

Abstract
Neuroinflammation contributes to neuronal deficits in neurodegenerative CNS (central nervous system) autoimmune diseases, such as multiple sclerosis and uveitis. The major goal of most treatment modalities for CNS autoimmune diseases is to limit inflammatory responses in the CNS; immune-suppressive drugs are the therapy of choice. However, lifelong immunosuppression increases the occurrence of infections, nephrotoxicity, malignancies, cataractogenesis, and glaucoma, which can greatly impair quality of life for the patient. Biologics that target pathogenic T cells is an alternative approach that is gaining wide acceptance as indicated by the popularity of a variety of Food and Drug Administration (FDA)-approved anti-inflammatory compounds and humanized antibodies such as Zenapax, Etanercept, Remicade, anti-ICAM, rapamycin, or tacrolimus. B cells are also potential therapeutic targets because they provide costimulatory signals that activate pathogenic T cells and secrete cytokines that promote autoimmune pathology. B cells also produce autoreactive antibodies implicated in several organ-specific and systemic autoimmune diseases including lupus erythematosus, Graves’ disease, and Hashimoto’s thyroiditis. On the other hand, recent studies have led to the discovery of several regulatory B-cell (Breg) populations that suppress immune responses and autoimmune diseases. In this review, we present a brief overview of Breg phenotypes and in particular, the newly discovered IL35-producing regulatory B cell (i35-Breg). We discuss the critical roles played by i35-Bregs in regulating autoimmune diseases and the potential use of adoptive Breg therapy in CNS autoimmune diseases.
Keywords: interleukin 35, interleukin 10, regulatory B cell or Breg, interleukin 35-producing Breg or i35-Breg, autoimmunity, experimental autoimmune uveitis
I. INTRODUCTION
Central nervous system (CNS) autoimmune diseases such as uveitis and multiple sclerosis (MS) are characterized by repeated cycles of remission and recurrent inflammation and are often associated with the presence of B cells, T cells, and macrophages in various regions of the brain, spinal cord, or uvea.1,2 Perivascular accumulations of lymphocytes surrounding venules in the affected tissues led to the view that T cells may play a central role in etiology of these diseases. Experimental evidence for the involvement of T cells in CNS autoimmune diseases have come from studies of experimental autoimmune uveitis (EAU) and experimental autoimmune encephalomyelitis (EAE), animal models of uveitis, and MS, respectively.1–6 Th17 cells and type-17 signature cytokines such as interleukin 17 (IL17), IL22, IL23, and granulocyte-macrophage colony-stimulating factor (GM-CSF) have been implicated in the pathogenesis of these autoimmune diseases, fueling interest in developing biologics that can be used to target T cells and signal transduction pathways that regulate Th17 development and effector functions.7–10 However, Phase II clinical trials of humanized monoclonal antibodies such as ustekinumab and briakinumab (subunit of IL12 and IL23), brodalumab (anti-IL17RA), or secukinumab and ixekizumab (anti-IL17A) have resulted in mixed results and fallen short of expectations.11 An important exception is rituximab (Rituxan or Zytux), which targets the B-lymphocyte-cell-surface protein CD20.12 In fact, B-cell depletion using rituzimab has been effective for the treatment of rheumatoid arthritis.12 However, the efficacy of rituximab was subsequently shown to derive in part from the expansion of rare regulatory B-cell (Breg) populations, and disease suppression is related to secretion of the anti-inflammatory cytokine IL10 by the Bregs.13–15 In this review, we provide a brief overview of B-cell and regulatory Breg development, with particular focus on interleukin 35–producing regulatory B cells (i35-Breg) and their involvement in regulating inflammation in the brain and retina.
