

Abstract
Background
There is a strong evidence linking inflammation and the development of pancreatic ductal adenocarcinoma (PDAC). Cyclooxygenase-2 COX-2 and COX-2-derived PGE2 are overexpressed in human and murine PDAC. Several studies demonstrated an important role of COX-2-derived PGE2 in tumor-stroma interactions; however, the direct growth effects of PGE2 on PDAC cells is less well defined. Our aim was to investigate the effects of PGE2 on PDAC cell growth and to characterize the underlying mechanisms.
Methods
Human PDAC cell lines Panc-1 and MIA PaCa-2 were treated with PGE2 in varying doses (0–10 μM). Effects on the phosphorylation of ERK1/2 were evaluated by Western blot. Colony formation was observed for cells treated with PGE2 for 11 days. DNA synthesis was determined by [3H]-thymidine incorporation assay. Gene expression of EP2/EP4 receptors and their correlation with survival in patients with PDAC was assessed using the RNA-Seq dataset from The Cancer Genome Atlas (TCGA) Research Network.
Results
PGE2 decreased the size and number of colonies in Panc-1, but not MIA PaCa-2 cells. In the Panc-1 cells, PGE2 activated PKA/CREB and decreased phosphorylation of ERK1/2, which was reversed by an EP4 receptor antagonist, while a EP2 receptor antagonist had no effect. In contrast, in MIA PaCa-2 cells, PGE2 had no effect on ERK1/2 phosphorylation. Treatment of both Panc-1 and MIA PaCa-2 cells with forskolin/IBMX decreased ERK1/2 phosphorylation. Finally, PGE2 decreased DNA synthesis only in Panc-1 cells, which was reversed by an EP4 receptor antagonist. In human PDAC, high EP2 and low EP4 gene expression was correlated to worse median overall survival (15.6 versus 20.8 months, log rank p=0.017).
Conclusion
Our study provides evidence that PGE2 can inhibit directly PDAC cell growth through a EP4-mediated mechanism. Together with our gene expression and survival analysis, this observation suggests a protective role of EP4 receptors in human PDAC that express EP receptors.
Introduction
As in many other malignancies, inflammation has been linked to pancreatic ductal adenocarcinoma (PDAC) as being pro-tumorigenic and a major contributor not only to cancer development, but also to maintaining a tumor environment that further enhances tumor growth and invasion [1,2]. Studies have shown that cyclooxygenase 2 (COX-2) is overexpressed in various malignancies [3–8] and that the mechanism by which COX-2 enhances cancer development includes conversion of the omega-6 polyunsaturated fatty acid arachidonic acid (20:4w6) into 2-series prostanoids. The most abundant 2-series prostanoid in many malignancies is prostaglandin E2 (PGE2) [9]. Similar to COX-2 overexpression [10], PGE2 was found to be increased [11] and to promote tumor growth and invasiveness in PDAC [12,13] and other malignancies [14–18]. Selective COX-2-inhibitors and microRNA-143 showed the capacity to inhibit growth of COX-2-expressing PDAC by effecting PGE2 synthesis [19–21]. Moreover, selective COX-2-inhibitors can attenuate the progression of pancreatic intraepithelial neoplasia, which are known precursor lesions of PDAC [22].
The cellular effects of PGE2 are mediated via membranous E-type prostaglandin (EP) receptors, G protein-coupled receptors, which are classified into four subtypes, named EP receptors 1-4 (EP1-4) [23,24]. The EP1 receptor couples to Gαq and signals through inositol trisphosphate (IP3) and calcium (Ca2+)-dependent pathways. The EP3 receptor is coupled to Gαi, inhibits adenylyl cyclase and decreases intracellular levels of cAMP, while EP2 and EP4, both linked to Gαs, stimulate adenylate cyclase and increase cAMP leading to activation of protein kinase A (PKA) and exchange protein activated by cAMP (Epac). In addition, EP4 but not EP2 can also signal through Gαi leading to inhibition of (Gαi-sensitive) adenylyl cyclase isoforms [25]. In cancer, EP1, but even more EP2 and EP4, have been associated to tumor progression and invasiveness [26–29]. In PDAC, PGE2, acting through EP2 and EP4, enhances stromal vascularization by increasing the production of vascular endothelial growth factor (VEGF) [30] in cancer cells and promotes fibrosis through activation of pancreatic stellate cells [31]. As a pro-inflammatory mediator, PGE2 also increases T cell invasion [18] and matrix metalloproteinase-9 (MMP-9) [13] in the tumor-microenvironment, and promotes resistance to chemotherapy by interleukin-1 beta-expression from mononuclear cells [32]. Overall, the pro-tumorigenic effects of the PGE2/EP-receptor signaling axis involve modulating the tumor environment. The direct impact of PGE2 on PDAC cell growth is less well understood. There is limited evidence for an anti-apoptotic effect through COX-2-derived PGE2 in PDAC cells [33]. We have reported previously EP receptor expression in a panel of human PDAC cell lines [34]. Panc-1 was found to have high mRNA expression of EP2 and EP4 receptors, while MIA PaCa-2 cells display relatively low levels of both receptors. Our aim of the present study was to investigate the direct effects of PGE2 on cell growth of several tumor cell lines of PDAC. We chose Panc-1 and MIA PaCa-2 cells, because they represent two human PDAC cell lines with different expression levels of EP2/EP4 receptors. We found that PGE2 has a direct growth inhibitory effect in human PDAC cell lines in vitro through an EP4-mediated mechanism.
