the SOD1-G93A mouse model of ALS using a novel knock-in mouse model. Knock-in mice bearing an EAAT2-D504N point mutation were crossed with SOD1-G93A mice and assessed for ALS-like disease progression. Lifespan, onset, and duration of disease were primarily assessed. Grip strength, accelerating rotarod stamina, diaphragm innervation, and motor neuron survival were also studied.
MATERIALS AND METHODS
Animals and behavioral analysis
Transgenic mice expressing the human SOD1-G93A transgene (B6.Cg-Tg(SOD1*G93A)1Gur/J; Stock No: 004435, Jackson Labs) were bred in house in accordance with IACUC SOP protocols. Copy number of the human transgene was assessed by qPCR (primers: GGG AAG CTG TTG TCC CAA G and CAA GGG GAG GTA AAA GAG AGC) and normalized to an internal control (primers: CAC GTG GGC TCC AGC ATT and TCA CCA GTC ATT TCT GCC TTT G). SOD1-G93A mice of our colony typically become symptomatic (defined as onset of weight loss) after 100 days of age (116.8 ± 6.1 for females, 109.3 ± 5.2 for males) and reach end-stage (defined as being unable to right itself within 30 s of being placed on its side) around 160–170 days of age (168.2 ± 3.8 for females, 160.1 ± 2.7 for males). Mice were considered pre-symptomatic at P90, symptomatic at P120, and late-symptomatic at P140 (for CMAP analysis, after which signals are too low for reliable identification) and P150 (for histologic and homogenate protein analysis).
An EAAT2-D504N knock-in mouse on a C57BL/6J background was generated in conjunction with ingenious Targeting Laboratory, Inc. (Ronkonkoma, NY). Briefly, a targeting vector containing a left arm homology domain, partial EAAT2 sequence with the point mutation, a neo cassette between FRT sites, and a short right arm homology domain was transfected into C57BL/6NTac (Taconic) embryonic stem cells. Transfected cells were selected with G418 antibiotic and microinjected into Balb/c blastocysts. Resulting chimeras with a primarily black coat color were mated to FLP mice (B6.Cg-Tg(ACTFLPe)9205Dym/J, Jackson stock #005703) to remove the Neo cassette. Mice were then bred in house to remove the FLP gene. Removal of the Neo cassette, FLP transgene, and presence of the point mutation were confirmed by PCR and sequencing (using primers: CTT GCT GGT TCT AGC AGC ACT CTG and GAA AGT ATA GGA ACT TCG CGA CAC GGA C). EAAT2 mutation status in each mouse was determined by PCR (primers: GAC CAT CTC ACT CTG CAT CCA G and GGA AGA CAG CCA ATT AAA CTT TTA TAT GAGC), where EAAT2-D504 mice had a slightly longer sequence due to the presence of a remaining FRT site.
Mice homozygous for wild-type EAAT2 are denoted as EAAT2WT/WT, mice homozygous for caspase-3-resistant EAAT2 (two copies of EAAT2-D504N) are denoted as EAAT2CR/CR, and heterozygous mice are denoted as EAAT2WT/CR. Male EAAT2WT/CR mice carrying the SOD1-G93A mutation were bred with female heterozygous EAAT2WT/CR mice to generate SOD1-G93A mice and non-ALS mice with EAAT2WT/WT, EAAT2WT/CR or EAAT2CR/CR. To avoid confounders from substrain differences, a colony was derived from SOD1-G93A mice were crossed with an EAAT2-D504N founder. Closely related pups were used for all experiments (littermates were not feasible as only one out of eight pups were homozygous for EAAT2-WT or -D504N with or without SOD1-G93A).
The behavior of non-ALS mice with EAAT2WT/CR or EAAT2WT/CR was compared to assess the mutation’s effects in normal mice and identify any potential confounders (N = 5 females and 5 males per group). The behavior and lifespan of SOD1-G93A mice was assessed to determine the effects of the EAAT mutation on disease progression (N = 10 females and 10 males per group, with 11 male EAAT2WT/CR mice). Mice assessed for behavior and lifespan were weighed and assessed for neurological score at least twice weekly from 8 weeks of age. The lifespan of one mouse was more than 2 standard deviations away from the mean of its group (by gender and genotype), so it was excluded from lifespan and behavioral analysis as an outlier. Forelimb and hindlimb grip strength was measured twice weekly (Columbus Instruments) and mice were tested on an accelerating rotarod (UgoBasile, Varese, Italy) once per week until end stage.
All mice were housed in accordance with Thomas Jefferson University Institutional Animal Care and Use Committee (IACUC) and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Immunohistochemistry
HEK 293T cells were transfected with EAAT2-WT or EAAT2-D505N using lipofectamine LTE (Thermo Fisher, Waltham, MA). Cells were fixed and stained with anti-EAAT2 (custom polyclonal rabbit raised against amino acids 518-556 or commercial polyclonal rabbit raised against C-terminus domain, Millipore #AB1783,) 48 h later, as previously described (Foran et al., 2011).
Caspase-3 cleavage
Caspase-3 cleavage of mouse hippocampal homogenate was performed as previously described (Boston-Howes, 2006). Briefly, 1.25 μg of homogenate was suspended in 50 μl of caspase-3 cleavage buffer with 1 μl freshly prepared DTT (20 mM, final concentration) and 200 ng of recombinant active Caspase-3 (cat.# 556472, BD Pharminogen, San Jose, CA). Reactions were incubated for 6 hours and the reaction was stopped by the addition of sample buffer and 10 min of boiling. Caspase-3 inhibitor Ac-DEVD-CHO (0.2 μg/μl; BD Bioscience cat.#556485) was added to the reaction.
Western blots
Spinal cords were collected and snap frozen in liquid nitrogen before storage at −80°C. Cords were homogenized in 30 volumes of modified radioimmunoprecipitaiton assay (RIPA) buffer with 1% SDS and cOmplete™ Mini Protease Inhibitor cocktail (Sigmal Aldrich, St. Louis, MO) using a Teflon-glass homogenizer at 600 rpm (10 strokes). Homogenates were incubated for 10 min at room temperature and then briefly sonicated. Unbroken cells were removed by centrifugation (2,000 × g for 5 min). Homogenates were boiled with Laemmli sample buffer for 10 minutes to generate samples at 1 μg/μl.