II. B-CELL DEVELOPMENT
The B-lymphocyte developmental program is initiated in the bone marrow from hematopoietic precursor cells derived from the fetal liver (Fig. 1).16 The B cell develops from the pro-B cell (CD19+CD34+CD10+IgM−) into the pre-B cell (CD19+CD34−CD10+IgM−) following induction of recombination-activating genes (RAG1 and RAG2) and production of the heavy chain immunoglobulin (Ig).17 The immature B cell with a functional pre-B B-cell receptor (BCR) emerges following association of the Ig μ chains with invariable surrogate light chains (λC and Vpre-B9).18 Immature B cells that do not express a functional BCR undergo developmental arrest and are subjected to receptor editing to produce a new receptor. Before leaving the bone marrow, potentially autoreactive immature B cells that react strongly to self-antigens are eliminated by central tolerance mechanisms while positively selected immature B cells, with requisite affinity to self-antigens, switch-off expression of their RAG1 and RAG2 genes following generation of the functional receptor.19 The immature B cells first seed the blood as transitional T1 B cells (IgM+CD10+) and then proceed into the lymphoid follicles of the spleen for further maturation into transitional T2 cells (IgM+IgD+CD10+CD23+).20 Final maturation of the transitional T2 cells into mature naïve B cells (IgM+IgD+CD10−) occurs in the spleen. To prevent any possibility of autoimmunity, immature transitional T2 cells are further subjected to peripheral tolerance mechanisms that delete or render potentially autoreactive B cells anergic21 (Fig. 1). Several transcription factors including EA2, EBF, and Pax5 play essential roles in B-cell differentiation and commitment to the plethora of highly diverse conventional follicular (B2), marginal zone (MZ), B1 or Breg phenotypes; a great deal is now known about these distinct B-lymphocyte phenotypes and subsets.22
FIG. 1.
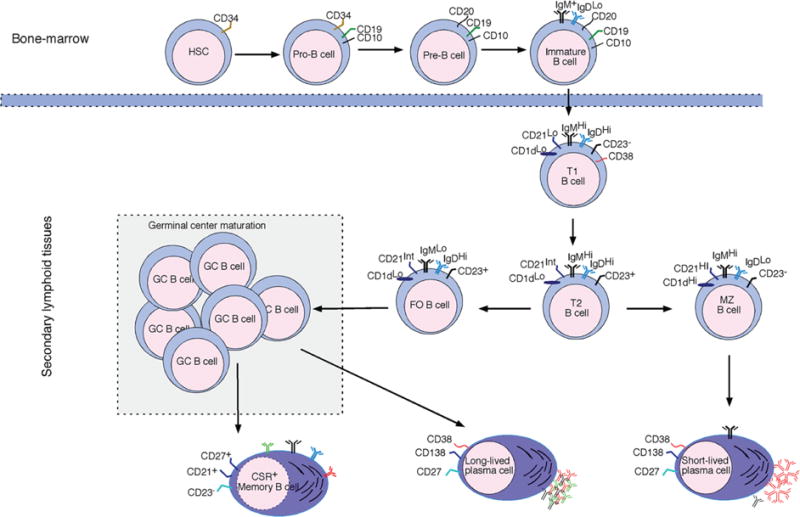
Sequential development of B cells in the bone marrow and maturation in the spleen. Differential expression of cell-surface markers has allowed delineation of the various B-cell phenotypes that emerge as the hematopoietic precursor B cells derived from fetal liver progress from the pro-B cells following induction (RAG1 and RAG2) and production of the heavy chain IG. Immature B cells exit the bone marrow after subjection to central tolerance mechanisms that eliminate autoreactive immature B cells. After leaving the bone marrow, T1 and T2 immature B cells are further subjected to peripheral tolerance mechanisms before full maturation into MZ, follicular, or plasma B cells. (RAG) Recombination-activating gene; (T1) transitional 1; (T2) transitional 2; (MZ) marginal zone; (FO) follicular-type-B cell.