Materials and Methods
Cell Culture
The human PDAC cell lines Panc-1 (CRL-1469) and MIA PaCa-2 (MP2; CRL-1420), both of which harbor mutations in Kras and p53, were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown in Dulbecco’s modified Eagle Medium (Life technologies, Carlsbad, CA) containing 25 mM D-glucose, 4 mM glutamine, 1 mM sodium pyruvate, 1% Penicillin G, and 10% fetal bovine serum (FBS). The cells were kept at 37 °C in a humidified atmosphere containing 10% CO2.
Colony Formation Assay
To monitor human PDAC cell growth in the presence or absence of PGE2, colony growth of Panc-1 and MP2 was recorded over 10–12 days. Briefly, 250 cells of each cell line were seeded onto 6 cm dishes containing DMEM with 10% FBS (in triplicates). After 24 h, cells were cultured in serum-free medium for 6 h. Cells were then exposed to PGE2 (Cayman Chemical Company, Ann Arbor, MI) (0–10 μM) in medium containing 3% FBS. PGE2 was added every 24 h. After 11 days, cells were fixed with 10% buffered formalin phosphate and stained with Giemsa. Images were taken with FujiFilm ImageQuant LAS 4000 (GE Healthcare Sciences, Pittsburgh, PA) and analysis performed with the open-source, cell image analysis software CellProfiler, version 2.1.1 [35].
[3H]-Thymidine Incorporation Assay
Cells were treated with the indicated compounds overnight. Treatment groups were as follows (all agents were obtained from Cayman Chemical Company): vehicle control, PGE2 (1 μM), EP4 receptor antagonist L-161,982 (1 μM), EP2 receptor antagonist PF-04418948 (10 μM), forskolin (10 μM) and IBMX (100 μM), and various combinations. The next day, [3H]-thymidine (0.25 μCi/ml) was added to each well and incubated for 6 h. Then, the medium containing the radioactive label was removed and the cells washed twice with PBS. Cells were fixed with trichloroacetic acid (5%), washed with 70% ethanol, and lysed with 0.1 N NaOH before transferred to a liquid scintillation counter.
Western blotting
Panc-1 and MP2 cells were seeded to 6-well plates (4 × 105 cells per well) and after 24 h serum-starved with serum-free DMEM overnight. Treatment was performed by adding the indicated compounds in serum-free medium. Cells were lysed and protein concentration measured by the BCA Protein Assay Kit from Thermo Scientific (Rockford, IL). Equal protein amounts were added to the Mini-Protean TGX Precast Gels from Bio-Rad Laboratories (Hercules, CA). Protein transfer to supported nitrocellulose membranes (Bio-Rad, Hercules, CA) was performed using transfer buffer containing 20 % methanol. Membranes were blocked using 5% skim milk, washed with 1 % tween-TBS (Tris buffered saline), and incubated with the primary antibody overnight. The following primary antibodies were used (all from Cell Signaling Technologies; Danvers, MA): phospho-ERK (phospho-p44/42 Erk1/2, Thr202/Tyr204), total ERK (p44/42), phospho-CREB (Ser133), and GAPDH. Membranes were incubated with the secondary antibodies, and proteins were visualized using the enhanced chemiluminescent substrate kit (SuperSignal West Pico Chemiluminescent Substrate or SuperSignal West Femto Maximum Sensitivity Substrate; Thermo Scientific; Rockford, IL). Images were taken by the ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA).
EP2 and EP4 gene expression analysis in human PaCa
Gene expression for EP2 (PTGER2), EP4 (PTGER4), and membrane-associated Prostaglandin E Synthase-2 (PTGES2) on patient samples was obtained from the publicly available pancreatic adenocarcinoma gene expression by RNAseq (TCGA_PAAD_exp_HiSeqV2) dataset from The Cancer Genome Atlas (TCGA). Fully processed data on gene expression and clinico-pathologic information from the TCGA dataset were obtained using the UC Santa Cruz (UCSC) Cancer Genomics Browser. Briefly, for the wrangling procedure, Level_3 Data (file names: *.rsem.genes.normalized_results) were downloaded from the TCGA Data Coordination Center, log2(x+1) transformed, and processed at UCSC into cgData repository by the Cancer Browser Team (cgData TCGAscript RNAseq processed on 2015-01-27). Gene-level transcription estimates were based on RSEM (RNA-Seq by Expectation Maximization) software package. For patient dichotomization, k-means clustering was performed, whereby each tumor was segregated into low or high expression groups based on its relationship to the nearest of 2 means. Statistical analyses were performed using SPSS 23.0 (IBM, Armonk, NY) software. Fisher Exact tests were used to evaluate the relationship between dichotomized patient groups and clinical variables. Kaplan-Meier survival curves were evaluated by log-rank test. The level of significance for all statistical tests was defined as α=0.05.