Samples were run on Mini-PROTEAN Precast Gels (Bio-Rad, Hercules, CA) and transferred at 90 V for 60 min on PVDF membranes. A custom polyclonal rabbit anti-EAAT2 antibody directed towards amino acids 518-536 (ABR518-536) was used at 1:10,000 and a polyclonal rabbit anti-SUMO-1 antibody (#4930, Cell Signaling, Danvers, MA) was used at 1:250.
Compound Muscle Action Potential (CMAP) recordings
After mice were anesthetized with isofluorane, stimulating needle electrodes were placed in the neck along the phrenic nerve, as described by Li et. al. (K. Li et al., 2015). A ground needle electrode was placed in the tail, a reference needle electrode was placed subcutaneously in the contralateral abdomen, and a recording surface strip was placed on the costal margin of the hemi-diaphragm, ipsilateral to stimulation. Stimulation pulses of 0.5 ms and up to 6 mV amplitude were injected via the stimulating electrode and the resulting CMAP was recorded using an ADI Powerlab 8/30 stimulator and BioAMP amplifier (ADInstruments, Colorado Springs, CO), followed by computer-assisted data analysis (Scope 3.5.6, ADInstruments). The baseline to peak CMAP amplitude was measured and averaged over at least 10 pulses. Non-ALS mice with EAAT2WT/WT, EAAT2WT/CR or EAAT2CR/CR were assessed at P90 (N = 16, 8, 14 respectively), P120 (N = 20, 12, 19) and P150 (N = 8, 8, 8) to identify potential confounders. Mice without SOD1-G93A and SOD1-G93A mice with EAAT2WT/WT, EAAT2WT/CR or EAAT2CR/CR were assessed at P90 (N = 42, 14, 15, 14, respectively), P120 (N = 51, 18, 29, 28) and P150 (N = 34, 10, 14, 17).
Motor neuron counts
Mice were anesthetized with ketamine (120 mg/kg) and xylazine (5 mg/kg), then transcardially perfused with DPBS, followed by 4% paraformaldehyde in DPBS. Spinal cords were dissected, fixed in 4% paraformaldehyde overnight, and cryopreserved in 30% sucrose for at least 3 days. The lumbar cords were embedded in optical cutting temperature (OCT) freezing medium and tissues were sectioned at 40 μm thickness for motor neuron counts. Tissue slices were collected on ColorFrost Plus slides (Fisherbrand) and stored at −20°C. Before staining, slides were dried on a 30°C heating block for 20 min. and dipped in ddH2O to remove TissueTek. Slides were dipped in 70% ethanol for 4 min., twice, then in thionin staining solution for 3 min. Slides were rinsed in deionized water, put in differentiation solution for 2 min., 95% ethanol for 1 min., 100% ethanol for 1 min. twice, and HemoD for 5 min. twice. Coverslips were added with permount. Staining solution was composed of 2.5 g Thionin, 1.5 L deionized water, 2 ml glacial acetic acid, and 7 g sodium acetate. Differentiation solution was composed of 1 L 70% ethanol and 1 ml glacial acetic acid. Two female and 2 male mice per group were assessed.
Statistics
SPSS Statistics software, version 23 (IBM, Armonk, NY) was used for all statistical analysis. Behavioral analysis, CMAP strength, and motor neuron counts were compared using ANOVA analyses with gender as a covariate, using the LSD post-hoc test. Kaplan-Meier analysis was used to compare development of onset, symptoms, and end-stage, with groups stratified by gender. The Log-Rank post-hoc was used so early and late events would be weighted equally.
RESULTS
Generation of EAAT2-D504N Mouse
We previously showed that caspase-3 cleavage leads to accumulation of a sumoylated C-terminal fragment of EAAT2, termed CTE-SUMO1, and that expression of the fragment by astrocytes causes those cells to become toxic to motor neurons (Foran et al., 2011; Gibb et al., 2007). In this study, we sought to prevent the toxicity of the EAAT2-derived CTE-SUMO1 fragment in vivo by genetically inhibiting its production. EAAT2 contains a unique caspase-3 consensus sequence, DTID (amino acids 501-504 in mouse EAAT2 or 502-505 in its human counterpart) and a D505N mutation (in the human sequence) that inhibits caspase-3 cleavage was previously generated (Boston-Howes et al., 2006). The effect of caspase-3 cleavage of EAAT2 on the development and progression of ALS was studied with a novel mouse model of ALS bearing a point mutation in the EAAT2 gene.
The effect of the mutation was first tested in vitro for EAAT2 cellular localization pattern and activity. There was no difference in the plasma membrane localization of transfected EAAT2-D505N (human plasmid) versus EAAT2-WT in HEK 293T cells (Fig. 1A and B), which do not normally express EAAT2 and are incapable of localization-altering sumoylation of EAAT2 (Foran et al., 2014; Sullivan et al., 2004). We previously reported that EAAT2-WT and EAAT2-D505N do not exhibit statistically different affinities for glutamate (20.2 ± 4.5 μM for EAAT2-WT vs. 17.7 ± 5 μM for EAAT2-D505N) (Boston-Howes et al., 2006). Similarly, no statistically significant change in glutamate uptake velocity was observed (23 ± 4 nmol/mg protein/15 min for EAAT2-WT versus 25 ± 3 nmol/g protein/15 min), indicating that the D505N mutation in the C-terminus of EAAT2 does not significantly affect transport kinetics (Boston-Howes et al., 2006; Yernool et al., 2004). The lack of a difference in plasma membrane localization and glutamate uptake prompted the generation of a mouse model with the EAAT2-D504N mutation.
Fig. 1. Generation of EAAT2-D504N mice.
