

SUMMARY
Despite antiretroviral therapy, HIV-1 persists in memory CD4+ T cells creating a barrier to cure. The majority of HIV-1 proviruses are defective and considered clinically irrelevant. Using cells from HIV-1-infected individuals and reconstructed patient-derived defective proviruses, we show that defective proviruses can be transcribed into RNAs that are spliced and translated. Proviruses with defective major splice donors (MSDs) can activate novel splice sites to produce HIV-1 transcripts and cells with these proviruses can be recognized by HIV-1-specific cytotoxic T lymphocytes (CTLs). Further, cells with proviruses containing lethal mutations upstream of CTL epitopes can also be recognized by CTLs potentially through aberrant translation. Thus, CTLs may change the landscape of HIV-1 proviruses by preferential targeting cells with specific types of defective proviruses. Additionally, the expression of defective proviruses will need to be considered in the measurement of HIV-1 latency reversal.
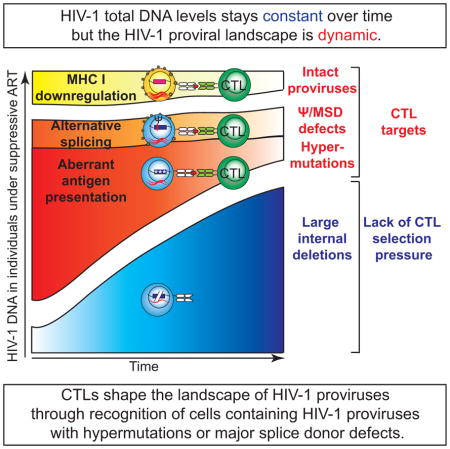
Graphical Abstract
INTRODUCTION
Despite effective antiretroviral therapy (ART), HIV-1 persists in a latent reservoir in resting memory CD4+ T cells (rCD4s). This reservoir is a major barrier to cure (Chun et al., 1997; Finzi et al., 1997; Wong et al., 1997). In HIV-1-infected individuals on ART, ~300/106 rCD4s contain HIV-1 proviruses (Eriksson et al., 2013), but only ~1/106 contains inducible replication-competent proviruses (Crooks et al., 2015; Siliciano et al., 2003). Most of HIV-1 proviruses are defective due to G→A hypermutations introduced by the enzyme apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC)-3G, large internal deletions, packaging signal (Ψ) deletions, major splice donor (MSD) mutations, and inactivating point mutations (Bruner et al., 2016; Ho et al., 2013). However, these defective HIV-1 proviruses (HIV-1def) may have the potential to express HIV-1 genes because of intact LTR promoter function, integration into active transcription units, and the lack of promoter CpG methylation (Ho et al., 2013).
In latently infected cells, HIV-1 proviruses with functional promoters are transcriptionally inactive because the cells lack active forms of key cellular factors and the Tat positive feedback loop and its cofactors (Bohnlein et al., 1988; Jones and Peterlin, 1994; Tyagi et al., 2010; Weinberger et al., 2005; West et al., 2001). Further, HIV-1 may be silenced through repressive histone modifications (Coull et al., 2000; Kauder et al., 2009; Van Lint et al., 1996). Latent proviruses are not affected by ART, and the lack of viral protein production conceals the latently infected cells from immune surveillance. Furthermore, some HIV-1-infected cells may proliferate in response to antigen (Douek et al., 2002) or homeostatic signals (Chomont et al., 2009). Clonal expansion of infected cells may also be the result of integration into host genes involved in cellular proliferation (Cohn et al., 2015; Maldarelli et al., 2014; Simonetti et al., 2016; Wagner et al., 2014). Therefore, the frequency of clonally expanded HIV-1-infected cells increases over time even in the setting of prolonged suppressive ART. However, the total frequency of HIV-1-infected cells remains unchanged over time (Besson et al., 2014), and the frequency of infected cells containing inducible replication-competent proviruses decays minimally over time (Crooks et al., 2015; Siliciano et al., 2003). Based on the discrepancy between stable levels of infected cells and increasing frequency of clonally expanded HIV-1-infected cells, we propose that the frequency of cells harboring a subset of HIV-1 proviruses may decrease under suppressive ART. Understanding the dynamics of infected cell populations may contribute to the development of cure strategies.
One of the major HIV-1 cure strategies is the shock-and-kill strategy (Deeks, 2012), which reverses HIV-1 latency and exposes infected cells to immune clearance. New rounds of infection will presumably be blocked by concurrent ART. Latency reversal can be measured by detecting changes in levels of cell-associated (CA-) or plasma HIV-1 RNA (Archin et al., 2012; Elliott et al., 2014; Elliott et al., 2015; Leth et al., 2016; Rasmussen et al., 2014; Sogaard et al., 2015). In some in vivo studies testing the shock-and-kill strategy, proviral DNA levels decreased by up to 40% (Leth et al., 2016). Given that >88% of the HIV-1 proviruses are defective (Bruner et al., 2016; Ho et al., 2013), it is unclear how proviral DNA levels can decrease by such a large amount. Thus, we propose that a subset of HIV-1def may be eliminated during the shock-and-kill strategy.
Elimination of HIV-1-infected cells relies on cytotoxic T lymphocyte (CTL) killing (Borrow et al., 1994; Koup et al., 1994; Schmitz et al., 1999; Walker et al., 1987). However, CTL mutations may develop rapidly (Borrow et al., 1997). Further, HIV-1-specific CTLs frequently show an exhausted phenotype and diminished function (Day et al., 2006; Petrovas et al., 2006; Trautmann et al., 2006) in HIV-1-infected individuals on ART during chronic infection. Without appropriate prestimulation, CTLs may not kill HIV-1-infected cells upon latency reversal (Deng et al., 2015; Shan et al., 2012). However, it has been proposed that CTLs contribute to viral suppression during chronic infection under ART (Cartwright et al., 2016). Given the potential of HIV-1def to be expressed, the presence of a subset of HIV-1 proviruses which may decay over time, and the pivotal role of CTL in eliminating HIV-1-infected cells, we hypothesize that CTLs can target a subset of HIV-1def and therefore actively shape the HIV-1 proviral landscape.
Here we show that HIV-1def can be transcribed and translated in vitro and ex vivo, complicating the measurement of the shock-and-kill strategy using HIV-1 RNA- and protein-based methods. We propose that cells containing a subset of HIV-1def can be targeted by CTLs, and thus CTL selection pressure may shape the landscape of HIV-1 proviruses.
RESULTS
HIV-1 proviral landscape may change over time
Given that the frequency of HIV-1-infected, clonally expanded CD4+ T cells increases over time (Maldarelli et al., 2014; Wagner et al., 2014) but HIV-1 DNA levels remain constant (Besson et al., 2014), we examined changes in the HIV-1 proviral landscape over time to understand which subsets of HIV-1 proviruses may change. We analyzed data from 2 previous studies of HIV-1-infected individuals using limiting dilution HIV-1 full-length proviral sequencing (Bruner et al., 2016; Ho et al., 2013)(Figure 1). We found that although the proportion of intact proviruses (Figure 1A) and proviruses with Ψ deletions (HIV-1dMSD)(Figure 1B) did not change significantly (potentially related to their low frequency during infection), the proportion of hypermutated HIV-1 proviruses (HIV-1hypermut) correlated inversely with duration of infection (Figure 1C), while the proportion of proviruses with large internal deletions (HIV-1del) correlated positively with duration of infection (Figure 1D). While HIV-1hypermut can accumulate rapidly during acute infection as shown by the observation that individuals treated during acute infection have a higher frequency of HIV-1hypermut than individuals treated during chronic infection (Bruner et al., 2016), interpretation of this result is complicated by the fact that patients treated during acute infection in this cohort have a significantly shorter duration of infection than patients treated during chronic infection (Figure S1). In a separate cohort of individuals treated during chronic infection, there was a high frequency of HIV-1hypermut (Figure 1C). Thus, the higher frequency of HIV-1hypermut in individuals treated during acute infection may reflect not only the higher activity of the interferon-induced restriction factors APOBEC during acute infection (Bruner et al., 2016) but also the fact that cells containing HIV-1hypermut and cells containing HIV-1del may proliferate, or be eliminated at different rates. In other words, the HIV-1 proviral landscape may be dynamic.
Figure 1. The landscape of HIV-1 proviruses may be dynamic.

Correlation between the duration from diagnosis to HIV-1 proviral landscape analysis (enrollment) and the proportion of different subsets of HIV-1 proviruses containing (A) intact genome, (B) Ψ/MSD deletions/mutations, (C) hypermutations, (D) large internal deletions. r and P values are calculated by Pearson correlation. See also Figure S1.
