

Abstract
Cell surface p32, the target of LyP-1 homing peptide, is upregulated in tumors and atherosclerotic plaques and has been widely used as a receptor for systemic delivery of payloads. Here we identified an improved LyP-1 mimicking peptide (TT1, CKRGARSTC). We used this peptide in a fluorescence polarization–based high-throughput screening of a 50,000-compound chemical library and identified a panel of compounds that bind p32 with low micromolar affinity. Among the hits identified in the screen, two compounds were shown to specifically bind to p32 in multiple assays. One of these compounds was chosen for an in vivo study. Nanoparticles surface-functionalized with this compound specifically adhered to surfaces coated with recombinant p32 and, when injected intravenously, homed to p32-expressing breast tumors in mice. This compound provides a lead for the development of p32-targeted affinity ligands that circumvent some of the limitations of peptide-based probes in guided drug delivery.
Keywords: Peptides, High-throughput screening, Nanoparticles, Cancer, Drug Delivery
Graphical Abstract
We identified an improved p32-targeted tumor homing peptide (TT1, CKRGARSTC) and used screening of a chemical library to identify compounds that bind to p32 at low micromolar affinity. This study provides a starting point for the development of p32-targeted affinity ligands that circumvent some of the limitations of peptide-based probes in guided drug delivery.
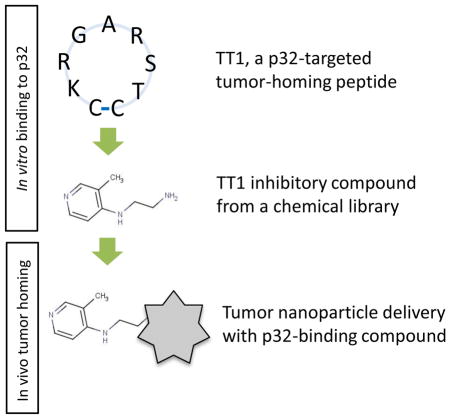
Targeted drug delivery into tumors remains a major goal in cancer drug development. The aim is to achieve effects similar to topical drug administration: high tumor accumulation and reduction of drug burden in non-target tissues. Drug selectivity can be improved by coupling a drug to a targeting moiety such as an antibody, aptamer, or peptide. Such synaphic (affinity-based) targeting relies on overexpression of specific receptor molecules in a systemically accessible compartment of the chosen target tissue [1].
LyP-1 (CGNKRTRGC) is a widely used tumor-homing peptide originally identified by in vivo phage display [2]. This peptide homes to tumors by binding to p32, a mitochondrial chaperone protein important in the maintenance of oxidative phosphorylation. In tumor endothelial cells (blood and lymphatic), tumor cells, and tumor macrophages, p32 is also expressed at the cell surface, making it a tumor-specific target [2,3]. LyP-1 has been used for targeting of drugs and nanoparticles in breast cancer [4,5] and metastatic tumors in lymph nodes [6,7]. Interestingly, LyP-1 has a proapoptotic/cytotoxic activity on cultured malignant cells and it inhibits growth of certain breast tumor xenografts in vivo [8]. LyP-1 peptide has applications beyond tumor targeting – it also homes to atherosclerotic plaques and penetrates into their interior [9].
Whereas peptides are useful for targeted delivery of drugs in vivo, they have to be administered parenterally, and are susceptible to degradation by blood and tissue proteases [10]. The stability of peptides can be improved without loss of activity by modifications such as introduction of reduced peptide bonds or N-methylated amino acids [11]. A methylated version of the tumor-homing peptide CREKA circulates longer and homes to tumors more effectively than the unmodified peptide [12–14]. There are also some low molecular weight affinity ligands, such as folate [15] and cobalamin [16] that are not peptides. Such alternatives to peptide ligands may have advantages such as greater stability and potential to be orally active [17,18].
Here, we use a multi-step approach to develop in vivo targeting ligands for the tumor-homing LyP-1 peptide receptor, p32 (Figure 1). First, we screen peptide phage libraries on purified p32 protein to identify peptides that have more favorable p32 binding properties than LyP-1. Second, we use a fluorescence polarization-based screening assay to identify low molecular weight compounds that compete with p32-binding peptides. Third, we show that one of the low molecular weight compounds from the screen, when coupled to nanoparticles, acts as an affinity ligand capable of p32-directed payload delivery in vitro and in vivo.