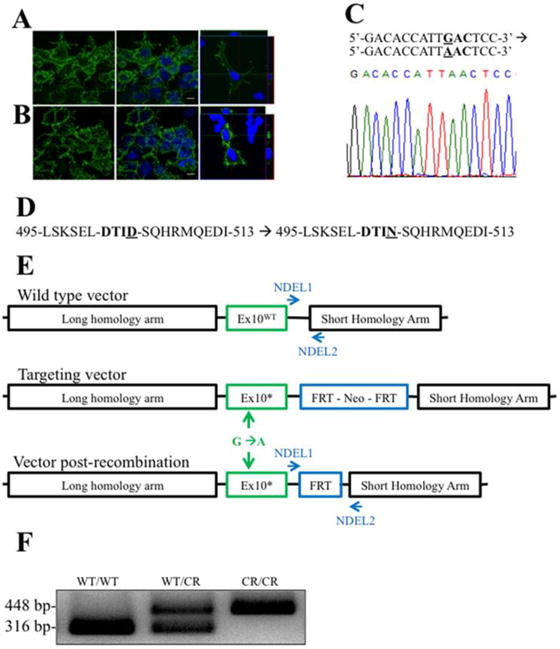
(A) EAAT2-WT and (B) EAAT2-D505N comparably localize to the plasma membrane of transfected HEK cells. (C) A single nucleotide point mutation was introduced into the Slc1a2 nucleotide sequence mutation in exon 10 of the Slc1a2 gene, which codes for EAAT2 in rodents. A segment of the sequenced DNA confirming the point mutation is shown. (D) This generates an aspartate to asparagine mutation in the — DTID- caspase-3 consensus sequence of EAAT2. (E) To introduce the point mutation, a targeting vector with the point mutation and an FRT-flanked neomycin resistance cassette (to enable selection of recombined genes) between long and short homology arms was generated. Mice were crossed with FLP mice to recombine the FRT sites, removing the cassette except for one remaining FRT site. (F) The remaining FRT site enabled genotype to be determined by PCR: the wild-type allele is 316 bp and the mutant allele (with the FRT site) is 448 bp, while a mutant allele without recombination is too long to amplify.
To generate the knock-in mouse, a GAC codon was mutated to AAC in the Slc1a2 gene and confirmed by sequencing, leading to an aspartate to asparagine point mutation at amino acid 504 of EAAT2 (Fig. 1C and D). The neomycin-selection cassette was removed by crossing mice with FLP mice, causing recombination of flanking FRT sites (Fig. 1E). One FRT site remained in an intron, enabling detection of the mutation by PCR with primers (NDEL1 and NDEL2) flanking the site (Fig. 1F). EAAT2-D504N is used to refer to the mutation in both heterozygous and homozygous mutant mice, while genotypes are denoted as EAAT2WT/WT (wild-type (WT)), EAAT2WT/CR (heterozygous wild type- caspase-3 resistant (CR)), and EAAT2CR/CR (homozygous caspase-resistant EAAT2-D504N).
Caspase-3 cleavage of EAAT2 was assessed in homogenates of hippocampal tissue, which is high in EAAT2 expression (Lehre et al., 1995), from both EAAT2WT/WT and EAAT2CR/CR mice. After incubation for 6 h with recombinant active caspase-3, cleavage was quantified as disappearance of the main EAAT2 monomeric and multimeric bands, when the blot was probed with an anti-EAAT2 antibody directed against an epitope in the C-terminus domain of EAAT2, downstream the caspase-3 consensus site (ABR518-536) (Boston-Howes et al., 2006). Significant cleavage was seen only in homogenates of EAAT2WT/WT mice (Fig. 2A), demonstrating that the D504N mutation inhibits caspase-3 cleavage of EAAT2 (Boston-Howes et al., 2006). Loss of EAAT2 immunoreactivity by active caspase-3 was also prevented by adding to the reaction mixture the caspase-3 inhibitor Ac-DEVD-CHO, indicating that caspase-3 cleavage was indeed responsible for loss of EAAT2 full length immunoreactivity (Fig. 2A) (Boston-Howes et al., 2006).
Fig. 2. EAAT2-D504N inhibits caspase-3 cleavage and CTE-SUMO1 formation.
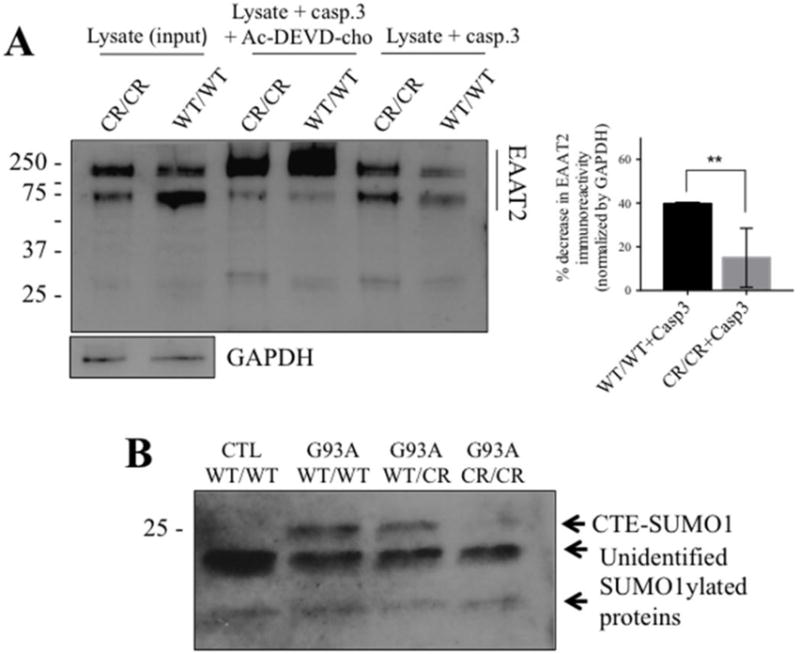
(A) Recombinant active caspase-3 cleaves EAAT2WT/WT in spinal cord homogenates but cleavage is inhibited in spinal cord homogenates of EAAT2CR/CR mice (custom ABR518-536 at 1:10,000). One blot is shown to represent the effect of caspase-3 cleavage on EAAT2 immunoreactivity in the presence or absence of caspase-3 inhibitor together with quantification of the effect. EAAT2 content is normalized to GAPDH immunoreactivity in the input lysate samples. Cleavage was quantified as disappearance of EAAT2-WT or EAAT2-CR monomer and oligomer immunoreactivity in the presence of active caspase-3 from 3 different cleavage reactions. ** p<0.005, two-tailed t-test. (B) CTE-SUMO1 is seen at ~25 kDa in spinal cord homogenates of SOD1-G93A mice (G93A) expressing EAAT2WT/WT or EAAT2WT/CR, but significantly decreased in SOD1-G93A mice expressing EAAT2CR/CR (Cell Signaling #4930 anti-SUMO1 at 1:250). It is also not seen in spinal cord homogenates of control non-ALS EAAT2WT/WT control mice. Two unidentified lower molecular weight SUMO1 immunopositive bands are shown as evidence of equal protein loading among the different lanes of the blot.