A. Marginal Zone B Lymphocytes
Immature B cells that have successfully undergone positive and negative selection progress through transient transitional (T1 and T2) B-cell stages. Transitional (T1 and T2) B cells can mature into follicular-type-I (FO-I) B cells in a BCR and Bruton’s tyrosine kinase (Btk)-dependent manner or into follicular-type-II (FO-II) B cells in a B-cell-activating factor (BAF)-Btk-independent manner. The least mature transitional B cells are the T1 B cells, and soon after leaving the bone marrow, they enter the lumen of the red pulp or MZ venule with fenestrated architecture that promotes access to Notch2/DL1 (δ-like 1) inductive signals.23 Interactions with DL1+ vascular endothelial cells promote their retention in the MZ and maturation into MZ B cells.24,25 MZ B cells are capable of responding to both T-dependent and T-independent antigens (Ags), and their growth or expansion is highly dependent on Notch2 signals. Localization of MZ B cells at the MZ, red pulp junction, and near the blood-rich marginal sinus contributes to their diverse responses during host defense against blood-borne pathogens. While stimuli such as the polysaccharides of encapsulated bacteria elicit Ag-specific B-cell responses, LPS, CpG, or poly-IC induce polyclonal B-cell activation via Toll-like receptors. Their canonical Ig receptors allow rapid short-lived antibody responses to viruses and are critical for protective immunity against encapsulated bacteria that cause human pneumonia, septicemia, and meningitis.
B. B1 B Lymphocytes
Like MZ B cells, the B-1 (B1a and B1b) subset plays important roles in immunity to viruses and Gram-positive and Gram-negative bacteria. They are thought to derive from mouse fetal-liver hematopoietic stem cells but reside primarily in the peritoneal cavity and gut-associated lymphoid tissues. The B1 subset is divided into two subtypes based on CD5 expression. The B1a subtype is characterized by production of natural antibodies that provide innate protection against bacterial infections in naïve hosts, whereas B1b cells produce long-term adaptive antibody responses to polysaccharides and other TI–T2 Ags during infection.26,27 Notwithstanding their distinct anatomic localization, MZ and B-1 B cells have overlapping functions. Because they interact with a wide array of pathogens (T-dependent and T-independent Ags), they contribute to rapid innate-like responses. However, they also exhibit a relatively low threshold for Ag activation, making them more likely to produce autoreactive Abs that might contribute to autoimmune pathology.
C. Conventional or Follicular (B2) B Cells
The majority of transitional B cells are the more mature IgD+CD23+ T2 B cells. In contrast to T1 B cells, T2 cells are not retained in the MZ because they do not have adequate access to the DL1+ endothelial cells and Notch2 signals.23,28 They readily enter lymphoid follicles where they undergo further maturation, acquire the capacity to recirculate, and eventually become mature follicular B cells also known as conventional or B2 cells.29 T2 B cells are thought to mature into FO-I B cells if they are high-affinity self-reactive B cells. On the other hand, low-affinity self-reactive T2 B cells can become recirculating mature FO-II B cells if the MZ compartment is completely full with MZ B cells.25 It is thought that FO-II B cells replenish or reconstitute the MZ pool during diseases states that deplete MZ B cells. The bulk of B cells in the peripheral circulation are FO B cells and comprise >95% of the B cells in peripheral lymph nodes. They interact and present Ag to T cells at the edge of the follicles, between the T- and B-cell zones, and signals emanating from CD40/CD40L interactions and T-cell-secreted cytokines induce them to proliferate and differentiate into short-lived antibody-secreting extra-follicular B cells. Follicular B cells that subsequently enter follicles collaborate with follicular T-helper (TFH) cells to form germinal centers and undergo further clonal expansion. Repeated interactions with the same Ag presented by follicular dendritic cells facilitate the activation of somatic hypermutation mechanisms and the generation of antibodies of high affinity/avidity. These cells ultimately undergo terminal differentiation into memory B cells (CD19+CD27+ or long-lived Ig-secreting CD138+ plasma cells. Upon encounter with a cognate Ag in extrafollicular lymphoid organs or bone marrow, recirculating follicular cells can undergo isotype switching and begin secreting different classes of antibodies (IgM, IgG, IgA, and IgE). Although the switched mature B cells share the same Ag specificity, interactions of the various constant regions with specific Ig Fc receptors or complement proteins confer additional effector functions. These Ag-specific recirculating B cells differ from T-independent B cells in some important ways. For example, T-independent B cells are unable to undergo somatic hypermutation, class switch recombination, or memory cell generation.