Figure 1. Multistep development of systemic homing compounds using reiterative biopanning and FP-based screening.

A) In vivo phage display is used for mapping of systemically accessible diversity of vascular beds. It yields primary homing peptides that can be used for biochemical identification of binding partners, “receptors” (denoted as a red square with cavity). B) In vitro biopanning on purified receptors is used for identification of secondary receptors of improved binding properties. C) Fluorescence polarization assays is used for high-throughput screening of compounds that bind to peptide binding site on target receptors. Top: when the rapidly rotating FAM-labeled peptide binds to its larger partner protein, its rotation approximates the slower rotation of this larger protein, so the polarization of the excitation light is retained in the emitted light (light green emission thunderbolts aligned with light blue excitation thunderbolts). Below: in the presence of competitor compound, an increased concentration of free fast-rotating FAM-labeled peptide is seen as decrease in fluorescence polarization (divergent excitation thunderbolts)
To identify low molecular weight mimics of p32-binding homing peptides, we decided to use fluorescence polarization (FP)-based assay, which is a solution-based, homogeneous technique requiring no immobilization or separation of reaction components [19,20] (Figure 1C). As a target, we used recombinant 6x-His-tagged p32, which according to sedimentation velocity analysis was in the expected trimeric form ([21]; Figure S1). The affinity of synthetic LyP-1 peptide for p32 protein in FP assay buffer, >23μM (Figure 3A), was suboptimal for a FP-based interaction assay. To identify p32 binding peptides that bind p32 better than LyP-1, we subjected random heptapeptide T7 phage libraries to in vitro biopanning on purified p32 protein. After three rounds of biopanning the selected phage pool showed a robust increase in p32 binding (2060-fold binding over control CG7C phage and 1560-fold for another control, an X7 library; Figure 2A and Figure S2A). Sequencing of peptide-encoding phage DNA inserts showed that the bound phage pool had converged to display a RGXRS pentapeptide consensus motif (Figure 2B and Figure S2B). The binding of RGXRS-displaying phage pool to p32 was specific, as the pool showed no specific binding to a control protein, b1b2 domain of neuropilin-1 (NRP-1), known to bind basic peptides and used for affinity targeting of payloads [22].. Phage clones displaying 3 candidate peptides from highly enriched phage clones (CKRGARSTC, CKRGNRSMC, and CTRGSRSKC) showed greatly improved binding to p32 compared to LyP-1 phage (Figure 2C and Figure S3). The phage displaying the CKRGARSTC peptide (designated TT1), which showed the strongest binding to p32, was chosen for FP-based screening of compound libraries.
Figure 3. FP- and phage binding- based validation of p32 binding peptides and hit compounds.

A) Fluorescence polarization (FP) measurement of binding affinity of FAM-LyP-1 and FAM-TT1 peptides to p32 protein, and dissociation constants (KD) based on binding curves fitted on Michaelis-Menten kinetics. Measurements were performed with PheraStar FS plate reader (BMG Labtech, Ortenberg, Germany) on 384-well plate. B) Affinity of the 7 hit compounds in dose-dependent (8.25 μM to 100 μM) inhibition of FAM-TT1 binding to p32 in FP assay. C) Assessment of hit compound binding to a non-target secondary protein, NRP-1 to determine the specificity of the hit compounds in FP assay. A known NRP-1 binding peptide, RPARPAR, was used as a control. D) Effect of the hit compounds on the TT1 phage binding to p32 protein. TT1 phage binding to p32 in the presence of 100 μM hit compounds was determined in ELISA-type of binding assay.Library compounds can be converted into Pubchem Compound identifiers using the utility by Chembridge: http://www.chembridge.com/conversion_tool/index.php. Statistical analysis was performed by one-way ANOVA. Significance of differences between individual data points was determined by paired t-test. *P < 0.05, **P < 0.005 and ***P < 0.001. Each data point presents average ± S.D., n=4.
Figure 2. In vitro identification and characterization of novel p32 binding peptides.