To eliminate the possibility that the mutation could cause changes in the motor system of non-ALS mice, EAAT2WT/WT and EAAT2CR/CR mice were tracked for at least 200 days, longer than the age of end-stage of any studied SOD1-G93A mice. Observation was focused on growth and motor phenotypes that might overlap with ALS symptoms. Though mice grew significantly between time points (p = 0.001) and males were heavier than females (p = 0.049), EAAT2 genotype did not affect the growth, as no significant differences were seen in weight at P90 (ALS onset), P120 (ALS symptomatic stage), nor P150 (ALS late symptomatic stage) (Supp. Table 1). Similarly, no significant differences were measured in hindlimb grip strength, forelimb grip strength, nor rotarod endurance analyses at those time points.
To assess functional muscle innervation by phrenic motor neurons, diaphragm compound muscular action potential (CMAP) amplitude following phrenic nerve stimulation was measured in non-ALS mice with each EAAT2 genotype. There was no statistically significant difference in CMAP amplitude amongst EAAT2WT/WT, EAAT2WT/CR, and EAAT2CR/CR mice at P90, P120, nor P140 (Supp. Table 2). The EAAT2-D504N mutation does not appear to have an effect on the motor function of wild-type mice, as no differences were observed in growth, grip strength, nor CMAP amplitude.
Generation of SOD1-G93A mice with EAAT2-D504N mutation
SOD1-G93A mice were crossed with EAAT2WT/CR mice to generate a novel bigenic model to determine the impact of EAAT2 cleavage on the development and progression of ALS. Twenty mice (10 female and 10 male) expressing SOD1-G93A and EAAT2WT/WT, EAAT2WT/CR, or EAAT2CR/CR were tracked for weight, symptom development, and survival. To ensure that observed differences were due to the EAAT2 mutation and not to changes in SOD1-G93A transgene expression levels, copy number was determined for all assessed mice by qPCR of mutant hSOD1-G93A, and all were found to fall within one CT (Supp. Table 3). Furthermore, copy number was not significantly different between the different EAAT2 groups (p=0.089).
We previously showed that CTE-SUMO1 accumulates in spinal cord homogenates with disease course in SOD1-G93A mice (Gibb et al., 2007). To demonstrate that EAAT2-D504N inhibits caspase-3 cleavage and thus formation of CTE-SUMO1 in vivo, accumulation of the modified fragment was compared in spinal cord homogenates of non-ALS mice and SOD1-G93A mice with each EAAT2 genotype. The presence of a SUMO1-positive band corresponding to CTE-SUMO1 fragment (Gibb et al., 2007) at approximately 25 kDa was significantly different depending on genotype (p = 0.023; Fig. 2B). Relative to the strong signal observed in SOD1-G93A mice with EAAT2WT/WT, the band was absent in non-SOD1-G93A mice (p = 0.007), slightly decreased in SOD1-G93A mice with EAAT2WT/CR mice (p = 0.124), and significantly decreased in SOD1-G93A mice with EAAT2CR/CR (p = 0.016), indicating that its formation is specific to SOD1-G93A expression and inhibited by the EAAT2-D504N mutation. The formation of the SUMO-1-positive 25 kDa was thus a result of caspase-3 cleavage of EAAT2 in the SOD1-G93A mouse.
EAAT2-D504N mutation confers protection to SOD1-G93A mice
Lifespan significantly differed between the EAAT2WT/WT, EAAT2WT/CR, and EAAT2CR/CR groups (p = 0.029), as assessed with Kaplan-Meier analysis (stratified by gender and using the Log-Rank post-hoc test). Average lifespans were 168.2 ± 3.8, 174.6 ± 4.6, and 176.0 ± 4.1 days for the female mice and 160.1 ± 2.7, 169.6 ± 3.4, and 167.5 ± 4.0 days for the male mice (Fig. 3A). To assess the relevance of the mutation, heterozygous and homozygous EAAT2 mutant mice were then combined into a single group as their lifespans were comparable. In this manner, an extension in lifespan of 4.2% for females and 5.3% for males due to the EAAT2-D504N mutation was observed.
Fig. 3. EAAT2-D504N protects SOD1-G93A mice.
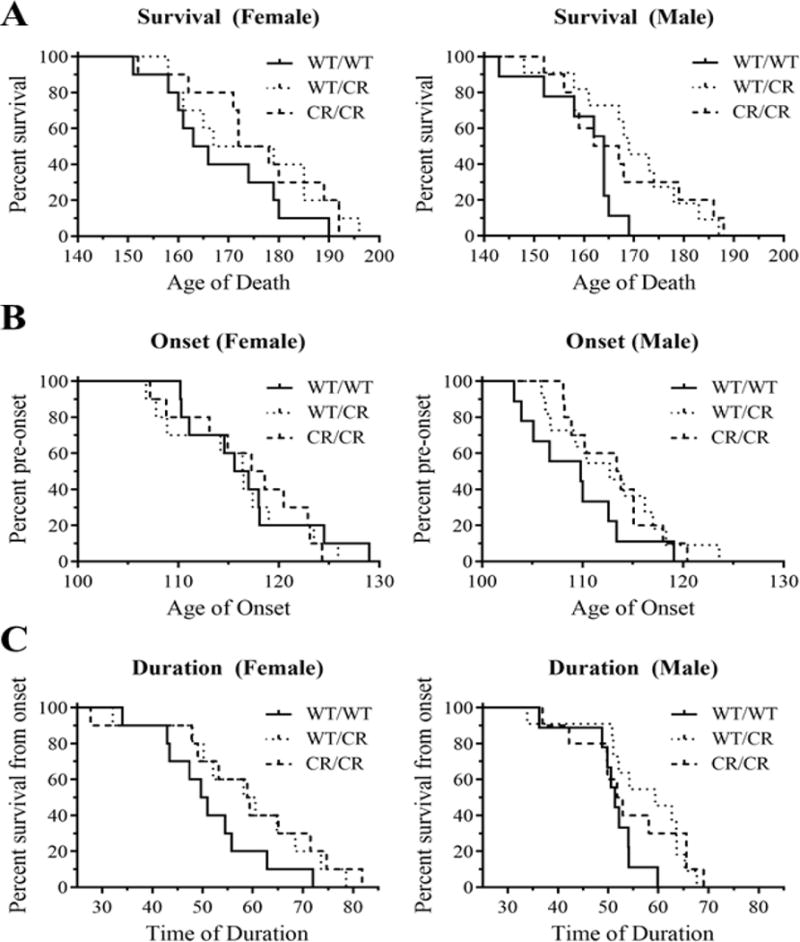
(A) The lifespan of SOD1-G93A mice was significantly extended in mice with the EAAT2-D504N mutation. (B) Onset of disease, as determined by age at peak weight, however, was unaltered in EAAT2 mutant mice. (C) The duration of disease, measured from age of onset to age of end stage was significantly delayed in EAAT2-D504N mice.