Results
PGE2 decreased colony growth of Panc-1 but not MIA PaCa-2 cells in vitro
The effect of PGE2 (0–10 μM) on cell growth was assessed by colony formation assays. PGE2 decreased dose-dependently the numbers of Panc-1 colonies (PGE2 1 μM vs. 0 μM: 74±10 vs. 96±3 colonies; p<0.001; Fig. 1A). Similar results were seen when comparing the total area covered by Panc-1 colonies, although significance was only observed at 10 μM PGE2 (PGE2 10 μM vs. 0 μM: 4,401±779 vs. 14,635 ±1,962 pixel/dish; p<0.05). PGE2 had no significant effect on MIA PaCa-2 colony formation (Fig. 1B), as measured by colony numbers and total colony area (e.g. PGE2 10 μM vs. 0 μM: 37,663±14,749 vs. 47,625±9,452 pixel/dish; p>0.05).
Figure 1.

PGE2 leads to decreased cell growth of Panc-1 cells
Colony formation assay was performed by daily addition of PGE2 to the medium of Panc-1 (A) and MIA PaCa-2 cells (B). Colony formation was assessed by quantifying the numbers of colonies and the area covered by the colonies (*p<0.05, **p<0.001 vs. 0 μM). Representative images of colony growth to the right.
PGE2 inhibited phosphorylation of ERK1/2 in Panc-1 cells via protein kinase A
To determine the signaling pathways underlying the growth-inhibitory effects of PGE2 in Panc-1 cells, we first evaluated the extracellular-signal regulated kinase (ERK1/2) pathway, a major growth-stimulating module in PDAC [35,36]. In Panc-1 cells, PGE2 at 1 and 10 μM decreased time-dependently phosphorylation of ERK1/2 (Fig. 2A). A significant decrease in pERK1/2 was seen already at 15 minutes and disappeared after 60 minutes. PGE2 had no significant effect on pERK1/2 in MIA PaCa-2 cells (Fig. 2A). PGE2 binds to EP receptors, which can activate protein kinase A (PKA). To assess whether the effect of PGE2 on ERK1/2 phosphorylation was mediated by PKA, we first used forskolin, which activates directly adenylyl cyclase with a subsequent increase in intracellular cAMP levels leading to activation of PKA [37]. In both Panc-1 and MIA PaCa-2 cells, forskolin in the presence of IBMX, a competitive, non-selective phosphodiesterase inhibitor that decreases the breakdown of cAMP [38], decreased pERK1/2 to almost non-detectable levels (Fig. 2B). Second, we used the pharmacologic PKA inhibitor H89. Exposure of Panc-1 and MIA PaCa-2 cells to H89 alone increased pERK1/2. In addition, H89 at 5 μM reversed completely the inhibitory effects of PGE2 on pERK1/2 in Panc-1 cells (Fig. 2C). In MIA PaCa-2 cells, the combination of H89 and PGE2 had a similar effect on ERK1/2 phosphorylation as H89 alone (Fig. 2C). Inhibition of PKA by H89 at 5 μM was confirmed by evaluating levels of phosphorylated CREB (cAMP response element-binding protein), a downstream target of PKA [39]. Exposure of Panc-1 cells to PGE2 led to activation of CREB, which was attenuated by 5 μM H89 (Fig. 2C).
Figure 2.
