

Abstract
Lithium (Li+) is a drug widely employed for treating bipolar disorder, however the mechanism of action is not known. Here we study the effects of Li+ in cultured hippocampal neurons on a synaptic complex consisting of δ-catenin, a protein associated with cadherins whose mutation is linked to autism, and GRIP, an AMPA receptor (AMPAR) scaffolding protein, and the AMPAR subunit, GluA2. We show that Li+ elevates the level of δ-catenin in cultured neurons. δ-catenin binds to the ABP and GRIP proteins, which are synaptic scaffolds for GluA2. We show that Li+ increases the levels of GRIP and GluA2, consistent with Li+-induced elevation of δ-catenin. Using GluA2 mutants, we show that the increase in surface level of GluA2 requires GluA2 interaction with GRIP. The amplitude but not the frequency of mEPSCs was also increased by Li+ in cultured hippocampal neurons, confirming a functional effect and consistent with AMPAR stabilization at synapses. Furthermore, animals fed with Li+ show elevated synaptic levels of δ-catenin, GRIP, and GluA2 in the hippocampus, also consistent with the findings in cultured neurons. This work supports a model in which Li+ stabilizes δ-catenin, thus elevating a complex consisting of δ-catenin, GRIP and AMPARs in synapses of hippocampal neurons. Thus, the work suggests a mechanism by which Li+ can alter brain synaptic function that may be relevant to its pharmacologic action in treatment of neurological disease.
Introduction
The efficacy of lithium (Li+) as an antimanic and antidepressant drug makes it frequently employed for treatment of Bipolar Disorder (BD), an illness characterized by recurrent episodes of mania and depression (Geddes et al, 2004). Although Li+ has been used for treatment of BD for years, its mechanism of action is not established. Li+ has been proposed to alter electrolyte balance, monoamine neurotransmitter release, adenylyl cyclase function, phosphoinositides and PKC, and arachadonic acid metabolism (Marmol, 2008). Significantly, there is strong evidence that Li+ acts as a mood stabilizer through its inhibition of GSK3β (Glycogen synthase kinase 3 beta) since this inhibition has been shown to counteract symptoms of depression and mania (Chuang and Manji, 2007; O’Brien and Klein, 2009). GSK3β, the predominant isoform of GSK3 found in the brain, is a serine/threonine kinase that plays an important role in several cellular processes (Jope and Johnson, 2004). Li+ reduces GSK3β activity by elevating the Akt-dependent phosphorylation of GSK3β autoinhibitory residue, serine 9 (Zhang et al, 2003) or by competing with magnesium for enzyme binding (Ryves and Harwood, 2001). Because GSK3β has been linked to cognitive processes, GSK3β inhibitors such as Li+ may be useful therapeutic interventions for brain disorders that have a cognitive basis including bipolar disorder, depression, and schizophrenia (O’Leary and Nolan, 2015). However, the mechanisms underlying the pro-cognitive effects of GSK3β inhibitors are not yet understood.
Regulation of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid,) receptors (AMPARs) has been proposed as a means for treating depression and mania (Li et al, 2001; O’Neill et al, 2004). AMPARs are glutamate-gated ion channels that consist of tetramers of subunits GluA1-4 (Hollmann and Heinemann, 1994). One action of Li+ that is dependent on GSK3β inhibition is the ability to regulate protein stability (O’Leary et al, 2015; Xu et al, 2009), suggesting that therapeutic activity of Li+ could depend on protein stabilization, in particular, stabilization of synaptic proteins. Indeed, the Li+-mediated inhibition of GSK3β elevates catenins, which are armadillo (ARM) repeat proteins that bind to the intracellular domain of cadherins at adherens junctions and that contribute to the architecture of synapses (Takeichi, 1988). β-catenin links cadherins to the actin cytoskeleton (Valenta et al, 2012) and binding of Wnt to its receptor, frizzled, induces GSK3β-induced phosphorylation of