Growth was similar between the three EAAT2 genotypes; there was no difference in weight at P90 (p = 0.971), P120 (p = 0.914), nor P150 (p = 0.068), though males were significantly heavier than females at all ages (p < 0.001). Age of onset, as defined as the age of peak weight, did not differ between groups (p = 0.786; Fig. 3B).
Duration, defined by the difference between the age of onset and death, was significantly different between the three groups with each EAAT2 genotype (p = 0.022), confirming that the EAAT2-cleavage pathway is most relevant post-onset (Gibb et al., 2007). Disease duration for EAAT2WT/WT, EAAT2WT/CR, and EAAT2CR/CR was 51.4 ± 3.4, 58.7 ± 4.3, and 58.9 ± 4.9 days for females, respectively, and 50.8 ± 2.1, 56.8 ±2.9, and 54.2 ± 3.3 days for males (Fig. 3C). This represents an extension of progression time of 14.5% for females with at least one allele with the EAAT2-D504N mutation, and 9.3% for males.
EAAT2-D504N mutation delays loss of grip strength in SOD1-G93A mice
Behavioral assessments were conducted in addition to onset, progression, and end stage analysis to assess the degeneration of the motor system again using Kaplan-Meier analysis (stratified by gender, using the Log-Rank post-hoc test). Hindlimb and forelimb grip strengths were measured twice weekly and accelerating rotarod endurance was assessed once per week. Early symptoms appeared later in EAAT2-D504N mice. There was a delay in loss of 20% of maximum hindlimb grip strength (p = 0.008), which trended towards being even more delayed in females (p = 0.059; Fig. 4A). Post-hoc analysis revealed a delay in heterozygous (p = 0.003) and homozygous (p = 0.022) EAAT2-D504N mice. Though later in the disease course, there was also a delay in onset of loss of maximum forelimb grip strength (p = 0.003; Fig. 4B), again observed in both heterozygous (p = 0.002) and homozygous (p = 0.005) EAAT2-D504N mice as compared to EAAT2WT/WT mice. Both measures indicate that the early loss of motor neuron strength in SOD1-G93A mice was delayed when EAAT2-D504N was present.
Fig. 4. Development of early symptoms is delayed.
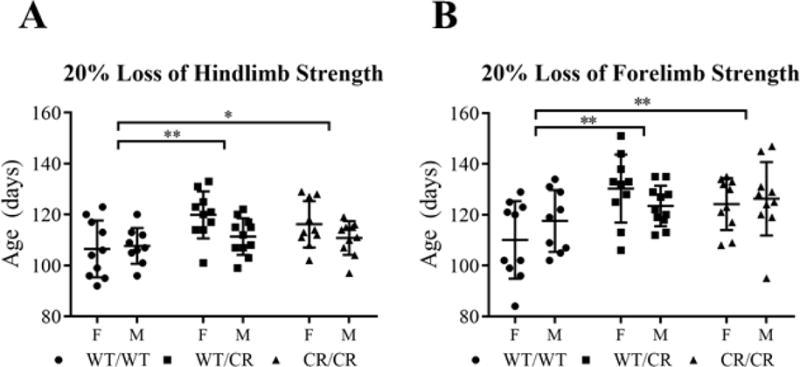
(A) Hindlimb weakness (as measured by 20% loss of grip strength) begins significantly later in EAAT2WT/CR and EAAT2CR/CR mice, relative to EAAT2WT/WT mice. (B) Forelimb weakness also began significantly later in EAAT2WT/CR and EAAT2CR/CR mice, relative to EAAT2WT/WT mice.
Mice were also tested on the accelerating rotarod as an additional measure of motor function, though it is considered a better measure of coordination than strength. Loss of ability to remain on the accelerating rotarod, as indicated by a decline of 50% from the maximum time, was not affected by EAAT2 genotype (p = 0.859). Gender did affect the development of decreased grip strength nor rotarod stamina in the different genotypes, indicating lack of gender specificity for the changes. Later measures of symptom development were not statistically different. No delay in the loss of 80% of hindlimb grip strength (p = 0.785), 80% of forelimb grip strength (p = 0.425), nor 80% loss of accelerating rotarod stamina (p = 0.732) was observed.
Diaphragm CMAP recordings and motor neuron counts
Functional diaphragm innervation by phrenic motor neurons of the cervical spinal cord was measured in a subset of mice at P90 (pre-symptomatic), P120 (symptomatic), and P140 (late symptomatic). Around the age of onset (P90), there was no statistical difference between non-ALS mice and SOD1-G93A mice with any EAAT2 genotype (p = 0.132; Fig. 5A–B). By early and late symptomatic stages (at P120 and P140), all SOD1-G93A groups had reduced CMAP amplitudes when compared to non-SOD1 mice (p ≤ 0.001 for all EAAT2 genotypes; Fig. 5A–B). At P140, however, the phrenic nerve CMAPs of SOD1-G93A mice expressing EAAT2WT/CR and EAAT2CR/CR trended towards higher amplitudes than the CMAPs of SOD1-G93A mice expressing EAAT2WT/WT, though the increases were not statistically significant (p = 0.158 and 0.148, respectively with LSD post-hoc test). A difference may have been difficult to demonstrate significantly due to variability in measurements across mice.
Fig. 5. No protection was seen to functional diaphragm innervation nor motor neuron survival.