D. Regulatory B Cells
Besides the production of autoantibodies, B cells have other functions including antigen presentation and activation of T cells, expression of costimulatory molecules, and secretion of effector cytokines. Similar to T cells, B cells can suppress immune responses and inhibit inflammation. B-lymphocyte populations that mediate cellular immune suppression in vitro and in vivo are collectively referred to as Bregs and have been reported in humans and mice.30 However, they are a diverse group of B cells and there is no unique marker, or set of markers, that exclusively identifies the Breg.30 Although the exact mechanisms underlying suppressive actions of Breg cells remain an area of intense investigation, a variety of cytokines produced by Bregs have been implicated, with IL10 being the most studied.31 B1a cells (B220+CD5+) in the mouse peritoneal cavity were one of the first subsets to be identified as a source of IL10, and subsequent studies revealed the generation of IL10-producing CD1d+ B cells in gut-associated lymphoid tissues during chronic intestinal inflammation.32–34 Other B-cell types that produce IL10 include splenic MZ B cells (CD19+CD21+CD23−CD24+IgM+CD1d+) and T2-MZP (CD19+CD21+CD23+CD24+IgM+CD1d+) cells (Fig. 2). Bregs that are functionally defined mainly by their competency to produce and secrete IL10 following appropriate stimulation are denoted as B10 cells.31,35 However, it is now clear that Bregs can suppress immune responses through other mechanisms besides IL10 production. Two recent reports described a novel i35-Breg population that mediates suppression of CNS inflammatory diseases though IL3536,37 or secretion of IL35 and IL10.37 Because Bregs comprise many phenotypically distinct B-cell lineages and appear to be identified by the production of the anti-inflammatory cytokines IL10 or IL35, it could be argued that Bregs are not truly a distinct lineage but rather possess a common effector function that can be induced by any B cells. However, the i35-Breg population is unique in that it is produced only by CD138+ plasma cells, and stringent regulation of the coexpression of Ebi3 and IL12p35 by this B-cell type suggests that it is a distinct B-cell lineage. Here, we highlight the current knowledge on the enigmatic i35-Breg and its use in the treatment of autoimmune diseases.
FIG. 2.
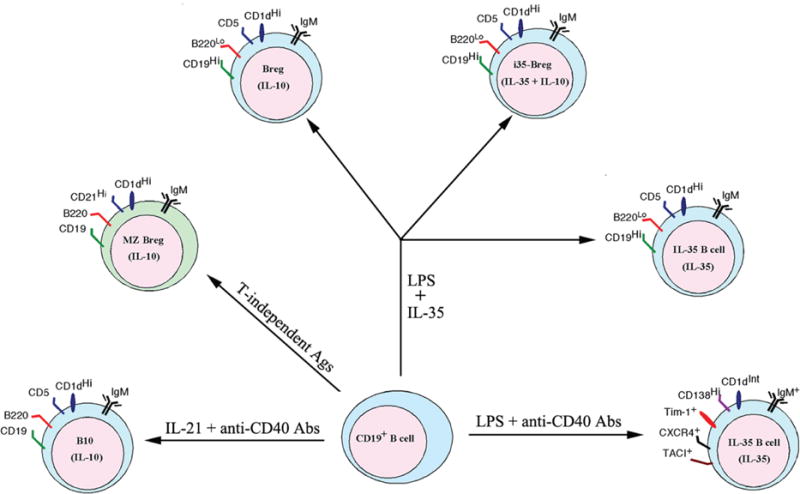
Phenotypes of presently described Breg subsets. Prior activation by TLR and/or BCR signals appears to be required for generation of IL10-producing, IL35-producing, and IL10-/IL35-producing Bregs expressing overlapping cell-surface markers shared by Bregs and conventional B cells.