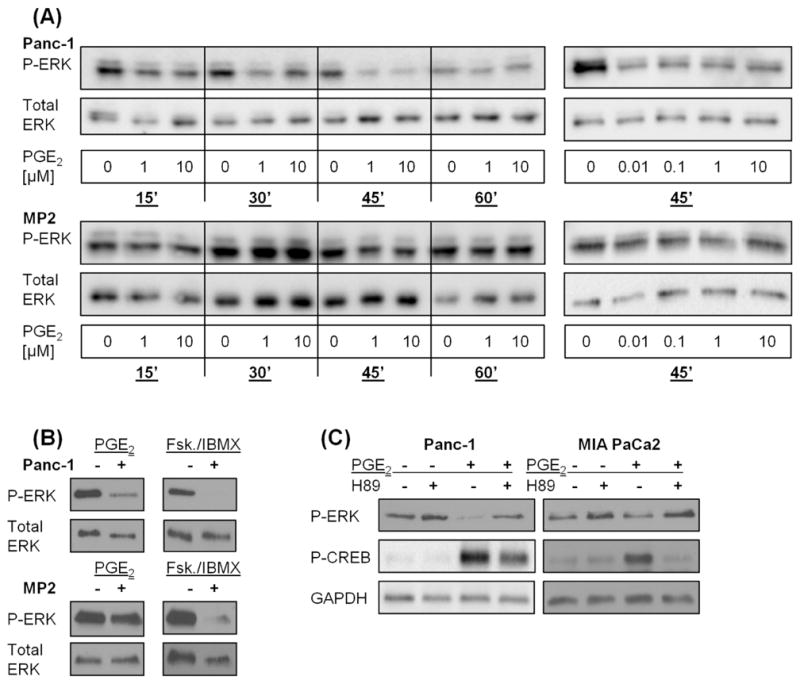
PGE2 decreased ERK phosphorylation in Panc-1 cells through PKA
A) Panc-1 and MIA PaCa-2 cells were exposed to PGE2 for various time intervals and doses and ERK1/2 (Thr202/Tyr204) phosphorylation was evaluated. Total ERK was used as loading control. B) Panc-1 and MIA PaCa-2 cells were exposed to PGE2 or forskolin/IBMX and ERK1/2 (Thr202/Tyr204) phosphorylation was evaluated. Total ERK was used as loading control. C) Panc-1 and MIA PaCa-2 cells were exposed to PGE2 in the presence or absence of the PKA inhibitor H89 and ERK1/2 (Thr202/Tyr204) phosphorylation was evaluated. GAPDH was used as loading control. P-CREB was used as a measure for PKA activation.
The inhibitory effect of PGE2 on ERK1/2 phosphorylation was mediated by the EP4 receptor
To determine which EP receptor(s) mediate(s) the effects of PGE2 in Panc-1 cells, we used pharmacologic receptor antagonists. We have reported previously that Panc-1 cells express high levels of EP4 and EP2, while MIA PaCa-2 displayed relative low levels of both receptors [34]. In Panc-1 cells, the selective EP4 receptor antagonist L-161,982 increased phosphorylation of ERK1/2. In addition, L-161,982 reversed completely the inhibitory effects of PGE2 on ERK1/2 phosphorylation (Fig. 3). In contrast, the selective EP2 receptor antagonist PF-04418948 had no effect on PGE2-mediated inhibition of ERK1/2 phosphorylation (Fig. 3). Moreover, in MIA PaCa-2, neither the EP4 nor the EP2 receptor antagonist affected ERK1/2 phosphorylation (Fig. 3).
Figure 3.
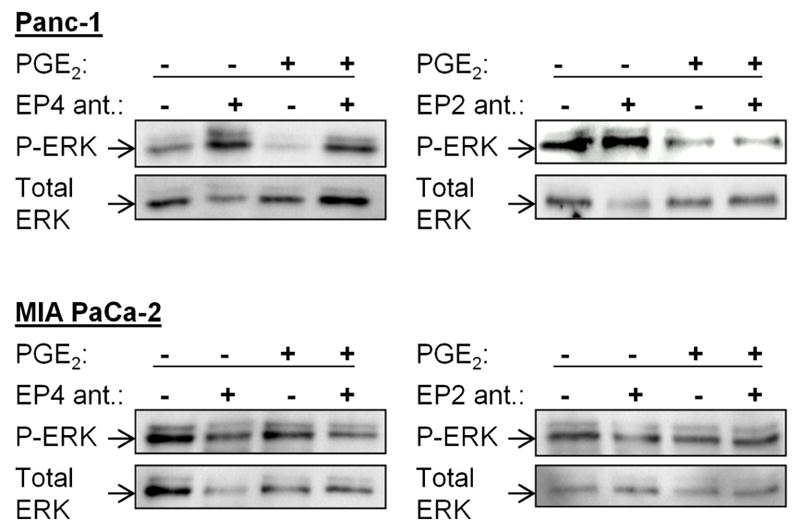
PGE2 induced ERK1/2 phosphorylation via the EP4 receptor
Panc-1 and MIA PaCa-2 cells were exposed to PGE2 in the presence or absence of EP4 (1 μM) and EP2 (10 μM) receptor antagonists and ERK1/2 (Thr202/Tyr204) phosphorylation was evaluated. Total ERK was used as loading control.
PGE2 decreased DNA synthesis in Panc-1 cells through the EP4 receptor
Having demonstrated that PGE2 inhibited ERK1/2 phosphorylation through EP4-mediated activation of PKA in Panc-1 cells, we sought to determine the effects of PKA on cell growth using thymidine incorporation assays. Exposure of Panc-1 cells to PGE2 (1 μM) significantly decreased DNA synthesis. This effect was reversed by the EP4 receptor antagonist, while the EP2 receptor antagonist had no effect (Fig. 4). Activation of PKA by forskolin and IBMX resulted in a strong decrease of DNA synthesis in Panc-1 cells. Addition of the EP4 receptor antagonist to forskolin/IBMX significantly increased thymidine incorporation (Fig. 4). Again, PGE2 had no effect on DNA synthesis in MIA PaCa-2 cells. In addition, EP2 and EP4 receptor antagonists failed to modulate DNA synthesis in MIA PaCa-2 cells, while forskolin/IBMX significantly decreased thymidine incorporation (Fig. 4).