A) CX7C peptide library was used for in vitro selection on hexahistidine-tagged recombinant p32 protein immobilized on Ni-NTA magnetic beads (Qiagen, Hilden, Germany). Binding of phage particles in each selection round is expressed fold control phage displaying heptaglycine peptide (G7). B) Representative peptide sequences recovered after three rounds of ex vivo selection. TT1 peptide (CKRGARSTC) was present 3 times among 30 sequenced phage clones. Note emergence of RGXRS consensus motif in selected peptide pool. C) Binding of TT1, CKRGNRSMC, CTRGSRSKC and LyP-1 phage to immobilized p32 protein. The data are representative of four independent binding experiments. Binding is expressed as fold over control phage displaying polyglycine heptapeptide (G7). Statistical analysis was performed by one-way ANOVA (F: 15.2; F crit 3.4) Error bars indicate s.e.m.; triple asterisk, p<0.001, n=4..
We next optimized the FP-based assay conditions to study the interaction of FAM-labeled TT1 peptide with p32 protein. The composition of binding buffer had profound influence on TT1-p32 binding; the Kd in phosphate buffered saline (PBS) was considerably higher (14 μM) than in BIS-TRIS or HEPES buffers (2.3 μM and 1.6 μM, respectively), suggesting that the higher salt concentration in PBS decreases peptide binding (Figure 3A, Figure S4). FAM-TT1 binding to p32 was inhibited in a dose-dependent manner by unlabeled TT1 and LyP-1, but not with the RPARPAR peptide. RPARPAR binds to NRP-1, a receptor with specificity somewhat similar to that of p32, as both proteins favor positively charged ligands ([22]; Figure S5). The specificity of interaction of TT1 and p32 was further demonstrated by lack of binding of TT1 to b1b2 domain of NRP-1 (Figure S6). Thus, TT1 interaction with p32 is specific, and the mutual inhibition of the binding of one peptide by the other suggests that TT1 and LyP-1 bind to the same site on the p32 protein.
After establishing and validating the FP assay, we screened a proprietary library of 50,000 compounds (Sanford-Burnham-Prebys Medical Discovery Institute, La Jolla, USA) in 1536-well format for molecules capable of inhibiting the interaction of TT1 peptide with p32. The compounds in this library have been selected to provide 2D diversity for extensive pharmacophore coverage for primary screening and to offer the most attractive features found in lead- and drug-like compounds. The screening was robust as we obtained the following assay performance parameters of Z′ = 0.68 ±0.05 (s.e.m.), S/B = 4.07 ± 0.2 (s.e.m.), and CV = 5.6%. The NIH defines a robust assay [23] as one with Z′ >0.5 [24], S/B > 4 and CV<10%. This screen led to identification of 7 hit compounds (Supplementary Material Table S2) that at 100 μM concentration caused greater than 20% inhibition of the TT1-p32 interaction. Six out of the 7 compounds showed dose-dependent inhibition of FAM-TT1 binding to p32 (Figure 3B). At the highest concentration used (100 μM), the inhibition ranged from 50 to 80%. Compound #54778828 was only marginally active, and was not studied further. Compound #6367577 was less effective than the other active compounds. We next used an assay based on the binding of the RPARPAR peptide to NRP-1 to study the effect of the 6 active compounds on a different, but somewhat similar peptide-protein interaction. Compounds #4014008, #9132086, #88361068, and #7933989 had no effect in this assay; the polarization decrease seen at the highest compound concentrations was no greater than that caused by the DMSO solvent (Figure 3C). The FP level in the presence of compound #6367577 at the highest concentration had a negative value, indicating that the compound itself is a fluorophore. Therefore, compound #6367577 was excluded from further studies.
Inhibition of TT1 phage binding to p32 was used to confirm the activity of the remaining 5 compounds. At 100 μM, compounds #4014008 and #7933989 inhibited TT1 phage binding to p32 by 50% and 75%, respectively (Figure 3D). This agreed with the results of the FP assay measuring TT1 binding to p32 (Figure 4A), where #4014008 showed 75% and #7933989 80% inhibition. Compounds #88361068, #9132086, and #91984046 inhibited TT1 phage binding to p32 only by 20%, whereas in the FP assay had given 65% to 75% inhibition (Figure 3A). Peptides are displayed on the T7 bacteriophage at about 200 copies per particle [25] and the multivalent phage binding to immobilized p32 is likely to be less susceptible to inhibition by a free competitor than the TT1 binding to p32. Thus, the affinity of these latter 3 compounds for p32 may be low. To further confirm interaction of p32 with the most promising 2 compounds, #4014008 and #7933989, we used NMR analysis. While the biochemical assays we used are robust, they are not immune to occasional false positives [31]. To validate the binding of the most promising 2 compounds, #4014008 and #7933989, we used a direct binding assay based on NMR spectroscopy, which monitors chemical shift perturbations a ligand induces to the resonances of a protein. As control, we used the TT1 peptide. Under the same experimental conditions, the TT1 peptide caused significant perturbations in the 1D 1H-aliph spectra [26] with 5 μM p32 concentration. Both compounds #4014008 and #7933989 caused similar perturbations indicating binding to p32 similar to that of TT1 (Figure 4).