E. i35-Bregs
Recent studies have revealed that suppressive activity of B cells requires prior activation by Toll-like receptor (TLR). Mice with targeted deletion of TLR4 in B cells developed exacerbated EAE, underscoring the critical for B-cell-mediated suppression of an autoimmune disease.36,38 Interestingly, naïve B cells can produce IL10 upon activation by lipopolysaccharide (LPS) but costimulation with LPS plus anti-CD40 switched off IL10 production while inducing IL35 production.36 At approximately the same time, we genetically engineered a heterodimeric IL35 (rIL35) using a bicistronic vector.37 Before these studies, it was thought that the secretion of IL35 was restricted to regulatory T cells (Tregs).39 Screening a wide variety of hematopoietic and lymphoid cell types to determine other potential targets of IL35 effects led to the discovery that IL35 inhibits the proliferation of CD19+B220hiCD5− B cells while inducing IL10-producing CD5+CD19+B220lo Bregs to produce IL35.37 It is notable that following LPS-driven B-cell activation in mice, ~8% of the IL10-producing B cells in the mouse spleen also produced IL35. Culturing the Bregs in the presence of rIL35 increased the level of the IL35-producing B cells to ~35%, with ~18% of these cells coproducing IL10 and IL35.37 These observations suggest that IL10- and IL35-producing Breg cells may either be overlapping Breg subsets or Breg cells at different stages of development. These observations provided direct evidence that IL35 can induce the conversion of conventional B cells or B10 cells into the novel IL35-producing B cells named i35-Breg.37
III. IL35 SIGNALING MECHANISM IN Breg CELLS
The IL12 family of cytokines has emerged as important regulators of hematopoietic cell lineage commitment, differentiation, and function. The family is comprised of four heterodimeric cytokines: IL12 (IL12p35/IL12p40), IL23 (IL23p19/IL12p40), IL27 (IL27p28/Ebi3), and IL35 (IL12p35/Ebi3). Each member is composed of an α subunit, with a helical structure similar to type-1 cytokines such as IL6, and a β subunit structurally related to the soluble IL6 receptor (IL6Rα).40 Chain-pairing promiscuity is a distinctive feature that accounts for their involvement in many aspects of host immunity.41 Some members (IL12 and IL23) are immunostimulatory and induce autoimmune pathology, but IL35 suppresses cellular immune responses and mitigates autoimmune diseases.41,42 In T cells, the IL35 signal is mediated through receptors comprising IL12Rβ2/gp130, IL12Rβ2/IL12Rβ2, or gp130/gp130.42 However, it is not clear which of these is the high-affinity IL35 receptor that activates receptor-associated JAK1 and JAK2. On the other hand, the IL35 receptor identified in B cells comprises IL12Rβ2 and IL27Rα, but analysis of IL35-receptor usage in B cells did not examine whether the IL12Rβ2/IL12Rβ2 or IL27Rα/IL27Rα homodimer is also used.37 Nonetheless, in B cells, signaling downstream from the IL35 receptor activates STAT1 and STAT3. In T cells, signaling through IL12Rβ2/gp130 induces STAT1 and STAT4 phosphorylation, whereas signaling through IL12Rβ2 or gp130 homodimers activates STAT4 or STAT1 only, respectively.41,42 It is interesting that gp130 homodimerization, as a functional receptor signaling pathway, has only been observed in viral IL6 signaling or during IL6 trans-signaling, where IL6 complexes with soluble IL6R.43,44 It is of note that in the later cases, both STAT1 and STAT3 is activated, in stark contrast to the activation of only STAT1 or STAT4 by IL35 noted in T cells.45,46 Nonetheless, it is still unclear whether difference in receptor and STAT use in response to IL35 derives from intrinsic differences between IL35 signaling mechanisms of T and B cells or due to differences in IL35 preparations used for the analyses.