Figure 4.

PGE2 decreased DNA synthesis in Panc-1 cells
Panc-1 and MIA PaCa-2 cells were exposed to PGE2 in the presence or absence of EP4 (left panel) and EP2 (middle panel) receptor antagonists or to forskolin/IBMX in the absence of presence of EP4 receptor antagonist (right panel). DNA synthesis was measured by [3H]-thymidine incorporation. *p<0.05
High EP2 and low EP4 gene expression was correlated to worse overall median survival in patients with PDAC
Because our in vitro data suggested a potential protective (inhibitory) effect of EP4 receptors on PDAC growth, we sought to evaluate the expression of EP4 receptors and their correlation to survival of patients with PDAC. Gene expression patterns of PTGER2/4 in PDAC patient samples and their correlations with clinicopathologic variables were explored using a publicly-available RNA-Seq dataset (N=183) from The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/). We restricted our analysis to neoplasms with a histologic phenotype of adenocarcinoma and a tumor cell content of greater than 50% (N=77) to evaluate tumor cell-specific gene expression to the relative exclusion of accompanying non-malignant cell types (i.e., benign pancreas, stroma, inflammation). Tumors were segregated into groups of high versus low expression of PTGER2 or PTGER4 expression by K-means clustering. High versus low PTGER2 expression showed no significant correlation with survival (median overall survival 19.5 vs. 20.8 months, log rank P=0.34, data not shown), while high versus low PTGER4 expression nearly reached significance (median overall survival 22.7 vs. 19.5 months, log rank P=0.06, data not shown). Stratification based on combinations of PTGER2 and PTGER4 (Groups 1–4, Fig. 5A) revealed that neoplasms with high PTGER2/low PTGER4 (Group 3) were strongly correlated with worse overall survival relative to other subgroups (median overall survival 15.6 vs. 20.8 months, log rank P=0.017, Fig. 5B). No significant correlations were noted between high PTGER2/low PTGER4 and various clinicopathologic factors (table).
Figure 5.

PTGER expression in human PDAC correlates with overall survival.
A) Heatmap of PTGER2, PTGER4 and PTGES2 gene expression for patient neoplasms from RNA-seq of TCGA PDAC of tumors based on PTGER2 and PTGER4 identifies four distinct patterns of expression (group 1: both high; group 2: PTGER2 low, PTGER4 high; group 3: PTGER2 high, PTGER4 low; group 4: both low). (B Kaplan-Meier plots showing reduced overall survival for patient group 3 (solid line) versus all other groups combined (dotted line) (log rank P=0.017).
Table.
Clinicopathologic characteristics of TCGA PDA dataset (tumor content ≥ 50%) and correlation with dichotomized groups based on PTGER2 and PTGER4 expression
| Clinicopathologic variable | High PTGER2 + Low PTGER4 | All other combinations | |||
|---|---|---|---|---|---|
|
| |||||
| N | % | N | % | P valuea | |
|
| |||||
| Age (yrs) | |||||
| ≤ 60 | 8 | 38.1 | 21 | 37.5 | 1.00 |
| > 60 | 13 | 61.9 | 35 | 62.5 | |
|
| |||||
| Sex | |||||
| Female | 8 | 38.1 | 30 | 53.6 | 0.31 |
| Male | 13 | 61.9 | 26 | 46.4 | |
|
| |||||
| Tumor size (cm) | |||||
| ≤ 3 | 7 | 33.3 | 23 | 41.1 | 0.61 |
| > 3 | 14 | 66.7 | 33 | 58.9 | |
|
| |||||
| Tumor grade | |||||
| Low (G1+G2) | 17 | 81.0 | 43 | 76.8 | 0.77 |
| High (G3+G4) | 4 | 19.0 | 13 | 23.2 | |
|
| |||||
| T-stage | |||||
| T1 + T2 | 2 | 9.5 | 9 | 16.4 | 0.72 |
| T3 | 19 | 90.5 | 47 | 83.6 | |
|
| |||||
| N-stage | |||||
| N0 | 5 | 23.8 | 15 | 27.3 | 1.00 |
| N1 | 16 | 76.2 | 41 | 72.7 | |
|
| |||||
| Margin status | |||||
| Negative (or unknown) | 11 | 52.4 | 42 | 76.4 | 0.09 |
| Positive (any) | 10 | 47.6 | 14 | 23.6 | |
Discussion
The cancer-promoting effects of COX-2 and COX-2 -derived PGE2 have been described in several studies [3–8,12,13]. In PDAC, PGE2 can stimulate COX-2 expression, constituting a positive feedback loop, which maintains a high production of prostanoids [36]. Further, PGE2 promotes angiogenesis through VEGF [30], fibrosis through activation of pancreatic stellate cells [31], and inflammation [13], all of which impact the tumor microenvironment, thereby emphasizing the crucial role it has on PDAC progression.