β-catenin, leading to β-catenin ubiquitination and degradation (Metcalfe and Bienz, 2011). Inhibition of GSK3β by Li+ stabilizes β-catenin and elevates its levels (Hedgepeth et al, 1997). Significantly, GSK3β also phosphorylates and regulates a closely related protein, δ-catenin (Bareiss et al, 2010; Oh et al, 2009). δ-catenin (also called neural plakophilin-related armadillo protein, NPRAP) is a neuron-specific catenin that also binds the cadherin intracellular domain, proximal to the plasma membrane (Kosik et al, 2005; Paffenholz and Franke, 1997). The ARM domains of δ-catenin mediate interaction with N-cadherin (Silverman et al, 2007) and the carboxyl terminus of δ-catenin binds to PDZ domains of two related scaffolds, AMPAR-binding protein (ABP) and glutamate receptor-interaction protein (GRIP) (Kosik et al, 2005; Silverman et al, 2007; Yuan et al, 2015) (Fig. 1a). ABP and GRIP are multi-PDZ domain components of the postsynaptic density (PSD), whose PDZ domains in turn bind the C-termini of the GluA2 and GluA3 AMPARs (Dong et al, 1997; Srivastava et al, 1998) (Fig. 1a). Thus, δ-catenin forms a bridge at synapses between cadherins and the ABP and GRIP scaffolds and associated GluA2/3-containing AMPARs (Silverman et al, 2007; Yuan et al, 2015) (Fig. 1a). Furthermore, δ-catenin promotes dendritic morphogenesis via Akt1-mediated phosphorylation (Kim et al, 2008). Significantly, the loss of δ-catenin gene is associated with the severe mental retardation found in the Cri du chat syndrome (Medina et al, 2000). δ-catenin knockout (KO) mice show impairments in hippocampal synaptic plasticity, including short-term and long-term plasticity, which is strongly associated with severe defects in cognitive function (Israely et al, 2004). Furthermore, N-cadherin, PSD-95 (post synaptic density-95), and GluA2 are significantly reduced, contributing to spine loss in the hippocampal synapse of the mutant animal (Israely et al, 2004; Restituito et al, 2011; Yuan et al, 2015). A recent study shows that loss of δ-catenin function by missense mutations causes severe autism (Turner et al, 2015). Taken together, δ-catenin is a critical regulator of spine architecture and brain function (Fig. 1a). Therefore, an increase in δ-catenin at synapses by Li+-dependent inhibition of GSK3β could stabilize synaptic GluA2/3 and spine integrity, which could be beneficial for neurological disorders, including BD and autism.
Figure 1. Li+ increases synaptic GluA2 and its associated scaffolding protein in cultured hippocampal neurons.
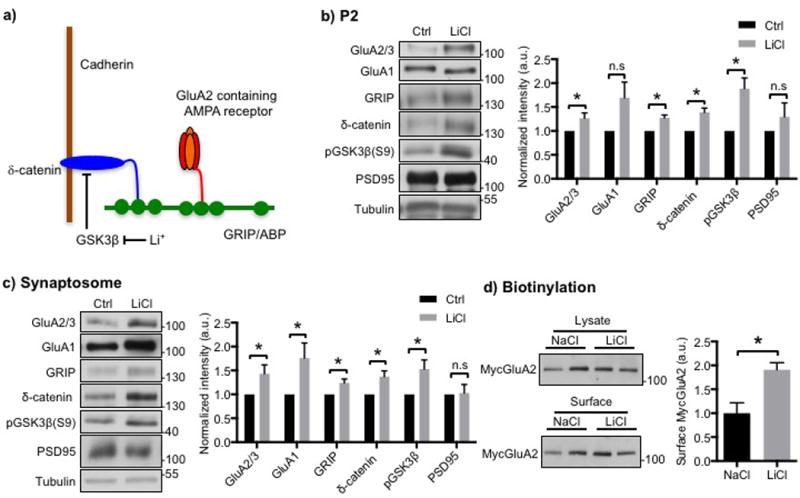
a) Model for the regulation of synaptic GluA2 by Li+. The GluA2 AMPA receptor subunit is anchored at the synaptic membrane by an interaction of its C-terminus with the fifth PDZ domain of the scaffolding protein, GRIP, and the related ABP protein. The second PDZ domain of GRIP associates with the C-terminus of δ-catenin. δ-catenin binds to the juxtamembrane region of cadherin, which is a synaptic cell adhesion protein. Phosphorylation of δ-catenin by GSK3 leads to the degradation of δ-catenin, which disrupts the δ-catenin-GRIP scaffold at the synapse and lowers synaptic levels of GluA2. Inhibition of GSK3 by Li+ stabilizes δ-catenin, which elevates GRIP and GluA2 at the synapse. See Silverman et al., (2007) and the text for details. Representative immunoblots and quantitative analysis of b) P2 membrane fraction and c) synaptosome fraction from cultured hippocampal neurons treated with NaCl (control) or LiCl showing that LiCl treatment increased GluA2/3, GRIP, and δ-catenin levels (n=5 and 10 experiments in b) and c), respectively, *p<0.05 and **p<0.01, unpaired two-tailed Student’s t tests). GSK3β inhibition by LiCl was identified by an increase in its phosphorylation. d) Representative immunoblots of surface biotinylation and a summary graph for cultured hippocampal neurons treated with NaCl or LiCl showing that LiCl treatment increased surface GluA2/3 levels (n=4 experiments, *p<0.05, unpaired two-tailed Student’s t tests).