HIV-1def can be transcribed in vitro
To examine whether cells containing HIV-1def can be potential targets for CTLs, we first examined whether HIV-1def can be transcribed. We reconstructed 9 HIV-1def identified from 6 HIV-1-infected individuals on ART (Bruner et al., 2016; Ho et al., 2013)(Table S1–S4, Figure S2). We transfected activated primary CD4+ T cells with patient-derived proviruses in the presence of enfuvirtide to assess how proviruses containing a defective genome can be transcriptionally expressed. Enfuvirtide was chosen to block viral entry and to more precisely measure LTR-driven CA-HIV-1 RNA expression and avoid detection of HIV-1 RNA from any incoming virions. Of note, the expression level of HIV-1 proviral plasmids may be higher than that in vivo due to the lack of the influence of read-through transcription, epigenetic silencing and integration site bias. After DNase treatment and oligo-dT-primed cDNA synthesis, we measured CA-HIV-1 RNA by qRT-PCR using a HIV-1-RNA-specific primer/probe set specific for polyadenylated HIV-1 RNA to measure both spliced and unspliced HIV-1 RNA and avoid the amplification of plasmid DNA (Shan et al., 2013). Additionally, qRT-PCR products were sequenced to exclude nonspecific amplifications. Surprisingly, we found that most HIV-1def can be transcribed (Figure S3). HIV-1 RNA expression was detected from proviruses with Ψ/MSD defects, hypermutations, large internal deletions, and nonsense mutations. The only clone for which no HIV-1 RNA was detected was the HIV-1hypermut clone 2G10 which contains qRT-PCR primer/probe mismatches (red asterisks on Figure S3). HIV-1 RNA expression from this hypermutated clone can be detected using gel electrophoresis and custom primer/probes matching the relevant hypermutated nucleotides (Figure S4A–B). This demonstrates that HIV-1def, even proviruses with a hypermutated LTR involving mutated major transcription factor binding sites (Figure S4C), can be transcribed.
Although the above studies indicate that HIV-1def can be transcribed, many HIV-1def contain Ψ/MSD defects. The production of essential HIV-1 proteins requires mRNA splicing (Chang and Sharp, 1989; Feinberg et al., 1986; Guatelli et al., 1990; Schwartz et al., 1990a). Measurement of spliced HIV-1 RNA is used in some assays for the latent reservoir (Procopio et al., 2015). However, it remains unclear whether HIV-1def containing defective splice donor and acceptor sites can produce spliced transcripts. We used primers to detect singly spliced HIV-1 mRNA (Shan et al., 2013) and multiply spliced HIV-1 RNA (Massanella et al., 2015; Procopio et al., 2015)(Table S5). Interestingly, we found that spliced HIV-1 RNAs were detectable despite defective MSD (Figure S3B–D). This suggests that alternative splicing may bypass the MSD defects to produce spliced HIV-1 RNA (see below). As expected, clones containing deletions spanning the primer/probe binding sites were not amplified (blue asterisks, Figure S3B–D). Clones containing mismatched primer/probe binding sites (red asterisks, Figure S3B–D) were either not amplified or were detected at a lower level in the nested amplification (Figure S3D). Overall, our data show that even HIV-1dMSD can still produce readily detectable spliced HIV-1 RNA.
Induction of HIV-1 RNA release into cell supernatants is also used as a measure of functional proviruses in some assays (Cillo et al., 2014). To understand whether HIV-1def can produce virions, we measured polyadenylated HIV-1 RNA from culture supernatant after RNase treatment to eliminate RNA not protected within the virion (Figure S3E). We found that the detection of supernatant HIV-1 RNA more precisely reflects viral particle production which requires intact tat, rev and gag genes (Table S3) because the production of viral particles requires the structural protein Gag and Rev-mediated nuclear export of Gag-encoding unspliced HIV-1 RNA into the cytoplasm (Malim et al., 1989). With a defective Ψ, genomic HIV-1 RNA was packaged at a much lower efficiency (Lever et al., 1989) but was not completely undetectable (Figure S3). Thus, supernatant HIV-1 RNA detection may more precisely reflect the presence of intact structural (such as Gag) and regulatory elements (such as Rev), although HIV-1dMSD may still be detected. Overall, our finding suggests that both spliced and unspliced HIV-1 RNA produced by HIV-1def can be detected by assays currently used to measure HIV-1 RNA expression.
HIV-1def can bypass defects in MSD using novel alternative splice sites
While HIV-1 proviruses can utilize alternative splice sites to produce spliced transcripts containing coding regions of essential viral proteins such as Tat and Rev (Guatelli et al., 1990; Ocwieja et al., 2012; Purcell and Martin, 1993; Schwartz et al., 1990a), the MSD is critical for the production of functional HIV-1 splice variants. It remains unclear how patient-derived proviruses containing extensive and previously unreported MSD defects are able to produce splice variants encoding essential viral proteins. We sequenced spliced RNAs and detected the use of known alternative splice donor sites (SDSs)(light blue in Figure 2) as previously described (Ocwieja et al., 2012). Surprisingly, we identified novel SDS (DNovel1–4)(Figure 2A–F) which can splice into either canonical splice acceptor sites (SASs) or a previously unreported novel SAS (ANovel, Figure 2B). These previously unreported splice variants provide insight into the transcriptional landscape of HIV-1 proviruses. First, novel splicing reactions into canonical SASs (Figure 2B–F) indicate that HIV-1 RNAs encoding Tat and Rev can be produced despite a defective MSD. Second, alternative SDS sequence such as AC|GG (DNovel1, Figure 2B) and TA|GT (mutated MSD, Figure 2E) can be activated, suggesting that HIV-1 splicing may not be restricted by typical cellular splicing patterns. Third, DNovel2 uses the ATGG|GT site at the gag start codon and was identified in RNAs produced from 3 different clones (45E6, 39G2, 42B6)(Figure 2B–D) reconstructed from 3 different individuals. This reveals a previously unreported SDS hotspot. The novel splice product contains the ATG start codon, suggesting that it may produce aberrant proteins. Overall, we found that proviruses containing defective MSD can activate alternative SDSs and splice into canonical SASs to produce RNA splice variants encoding essential viral proteins.
Figure 2. HIV-1 proviruses can bypass MSD defects by activating novel SDSs and splicing into canonical SASs in vitro and ex vivo.

(A) Spliced HIV-1 RNA transcripts from reconstructed patient-derived proviruses. Numbers on the right indicate the number of isolates from cloning of PCR products. (B–F) Sequences of canonical (Ocwieja et al., 2012; Schwartz et al., 1990a), known alternative (Ocwieja et al., 2012), and novel splice sites. (G) Spliced HIV-1 RNA from patient resting CD4+ T cells upon ex vivo activation. (H) Sequences of novel splice sites in participant 85. Purple box, short sequence repeats likely related to the 245 bp deletion encompassing MSD. See also Table S1–S5, Figures S2–S6.
HIV-1def can be transcribed ex vivo
To examine whether HIV-1dMSD can be transcribed ex vivo, we isolated rCD4s from HIV-1-infected individuals on ART. We sequenced spliced HIV-1 RNA products from these CD4+ T cells following activation with anti-CD3/CD28 in the presence of enfuvirtide. While most spliced HIV-1 RNAs utilize canonical splice sites (Figure 2G), we identified one novel spliced product from participant 85 (Figure 2H). This HIV-1 RNA transcript contains a deletion (HXB2 nt 707–952) encompassing both the MSD and the gag start site and activates a novel SDS at nt 1,066 allowing splicing into a canonical SAS at nt 5,975 (Figure 2H). The presence of short sequence repeats at the junction of the large internal deletion is characteristic of proviral HIV-1 deletions which arise from copy choice recombination (Ho et al., 2013). Together these results suggest that HIV-1def containing a defective MSD can be transcribed and produce HIV-1 RNAs with novel splicing patterns ex vivo.
Hypermutated HIV-1 RNA has been identified in HIV-1-infected individuals (Barton et al., 2016; Kearney et al., 2015). To examine the proportion of HIV-1 RNA that are produced from HIV-1hypermut, we isolated rCD4s from HIV-1-infected individuals on ART and measured CA-HIV-1 gag RNA in the resting state and upon anti-CD3/CD28 activation in the presence of enfuvirtide. Genomic DNA was removed by DNase treatment of RNA samples. We measured the amount of HIV-1 gag RNA by qRT-PCR and determined the proportion of hypermutated HIV-1 gag RNA containing a mutated start codon or premature stop codons using targeted gag deep sequencing (Figure 3A). A full-length gag RNA read containing either mutated start codon or in frame stop codon in the hypermutation hotspots (tryptophan coding TGG) was counted as 1 hypermutated read containing lethal mutations. A significant proportion of CA-gag RNAs contained lethal nonsense mutations characteristic of APOGBEC-mediated G→A hypermutations, both in the resting state (0–56%)(Figure 3B) and the activated state (5–29%)(Figure 3C), accounting for ~100–100,000 copies for HIV-1 RNA/106 cells (Figure 3D–E). Therefore, measurement of the latent reservoir by assays for CA-HIV-1 RNA may be complicated by the expression of defective HIV-1 RNAs.
Figure 3. HIV-1hypermut can be transcribed ex vivo.