IV. Breg THERAPY
IL35 suppresses lymphocyte proliferation and effector functions by inducing the expansion of IL35-producing regulatory T cells (iTR3547) and Bregs.37 Demonstration that rIL35 can induce ex-vivo conversion of mouse or human B cells into IL35 and IL10-producing Breg cells led us to investigate whether ex-vivo-generated Bregs and i35-Bregs can be used to treat an organ-specific autoimmune disease such as uveitis. EAU is the animal model of human uveitis and is induced in mice by immunization with interphotoreceptor retinoid-binding protein (IRBP) in Complete Freund’s adjuvant (CFA).48 Onset of EAU occurs typically between 12 and 14 days, with the disease peaking between days 16 and 21. The mice develop severe inflammation characterized by papilledema, retinal vasculitis, retinal folds, and infiltration of inflammatory cells into the vitreous.10 Treatment of mice with rIL35 conferred protection from ocular pathology by inducing expansion of Bregs and i35-Bregs in the spleen and lymph nodes, and the suppression of uveitis was accompanied by inhibition of pathogenic Th17 cells while promoting the expansion of Treg cells.37 Adoptive transfer of ex-vivo-generated Bregs also suppressed EAU by inducing endogenous expansion of Breg, Tregs, and i35-Bregs while inhibiting Th17 expansion. It is also remarkable that mice that lack IL35 or are defective in IL35-signaling develop exacerbated uveitis, with reduced capacity to produce i35-Breg and IL10-producing Bregs, further underscoring the critical roles of Bregs in regulating intraocular inflammation.37 Taken together, IL35 produced by i35-Bregs may have dual effects on host immunity. By inhibiting the expansion of Th17 cells, IL35 might be involved in preventing the activation of pathological immune responses mediated by this pathogenic T-cell subset. Its expansion of Breg, Tregs, and i35-Bregs in the spleen and lymph nodes may serve to limit the duration and intensity of immune responses that promote the development of autoimmune diseases. However, the extent to which the anti-inflammatory effect derives from intrinsic effects of Bregs or the anti-inflammatory cytokines that they produce is still unclear.
The role of IL35-producing Bregs has also been established in the EAE model. EAE was induced in wild-type (WT) mice or bone marrow chimeric mice reconstituted with IL12p35−/−, Ebi3−/−, IL12p40−/−, or IL27p28−/− B cells by immunization with MOG35–55 peptide in CFA. In contrast to mice reconstituted with IL12p40−/− or IL27p28−/− B cells, mice that received IL12p35−/− or Ebi3−/− B cells developed exacerbated EAE, indicating that provision of IL35 and i35-Breg is required for recovery from EAE.36 Similar to observations in the EAU model, mice with loss of IL35 expression in the B-cell compartment could not recover from EAE.36 On the other hand, mice that lack IL35 or are defective in IL35-signaling were more resistant to infection with the intracellular bacterial pathogen Salmonella enterica serovar Typhimurium. Compared to control mice, they exhibited superior containment of bacterial growth and prolonged survival after the primary infection.36 The observed effects in the EAE and bacteria infection studies were attributed to the expansion of IL35- and IL10-producing plasma cells exhibiting the IgM+CD138hiTACI+CXCR4+ CD1dintTim1int phenotype.
V. CONCLUSION
The recent discoveries of B cells that produce the anti-inflammatory cytokine IL35 expand the repertoire of Breg subsets that can be exploited therapeutically and suggests that additional Breg subsets will probably be identified in the future. Bregs are relatively rare, comprising <3% of total B cells in mice and humans, and there are significant scientific and therapeutic interests to discover factors that regulate the generation and induction of Bregs. The physiological inducers of IL10- and IL35-producing Bregs are still unknown. With regard to the IL35-producing Breg or i35-Breg subset, it remains to be determined whether this comprises several subtypes that can be generated in response to distinct physiological inducers. It is notable that stimulation of B cells by LPS induces the expansion of IL10-producing Bregs, whereas costimulation with LPS and anti-CD40 Abs promotes the expansion of IL35-producing Bregs, suggesting that generation of i35-Bregs may have obligatory requirement of T-helper cells.36 These observations also beg the question as to whether i35-Bregs and IL10-producing Breg cells are overlapping subsets or exist as distinct Breg populations at different stages of B-cell development. In fact, many other basic questions regarding the roles of TLR, CD40L, and cytokines such as IL21 and IL35 in the induction of Bregs still remain. For example, do these factors induce de novo differentiation or conversion of conventional B cells into the Breg phenotypes or do they merely expand pre-existing B10 and i35-Bregs populations? Does the same cell coordinately express the two subunits of IL35 or can they be expressed as individual IL12p35 and Ebi3 subunits, which then associate extracellularly to form the functional IL35? What factors regulate the stability of the non-covalently linked IL35 (p35 and Ebi3) heterodimer? What factors regulate their dissociation to allow termination of their inhibitory activities? Notwithstanding the fact that there may be more questions than answers, the discovery that IL35 induces the conversion of human/mouse B cells into Bregs allows ex-vivo production of large amounts of Bregs for immunotherapy. It would also undoubtedly facilitate elucidation of the roles of Bregs and i35-Bregs in the regulation of autoimmune diseases.