Here, we analyze effects of Li+ on the levels of synaptic GluA2 and GRIP anchored at synapses via a scaffolding complex containing δ-catenin in hippocampal neurons. We show that consistent with Li+ inhibition of GSK3β-dependent degradation of δ-catenin, Li+ elevates the levels of a synaptic protein complex consisting of δ-catenin and the GRIP scaffold. GluA2 is also elevated in these complexes, dependent upon its ability to bind to GRIP. Moreover, we confirm that these Li+-mediated synaptic changes also occur in vivo. This work thus demonstrates a Li+-dependent modification of synapses that may underlie the mechanism of its therapeutic action.
Materials and Methods
Hippocampal neuron culture
Primary rat hippocampal neuron cultures were prepared by a previously described protocol (Osten et al, 1998; Restituito et al, 2011). Animal experiments were conducted in compliance with the Institutional Animal Care and Use Committee at the New York University School of Medicine.
Mice and lithium treatment
C56BL/6J mice were kept under 12hrs light and 12hrs dark with food and water ad libitum. Li+ animals received standard chow containing 0.17% w/w lithium carbonate (Harlan Teklad, Madison, WI) ad libitum for 7 days. Control animals were fed with standard chow. To determine serum Li+ levels, blood was collected after animals were fed with Li+ chow for 7 days, and Li+ levels were determined using the VITROS Li+ Slides and the VITROS Chemistry Products Calibrator Kit 1 (Ortho-Clinical Diagnostics, Inc. Rochester, NY) at New York University Langone Medical Center Clinical Chemistry Laboratory. Animal experiments were conducted in compliance with the Institutional Animal Care and Use Committee at the New York University School of Medicine.
MycGluA2 expressed Sindbis virus preparation and infection
MycGluA2, which is the GluA2 AMPAR subunit with an N-terminal myc epitope tag, was expressed from Sindbis virus that was generated and used as described previously (Osten et al, 2000).
PICK1 knockdown in rat hippocampal cell cultures
For PICK1 knockdown in rat hippocampal cultures, at 7–10 DIV, neurons were infected by a lentivirus containing a vector plasmid (pTrip vector) with an inserted siRNA sequence (TGGATGTGAAGTTTGAGTA) that specifically silences PICK1 expression. As a control, neurons were infected with a lentivirus containing the vector alone.
Pharmacological agents for immunocytochemical studies
In order to assess the effects of LiCl on surface GluA2, 2mM LiCl was added to growth medium one hour after MycGluA2 expressed sindbis virus infection. 2mM NaCl was used as a control. Neurons were incubated in the presence of NaCl and LiCl for 16–18 hours before performing live staining or preparing cell extracts.
Immunocytochemistry
Acquisition and analysis of images of immune fluorescent labeled neurons were performed using a Nikon (Tokyo, Japan) PCM2000 confocal microscope and the Simple PCI imaging software (C-Imaging Systems) as described previously (Osten et al, 2000). Live staining for surface MycGluA2 was carried out as described previously (Osten et al, 2000). In brief, 16–18 hrs after infection with sindbis virus expression of MycGluA2 and treatment with NaCl or LiCl, neurons were incubated with anti-Myc 9E10 monoclonal antibody (Santa Cruz, 4μg/ml) for 15 min at 37°C to label surface MycGluA2 receptors. After live staining, neurons were fixed with 4% paraformaldehyde for 10 min and then permeabilized with 0.2% Triton × solution in PBS for 5 min at room temp. Subsequently, the neurons were treated with polyclonal primary goat anti-myc antibody (Bethyl laboratories, 1μg/ml) for 1 hour at RT in order to label the total, intracellular receptor population. After washing with PBS, neurons were incubated with secondary antibody (Carroll et al, 1999; Lu and Ziff, 2005). Neurons were selected for quantitation based on images in which the intracellular fluorescent stain was selectively displayed, and the surface fluorescence was then displayed and quantitated. Data acquisition was thresholded only to the extent required to eliminate background signal not directly associated with cells. For a given experiment, the ratio of surface intensity to cell area was determined for each scan and these values were averaged for each group, and the experimental group average was normalized to the control group average, the normalized experimental values for the different experiments were then averaged.