Transcription of HIV-1hypermut in vivo from rCD4s and upon ex vivo anti-CD3/CD28 activation. Resting CD4+ T cells from ART-suppressed individuals were treated with or without anti-CD3/CD28 activation for 3 days in the presence of enfuvirtide. Levels of non-hypermutated and hypermutated HIV-1 gag RNA were measured as the frequency of total HIV-1 gag (by qRT-PCR) times the proportion of hypermutated HIV-1 gag RNA (as measured by targeted gag deep sequencing). (A) Location of hypermutation hotspots (TGG) which will cause lethal missense start codon mutation or lethal nonsense mutations. (B–C) Proportion of hypermutated HIV-1 RNA. Green, nonhypermutated HIV-1 RNA. Red, hypermutated HIV-1 RNA containing lethal mutations. (D–E) Frequency of hypermutated HIV-1 RNA in resting and activated CD4+ T cells. Data represent mean ± SEM.
Reconstructed patient-derived HIV-1def can be translated
Although HIV-1def can be transcribed, it remains unclear whether the resulting RNAs can be translated into viral proteins. We transfected activated primary CD4+ T cells with defective proviral plasmids and measured intracellular Gag protein by flow cytometry (Figure S5A). Gag protein was readily detected in cells transfected with HIV-1dMSD (45E6, 39G2, 42B6) and point mutations elsewhere in the genome (19B3), but not from HIV-1hypermut and HIV-1del (Figure S5A).
To examine whether HIV-1def can support the packaging and release of virus particles into the supernatant, we measured extracellular Gag protein in culture supernatants using a p24 enzyme-linked immunosorbent assay (ELISA)(Figure S5B) over 7 days. Consistent with the intracellular Gag staining results, Gag protein could be detected in supernatants of cells transfected with HIV-1dMSD and clones a clone containing point mutations, but not from HIV-1hypermut or HIV-1del (Figure S5B). Interestingly, none of these HIV-1def can express sufficient level of Nef as shown by inability to downregulate CD4 expression (Figure S6). Together, these results indicate that cells carrying HIV-1dMSD can release Gag protein into the supernatant, potentially complicating measurement of the latent reservoir by assays that detect release of p24 antigen into the supernatant.
Cells carrying HIV-1def can be recognized by CTLs in vitro
The translation of Gag, and potentially other viral gene products, from HIV-1def could comprise a source of antigens capable of stimulating adaptive immune responses. Despite the presence of premature stop codons and deletions, HIV-1def may produce defective ribosomal products (DRiPs) that are presented on major histocompatibility complex (MHC) class I using alternative translation pathways (Goldwich et al., 2008; Yewdell et al., 1996). To examine whether cells with HIV-1def can be targeted by HIV-1-specific CTLs, we co-cultured activated CD4+ T cells transfected with HIV-1def with autologous CTL clones specific for 2 Gag (Figure 4A–B) and 2 Nef (Figure 4C–D) epitopes from 2 HIV-1-infected individuals on ART, as well as a cytomegalovirus (CMV)-specific clone as a negative control (Figure 4E). CTL recognition was assessed by CD107a expression as a measure of degranulation (Figure S7A) using previously optimized conditions (Jones et al., 2012). As a positive control, we used autologous CD4+ T cells pulsed with corresponding peptides, which stimulated robust CTL degranulation. As a negative control, we used vector-transfected CD4+ T cells which showed minimal CTL recognition, reflecting baseline CTL recognition resulting from the expression of autologous HIV-1 proviruses (Figure 4A, white bars). Cells transfected with HIV-1def 45E6 and 19B3, which showed Gag production (Figure S5A), were recognized by the B27-Gag-IK9-specific CTL clone (Figure 4A, green bars). Surprisingly, cells transfected with the HIV-1hypermut 31G4 which has a missense Gag start codon mutation 5′ to the B27-Gag-IK9 epitope and a premature stop 5′ to the B07-Gag-HA9 epitope (Figure 4AB, red bars) were still recognized by respective CTL clones despite no detectable Gag protein expression by flow cytometry (Figure S5A). In addition, cells transfected with 19B3 containing premature stop codons 5′ to the Cw8-Nef-AL9 epitope were recognized by the CTL clone (Figure 4C, red bar). 45E6 did not induce significant levels of CTL recognition likely because of the lack of sufficient Nef expression (Figure S6) due to less efficient alternative splicing. The level of CTL recognition of cells transfected with HIV-1def is comparable to or higher than that of cells transfected with the NL4–3 reference plasmid. Cells transfected with HIV-1def containing mismatches or deletions encompassing the targeted epitope were not recognized (Figure 4A–D, gray and dark blue bar), indicating the specificity of this assay. Cells transfected with HIV-1del despite the presence of an intact epitope (48C8 and E44E11, Figure 5A, 5C–D, light blue bars) were not recognized, likely reflecting the effect of an insufficient level of protein production or antigen presentation. For example, deletion of the tat and rev genes in 48C8 is likely to reduce gene expression and export of unspliced mRNA containing the gag gene. Together, these results show that cells with proviruses containing Ψ defects and hypermutations may induce CTL recognition.
Figure 4. Cells containing HIV-1def can be recognized by HIV-1-specific CTLs.

Flow cytometry showing %degranulated CTLs (CD8+CD107a+) from 2 Gag-specific clones (A–B), 2 Nef-specific clones (C–D) and a CMV-specific clone (E) upon co-culture with autologous CD4+ T cells transfected with HIV-1def plasmids. NL4-3 has an anchor residue mutation (K162Q) at the IK9 epitope making it not susceptible to CTL recognition (A). Each target was tested in independent quadruplets shown in mean ± SEM. P values were calculated by one-way ANOVA with Dunnett’s multiple comparison test. *P<0.05, **P<0.01, ***P<0.001, **** P<0.0001. See also Figure S7.
Figure 5. Cells containing HIV-1def competes with cells containing intact proviruses for CTL recognition.

(A) Negative control using CD4+ T cells transfected with the vector plasmid. (B) Positive control using CD4+ T cells transfected with the vector plasmid and loaded with cognate peptide. (C–E) Cold-target inhibition assay showing HIV-1-specific CTL recognition of cells containing HIV-1def. Six paired replicates per condition were shown. P values were calculated by paired Student t-test. See also Figure S7.
We next used a competition assay analogous to the classic “cold-target” inhibition assays to examine HIV-1-specific CTL recognition of cells containing HIV-1def. Cold-target inhibition was initially used to determine the antigenic similarity of target cells with respect to CTL recognition (Schick and Berke, 1979). “Cold” (unlabeled) targets classically inhibit the CTL-mediated lysis of 51Cr-labeled “hot” targets if cold and hot targets are antigenically similar. We tested whether cells containing HIV-1def (cold targets) may impair CTL killing of cells containing intact proviruses (real targets)(Figure S7). CD4+ T cells autologous to the CTL clones were activated and either transfected with plasmids encoding HIV-1def known to support CTL recognition (45E6, 31G4, 19B3)(Figure 4A) to serve as “cold targets”, or infected with replication-competent HIV-1LAI to serve as “real targets”. Real targets and cold targets were co-cultured with the autologous HIV-1-Gag-IK9-specific CTL clone at effector CTL : real target : cold target ratios of 1 : 20 : 800 and 1 : 100 : 4000 (input cell number)(Figure S7B). The ratio of infected real targets versus transfected cold targets is approximately 1:12 after accounting for infection and transfection efficiency (Figure S7). Cells transfected with empty vector did not impair CTL killing of the real targets (Figure 5A), while vector-transfected cold targets that had been pulsed with cognate Gag-IK9 peptide significantly impaired the elimination of HIV-1-infected real targets (Figure 5B). We observed significant cold-target inhibition indicating CTL recognition of the HIV-1dMSD 45E6 clone (Figure 5C) capable of Gag protein expression (Figure S5A). Surprisingly, we showed cold-target inhibition by the HIV-1hypermut 31G4 clone which contains a mutated gag start codon 5′ to the CTL epitope recognition site (Figure 5D), confirming that this hypermutated clone can be recognized by CTL. We did not observe significant CTL recognition of the mutated 19B3 clone (Figure 5E), probably related to marginal recognition of cells transfected with 19B3 (Figure 4A). Thus, this competition assay confirms that cells containing HIV-1dMSD and HIV-1hypermut can be recognized by CTLs.