ABBREVIATIONS
- IL-35
Interleukin 35
- IL-10
Interleukin 10
- Breg
regulatory B cell
- i35-Breg
IL-35-producing regulatory B cell
- CNS
central nervous system
- EAU
experimental autoimmune uveitis
- EAE
experimental autoimmune encephalomyelitis
- MS
multiple sclerosis
- STAT
signal transducer and activator of transcription
- MZ
marginal zone
- FO
follicular
References
- 1.Caspi RR. A look at autoimmunity and inflammation in the eye. J Clin Invest. 2010;120(9):3073–3083. doi: 10.1172/JCI42440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol. 2009;9(6):440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- 3.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. T(H)17 cells contribute to uveitis and scleritis and are expanded by IL-2 and in hibited by IL-27/STAT1. Nat Med. 2007;13(6):711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 4.Caspi RR. Understanding autoimmune uveitis through animal models. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 2011;52(3):1872–1879. doi: 10.1167/iovs.10-6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248(1):205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simmons SB, Pierson ER, Lee SY, Goverman JM. Modeling the heterogeneity of multiple sclerosis in animals. Trends Immunol. 2013;34(8):410–422. doi: 10.1016/j.it.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–63. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol. 2008;180(9):6070–76. doi: 10.4049/jimmunol.180.9.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardol DM, Drake CG. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179(7):4313–17. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 10.Oh HM, Yu CR, Lee Y, Chan CC, Maminishkis A, Egwuagu CE. Autoreactive memory CD4+ T lymphocytes that mediate chronic uveitis reside in the bone marrow through STAT3-dependent mechanisms. J Immunol. 2011;187(6):3338–3346. doi: 10.4049/jimmunol.1004019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–81. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 13.Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev. 2008;224:201–14. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 14.Bouaziz JD, Yanaba K, Venturi GM, Wang Y, Tisch RM, Poe JC, Tedder TF. Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc Natl Acad Sci USA. 2007;104(52):20878–883. doi: 10.1073/pnas.0709205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pers JO, Daridon C, Bendaoud B, Devauchelle V, Berthou C, Saraux A, Youinou P. B-cell depletion and repopulation in autoimmune diseases. Clin Rev Allergy Immunol. 2008;34(1):50–5. doi: 10.1007/s12016-007-8015-4. [DOI] [PubMed] [Google Scholar]
- 16.Osmond DG. B cell development in the bone marrow. Semin Immunol. 1990;2(3):173–80. [PubMed] [Google Scholar]
- 17.Schatz DG, Baltimore D. Uncovering the V(D)J recombinase. Cell. 2004;116(2 Suppl):S103–6. doi: 10.1016/s0092-8674(04)00042-x. 2 p following S6. [DOI] [PubMed] [Google Scholar]
- 18.Martensson IL, Keenan RA, Licence S. The pre-B-cell receptor. Current opinion in immunology. 2007;19(2):137–42. doi: 10.1016/j.coi.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 19.von Boehmer H, Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol. 2010;11(1):14–20. doi: 10.1038/ni.1794. [DOI] [PubMed] [Google Scholar]
- 20.Engel P, Gomez-Puerta JA, Ramos-Casals M, Lozano F, Bosch X. Therapeutic targeting of B cells for rheumatic autoimmune diseases. Pharmacol Rev. 2011;63(1):127–56. doi: 10.1124/pr.109.002006. [DOI] [PubMed] [Google Scholar]
- 21.Pillai S, Cariappa A. The bone marrow perisinusoidal niche for recirculating B cells and the positive selection of bone marrow-derived B lymphocytes. Immunol Cell Biol. 2009;87(1):16–9. doi: 10.1038/icb.2008.89. [DOI] [PubMed] [Google Scholar]
- 22.Bartholdy B, Matthias P. Transcriptional control of B cell development and function. Gene. 2004;327(1):1–23. doi: 10.1016/j.gene.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Tan JB, Visan I, Yuan JS, Guidos CJ. Requirement for Notch1 signals at sequential early stages of intrathymic T cell development. Nat Immunol. 2005;6(7):671–9. doi: 10.1038/ni1217. [DOI] [PubMed] [Google Scholar]
- 24.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science. 2002;297(5580):409–12. doi: 10.1126/science.1071632. [DOI] [PubMed] [Google Scholar]