Figure 4. NMR based evaluation of binding of compounds 4014008 and 7933989 to P32 protein.

The 1D 1H-aliph spectra of 5 μM P32 in the absence (black) and presence of compounds #4014008 (A) and #7933989 (B) (red for 100 μM and blue for 500 μM) were collected. As a control, the spectrum of p32 in the presence of the known peptide binder, TT1 at 70 μM, was also collected (green). In presence of both compounds there is a shift in the peak at around −0.3 ppm (dashed line) and the concomitant appearance of a shoulder peak (indicated by arrows) similar to what observed in presence of the reference peptide TT1.
The data described above indicate that compounds #4014008 and #7933989 interact specifically with the homing peptide binding site on the p32 protein and represent potential affinity ligands for p32-directed payload delivery. We chose compound #401008 for further studies because its structure is more appealing as lead molecule than that of #7933989; #4014008 is more amenable to further SAR studies than #7933989, and an amine group in #401008 provides a readily available coupling group. FP-based FAM-TT1 displacement assay demonstrated that #4014008 interacts with the p32 specifically albeit at affinity that is too low to measure reliably (>50μM). On nanoparticles, even weak small molecule targeting ligands can significantly enhance target-specific avidity (by up to 4 orders of magnitude) through multivalent interactions. Furthermore, low affinity targeting ligands may be used to avoid the “affinity site barrier” (getting trapped near the site of entry into the tumor), resulting in better tissue distribution [32]. Silver nanoparticles (AgNP) coated with neutravidin [27] and functionalized with biotinylated compound #4014008 (Figure 5A) showed specific in vitro binding to immobilized recombinant p32 protein (Figure 5B). In contrast, the #4014008AgNPs showed only background binding to a non-target recombinant protein. To evaluate #4014008 as an in vivo targeting ligand, we used iron oxide nanoparticles, dubbed “nanoworms” (NWs; Figure 5C). These paramagnetic nanoparticles are PEGylated to extend blood half-life, and have because of their elongated shape more effective targeting properties than spherical nanoparticles [13,28]. MCF10Ca1A breast tumor cells express cell surface p32 in vitro and p32-binding peptides home to orthotopic MCF10Ca1A xenograft tumors that are strongly positive for the presence of systemically accessible p32 [29]. NWs functionalized with #4014008 showed robust homing to the blood vessels in MCF10Ca1A tumors (Figure S 7A–C). The pattern was similar to what was seen with TT1-NWs (Figure S 7 D–F). Among normal tissues, some #4014008-NW uptake was seen in organs of the reticuloendothelial system (liver and spleen), which non-specifically scavenge nanoparticles, and in the lungs, but not in other organs studied (Figure S 8). MCF10Ca1A breast tumor xenografts express abundant p32 in blood vessels, and p32 immunoreactivity showed overlap with #4014008 functionalized NW signal (Figure 6D–F). : As a specificity control, tumor mice were injected with FAM-NW functionalized with a control peptide. In case of control nanoparticles only a residual vascular signal was seen in tumor (Figure S 9 and 10). These studies show that functionalization with #4014008 renders nanoparticles selective for p32 binding in vitro and in vivo.
Figure 5. Nanoparticles functionalized with #4014008 bind to p32 protein and home to p32-positive breast tumors.