Cells carrying HIV-1hypermut can be recognized by CTLs ex vivo
To examine whether cells containing HIV-1hypermut can be recognized by CTLs ex vivo, we tested whether the amount of HIV-1 RNA containing intact CTL epitope but a lethal mutation 5′ to the CTL recognition epitope would decrease upon CTL co-culture. We examined human leukocyte antigen (HLA) types of the study participants and identified known CTL epitopes using a previously described method (Deng et al., 2015). We isolated rCD4s from HIV-1-infected individuals on ART and activated these cells with anti-CD3/CD28 for 3 days in the presence of enfuvirtide to induce HIV-1 RNA expression. We also stimulated autologous CD8+ T cells using a Gag consensus peptide mixture, SL9 (a HLA-A2-restricted Gag epitope), and a CMV peptide mixture in the presence of autologous peripheral blood mononuclear cells for 6 days. We then isolated prestimulated autologous CD8+ T cells, co-cultured them with autologous activated CD4+ T cells and measured the amount of hypermutated CA-HIV-1 RNA (Figure 6). We found that the proportion (Figure 6A–C) and amount (Figure 6D) of HIV-1 RNA containing lethal hypermutations (red, Figure 6A–C) decreased strikingly upon co-culture with HIV-1-specific CD8+ T cells relative to coculture with CMV-stimulated CD8+ T cells, indicating that cells containing hypermutated HIV-1 RNA may be eliminated by antigen-specific CTLs. We next examined the proportion of HIV-1 RNA with and without translation-terminating mutations 5′ to the CTL epitope. The proportion of HIV-1 RNA containing escape mutations at the indicated dominant CTL epitopes without upstream lethal mutations (light green, Figure 6E–G) increased upon HIV-1-specific CD8+ T cell co-culture, indicating the selection pressure of HIV-1-specific CD8+ T cells against cells containing HIV-1 with wild-type CTL epitopes. Strikingly, we found that HIV-1 RNA containing lethal hypermutations 5′ to the CTL epitope, even those containing escaped epitopes (pink, Figure 6E–G), decreased dramatically upon co-culture with HIV-1-specific CD8+ T cells. Together these results strongly suggest that cells containing hypermutated HIV-1 RNA can be eliminated by HIV-1-specific CTLs ex vivo.
Figure 6. Cells containing HIV-1hypermut can be recognized by CD8+ T cells ex vivo.

Resting CD4+ T cells were activated with anti-CD3/CD28 in the presence of enfuvirtide and co-cultured with autologous CD8+ T cells prestimulated with a Gag peptide mixture, a CMV peptide mixture or a HLA-A2-restricted SL9 peptide. Targeted gag RNA deep sequencing and qRT-PCR was used to measure the proportion and the frequency of HIV-1 RNA containing lethal hypermutations. (A–D) Proportion (A–C) and frequency (D) of HIV-1 RNA containing lethal hypermutations from CD4+ T cells upon co-culture with autologous CMV- (A), Gag- (B), SL9-(C) prestimulated CD8+ T cells. (E–G) Proportion of HIV-1 RNA with or without lethal hypermutations 5′ to wild-type or escaped CTL epitopes upon co-culture with autologous CMV- (E), Gag- (F), and SL9- (G) prestimulated CD8+ T cells. Data represent mean ± SEM. *, P value <0.05 by two-tailed Student t-test.
Overall, we showed that different subsets of HIV-1def may encounter different levels of CTL selection pressure (Figure 7). Cells containing HIV-1dMSD and HIV-1hypermut are potential targets for CTLs. Cells containing HIV-1dMSD exist at a low frequency (<7%)(Bruner et al., 2016; Ho et al., 2013), making it hard to see a change in frequency over time. However, cells containing HIV-1hypermut account for a significant proportion of infected cells (>15%)(Bruner et al., 2016; Ho et al., 2013) and may be recognized by CTLs. Cells containing HIV-1del may not be recognized by CTLs, and therefore may have a better chance to persist over time due to a lack of CTL selection pressure. The dynamic nature of the proviral landscape may provide insights into effective eradication strategies.
Figure 7. Proposed model of the scope of potential targets of HIV-1-specific CTLs compared with the size of the latent reservoir.

The provirus population includes intact (yellow and pink) and defective (blue) proviruses as described (Ho et al., 2013). Following reversal of latency by endogenous stimuli or latency reversing agents, cells with certain types of HIV-1def may express HIV-1 RNA and protein and may become targets for HIV-1-specific CTLs.
DISCUSSION
The studies presented here redefine the scope of potential targets for HIV-1-specific CTLs. We propose that both CTL selection pressure and clonal expansion shape the HIV-1 proviral landscape. HIV-1def account for the vast majority of proviruses in rCD4s of HIV-1-infected individuals on ART. These HIV-1def were generally considered harmless and irrelevant to cure strategies. We showed that HIV-1def can be transcribed into RNAs that are spliced and translated. The level of HIV-1def gene expression can be lower than that of intact proviruses due to decreased LTR function and insufficient Tat expression. However, cells containing HIV-1def can be recognized by CTLs specific for epitopes downstream of a mutated start codon or premature stop codons. These cryptic epitopes may be presented through various forms of non-canonical translation including the use of defective ribosomal products (Goldwich et al., 2008; Yewdell et al., 1996), alternative reading frames (Cardinaud et al., 2004; Ho and Green, 2006), antisense transcripts (Bansal et al., 2010), alternative start codons (Malarkannan et al., 1999; Starck et al., 2012), and leaky ribosomal scanning (Bullock and Eisenlohr, 1996; Schwartz et al., 1990b). Our results are consistent with previous studies showing that cells harboring defective HIV-1 constructs containing premature stop codons in Gag (Casartelli et al., 2010) or presenting cryptic epitopes from alternative reading frames (Bansal et al., 2010; Maness et al., 2010) may induce CTL responses. Furthermore, HIV-1def may not downregulate MHC-I due to the lack of functional Nef. Thus, cells containing HIV-1def may present aberrant proteins more efficiently to CTLs. Given that HIV-1def greatly outnumber intact proviruses, and that HIV-1def were shown to be transcribed in vivo (Barton et al., 2016; Kearney et al., 2015), it is possible that HIV-1def may impact the repertoire and function of HIV-1-specific CTLs.
The role of HIV-1def in vivo remains to be determined. Unlike ex vivo experiments where CTLs and cells containing HIV-1 are cultured at defined ratios, the effector-to-target ratio in vivo may vary widely depending on anatomical location and other factors. CTLs may be excluded from some sites such as the germinal centers (Fukazawa et al., 2015). Interestingly, a recent study using a simian immunodeficiency virus (SIV)-infected macaque model (Borducci et al., 2016) showed that effective therapeutic vaccination decreased SIV DNA level by 2–3 logs, but the frequency of intact proviruses decreased only by ~1 log, suggesting immune clearance of cells containing defective proviruses. In addition, it has been shown that HIV-1 proviral DNA levels correlate with immune activation and exhaustion in HIV-1-infected individuals treated with suppressive ART (Hatano et al., 2013). Recent studies further imply that HIV-1def may induce immune activation (Imamichi et al., 2016). Given that the majority of the HIV-1 proviruses are defective, it is not unlikely that cells containing HIV-1def may induce immune activation and T cell exhaustion. Whether HIV-1def may distract CTL from eliminating the latent reservoir as decoys, may cause immune activation, or may boost CTL responses, should be explored in vivo.
Our results suggest that decreases in HIV-1 DNA levels may be possible from effective CTL-based interventions. On the other hand, our study demonstrates that proviruses with various defects can be detectable by CA- and supernatant HIV-1 RNA assays. Therefore, CA-HIV-1 RNA quantifications may serve as a surrogate for latency reversal and HIV-1 LTR function but not a precise reflection of the replication competence.