In our studies, we tested two human PDAC cell lines with different expression profiles of EP receptors [34]. While PGE2 attenuated colony formation, DNA synthesis, and ERK phosphorylation in Panc-1 cells, it had no effect on MIA PaCa-2 cells. These results may be explained by the differential expression levels of EP2 and EP4 receptors in Panc-1 and MIA PaCa-2 cells. Our group has described previously that expression levels of EP4 are multifold greater in Panc-1 than in MIA PaCa-2. EP2 is expressed at lower levels in both cell lines [34]. Our results provide evidence that the effects of PGE2 in Panc-1 cells are mediated by the EP4 receptor. First, an EP4 but not EP2 receptor antagonist attenuated the PGE2-induced decrease of pERK1/2 and DNA synthesis in Panc-1 cells. Second, PGE2 had no effect in MIA PaCa-2 cells, which have low expression of EP4 receptors. Consistent with that observation, direct stimulation of adenylyl cyclase with forskolin/IBMX in both cell lines, which enhances intracellular cAMP and subsequently activates PKA, increased ERK1/2 phosphorylation and DNA synthesis. This observation partially corroborates findings from another group [37].
Our observation is in contrast to other publications demonstrating that activation of G-coupled receptors with subsequent PKA activation promote growth of several malignancies [38–41]. Although the exact reasons for this discrepancy are unclear, differences in expression of EP receptors and distinct operative signaling networks in various cell lines might explain this finding. The proliferative effect of PKA-mediated activation of Ras-Raf-MEK-ERK can be regulated further by the GTPase Rap1, which is dependent on the absence or presence of B-Raf. While the presence of B-Raf leads to an enhancement of the Ras-Raf-MEK-ERK-pathway, in its absence, Rap1 will inhibit the Raf-1-MEK activation through Ras [42,43]. In addition, ERK1/2 regulation by Ras and Rap depend on the type of stimulation (acute vs. chronic) [44]. Our in vitro data suggest clearly a potentially protective, growth-inhibitory effect of EP4 receptors in human PDAC. This notion seemingly is in contrast to the available literature describing a tumor-promoting effect of COX-2 and COX-2-derived PGE2. To our knowledge, however, a detailed analysis of the expression of EP receptors in human PDAC and its correlation to survival has not been conducted before. It is theoretically plausible that PGE2 has a growth-inhibitory effect in a subgroup of human PDAC with high EP4 receptor expression (and, therefore, is correlated with a better prognosis). This hypothesis is supported in part by the finding from another group describing varying expression of EP4 receptors in human PDAC using a tissue microarray (TMA) containing 133 pancreatic tumor specimens along with 106-paired adjacent normal tissues [45]. Using the publicly available TCGA dataset of 77 human PDAC (with a tumor cell content of >50%), we were able to confirm varying gene expression levels of EP4 (and EP2) receptors. More importantly, the combination of high EP2 and low EP4 gene expression was significantly associated with a worse overall median survival. These data provide compelling evidence that EP4 receptors might have a protective effect in a subset of human PDAC. Moreover, our results emphasize the need to stratify human PDAC based on their expression of EP receptors to understand the effects of COX-2 and COX-2-derived PGE2 on PDAC growth. This is equally important for the interpretation of clinical trials testing selective and non-selective COX-2 inhibitors aimed at decreasing PGE2 levels.
In conclusion, we have shown that human PDAC cell lines respond differently to the major COX-2 metabolite, PGE2, and that PGE2 can decrease growth, DNA synthesis, and ERK1/2-activation in Panc-1 through a EP4-PKA mediated pathway. In addition, high EP2 and low EP4 gene expression correlated to worse overall median survival in human PDAC. Our results provide evidence that EP4 receptors might have a protective effect in a subset of human PDAC.