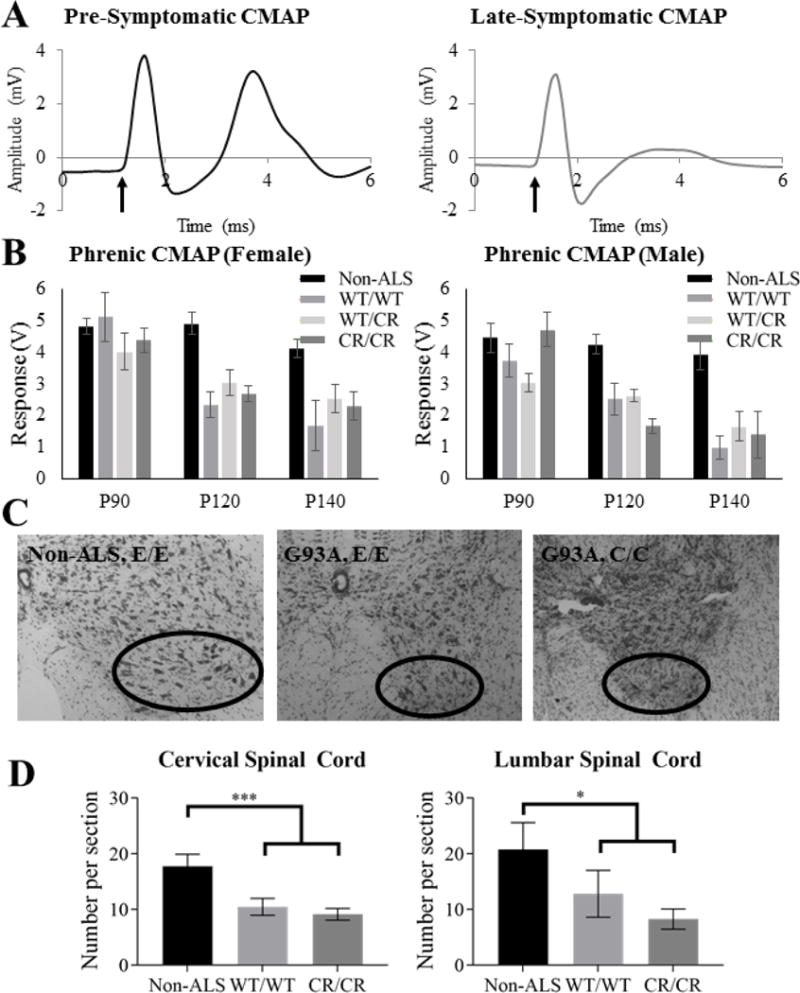
(A) Representative non-ALS (left) and diseased ALS (right) CMAP traces are shown. The arrow indicates the beginning of the phrenic nerve stimulus pulse and the second peak is the CMAP response measured at the ipsilateral hemi-diaphragm. (B) CMAP amplitudes of all SOD1-G93A groups were significantly lower than those of the non-ALS groups at P120 and P140, but not from one another. (C) Sample spinal cord ventral horns are shown. (D) While cervical motor neuron counts were significantly decreased in both ALS groups at P150, the EAAT2-D504N mutation offered no protection. Similarly, EAAT2WT/WT and EAAT2CR/CR groups of SOD1-G93A mice did not have significantly different motor neuron numbers in the earlier-affected lumbar spinal cord at P150, though both had significantly fewer than the non-ALS group.
As phrenic motor neurons are located in the cervical spinal cord, motor neuron counts in the cervical enlargement were determined at P150. While there was a decrease in motor neuron number as compared to wild type mice (p < 0.001 for EAAT2WT/WT and p < 0.001 for EAAT2CR/CR), no significant difference was measured between SOD1-G93A mice with EAATWT/WT or EAAT2CR/CR genotype (p = 0.504) (Fig. 5C–D). Similarly, no difference in motor neuron cell counts in the lumbar region at P150 was demonstrated (p = 0.269), while both differed significantly from the non-ALS control (p = 0.041 for EAAT2WT/WT and p = 0.003 for EAAT2CR/CR) (Fig. 5C–D).
DISCUSSION
EAAT2 is a critical astroglial transporter expressed throughout the CNS, clearing the majority of glutamate from the glutamatergic synaptic cleft (Lauriat and McInnes, 2007). Decreases have been reported in animal models of ALS and in some patient studies. The loss of expression and activity is thought to contribute to excitotoxicity in ALS, due to unmitigated glutamate signaling. Upregulation of EAAT2 in the spinal cord of SOD1-G93A mice by viral vector delivery, however, failed to protect motor neurons or extend lifespan (K. Li et al., 2015). Additionally, while protective in mice, treatment with ceftriaxone to increase EAAT2 expression in ALS patients failed to slow disease progression (Cudkowicz et al., 2014; Rothstein et al., 2005). It is possible that increased EAAT2 expression protects against excitotoxicity but also contributes to a secondary neurotoxic pathway that could occur in parallel.
In examining the decrease of EAAT2 in ALS, we noted that the transporter contains a unique caspase-3 consensus sequence, DTID (Boston-Howes et al., 2006). Activated in astrocytes in ALS, caspase-3 was shown to cleave EAAT2, generating a truncated transporter (trEAAT2) and a C-terminal end (CTE) that was found to be sumoylated (Boston-Howes et al., 2006; Foran et al., 2014, 2011; Gibb et al., 2007). The resulting modified fragment (CTE-SUMO1) accumulated in the spinal cord with disease progression and the fragment was not seen in non-disease-related tissues nor in other disease models (Gibb et al., 2007). Astrocytes expressing an artificial CTE-SUMO1 fragment were found to be toxic to motor neurons as compared to those expressing unmodified CTE (Foran et al., 2011). This toxicity was presumably conferred by one or more secreted factors, as astrocyte conditioned media from CTE-SUMO1 astrocytes was more toxic than that of CTE-expressing astrocytes.
We sought to assess the impact of this secondary pathway in vivo through the generation of a novel point mutation. The EAAT2-D504N mutation was designed to prevent caspase-3 cleavage and thus the generation of CTE-SUMO1 and its accumulation over disease progression. An EAAT2-D504N knock-in mouse was successfully generated on a C57Bl/6 background. The mutation successfully inhibited caspase-3 cleavage of EAAT2 in spinal cord homogenate, confirming the inhibition of cleavage in the consensus sequence and inhibition of CTE-SUMO1 formation, as it cannot be cleaved from the full-length protein. In vitro data demonstrated that the mutation has no effect on EAAT2 plasma membrane localization, glutamate affinity, nor glutamate uptake velocity. EAAT2-D504N had no effect on the growth and motor function of non-ALS mice over at least 200 days of age, as expected because CTE-SUMO1 generation via caspase-3 cleavage of EAAT2 was not seen in non-ALS tissue.