In conclusion, CTLs may shape the landscape of HIV-1 proviruses through differential negative selection pressure against cells with HIV-1dMSD and HIV-1hypermut. The scope of potential targets for CTLs may be greater than the size of the latent reservoir.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD3 antibody | BD Biosciences | 555336 RRID:AB_395742 |
| Anti-CD28 antibody | BD Biosciences | 555725 RRID:AB_396068 |
| Anti-Human CD3 Functional Grade Purified | eBioscience | 16-0037-85 RRID:AB_468855 |
| Anti-Human CD28 Functional Grade Purified | eBioscience | 16-0289-85 RRID:AB_468927 |
| Anti-HIV-1 core antibody clone KC57-PE | Beckman Coulter | 41116015 |
| BV421 Mouse Anti-Human CD4 Clone RPA-T4 | BD Biosciences | 562424 RRID:AB_11154417 |
| PE/Cy7 anti-human CD3 Antibody | Biolegend | 300420 RRID:AB_439781 |
| PerCP/Cy5.5 anti-human CD4 Antibody | Biolegend | 300530 RRID:AB_893322 |
| FITC Mouse Anti-Human CD8 | BD Biosciences | 347313 RRID:AB_400279 |
| PE anti-human CD107a (LAMP-1) Antibody | Biolegend | 328608 RRID:AB_1186040 |
| Bacterial and Virus Strains | ||
| HIV-1LAI | AIDS Reagent | 2522 |
| Biological Samples | ||
| Demographics of study participants, see Table S1 | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Enfurvitide | Genentech | NDC 0004-0381-40 |
| ProMix CMV Peptide Pool | ProImmune | PX-CMV |
| Gag PepMix HIV-1 | JPT Peptide Technologies | PM-HIV-CONB |
| SL9, HIV - 1 p17 (77 – 85) | Anaspec | AS-61526 |
| TRIzol | ThermoFisher | 15596-018 |
| TRIzol LS | ThermoFisher | 10296028 |
| RNase A | Qiagen | 158924 |
| Critical Commercial Assays | ||
| Human T Cell Nucleofector® Kit | Lonza | VPA-1002 |
| EasySep Direct Human CD4+ T cell kit | STEM CELL | 19662 |
| CD4 T cell isolation kit, human | Miltenyi Biotech | 130-096-533 |
| CD69 MicroBead Kit II, human | Miltenyi Biotech | 130-092-355 |
| CD25 MicroBeads, human | Miltenyi Biotech | 130-092-983 |
| HLA-DR MicroBeads, human | Miltenyi Biotech | 130-046-101 |
| Dynabeads Untouched CD8 Isolation Beads | ThermoFisher | 11348D |
| CD8 MicroBeads, human | Miltenyi | 130-045-201 |
| qScript Flex cDNA Synthesis Kit | Quanta Biosciences | 101414-112 |
| PerfeCTa qPCR ToughMix | Quanta Biosciences | 95139-250 |
| TaqMan Copy Number Reference Assay | ThermoFisher | 4403328 |
| Human Genomic DNA | Roche | 11691112001 |
| Dnase I, Amplification Grade | ThermoFisher | 18068015 |
| TA TOPO cloning kit | ThermoFisher | 45-0071 |
| Platinum Taq High Fidelity | ThermoFisher | 11304102 |
| HIV p24 ELISA | Perkin Elmer | NEK050B |
| IFN-γ secretion detection and enrichment kit | Miltenyi Biotech | 130-054-201 |
| Cytofix/Cytoperm | BD Biosciences | 554714 |
| CellTrace Far Red | ThermoFisher | C34564 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Primers and probes, see Table S5 | This paper | N/A |
| Recombinant DNA | ||
| pNL4-3 | AIDS Reagent | 114 |
| Patient-derived plasmids KF526141, KF526152, KF526217, KF526220, KF526245, KF526254, KF526298, KF526338, KX505621 |
Ho et al., Cell 2013 Bruner et al., Nature Medicine 2016 |
N/A |
| Software and Algorithms | ||
| MedCalc Statistical Software | MedCalc Software bvba, Ostend, Belgium | N/A |
| GraphPad Prism Software | GraphPad Software, La Jolla California USA | N/A |
| Other | ||
Listed in a separate file.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact Ya-Chi Ho (yho10@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study participants
Peripheral blood and leukapheresis samples were obtained from HIV-1-uninfected and ART-treated HIV-1-infected individuals who had undetectable plasma viral load for >6 months (Table S1). This study was approved by the Johns Hopkins University, and George Washington University Institutional Review Board. All participants were given written informed consent.
METHOD DETAILS
Reconstruction of patient-derived defective proviruses
Sequences of defective proviruses were obtained through pyrosequencing of the PCR product of near full-length limiting dilution nested PCR (Bruner et al., 2016; Ho et al., 2013). Defective proviral plasmids were reconstructed into the NL4-3 reference plasmid using de novo genome synthesis to avoid cloning of PCR-induced errors (Genscript)(Ho et al., 2013). The 5′ LTR sequences of clones 2G10, 48C8 and 4F12 are patient-derived. Sequences are available with GenBank accession numbers of KF526141, KF526152, KF526217, KF526220, KF526245, KF526254, KF526298, KF526338, and KX505621. The integrity of HIV-1 genomic region and primer/probe binding mismatches are listed in Table S2–S4.
Transfection of defective proviruses
Primary CD4+ T cells were activated with plate-coated anti-CD3/anti-CD28 antibody (BD Bioscience) for 2 days and subjected to Amaxa nucleofection (program T-020)(Lonza) with molecular equivalent of 5 μg of NL4-3 plasmid per 5 million CD4+ T cells per transfection. Transfected cells were cultured for 1 day in the presence of enfuvirtide and interleukin-2 (IL-2).
Activation and CD8+ T cell coculture of rCD4s from ART-suppressed participants
Resting CD4+ T cells from ART-suppressed participants were isolated using negative depletion (CD4+ T cell isolation kit, CD69 MicroBead Kit, CD25 MicroBead Kit, HLA-DR Microbead Kit, Miltenyi Biotec). Cells were activated with plate-bound anti-CD3/anti-CD28 antibody (BD Biosciences) for 3 days in the presence of 100 U/ml IL-2 and enfuvirtide to block new rounds of infection. Enfuvirtide was selected to block infection at the entry level to avoid detection of incoming viral RNA. Enfuvirtide is sufficient to block new rounds of infection within the time frame of experiment (data not shown).
CD8+ T cells were stimulated with CMV peptide pool (80 ng per peptide per ml, PX-CMV, ProImmune), group B concensus Gag peptide mixture (Gag PepMix HIV-1, 80 ng per peptide per ml, JPT Peptide Technologies), and SL9 (5 μg/ml (Shan et al., Immunity 2012), SLYNTVATL, AnaSpec) in the presence of IL-2 10 U/ml, enfuvirtide and CD4-depleted autologous peripheral blood mononuclear cell (PBMC) for 6 days. CD4-depleted autologous PBMC was used as antigen presenting cells to avoid nonspecific CD8+ T cell stimulation by autologous HIV-1 from CD4+ T cells. CD8+ T cells were then isolated by magnetic negative depletion (Dynabeads untouched CD8 T cell isolation kit, ThermoFisher) and co-cultured with activated CD4+ T cells at 1:1 ratio in the presence of IL-2–10 U/ml and enfuvirtide for 24 hours. CD8+ T cells were then removed by positive selection (Miltenyi CD8+ Microbeads positive selection). Cells were then pelleted and stored frozen in TRIzol.
Quantification of HIV-1 DNA, RNA and protein
RNA extraction, DNase treatment, cDNA synthesis
Pellets from 1 to 10 million cells were frozen in 1 ml TRIzol (Thermo Fisher Scientific). Culture supernatant was treated with RNase A (Qiagen)(400 μl culture supernatant treated with 1 μl (100 μg or 7 units) of RNase A at 37°C for 1 hour) and frozen in 1200 μl of TRIzol LS. HIV-1 RNA was isolated from the aqueous phase and genomic DNA was isolated from the organic phase of the TRizol separation. RNA samples were subjected to DNase treatment (DNase I, amplification grade, Thermo Fisher Scientific). cDNA was synthesized using oligo dT primers (qScript Flex cDNA kit, Quanta Biosciences).
Quantitative PCR
Quantitative real-time PCR from the DNA and cDNA was performed using absolute quantification method. qPCR was performed using 10 μl of PerfeCTa qPCR ToughMix (Quanta Biosciences), 600 nM of primers, 100 nM of probes, and 2 μl of template in a final reaction volume of 20 μl. Plasmid standards were serially diluted down to 2 copies per qPCR well for absolute quantification. qPCR cycles are: 95°C for 2 min then 50 cycles of 95°C for 15 sec and 60°C for 1 min on a ABI 7900HT Real-Time PCR System or 95°C for 30 second then 50 cycles of 95°C for 5 sec and 60°C for 30 seconds on a ViiA 7 Real-Time PCR System fast module (Thermo Fisher Scientific). For TILDA, nested qRT-PCR was performed using a specific protocol (Procopio et al., 2015). The number of cells per sample was measured by RNase P qPCR of the DNA (TaqMan Copy Number Reference Assay, Thermo Fisher Scientific) using human genome as standards (Human Genomic DNA, Roche). Sequences of primers and probes are listed in table S5. Samples from qRT-PCR were subjected to 1.5% agarose gel electropheresis and TA-TOPO cloning (Thermo Fisher Scientific) to confirm the size and sequence of the amplicon.
Targeted gag deep sequencing
To sequence HIV-1 gag RNA, RNA samples were extracted using TRIzol phase separation method, treated with DNase I, and synthesized into cDNA using oligo dT primers as described above. cDNA from clinical samples were subjected to outer and inner PCR using Platinum Taq High Fidelity polymerase (Thermo Fisher Scientific) containing 5 μl of 10X Buffer, 2 μl of 50 mM MgSO4, 1 μl of 10 mM dNTP mix, 1 μM of each primer, 0.25 μl of polymerase and 2 μl of templates in a final volume of 50 μl. From the outer PCR reaction product, 2 μl was used for inner PCR in a similar reaction mix. PCR cycles of outer (2,277 bp) and inner (1,593 bp) PCR are: 95°C 2 min, then 95°C 30 sec – 55°C 30 sec – 68°C 2 min 45 sec for 35 cycles, then 68°C for 5 min.