Surface Biotinylation and protein sample preparation and immunoblots
Surface biotinylation, P2 membrane, and synaptosome preparation from cultured neurons were performed according to a previous report (Restituito et al, 2011). For cell fractionation, cells were washed with PBS and collected in 1ml ice-cold solution A (0.32M sucrose, 1mM NaHCO3, 1mM MgCl2, 0.5mM CaCl2, 0.1mM PMSF and 1X protease inhibitor). Cells were dounce homogenized 10 times and centrifuged at 3000 rpm for 10 min. The supernatant was spun again at 14,000 rpm for 30 min to obtain the P2 membrane fraction. To prepare synaptosomes, the P2 membrane pellet was resuspended in 0.5ml solution B (0.32M sucrose, 1 mM NaHCO3). This homogenate was layered on top of a 2.5ml 1M sucrose, and 2.5ml 1.2M sucrose step sucrose gradient and centrifuged at 30,000rpm for 2 hrs. Purified synaptosomes were collected at the 1M and 1.2M sucrose interface by syringe aspiration. The synaptosomes were resuspended with 5 volumes of solution B and collected by centrifugation at 42000 rpm for 40 min. The synaptosome pellet was resuspended in 2% Tris buffer.
Hippocampal PSD fraction preparation from brain was performed as previously described (Kim et al, 2015). Equal amounts of protein were loaded on 10% SDS-PAGE gels and transferred to nitrocellulose or PVDF membranes. Membranes were blotted with GluA1 (Millipore, 1:5000), GluA2/3 (Millipore, 1:500), actin (Sigma, 1:5000), tubulin (Sigma, 1:5000), GRIP (Upstate, 1:1000), δ-catenin (BD biosciences, 1:250), β-catenin (Sigma, 1:1000), GSK3β (Cell Signaling, 1:1000), pGSK3β (S9) (Cell Signaling, 1:1000), PICK1 (NeuroMab, 1:1000), PSD95 (NeuroMab, 1:2000) and myc antibody (Bethyl laboratories, 1:1000) antibodies and developed with ECL (Perkin Elmer).
Miniature EPSCs recording
Cultured hippocampal neurons were visualized with an Olympus BX50WI microscope. For recording, neurons were transferred to a recording chamber at room temperature, superfused at 1.2ml/min with bicarbonate-buffered aCSF, containing (in mM): 124 NaCl; 3.7 KCl; 26 NaHCO3; 2.4 CaCl2; 1.3 MgSO4; 1.3 KH2PO4 and 10 glucose equilibrated with 95% O2/5% CO2. Whole-cell recordings were performed from neuron cultures using the Axopatch 200B (Molecular Devices, Sunnyvale, CA) and Digidata 1550B converter using Clampex 10.6 software. Neurons were held at −60mV and whole-cell currents were filtered at 5kHz, digitized at 10kHz. Pipette resistance was 2.8–6.4ΩM using the internal solutions contained (in mM): K-gluconate 120; KCl 20; MgCl2 2; HEPES 10; EGTA 1; Na2-ATP 2; Na3-GTP 0.3mM; pH7.3 with KOH, 273mOsm/kg. The series resistances were compensated by 40%. Recordings were discarded if the change of series resistances exceeded 20%. Electrodes were prepared using horizontal micropipette puller (P-97, Sutter, Novato, CA) from glass capillaries (Sutter). Miniature EPSCs were recorded in the presence of the 2μM tetrodotoxin (TTX) and 100μM picrotoxin. Miniature events were visually inspected, individually selected and analyzed in Clampfit 10.6 (Molecular Devices) using template matching and an amplitude threshold of 5pA.
Statistics
All statistical comparisons were performed with the GraphPad Prism6 software. Unpaired two-tailed Student’s t-tests were used in single comparisons. For multiple comparisons, we used one-way analysis of variance (ANOVA) followed by Fisher’s Least Significant Difference (LSD) test to determine statistical significance. Results were represented as a mean ± s.e.m. and p value<0.05 was considered statistically significant.