As hypothesized, the EAAT2-D504N mutation confers protection to the SOD1-G93A mouse model of ALS, although the extent of the effect is modest, suggesting that other pathological pathways remain at play. The mutation did not affect the onset of disease, as determined by the onset of weight loss, suggesting that the mutation is most impactful during disease progression rather than initial development. Homozygous EAAT2-D504N mice did not progress differently from heterozygous mice, suggesting a threshold effect wherein preventing only some of the cleavage is sufficient to confer protection from this neurotoxic pathway. However, this would be difficult to demonstrate experimentally because of the overall modest effect displayed by the homozygous genotype. Mice with one or two copies of the mutant allele had a modest extension of lifespan. As the majority of ALS cases are sporadic, treatment cannot begin until after disease onset, making extension of disease progression the more relevant measure of protection. More meaningfully, one or two copies of EAAT2-D504N caused an extension in progression time. The delay was also observed in the development of weakness, as mice with the EAAT2-D504N mutation lost 20% of both hindlimb and forelimb grip strength later than EAAT2WT/WT mice. EAAT2 mutations did not, however, alter the decline in weight loss. Notably, the decline in accelerating rotarod performance, diaphragm CMAP amplitude, and motor neuron number was also not improved in these mice, possibly due to insufficient sensitivity. Because the earlier losses in grip strength were delayed due to EAAT2-D504N expression, it is also possible that motor neurons were more preserved at earlier ages.
The modest protection and haplotype sufficiency suggest a secondary role for this pathway in ALS progression. As the D504N mutation inhibits caspase-3 cleavage of EAAT2, CTE-SUMO1 cannot be generated and cannot accumulate in astrocytes, inhibiting the downstream secretion of factors toxic to motor neurons. In non-mutant mice and in humans, however, the pathway might be exacerbated by increases in EAAT2 expression. Recent therapeutics have aimed to increase EAAT2 expression and/or function to decrease excitotoxicity. Libraries of compounds have been screened by a variety of methods for the ability to upregulate EAAT2 and some have shown promise in animal models of ALS (Benkler et al., 2015; Kong et al., 2014; Rothstein et al., 2005). Ceftriaxone, a blood-brain barrier permeable, FDA-approved antibiotic, was found by screening for upregulators of EAAT2 in organotypic rat spinal cord slices (Rothstein et al., 2005). Furthermore, it was successful in upregulating EAAT2 in mice, at delaying the loss of muscle strength and body weight, and extending the duration of disease in SOD1-G93A mice when administered beginning before or at onset (Rothstein et al., 2005). Unfortunately, in a Phase 3 clinical trial, ceftriaxone failed to decrease the functional decline nor delay survival in ALS patients (Cudkowicz et al., 2014). The efficacy of increasing EAAT2 expression, however, was not determined, so lack of clinical effect could be due to unsuccessful upregulation. Alternatively, a secondary pathway could prevent successful treatment: A potential cause for the discrepancy could be a concomitant increase in the CTE-SUMO1 pathway. While ceftriaxone might inhibit excitotoxicity by increasing EAAT2, that same increase might also cause the formation of additional astrocytic CTE-SUMO1. Studies of the impact of this mutation on ALS models treated with ceftriaxone or other EAAT2-increasing treatments could demonstrate additional protective effects.
Conclusions
Mutating the caspase-3 consensus sequence in EAAT2 prolongs disease progression, delays strength loss, and extends lifespan in the SOD1-G93A mouse model of ALS. While the extension of lifespan was modest, the mutation could confer additional protection with therapies designed to increase EAAT2 expression.
Supplementary Material
Highlights.
A novel bigenic SOD1-G93A mouse model carrying an EAAT2-D504N mutation was generated
The mutation delays ALS progression in SOD1-G93A mice
SOD1-G93A mice with the mutation have delayed loss of forelimb and hindlimb strength
EAAT2-D504N does not affect the onset of disease but extends survival in SOD1-G93A mice
Acknowledgments
Thank you to Ni Meng for grip strength measurements. LTR is the recipient of a Dubbs Scholar Fellowship Award.
Funding
This work was supported by the National Institutes of Health (grant number R01-NS044292 to DT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC. A Review of the Neural Mechanisms of Action and Clinical Efficiency of Riluzole in Treating Amyotrophic Lateral Sclerosis: What have we Learned in the Last Decade? CNS Neurosci Ther. 2011;17:4–31. doi: 10.1111/j.1755-5949.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkler C, Barhum Y, Ben-Zur T, Offen D. Multifactorial Gene Therapy Enhancing the Glutamate Uptake System and Reducing Oxidative Stress Delays Symptom Onset and Prolongs Survival in the SOD1-G93A ALS Mouse Model. J Mol Neurosci. 2015;58:46–58. doi: 10.1007/s12031-015-0695-2. [DOI] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V, Group, the A.S. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boston-Howes W, Gibb SL, Williams EO, Pasinelli P, Brown RH, Jr, Trotti D. Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J Biol Chem. 2006;281:14076–14084. doi: 10.1074/jbc.M600653200. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron. 1997;18:327–338. doi: 10.1016/S0896-6273(00)80272-X. [DOI] [PubMed] [Google Scholar]
- Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M, Garnes J, Dang V, Lievers J, Shoukat-Mumtaz U, Martinez R, Gai H, Blake R, Vaisberg E, Grskovic M, Johnson C, Irion S, Bright J, Cooper B, Nguyen L, Griswold-Prenner I, Javaherian A. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:355–364. doi: 10.1016/j.mcn.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillée S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Julien JP, Goldstein LSB, Cleveland DW. Wild-Type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Couratier P, Sindou P, Hugon J, Couratier P, Hugon J, Vallat JM, Dumas M. Cell culture evidence for neuronal degeneration in amyotrophic lateral sclerosis being linked to glutamate AMPA/kainate receptors. The Lancet. 1993;341:265–268. doi: 10.1016/0140-6736(93)92615-Z. [DOI] [PubMed] [Google Scholar]