(A) The compound coupled to fluorescent silver nanoparticles showed specific binding toward plate well bound p32 protein, relative to wells having a non-target control protein N3A. Increased concentration of silver nanoparticles caused a dose-dependent increase in fluorescent signal due to nanoparticle binding to the surface of the well. (B–D) Confocal imaging of tissue sections of MCF10Ca1A breast tumors from mice injected with FAM-#4014008 -NW. Red: p32; green: NW; blue: nuclei. Arrows point to vascular structures that show co-localization of nanoparticle and p32 signals. Representative fields from multiple sections of three independent mice are shown. Scale bars = 100 μm.
Our study suggests that following initial in vivo biopanning and peptide receptor identification, secondary in vitro phage screening on purified receptors may yield homing peptides possessing improved properties, in our case increased affinity for the tumor-associated p32 receptor over the original peptide. The increased affinity we obtained in this manner made it possible to screen for small molecular weight p32-binding compounds, for which the affinity of the original peptide was too low. Library screens using broadly diverse compounds, even those selected for “drug-like” properties generally do not typically yield high-affinity binders, especially for the target class of peptide-protein and protein-protein interactions inhibitors. Rather, moderate affinity lead compounds, such as #4014008, are identified and have to be subjected to further chemical optimization to obtain high-affinity binders. Importantly, we show that a lead compound with moderate affinity, such as #4014008, can be used for targeted delivery in vivo when rendered polyvalent through coupling to nanoparticles. It may greatly shorten the path to clinic in cases where multivalent presentation is feasible, or desirable, such as in nanomedicine. Of course, compound #4014008 can also serve as a lead for the development of conventional drug-like compounds by medicinal chemistry.
Our demonstration that the p32 receptor can be accessed with a drug-like compound has important implications. First, it shows a peptide used as a discovery tool for tumor delivery can be converted to a chemistry that is likely to expand what can be accomplished with peptides. For example, the sensitivity of peptides to proteolytic degradation would be circumvented. Second, these findings bring up the prospect of designing synaphically targeted drugs that are orally active. Lastly, the compounds identified through the peptide screening combined with subsequent drug screening may have inherent biological activities. As discussed elsewhere, peptides that bind to proteins generally bind at binding pockets for compounds that modify the activity of the protein [30]. As such, they are likely to have inherent biological activities. A prime example is the integrin-binding RGD peptides, probably the most frequently used peptide motif in the design of tumor-targeting drugs and nanoparticles [30]. These peptides inhibit integrin activity and drugs based on RGD are in the clinic and in clinical trials. The chemical compounds modeled after the p32-binding peptides may have similar potential.
Supplementary Material
Table 1.
Volumes and final concentrations of the reagents used in high-throughput fluorescence polarization screening of chemical compound library.
| Volume | Final concentration | |
|---|---|---|
| FAM-peptide | 1.5 μL | 10 nM |
| p32 protein | 1.5 μL | 6 μM |
| Library compounds | 37.5 nL | 25 μM |
Abbreviations used
- AgNP
Silver nanoparticles
- BSA
Bovine serum albumin
- DAPI
Diamidino-2-phenylindole
- DTT
Dithiothreitol
- ELISA
Enzyme-linked immunosorbent assay
- FAM
5(6)-carboxyfluorescein
- FP
Fluorescence polarization
- HPLC
High pressure liquid chromatography
- Kd
Dissociation constant
- NMR
Nuclear magnetic resonance
- NRP-1
Neuropilin-1 (NRP-1)
- NW
Iron oxide nanoworms
- PBS
Phospate buffered saline
- PBST
Phosphate buffered saline with 10% Tween20
- PEG
Polyethylene glycol
- Q-TOF
Quadrupole time-of-flight
- TFA
Trifluoro acetic acid
Footnotes
This work was supported by grants R01CA167174 (KNS) and R01CA152327 (ER) from the National Cancer Institute of NIH. LP was supported by Academy of Finland Post-doctoral Researchers grant. GBB was supported by CA121949 NIH T32 Fellowship, and TT was supported by Susan Komen for Cure Foundation career development award KG110704, European Research Council Starting Grant (GliomaDDS), a grant from European Regional Development Fund, Wellcome Trust International Fellowship (WT095077MA), by the Int. Research Staff Exchange Scheme (IRSES) grant CHIMERA (n° 609850), and by Norway Grants (EMP181). Authors would like to thank Dr. Cheng-Ting Ma and Dr. Fu-Yue Zeng for technical assistance in the development of the FP screening method and Dr. Tarmo Mölder and Dr. Sergei Kopanchuk for help with peptide affinity measurement. Dr. Fu-Yue Zeng is acknowledged for the analysis of high-throughput screening data, and Dr. Jani Saarela at Institute for Molecular Medicine Finland (FIMM) for assisting in FP measurements and data interpretation.