PacBio deep sequencing platform was used to sequence the full-length gag in one read so that multiple stop codons in different tryptophan residues of the same template is still called one read containing defective gag. In PacBio RS II single-molecule sequencing, amplicons were barcoded with a group of 10 bp indexes. Multiple samples were pooled together to generate a SMRT cell sequencing library following the Pacific Biosciences template preparation and sequencing-C4 user guide for 2 kb insert size using the Pacific Biosciences DNA template preparation kit. The resulting library was then run on a BioAnalyzer high-sensitivity DNA chip for size and concentration determination. The library was then sequenced on PacBio RSII. Circular consensus sequences (CCS) were generated from the single molecule sequence. The sequence reads from PacBio were demultiplexed using Fastx-Toolkit.
Analysis of deep sequencing data
The full-length gag region of HIV-1 DNA and RNA from HIV-1-infected individuals was amplified using nested PCR and subjected to PacBio deep sequencing. The presence of a missense mutation in the start codon (ATG→ATA) or a nonsense mutation in any of the tryptophan coding sequences (TGG→TAA, TAG or TGA) was counted as defective. A read containing multiple G→A mutations in the designated residues is counted as one read of mutated HIV-1 gag instead of multiple mutated reads.
PacBio CCS reads from each condition (resting and activated) from each individual were aligned to reference HIV-1 consensus B Gag sequence. CTL epitope analysis was performed as previously describe (Deng et al., 2015). A custom program was written using Perl scripts to identify and compute the frequency of all sequence variants that cause either a G-to-A mutated (ATG→ATA) start codon or any G-to-A mutation in the 9 tryptophan (TGG) residues. A read containing multiple G-to-A mutations in the designated residues is counted as one read of mutated HIV-1 gag instead of being counted as multiple mutated reads.
Quantification of HIV-1 translation
Intracellular Gag staining (clone KC57-PE, Beckman Coulter) and supernatant p24 ELISA (Perkin Elmer) were used to quantify HIV-1 Gag protein expression. CD4 expression was used to examine the function of Nef instead of using anti-Nef antibody to examine the expression level of Nef.
CTL clone generation and maintenance
CD8+ T cell responses in subjects were mapped by IFNγ-ELISPOT using 270 previously defined HIV-1 optimal CD8+ epitopes restricted by common HLA alleles. For each response, PBMCs were plated at 1×107 cells/well in a 24-well plate and stimulated with 10 μg/ml of peptide for 3 hours. T cells that had produced IFN-γ in response to this stimulation were enriched using the IFN-γ secretion detection and enrichment kit (Miltenyi Biotec) following the manufacturer’s instructions. These cells were plated at a series of dilutions in 96-well plates with feeder medium (RPMI 1640 supplemented with 10% FBS and PenStrep [RPMI-10]) with 1×106 cells/ml 5,000 rad irradiated PBMC + 50 U/ml IL-2 + 0.1 μg/ml each of anti-CD3 (OKT3, eBioscience), anti-CD28 (CD28.2, eBioscience). One month later, colonies were selected from the lowest dilution plate with positive wells (<1/5 of wells positive) and screened for responsiveness to peptide by IFN-γ ELISPOT. Positive clones were expanded bi-weekly with feeder medium. Clone specificities were confirmed by degranulation assay (CD107a flow cytometry) on the day prior to recognition/elimination assays. Clones were washed extensively prior to use in assays.
CTL recognition assay
Primary CD4+ T cells were enriched from PBMC by negative selection (EasySep, STEMCELL Technologies) and then activated for 24 hours with plate-bound anti-CD3 (OKT3, ebioscience) and 1 μg/ml anti-CD28 (CD28.2, eBioscience) in RPMI-10 supplemented with 50 U/ml IL-2. Activated CD4+ T cells were nucleofected with defective proviral plasmids and cultured in the presence of enfuvirtide for 1 day and then co-cultured with CTL clones in quadruplicate for 6 hours in the presence of PE-conjugated anti-CD107a antibody. CD107a gives the most dynamic readout of CTL recognition as previously optimized (Jones et al., 2012). Brefeldin A was added after the first 3 hours of stimulation at a concentration of 1 μg/ml. This condition is optimized based on the fact that CTL recognition and killing occurs within 1 hour. Cells were stained with fluorochrome-conjugated antibodies to CD3, CD4, and CD8, then fixed/permeabilized (Cytofix/cytoperm, cytoperm/wash system, BD Biosciences). Samples were analyzed on a Fortessa X20 instrument (BD Biosciences).
Cold-target inhibition
Leukapheresis material was obtained from participant OM5267, an individual from whom we had previously generated HIV-1- and CMV-specific T cell clones. CD4+ T cells were enriched from these PBMCs by negative selection (EasySep, Stemcell Technologies) and then activated with anti-CD3/CD28. The 1:20 and 1:100 ratio of effector: real target was determined by previous work (Jones et al., 2012). The ratio 1:40 of real target: cold target was chosen to reflect the infectivity of real targets at around 10% and the nucleofection efficiency of ~3% as shown by the Gag positivity in reference NL4-3 plasmid nucleofection (Figure 5A), which gives rise to an actual real target: cold target ratio of 1:12. This may still be an overestimation of the number of viable cold targets, as more cold-target cells die from nucleofection than real target cells which underwent spinoculation instead of the toxic nucleofection.
For real targets, a portion of activated CD4+ T cells were spinoculated with HIV-1LAI (NIH AIDS Reagent Program) 3 days prior to coculture and stained with CellTrace Far Red one day prior to coculture (Thermo Fisher Scientific). For cold targets, a separate portion of activated CD4+ T cells were nucleofected with pUC19 (vector control), 31G4, 45E6, or 19B3 one day prior to CTL co-culture. Vector + peptide cold targets were generated by pulsing vector-nucleofected cells with 1 μg/ml of IK9 peptide and washing. Cocultures were setup at the CTL: real target: cold target ratios indicated, and incubated for 4 hours. Cells were then stained with LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific), fluorochrome-conjugated antibodies to CD3, CD4, and CD8, fixed and permeabilized, and stained intracellulary with anti-HIV-Gag-PE (clone KC57, Beckman Coulter). Samples were analyzed on a Fortessa X20 instrument (BD Biosciences).
Analysis of HIV-1 proviral landscape
Full-length sequence analysis of HIV-1 proviruses from viral outgrowth negative wells (Ho et al., 2013) and rCD4s (Bruner et al., 2016) were obtained from two published independent cohorts. Since an average of 42% of defective proviruses with large internal deletions encompassing the 5′ end of the HIV-1 genome may be omitted in the Ho cohort due to the use of gag PCR for screening, we corrected the percentage of 5′ large internal deletions by addition of 42% of the potentially omitted proviral clones containing 5′ large internal deletions to the Ho cohort to make a comparison with the Bruner cohort (Bruner et al., 2016). Clones which have both hypermutations and large internal deletions at the same time were counted as large internal deletions, given the deleterious effect of the absence of essential segments of proviral genome. Definition of acute and chronic infection is defined as previously describe (Bruner et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Analyses were performed using MedCalc and Prism software (Graphpad). Statistical tests are indicated in the corresponding figure legend.
Supplementary Material
Figure S1. The duration of infection may contribute to the different frequency of hypermutated proviruses in individuals treated during acute and chronic infection.
The P value was calculated by two-tailed Student t-test. Associated with Figure 1.
Figure S2. Map of reconstructed patient-derived defective HIV-1 proviruses.
(A) Experimental scheme for isolation and reconstruction of patient-derived defective HIV-1 proviruses. (B) HIV-1 genome map and qRT-PCR primer binding sites. (C) Maps of reconstructed defective proviruses. (D) Status of HIV-1 genetic elements. (E) Status of qRT-PCR primer/probe binding sites. Associated with Figure 2.
Figure S3. Defective HIV-1 proviruses can be transcribed in vitro.
Activated primary CD4+ T cells were transfected with reconstructed patient-derived HIV-1 proviral plasmids. HIV-1 RNA was converted to cDNA using oligo-dT-based priming after DNase treatment. Levels of cell-associated polyadenylated HIV-1 RNA (A), singly spliced (B), multiply spliced (C) and preamplified multiply spliced HIV-1 RNA (D) in transfected activated primary CD4+ T cells were measured by qRT-PCR. HIV-1 RNA quantity was normalized to the number of cells as measured by RNaseP qPCR in the corresponding DNA component of the transfected cells. (E) Levels of supernatant HIV-1 RNA was measured by poly-adenylated HIV-1 RNA quantification. Culture supernatant was treated with RNaseA to remove virion-free RNA. Data represent mean ± SEM. Associated with Figure 2.
Figure S4. Hypermutated HIV-1 proviruses can be transcribed in vitro despite a mutated LTR promoter.
(A) Agarose gel electrophoresis (1.5%) of qRT-PCR products. Note that some clones which has sequence mismatch with the qPCR amplification probes (yellow asterisks) can be detected by gel electrophoresis. (B) Cell-associated and supernatant HIV-1 RNA level of 2G10 using customized qRT-PCR primer/probe to match the G→A hypermutations. Data represent mean ± SEM. (C) Sequence of NF-κB and Sp1 binding sites in HIV-1 LTR. Red, G→A hypermutations. Associated with Figure 2.