- Cudkowicz ME, Titus S, Kearney M, Yu H, Sherman A, Schoenfeld D, Hayden D, Shui A, Brooks B, Conwit R, Felsenstein D, Greenblatt DJ, Keroack M, Kissel JT, Miller R, Rosenfeld J, Rothstein JD, Simpson E, Tolkoff-Rubin N, Zinman L, Shefner JM. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:1083–1091. doi: 10.1016/S1474-4422(14)70222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifita JA, Williams KL, Sundaramoorthy V, Mccann EP, Nicholson GA, Atkin JD, Blair IP. A novel amyotrophic lateral sclerosis mutation in OPTN induces ER stress and Golgi fragmentation in vitro. Amyotroph Lateral Scler Front Degener. 2016:1–8. doi: 10.1080/21678421.2016.1218517. [DOI] [PubMed] [Google Scholar]
- Foran E, Bogush A, Goffredo M, Roncaglia P, Gustincich S, Pasinelli P, Trotti D. Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia. 2011;59:1719–1731. doi: 10.1002/glia.21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran E, Rosenblum L, Bogush A, Pasinelli P, Trotti D. Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia. 2014 doi: 10.1002/glia.22677. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran E, Trotti D. Glutamate Transporters and the Excitotoxic Path to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Antioxid. Redox Signal. 2009;11:1587–1602. doi: 10.1089/ars.2009.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb SL, Boston-Howes W, Lavina ZS, Gustincich S, Brown RH, Jr, Pasinelli P, Trotti D. A caspase-3-cleaved fragment of the glial glutamate transporter EAAT2 is sumoylated and targeted to promyelocytic leukemia nuclear bodies in mutant SOD1-linked amyotrophic lateral sclerosis. J Biol Chem. 2007;282:32480–32490. doi: 10.1074/jbc.M704314200. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17:383–395. doi: 10.1038/nrn.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Chang LC, Takahashi K, Liu Q, Schulte DA, Lai L, Ibabao B, Lin Y, Stouffer N, Mukhopadhyay CD, Xing X, Seyb KI, Cuny GD, Glicksman MA, Lin CLG. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest. 2014;124:1255–1267. doi: 10.1172/JCI66163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauriat TL, McInnes LA. EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders. Mol. Psychiatry. 2007;12:1065–1078. doi: 10.1038/sj.mp.4002065. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Hala TJ, Seetharam S, Poulsen DJ, Wright MC, Lepore AC. GLT1 overexpression in SOD1G93A mouse cervical spinal cord does not preserve diaphragm function or extend disease. Neurobiol Dis. 2015;78:12–23. doi: 10.1016/j.nbd.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Li Y, Balasubramanian U, Cohen D, Zhang PW, Mosmiller E, Sattler R, Maragakis NJ, Rothstein JD. A Comprehensive Library of Familial Human Amyotrophic Lateral Sclerosis Induced Pluripotent Stem Cells. PLOS ONE. 2015;10:e0118266. doi: 10.1371/journal.pone.0118266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LPW. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016;90:521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Marin B, Boumédiene F, Logroscino G, Couratier P, Babron M-C, Leutenegger AL, Copetti M, Preux P-M, Beghi E. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol. 2016:dyw061. doi: 10.1093/ije/dyw061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su Z, Gupta P, Fisher PB. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sen I, Nalini A, Joshi NB, Joshi PG. Cerebrospinal fluid from amyotrophic lateral sclerosis patients preferentially elevates intracellular calcium and toxicity in motor neurons via AMPA/kainate receptor. J Neurol Sci. 2005;235:45–54. doi: 10.1016/j.jns.2005.03.049. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Rauen T, Fischer F, Wießner M, Grewer C, Bicho A, Pow DV. Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: Implications for CNS glutamate homeostasis. Glia. 2004;45:155–169. doi: 10.1002/glia.10317. [DOI] [PubMed] [Google Scholar]
- Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M, Ince PG, Lin C, Miller RG, Mitsumoto H, Nicholson G, Ravits J, Shaw PJ, Swash M, Talbot K, Traynor BJ, Van den Berg LH, Veldink JH, Vucic S, Kiernan MC. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RAA, Võsa U, de Jong S, Robinson MR, Yang J, Fogh I, van Doormaal PT, Tazelaar GHP, Koppers M, Blokhuis AM, Sproviero W, Jones AR, Kenna KP, van Eijk KR, Harschnitz O, Schellevis RD, Brands WJ, Medic J, Menelaou A, Vajda A, Ticozzi N, Lin K, Rogelj B, Vrabec K, Ravnik-Glavač M, Koritnik B, Zidar J, Leonardis L, Grošelj LD, Millecamps S, Salachas F, Meininger V, de Carvalho M, Pinto S, Mora JS, Rojas-García R, Polak M, Chandran S, Colville S, Swingler R, Morrison KE, Shaw PJ, Hardy J, Orrell RW, Pittman A, Sidle K, Fratta P, Malaspina A, Topp S, Petri S, Abdulla S, Drepper C, Sendtner M, Meyer T, Ophoff RA, Staats KA, Wiedau-Pazos M, Lomen-Hoerth C, Van Deerlin VM, Trojanowski JQ, Elman L, McCluskey L, Basak AN, Tunca C, Hamzeiy H, Parman Y, Meitinger T, Lichtner P, Radivojkov-Blagojevic M, Andres CR, Maurel C, Bensimon G, Landwehrmeyer B, Brice A, Payan CAM, Saker-Delye S, Dürr A, Wood NW, Tittmann L, Lieb W, Franke A, Rietschel M, Cichon S, Nöthen MM, Amouyel P, Tzourio C, Dartigues J-F, Uitterlinden AG, Rivadeneira F, Estrada K, Hofman A, Curtis C, Blauw HM, van der Kooi AJ, de Visser M, Goris A, Weber M, Shaw CE, Smith BN, Pansarasa O, Cereda C, Del Bo R, Comi GP, D’Alfonso S, Bertolin C, Sorarù G, Mazzini L, Pensato V, Gellera C, Tiloca C, Ratti A, Calvo A, Moglia C, Brunetti M, Arcuti S, Capozzo R, Zecca C, Lunetta C, Penco S, Riva N, Padovani A, Filosto M, Muller B, Stuit RJ, PARALS Registry SLALOM Group SLAP Registry FALS Sequencing Consortium SLAGEN Consortium NNIPPS Study Group. Blair I, Zhang K, McCann EP, Fifita JA, Nicholson GA, Rowe DB, Pamphlett R, Kiernan MC, Grosskreutz J, Witte OW, Ringer T, Prell T, Stubendorff B, Kurth I, Hübner CA, Leigh PN, Casale F, Chio A, Beghi E, Pupillo E, Tortelli R, Logroscino G, Powell J, Ludolph AC, Weishaupt JH, Robberecht W, Van Damme P, Franke L, Pers TH, Brown RH, Glass JD, Landers JE, Hardiman O, Andersen PM, Corcia P, Vourc’h P, Silani V, Wray NR, Visscher PM, de Bakker PIW, van Es MA, Pasterkamp RJ, Lewis CM, Breen G, Al-Chalabi A, van den Berg LH, Veldink JH. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043–1048. doi: 10.1038/ng.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegorzewska I, Baloh RH. TDP-43-Based Animal Models of Neurodegeneration: New Insights into ALS Pathology and Pathophysiology. Neurodegener Dis. 2011;8:262–274. doi: 10.1159/000321547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Nalbandian A, Hsu CI, Li S, Llewellyn KJ, Mozaffar T, Kimonis VE, Weiss JH. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 2012;3:e374. doi: 10.1038/cddis.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.