Acknowledgments
Financial support: This work was supported by the NCI P01 CA163200 (to G.Eibl), NIDDK (CURE: Digestive Diseases Research Core Center Grant P30 DK41301 (to E. Rozengurt), the Hirshberg Foundation for Pancreatic Cancer Research, and the German Research Foundation (DFG) to A.I. Schmidt (SCHM 3246/1-1; salary).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Farrow B, Evers BM. Inflammation and the development of pancreatic cancer. Surg Oncol. 2002;10(4):153–69. doi: 10.1016/s0960-7404(02)00015-4. [DOI] [PubMed] [Google Scholar]
- 2.Sparmann A, Bar-Sagi D. Ras oncogene and inflammation: partners in crime. Cell Cycle. 2005;4(6):735–6. doi: 10.4161/cc.4.6.1714. [DOI] [PubMed] [Google Scholar]
- 3.Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58(22):4997–5001. [PubMed] [Google Scholar]
- 4.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183–8. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 5.Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, et al. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995;55(17):3785–9. [PubMed] [Google Scholar]
- 6.Gupta S, Srivastava M, Ahmad N, Bostwick DG, Mukhtar H. Over-expression of cyclooxygenase-2 in human prostate adenocarcinoma. Prostate. 2000;42(1):73–8. doi: 10.1002/(sici)1097-0045(20000101)42:1<73::aid-pros9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 7.Hwang D, Scollard D, Byrne J, Levine E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J Natl Cancer Inst. 1998;90(6):455–60. doi: 10.1093/jnci/90.6.455. [DOI] [PubMed] [Google Scholar]
- 8.Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, et al. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999;59(5):987–90. [PubMed] [Google Scholar]
- 9.Smith WL, Marnett LJ, Dewitt DL. Prostaglandin and thromboxane biosynthesis. Pharmacology & Therapeutics. 1991;49(3):153–79. doi: 10.1016/0163-7258(91)90054-p. [DOI] [PubMed] [Google Scholar]
- 10.Molina MA, Sitja-Arnau M, Lemoine MG, Frazier ML, Sinicrope FA. Increased cyclooxygenase-2 expression in human pancreatic carcinomas and cell lines: growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res. 1999;59(17):4356–62. [PubMed] [Google Scholar]
- 11.Hasan S, Satake M, Dawson DW, Funahashi H, Angst E, Go VLW, et al. Expression analysis of the prostaglandin E2 production pathway in human pancreatic cancers. Pancreas. 2008;37(2):121–7. doi: 10.1097/MPA.0b013e31816618ba. [DOI] [PubMed] [Google Scholar]
- 12.Pham H, Chen M, Li A, King J, Angst E, Dawson DW, et al. Loss of 15-hydroxyprostaglandin dehydrogenase increases prostaglandin E2 in pancreatic tumors. Pancreas. 2010;39(3):332–9. doi: 10.1097/MPA.0b013e3181baecbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bu X, Zhao C, Dai X. Involvement of COX-2/PGE(2) Pathway in the Upregulation of MMP-9 Expression in Pancreatic Cancer. Gastroenterol Res Pract. 2011;2011:214269. doi: 10.1155/2011/214269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276(21):18075–81. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 15.Liu X-H, Kirschenbaum A, Lu M, Yao S, Klausner A, Preston C, et al. Prostaglandin E(2) stimulates prostatic intraepithelial neoplasia cell growth through activation of the interleukin-6/GP130/STAT-3 signaling pathway. Biochem Biophys Res Commun. 2002;290(1):249–55. doi: 10.1006/bbrc.2001.6188. [DOI] [PubMed] [Google Scholar]
- 16.Dohadwala M, Batra RK, Luo J, Lin Y, Krysan K, Pold M, et al. Autocrine/paracrine prostaglandin E2 production by non-small cell lung cancer cells regulates matrix metalloproteinase-2 and CD44 in cyclooxygenase-2-dependent invasion. J Biol Chem. 2002;277(52):50828–33. doi: 10.1074/jbc.M210707200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nithipatikom K, Isbell MA, Lindholm PF, Kajdacsy-Balla A, Kaul S, Campell WB. Requirement of cyclooxygenase-2 expression and prostaglandins for human prostate cancer cell invasion. Clin Exp Metastasis. 2002;19(7):593–601. doi: 10.1023/a:1020915914376. [DOI] [PubMed] [Google Scholar]
- 18.Qian X, Gu L, Ning H, Zhang Y, Hsueh EC, Fu M, et al. Increased Th17 cells in the tumor microenvironment is mediated by IL-23 via tumor-secreted prostaglandin E2. J Immunol. 2013;190(11):5894–902. doi: 10.4049/jimmunol.1203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eibl G, Reber HA, Wente MN, Hines OJ. The selective cyclooxygenase-2 inhibitor nimesulide induces apoptosis in pancreatic cancer cells independent of COX-2. Pancreas. 2003;26(1):33–41. doi: 10.1097/00006676-200301000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA, et al. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65(3):982–90. [PubMed] [Google Scholar]
- 21.Pham H, Rodriguez CE, Donald GW, Hertzer KM, Jung XS, Chang H-H, et al. miR-143 decreases COX-2 mRNA stability and expression in pancreatic cancer cells. Biochem Biophys Res Commun. 2013;439(1):6–11. doi: 10.1016/j.bbrc.2013.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Funahashi H, Satake M, Dawson D, Huynh N-A, Reber HA, Hines OJ, et al. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67(15):7068–71. doi: 10.1158/0008-5472.CAN-07-0970. [DOI] [PubMed] [Google Scholar]