Author contribution
LP and TT designed and performed the experiments and wrote the manuscript. GBB performed experiments with silver nanoparticles. SS performed experiments with iron oxide nanoworms. VRK synthesized the peptides. ZS performed TT1 binding experiments. MP and BW carried out NMR experiments. KNS and ER designed the experiments and edited the manuscript. MY participated in development and interpretation of FP assay and helped to edit the manuscript.
References
- 1.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laakkonen P, Porkka K, Hoffman J, Ruoslahti E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med. 2002;8:751–755. doi: 10.1038/nm720. [DOI] [PubMed] [Google Scholar]
- 3.Fogal V, Zhang L, Krajewski S, Ruoslahti E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008;68:7210–7218. doi: 10.1158/0008-5472.CAN-07-6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karmali PP, Kotamraju VR, Kastantin M, Black M, Missirlis D, Tirrell M, Ruoslahti E. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine-Nanotechnology Biology and Medicine. 2009;5:73–82. doi: 10.1016/j.nano.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JH, von Maltzahn G, Xu MJ, Fogal V, Kotamraju VR, Ruoslahti E, Bhatia SN, Sailor MJ. Cooperative nanomaterial system to sensitize, target, and treat tumors. Proc Natl Acad Sci U S A. 2010;107:981–986. doi: 10.1073/pnas.0909565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo G, Yu X, Jin C, Yang F, Fu D, Long J, Xu J, Zhan C, Lu W. LyP-1-conjugated nanoparticles for targeting drug delivery to lymphatic metastatic tumors. Int J Pharm. 2010;385:150–156. doi: 10.1016/j.ijpharm.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 7.Yan Z, Wang F, Wen Z, Zhan C, Feng L, Liu Y, Wei X, Xie C, Lu W. LyP-1-conjugated PEGylated liposomes: A carrier system for targeted therapy of lymphatic metastatic tumor. J Controlled Release. 2012;157:118–125. doi: 10.1016/j.jconrel.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 8.Laakkonen P, Akerman ME, Biliran H, Yang M, Ferrer F, Karpanen T, Hoffman RM, Ruoslahti E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc Natl Acad Sci U S A. 2004;101:9381–9386. doi: 10.1073/pnas.0403317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamzah J, Kotamraju VR, Seo JW, Agemy L, Fogal V, Mahakian LM, Peters D, Roth L, Gagnon MKJ, Ferrara KW, Ruoslahti E. Specific penetration and accumulation of a homing peptide within atherosclerotic plaques of apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 2011;108:7154–7159. doi: 10.1073/pnas.1104540108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pernot M, Vanderesse R, Frochot C, Guillemin F, Barberi-Heyob M. Stability of peptides and therapeutic success in cancer. Expert Opin Drug Metab Toxicol. 2011;7:793–802. doi: 10.1517/17425255.2011.574126. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee J, Rechenmacher F, Kessler H. N-Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions. Angewandte Chemie-International Edition. 2013;52:254–269. doi: 10.1002/anie.201205674. [DOI] [PubMed] [Google Scholar]
- 12.Zanuy D, Flores-Ortega A, Jimenez AI, Calaza MI, Cativiela C, Nussinov R, Ruoslahti E, Aleman C. In Silico Molecular Engineering for a Targeted Replacement in a Tumor-Homing Peptide. J Phys Chem B. 2009;113:7879–7889. doi: 10.1021/jp9006119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agemy L, Sugahara KN, Kotamraju VR, Gujraty K, Girard OM, Kono Y, Mattrey RF, Park J, Sailor MJ, Jimenez AI, Cativiela C, Zanuy D, Sayago FJ, Aleman C, Nussinov R, Ruoslahti E. Nanoparticle-induced vascular blockade in human prostate cancer. Blood. 2010;116:2847–2856. doi: 10.1182/blood-2010-03-274258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanuy D, Sayago FJ, Revilla-Lopez G, Ballano G, Agemy L, Kotamraju VR, Jimenez AI, Cativiela C, Nussinov R, Sawvel AM, Stucky G, Ruoslahti E, Aleman C. Engineering strategy to improve peptide analogs: from structure-based computational design to tumor homing. J Comput Aided Mol Des. 2013;27:31–43. doi: 10.1007/s10822-012-9623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leamon C, Low P. Folate-mediated targeting: from diagnostics to drug and gene delivery. Drug Discov Today. 2001;6:44–51. doi: 10.1016/s1359-6446(00)01594-4. [DOI] [PubMed] [Google Scholar]