Figure S5. Defective HIV-1 proviruses can be translated in vitro.
(A) Intracellular Gag staining (B) supernatant p24 ELISA of primary activated CD4+ T cells transfected with defective patient-derived proviral plasmids in the presence of enfuvirtide. Data represent mean ± SEM. Open circles and dotted lines indicate limit of detection. Asociated with Figure 2.
Figure S6. Defective HIV-1 proviruses cannot downregulate CD4 expression in vitro.
CD4 expression level in primary activated CD4+ T cells transfected with defective patient-derived proviral plasmids in the presence of enfuvirtide. Associated with Figure 2.
Figure S7. Experimental scheme of CTL recognition in vitro.
(A) Representative flow cytometry plots for CD107a expression. (B) Experimental scheme for cold-target inhibition. (C–D) Representative flow cytometry plots. Gag+CD4low cells were gated to identify productively infected cells instead of cells containing incoming Gag without active viral protein production (Gag+CD4high). Gating was guided by the staining of negative controls. Associated with Figure 4 and 5.
Table S1. Demographics of study participants
Table S2. Splice sites of reconstructed patient-derived HIV-1 proviral plasmids*
Table S3. Genomic defects of reconstructed patient-derived HIV-1 proviral plasmids*
Table S4. Primer/probe mismatches of reconstructed patient-derived HIV-1 proviral plasmids*
Table S5. Primer and probe sequences
Acknowledgments
We thank all study participants. We thank Dr. Joel Blankson for helpful suggestions. We thank Christopher Avergas and Kevin Chesterton for HLA typing. This work was supported by the NIH 1R21AI118402-01, Martin Delaney CARE and DARE Collaboratories (NIH AI096113 and 1U19AI096109), ARCHE Collaborative Research Grant from the Foundation for AIDS Research (amfAR108165-50-RGRL), amfAR GeneratureCure initiative (amfAR109315-57-RGRL), Johns Hopkins Center for AIDS Research (CFAR)(P30AI094189) and DC-CFAR (P30AI087714), W. W. Smith Charitable Trust AIDS Research Grant, Gilead Sciences HIV Cure Research Grant, the Howard Hughes Medical Institute and the Bill and Melinda Gates Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
Y.-C.H., R.A.P, R.B.J., R.F.S. designed the experiments. Y.-C.H., R.A.P., R.B.J., K.M.B., A.R.M., A.S.T., S.H.H., S.K., P.C.Y. performed the experiments. M.P., E.H.-S., H.H. performed deep sequencing analysis. A.A.C., S.A.B., C.K., E.B. recruited study participants.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A, Carlson J, Yan J, Akinsiku OT, Schaefer M, Sabbaj S, Bet A, Levy DN, Heath S, Tang J, et al. CD8 T cell response and evolutionary pressure to HIV-1 cryptic epitopes derived from antisense transcription. J Exp Med. 2010;207:51–59. doi: 10.1084/jem.20092060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton K, Hiener B, Winckelmann A, Rasmussen TA, Shao W, Byth K, Lanfear R, Solomon A, McMahon J, Harrington S, et al. Broad activation of latent HIV-1 in vivo. Nat Commun. 2016;7:12731. doi: 10.1038/ncomms12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson GJ, Lalama CM, Bosch RJ, Gandhi RT, Bedison MA, Aga E, Riddler SA, McMahon DK, Hong F, Mellors JW. HIV-1 DNA decay dynamics in blood during more than a decade of suppressive antiretroviral therapy. Clin Infect Dis. 2014;59:1312–1321. doi: 10.1093/cid/ciu585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnlein E, Lowenthal JW, Siekevitz M, Ballard DW, Franza BR, Greene WC. The same inducible nuclear proteins regulates mitogen activation of both the interleukin-2 receptor-alpha gene and type 1 HIV. Cell. 1988;53:827–836. doi: 10.1016/0092-8674(88)90099-2. [DOI] [PubMed] [Google Scholar]
- Borducchi EN, Cabral C, Stephenson KE, Liu J, Abbink P, Ng’ang’a D, Nkolola JP, Brinkman AL, Peter L, Lee BC, et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature. 2016;540:284–287. doi: 10.1038/nature20583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, Lai J, Strain MC, Lada SM, Hoh R, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med. 2016;22:1043–1049. doi: 10.1038/nm.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock TN, Eisenlohr LC. Ribosomal scanning past the primary initiation codon as a mechanism for expression of CTL epitopes encoded in alternative reading frames. J Exp Med. 1996;184:1319–1329. doi: 10.1084/jem.184.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinaud S, Moris A, Fevrier M, Rohrlich PS, Weiss L, Langlade-Demoyen P, Lemonnier FA, Schwartz O, Habel A. Identification of cryptic MHC I-restricted epitopes encoded by HIV-1 alternative reading frames. J Exp Med. 2004;199:1053–1063. doi: 10.1084/jem.20031869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright EK, Spicer L, Smith SA, Lee D, Fast R, Paganini S, Lawson BO, Nega M, Easley K, Schmitz JE, et al. CD8(+) Lymphocytes Are Required for Maintaining Viral Suppression in SIV-Infected Macaques Treated with Short-Term Antiretroviral Therapy. Immunity. 2016;45:656–668. doi: 10.1016/j.immuni.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casartelli N, Guivel-Benhassine F, Bouziat R, Brandler S, Schwartz O, Moris A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J Exp Med. 2010;207:39–49. doi: 10.1084/jem.20091933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Sharp PA. Regulation by HIV Rev depends upon recognition of splice sites. Cell. 1989;59:789–795. doi: 10.1016/0092-8674(89)90602-8. [DOI] [PubMed] [Google Scholar]
- Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cillo AR, Sobolewski MD, Bosch RJ, Fyne E, Piatak M, Jr, Coffin JM, Mellors JW. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A. 2014;111:7078–7083. doi: 10.1073/pnas.1402873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, Hahn BH, Czartoski JL, McElrath MJ, Lehmann C, et al. HIV-1 integration landscape during latent and active infection. Cell. 2015;160:420–432. doi: 10.1016/j.cell.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks AM, Bateson R, Cope AB, Dahl NP, Griggs MK, Kuruc JD, Gay CL, Eron JJ, Margolis DM, Bosch RJ, Archin NM. Precise Quantitation of the Latent HIV-1 Reservoir: Implications for Eradication Strategies. J Infect Dis. 2015;212:1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- Deeks SG. HIV: Shock and kill. Nature. 2012;487:439–440. doi: 10.1038/487439a. [DOI] [PubMed] [Google Scholar]
- Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, Lai J, McHugh HL, Hao H, Zhang H, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517:381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, et al. Activation of HIV Transcription with Short-Course Vorinostat in HIV-Infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog. 2014;10:e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JH, McMahon JH, Chang CC, Lee SA, Hartogensis W, Bumpus N, Savic R, Roney J, Hoh R, Solomon A, et al. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV. 2015;2:e520–9. doi: 10.1016/S2352-3018(15)00226-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson S, Graf EH, Dahl V, Strain MC, Yukl SA, Lysenko ES, Bosch RJ, Lai J, Chioma S, Emad F, et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013;9:e1003174. doi: 10.1371/journal.ppat.1003174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg MB, Jarrett RF, Aldovini A, Gallo RC, Wong-Staal F. HTLV-III expression and production involve complex regulation at the levels of splicing and translation of viral RNA. Cell. 1986;46:807–817. doi: 10.1016/0092-8674(86)90062-0. [DOI] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, Hagen SI, Shoemaker R, Deleage C, Lucero C, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. 2015;21:132–139. doi: 10.1038/nm.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwich A, Hahn SS, Schreiber S, Meier S, Kampgen E, Wagner R, Lutz MB, Schubert U. Targeting HIV-1 Gag into the defective ribosomal product pathway enhances MHC class I antigen presentation and CD8+ T cell activation. J Immunol. 2008;180:372–382. doi: 10.4049/jimmunol.180.1.372. [DOI] [PubMed] [Google Scholar]
- Guatelli JC, Gingeras TR, Richman DD. Alternative splice acceptor utilization during human immunodeficiency virus type 1 infection of cultured cells. J Virol. 1990;64:4093–4098. doi: 10.1128/jvi.64.9.4093-4098.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano H, Jain V, Hunt PW, Lee TH, Sinclair E, Do TD, Hoh R, Martin JN, McCune JM, Hecht F, Busch MP, Deeks SG. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J Infect Dis. 2013;208:50–56. doi: 10.1093/infdis/jis630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho O, Green WR. Cytolytic CD8+ T cells directed against a cryptic epitope derived from a retroviral alternative reading frame confer disease protection. J Immunol. 2006;176:2470–2475. doi: 10.4049/jimmunol.176.4.2470. [DOI] [PubMed] [Google Scholar]
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamichi H, Dewar RL, Adelsberger JW, Rehm CA, O’Doherty U, Paxinos EE, Fauci AS, Lane HC. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc Natl Acad Sci U S A. 2016;113:8783–8788. doi: 10.1073/pnas.1609057113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Peterlin BM. Control of RNA initiation and elongation at the HIV-1 promoter. Annu Rev Biochem. 1994;63:717–743. doi: 10.1146/annurev.bi.63.070194.003441. [DOI] [PubMed] [Google Scholar]