- 25.Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. 2009;9(11):767–77. doi: 10.1038/nri2656. [DOI] [PubMed] [Google Scholar]
- 26.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity. 2004;21(3):379–90. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 27.Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, Tedder TF. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005;174(7):4389–99. doi: 10.4049/jimmunol.174.7.4389. [DOI] [PubMed] [Google Scholar]
- 28.Tan JB, Xu K, Cretegny K, Visan I, Yuan JS, Egan SE, Guidos CJ. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity. 2009;30(2):254–63. doi: 10.1016/j.immuni.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Allman D, Calamito M. Instructing B cell fates on the fringe. Immunity. 2009;30(2):175–7. doi: 10.1016/j.immuni.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–41. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 31.Tedder TF, Matsushita T. Regulatory B cells that produce IL-10: a breath of fresh air in allergic airway disease. The Journal of allergy and clinical immunology. 2010;125(5):1125–7. doi: 10.1016/j.jaci.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev. 2006;5(6):403–8. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16(2):219–30. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 34.O’Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur J Immunol. 1992;22(3):711–7. doi: 10.1002/eji.1830220314. [DOI] [PubMed] [Google Scholar]
- 35.Kalampokis I, Yoshizaki A, Tedder TF. IL-10-producing regulatory B cells (B10 cells) inautoimmune disease. Arthritis Res Ther. 2013;15(Suppl 1):S1. doi: 10.1186/ar3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, Li R, Jouneau L, Boudinot P, Wilantri S, Sakwa I, Miyazaki Y, Leech MD, McPherson RC, Wirtz S, Neurath M, Hoehlig K, Meinl E, Grützkau A, Grün JR, Horn K, Kühl AA, Dörner T, Bar-Or A, Kaufmann SH, Anderton SM, Fillatrea IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366–370. doi: 10.1038/nature12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, Wingfield PT, Kim SH, Egwuagu CE. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20(6):633–641. doi: 10.1038/nm.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fillatreau S. Novel regulatory functions for Toll-like receptor-activated B cells during intracellular bacterial infection. Immunol Rev. 2011;240(1):52–71. doi: 10.1111/j.1600-065X.2010.00991.x. [DOI] [PubMed] [Google Scholar]
- 39.Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, Finkelstein D, Forbes K, Workman CJ, Brown SA, Rehg JE, Jones ML, Ni HT, Artis D, Turk MJ, Vignali DA. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2012;11(12):1093–1101. doi: 10.1038/ni.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity. 2003;19(5):641–644. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 41.Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter CA, Drake CG, Murray PJ, Vignali DA. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. 2012;13(3):290–299. doi: 10.1038/ni.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adam N, Rabe B, Suthaus J, Grotzinger J, Rose-John S, Scheller J. Unraveling viral interleukin-6 binding to gp130 and activation of STAT-signaling pathways independently of the interleukin-6 receptor. J Virol. 2009;83(10):5117–5126. doi: 10.1128/JVI.01601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suthaus J, Adam N, Grotzinger J, Scheller J, Rose-John S. Viral Interleukin-6: Structure, pathophysiology and strategies of neutralization. Eur J Cell Biol. 2011;90(6–7):495–504. doi: 10.1016/j.ejcb.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 45.Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur J Cell Biol. 2011;90(6–7):484–494. doi: 10.1016/j.ejcb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 46.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochimica et biophysica acta. 2011;1813(5):878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 47.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450(7169):566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 48.Caspi RR. Experimental autoimmune uveoretinitis in the rat and mouse. Curr Protoc Immunol. 2003 doi: 10.1002/0471142735.im1506s53. Chapter 15: Unit 15 6. [DOI] [PubMed] [Google Scholar]