- 16.Waibel R, Treichler H, Schaefer NG, van Staveren DR, Mundwiler S, Kunze S, Kueenzi M, Alberto R, Nueesch J, Knuth A, Moch H, Schibli R, Schubiger PA. New derivatives of vitamin B12 show preferential targeting of tumors. Cancer Res. 2008;68:2904–2911. doi: 10.1158/0008-5472.CAN-07-6771. [DOI] [PubMed] [Google Scholar]
- 17.Friedman AD, Claypool SE, Liu R. The Smart Targeting of Nanoparticles. Curr Pharm Des. 2013;19:6315–6329. doi: 10.2174/13816128113199990375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas VH, Bhattachar S, Hitchingham L, Zocharski P, Naath M, Surendran N, Stoner CL, El-Kattan A. The road map to oral bioavailability: an industrial perspective. Expert Opinion on Drug Metabolism & Toxicology. 2006;2:591–608. doi: 10.1517/17425255.2.4.591. [DOI] [PubMed] [Google Scholar]
- 19.Owicki JC. Fluorescence polarization and anisotropy in high throughput screening: Perspectives and primer. Journal of Biomolecular Screening. 2000;5:297–306. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 20.Lea WA, Simeonov A. Fluorescence polarization assays in small molecule screening. Expert Opinion on Drug Discovery. 2011;6:17–32. doi: 10.1517/17460441.2011.537322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jha BK, Salunke DM, Datta K. Disulfide bond formation through Cys186 facilitates functionally relevant dimerization of trimeric hyaluronan-binding protein 1 (HABP1)/p32/gC1qR. European Journal of Biochemistry. 2002;269:298–306. doi: 10.1046/j.0014-2956.2001.02654.x. [DOI] [PubMed] [Google Scholar]
- 22.Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A. 2009;106:16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.NIH. PAR-14-284: High Throughput Screening (HTS) to Discover Chemical Probes. 2014 http://grants.nih.gov/grants/guide/pa-files/PAR-14-284.html.
- 24.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 25.Teesalu T, Sugahara KN, Ruoslahti E. Mapping of Vascular Zip Codes by Phage Display. Methods Enzymol. 2012;503:35–56. doi: 10.1016/B978-0-12-396962-0.00002-1. [DOI] [PubMed] [Google Scholar]
- 26.Barile E, Pellecchia M. NMR-Based Approaches for the Identification and Optimization of Inhibitors of Protein-Protein Interactions. Chem Rev. 2014;114:4749–4763. doi: 10.1021/cr500043b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braun GB, Friman T, Pang H, Pallaoro A, de Mendoza TH, Willmore AA, Kotamraju VR, Mann AP, She Z, Sugahara KN, Reich NO, Teesalu T, Ruoslahti E. Etchable plasmonic nanoparticle probes to image and quantify cellular internalization. Nature Materials. 2014;13:904–911. doi: 10.1038/nmat3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park J, von Maltzahn G, Zhang L, Derfus AM, Simberg D, Harris TJ, Ruoslahti E, Bhatia SN, Sailor MJ. Systematic Surface Engineering of Magnetic Nanoworms for in vivo Tumor Targeting. Small. 2009;5:694–700. doi: 10.1002/smll.200801789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agemy L, Kotamraju VR, Friedmann-Morvinski D, Sharma S, Sugahara KN, Ruoslahti E. Proapoptotic Peptide-Mediated Cancer Therapy Targeted to Cell Surface p32. Molecular Therapy. 2013;21:2195–2204. doi: 10.1038/mt.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruoslahti E. Peptides as Targeting Elements and Tissue Penetration Devices for Nanoparticles. Adv Mater. 2012;24:3747–3756. doi: 10.1002/adma.201200454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 32.Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31:1191–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.