- Jones RB, Garrison KE, Mujib S, Mihajlovic V, Aidarus N, Hunter DV, Martin E, John VM, Zhan W, Faruk NF, et al. HERV-K-specific T cells eliminate diverse HIV-1/2 and SIV primary isolates. J Clin Invest. 2012;122:4473–4489. doi: 10.1172/JCI64560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney MF, Wiegand A, Shao W, Coffin JM, Mellors JW, Lederman M, Gandhi RT, Keele BF, Li JZ. Origin of Rebound Plasma HIV Includes Cells with Identical Proviruses That Are Transcriptionally Active before Stopping of Antiretroviral Therapy. J Virol. 2015;90:1369–1376. doi: 10.1128/JVI.02139-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leth S, Schleimann MH, Nissen SK, Hojen JF, Olesen R, Graversen ME, Jorgensen S, Kjaer AS, Denton PW, Mork A, et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): a single-arm, phase 1B/2A trial. Lancet HIV. 2016;3:e463–72. doi: 10.1016/S2352-3018(16)30055-8. [DOI] [PubMed] [Google Scholar]
- Lever A, Gottlinger H, Haseltine W, Sodroski J. Identification of a sequence required for efficient packaging of human immunodeficiency virus type 1 RNA into virions. J Virol. 1989;63:4085–4087. doi: 10.1128/jvi.63.9.4085-4087.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkannan S, Horng T, Shih PP, Schwab S, Shastri N. Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity. 1999;10:681–690. doi: 10.1016/s1074-7613(00)80067-9. [DOI] [PubMed] [Google Scholar]
- Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, Coffin JM, Hughes SH. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014;345:179–183. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–257. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- Maness NJ, Wilson NA, Reed JS, Piaskowski SM, Sacha JB, Walsh AD, Thoryk E, Heidecker GJ, Citron MP, Liang X, et al. Robust, vaccine-induced CD8(+) T lymphocyte response against an out-of-frame epitope. J Immunol. 2010;184:67–72. doi: 10.4049/jimmunol.0903118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massanella M, Gianella S, Lada SM, Richman DD, Strain MC. Quantification of Total and 2-LTR (Long terminal repeat) HIV DNA, HIV RNA and Herpesvirus DNA in PBMCs. Bio-Protocol. 2015;5(11):e1492. doi: 10.21769/bioprotoc.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocwieja KE, Sherrill-Mix S, Mukherjee R, Custers-Allen R, David P, Brown M, Wang S, Link DR, Olson J, Travers K, Schadt E, Bushman FD. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012;40:10345–10355. doi: 10.1093/nar/gks753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, Koup RA. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procopio FA, Fromentin R, Kulpa DA, Brehm JH, Bebin AG, Strain MC, Richman DD, O’Doherty U, Palmer S, Hecht FM, et al. A Novel Assay to Measure the Magnitude of the Inducible Viral Reservoir in HIV-infected Individuals. EBioMedicine. 2015;2:872–881. doi: 10.1016/j.ebiom.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell DF, Martin MA. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J Virol. 1993;67:6365–6378. doi: 10.1128/jvi.67.11.6365-6378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. The Lancet HIV. 2014;1(1):e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- Schick B, Berke G. Competitive inhibition of cytotoxic T lymphocyte-target cell conjugation. A direct evaluation of membrane antigens involved in cell-mediated immunity. Transplantation. 1979;27:365–368. [PubMed] [Google Scholar]
- Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Felber BK, Benko DM, Fenyo EM, Pavlakis GN. Cloning and functional analysis of multiply spliced mRNA species of human immunodeficiency virus type 1. J Virol. 1990a;64:2519–2529. doi: 10.1128/jvi.64.6.2519-2529.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Felber BK, Fenyo EM, Pavlakis GN. Env and Vpu proteins of human immunodeficiency virus type 1 are produced from multiple bicistronic mRNAs. J Virol. 1990b;64:5448–5456. doi: 10.1128/jvi.64.11.5448-5456.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Rabi SA, Laird GM, Eisele E, Zhang H, Margolick JB, Siliciano RF. A novel PCR assay for quantification of HIV-1 RNA. J Virol. 2013;87:6521–6525. doi: 10.1128/JVI.00006-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- Simonetti FR, Sobolewski MD, Fyne E, Shao W, Spindler J, Hattori J, Anderson EM, Watters SA, Hill S, Wu X, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci U S A. 2016;113:1883–1888. doi: 10.1073/pnas.1522675113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015;11:e1005142. doi: 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck SR, Jiang V, Pavon-Eternod M, Prasad S, McCarthy B, Pan T, Shastri N. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science. 2012;336:1719–1723. doi: 10.1126/science.1220270. [DOI] [PubMed] [Google Scholar]
- Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014;345:570–573. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BD, Chakrabarti S, Moss B, Paradis TJ, Flynn T, Durno AG, Blumberg RS, Kaplan JC, Hirsch MS, Schooley RT. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature. 1987;328:345–348. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- West MJ, Lowe AD, Karn J. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J Virol. 2001;75:8524–8537. doi: 10.1128/JVI.75.18.8524-8537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol. 1996;157:1823–1826. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The duration of infection may contribute to the different frequency of hypermutated proviruses in individuals treated during acute and chronic infection.
The P value was calculated by two-tailed Student t-test. Associated with Figure 1.
Figure S2. Map of reconstructed patient-derived defective HIV-1 proviruses.
(A) Experimental scheme for isolation and reconstruction of patient-derived defective HIV-1 proviruses. (B) HIV-1 genome map and qRT-PCR primer binding sites. (C) Maps of reconstructed defective proviruses. (D) Status of HIV-1 genetic elements. (E) Status of qRT-PCR primer/probe binding sites. Associated with Figure 2.
Figure S3. Defective HIV-1 proviruses can be transcribed in vitro.
Activated primary CD4+ T cells were transfected with reconstructed patient-derived HIV-1 proviral plasmids. HIV-1 RNA was converted to cDNA using oligo-dT-based priming after DNase treatment. Levels of cell-associated polyadenylated HIV-1 RNA (A), singly spliced (B), multiply spliced (C) and preamplified multiply spliced HIV-1 RNA (D) in transfected activated primary CD4+ T cells were measured by qRT-PCR. HIV-1 RNA quantity was normalized to the number of cells as measured by RNaseP qPCR in the corresponding DNA component of the transfected cells. (E) Levels of supernatant HIV-1 RNA was measured by poly-adenylated HIV-1 RNA quantification. Culture supernatant was treated with RNaseA to remove virion-free RNA. Data represent mean ± SEM. Associated with Figure 2.
Figure S4. Hypermutated HIV-1 proviruses can be transcribed in vitro despite a mutated LTR promoter.
(A) Agarose gel electrophoresis (1.5%) of qRT-PCR products. Note that some clones which has sequence mismatch with the qPCR amplification probes (yellow asterisks) can be detected by gel electrophoresis. (B) Cell-associated and supernatant HIV-1 RNA level of 2G10 using customized qRT-PCR primer/probe to match the G→A hypermutations. Data represent mean ± SEM. (C) Sequence of NF-κB and Sp1 binding sites in HIV-1 LTR. Red, G→A hypermutations. Associated with Figure 2.
Figure S5. Defective HIV-1 proviruses can be translated in vitro.
(A) Intracellular Gag staining (B) supernatant p24 ELISA of primary activated CD4+ T cells transfected with defective patient-derived proviral plasmids in the presence of enfuvirtide. Data represent mean ± SEM. Open circles and dotted lines indicate limit of detection. Asociated with Figure 2.
Figure S6. Defective HIV-1 proviruses cannot downregulate CD4 expression in vitro.
CD4 expression level in primary activated CD4+ T cells transfected with defective patient-derived proviral plasmids in the presence of enfuvirtide. Associated with Figure 2.
Figure S7. Experimental scheme of CTL recognition in vitro.
(A) Representative flow cytometry plots for CD107a expression. (B) Experimental scheme for cold-target inhibition. (C–D) Representative flow cytometry plots. Gag+CD4low cells were gated to identify productively infected cells instead of cells containing incoming Gag without active viral protein production (Gag+CD4high). Gating was guided by the staining of negative controls. Associated with Figure 4 and 5.
Table S1. Demographics of study participants
Table S2. Splice sites of reconstructed patient-derived HIV-1 proviral plasmids*
Table S3. Genomic defects of reconstructed patient-derived HIV-1 proviral plasmids*
Table S4. Primer/probe mismatches of reconstructed patient-derived HIV-1 proviral plasmids*
Table S5. Primer and probe sequences