- 23.Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim Biophys Acta. 2015;1851(4):414–21. doi: 10.1016/j.bbalip.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Negishi M, Sugimoto Y, Ichikawa A. Molecular mechanisms of diverse actions of prostanoid receptors. Biochim Biophys Acta. 1995;1259(1):109–19. doi: 10.1016/0005-2760(95)00146-4. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The Prostanoid EP4 Receptor and Its Signaling Pathway. Pharmacol Rev. 2013;65(3):1010–52. doi: 10.1124/pr.112.007195. [DOI] [PubMed] [Google Scholar]
- 26.Chell S, Kaidi A, Williams AC, Paraskeva C. Mediators of PGE2 synthesis and signalling downstream of COX-2 represent potential targets for the prevention/treatment of colorectal cancer. Biochim Biophys Acta. 2006;1766(1):104–19. doi: 10.1016/j.bbcan.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Doherty GA, Byrne SM, Molloy ES, Malhotra V, Austin SC, Kay EW, et al. Proneoplastic effects of PGE2 mediated by EP4 receptor in colorectal cancer. BMC Cancer. 2009;9:207. doi: 10.1186/1471-2407-9-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujino H, Regan JW. Prostanoid receptors and phosphatidylinositol 3-kinase: a pathway to cancer? Trends Pharmacol Sci. 2003;24(7):335–40. doi: 10.1016/S0165-6147(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 29.Hull MA, Ko SCW, Hawcroft G. Prostaglandin EP receptors: targets for treatment and prevention of colorectal cancer? Mol Cancer Ther. 2004;3(8):1031–9. [PubMed] [Google Scholar]
- 30.Eibl G, Bruemmer D, Okada Y, Duffy JP, Law RE, Reber HA, et al. PGE(2) is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem Biophys Res Commun. 2003;306(4):887–97. doi: 10.1016/s0006-291x(03)01079-9. [DOI] [PubMed] [Google Scholar]
- 31.Charo C, Holla V, Arumugam T, Hwang R, Yang P, Dubois RN, et al. PGE(2) Regulates Pancreatic Stellate Cell Activity Via The EP4 Receptor. Pancreas. 2013;42(3):467–74. doi: 10.1097/MPA.0b013e318264d0f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angst E, Reber HA, Hines OJ, Eibl G. Mononuclear cell-derived interleukin-1 beta confers chemoresistance in pancreatic cancer cells by upregulation of cyclooxygenase-2. Surgery. 2008;144(1):57–65. doi: 10.1016/j.surg.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi H, Li A, Dawson DW, Hines OJ, Reber HA, Eibl G. Cyclooxygenase-2 confers growth advantage to syngeneic pancreatic cancer cells. Pancreas. 2011;40(3):453–9. doi: 10.1097/MPA.0b013e31820b9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang H-H, Young SH, Sinnett-Smith J, Chou CEN, Moro A, Hertzer KM, et al. Prostaglandin E2 activates the mTORC1 pathway through an EP4/cAMP/PKA- and EP1/Ca2+-mediated mechanism in the human pancreatic carcinoma cell line PANC-1. Am J Physiol Cell Physiol. 2015;309(10):C639–49. doi: 10.1152/ajpcell.00417.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology [Internet] 2006;7(10):1–11. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pino MS, Nawrocki ST, Cognetti F, Abruzzese JL, Xiong HQ, McConkey DJ. Prostaglandin E2 drives cyclooxygenase-2 expression via cyclic AMP response element activation in human pancreatic cancer cells. Cancer Biol Ther. 2005;4(11):1263–9. doi: 10.4161/cbt.4.11.2138. [DOI] [PubMed] [Google Scholar]
- 37.Boucher MJ, Duchesne C, Laine J, Morisset J, Rivard N. cAMP protection of pancreatic cancer cells against apoptosis induced by ERK inhibition. Biochem Biophys Res Commun. 2001;285(2):207–16. doi: 10.1006/bbrc.2001.5147. [DOI] [PubMed] [Google Scholar]
- 38.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7(2):79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 39.Rozengurt E, Guha S, Sinnett-Smith J. Gastrointestinal peptide signalling in health and disease. Eur J Surg Suppl. 2002;(587):23–38. [PubMed] [Google Scholar]
- 40.Allen LF, Lefkowitz RJ, Caron MG, Cotecchia S. G-protein-coupled receptor genes as protooncogenes: constitutively activating mutation of the alpha 1B-adrenergic receptor enhances mitogenesis and tumorigenicity. Proc Natl Acad Sci U S A. 1991;88(24):11354–8. doi: 10.1073/pnas.88.24.11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heasley LE. Autocrine and paracrine signaling through neuropeptide receptors in human cancer. Oncogene. 2001;20(13):1563–9. doi: 10.1038/sj.onc.1204183. [DOI] [PubMed] [Google Scholar]
- 42.Stork PJS, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12(6):258–66. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 43.Belcheva MM, Coscia CJ. Diversity of G protein-coupled receptor signaling pathways to ERK/MAP kinase. Neurosignals. 2002;11(1):34–44. doi: 10.1159/000057320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ. Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol. 2000;20(21):8069–83. doi: 10.1128/mcb.20.21.8069-8083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong J, Xie J, Bedolla R, Rivas P, Chakravarthy D, Freeman JW, et al. Combined targeting of STAT3/NF-κB/COX-2/EP4 for effective management of pancreatic cancer. Clin Cancer Res. 2014;20(5):1259–73. doi: 10.1158/1078-0432.CCR-13-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]