Results
Li+ increases the surface expression of GluA2 and its synaptic scaffolds in hippocampal neurons
Because Li+, acting as a GSK3β inhibitor, can increase the stability of catenin proteins (Oh et al, 2009), we hypothesized that Li+ would be able to elevate synaptic GluA2 as one aspect of its pro-cognitive effects by stabilizing δ-catenin, which is associated with GluA2 through synaptic scaffolds (Silverman et al, 2007). To analyze the effects of Li+ on GSK3β and the GluA2 scaffolding complex, we first treated DIV 14–17 cultured hippocampal neurons with 2 mM LiCl or 2mM NaCl for 18hrs and prepared the membrane fraction (P2), in which the synaptic scaffolding complex is enriched. Western blotting revealed that Li+ treatment significantly increased phosphorylation at serine 9 of GSK3β (pGSK3β), an indication of GSK3β inhibition (Bareiss et al, 2010; Oh et al, 2009) (Fig. 1b). Moreover, LiCl treatment induced a significant increase in the levels of the scaffolding complex components, GluA2/3, GRIP, and δ-catenin (Fig. 1b). We next purified synaptosomes to further analyze the effects of LiCl on synaptic proteins. The levels of these same proteins in synaptosomes also increased significantly in LiCl-treated hippocampal cultures compared with NaCl-treated neurons (Fig. 1c), confirming that the elevation took place at synapses. Notably, GluA1 levels were significantly increased in synaptosome with LiCl treatment (Fig. 1c), suggesting that LiCl-induced increased AMPARs would be GluA1/2 heteromers. Interestingly, the levels of PSD95, a second synaptic scaffold unrelated to GRIP, were not increased in the P2 and synaptosomal fractions (Fig. 1b, c) indicating specific of action of Li+. We next addressed whether Li+ can elevate GluA2 surface levels in cultured neurons. We expressed the wild-type GluA2 AMPAR subunit with an N-terminal Myc epitope tag (MycGluA2) via sindbis virus in cultured hippocampal neurons. Unlike endogenous GluA2, which is mainly expressed in GluA1/2 heteromers (Wenthold et al, 1996), virus-mediated GluA2 expression is predominately homomeric (Osten et al, 2000) and thus its trafficking properties can be ascribed unambiguously to this subunit. We infected neurons with a sindbis virus expressing MycGluA2. After treatment with either NaCl or LiCl for 18 hrs, we used biotinylation to determine surface MycGluA2 expression (Fig. 1d). Consistent with the increase of the GluA2-associated scaffolding protein, δ-catenin and GRIP, in LiCl-treated neurons (Fig. 1b–c), surface GluA2 levels were significantly elevated compared with those in control neurons (Fig. 1d). These results suggested that LiCl inhibited GSK3β, which is sufficient to elevate synaptic δ-catenin and GRIP levels, enhancing a scaffold for GluA2. This led to an increase in synaptic GluA1/2 in LiCl-treated hippocampal neurons, suggesting a potential role of Li+ in the action of therapeutics.
Binding of GluA2 to GRIP is required for synaptic GluA2 elevation by Li+
Following our observation that Li+ treatment elevated the GluA2 scaffolding protein, GRIP, we next asked if Li+ elevation of GluA2 depended on the interaction of GluA2 with GRIP (Dong et al, 1997). The GluA2-GRIP interaction depends upon binding of the PDZ ligand sequence of the GluA2 C-terminal domain (SVKI), to PDZ 5 of ABP or GRIP (Srivastava et al, 1998). This GluA2 PDZ ligand sequence also binds to the protein interacting with C-kinase (PICK1) PDZ domain (Dev et al, 1999). To deduce the role of GluA2 interaction with GRIP versus interaction with PICK1 in GluA2 surface elevation by Li+, we first expressed C-terminal domain mutants of GluA2 with altered PDZ binding abilities in hippocampal neurons in culture (Fig. 2). These mutants were: a) MycGluA2-SVKE, which is unable to bind to either GRIP or PICK1, and b) MycGluA2-AVKI, unable to bind to GRIP but able to bind to PICK1, and c) wild-type (WT) MycGluA2-SVKI, able to bind to both (Osten et al, 2000). Our model suggests that Li+ will not regulate surface levels of either mutant, since neither mutant can associate with the GRIP scaffolds, which is required for maintaining the receptor at the surface and is regulated by Li+ stabilization of δ-catenin. We stained living neurons with anti-Myc antibodies and quantitated surface GluA2 by immune fluorescence and confocal microscopy. Unlike the neurons expressing the WT MycGluA2-SVKI, for which LiCl elevated surface levels (Fig. 2a), the surface expression of the SVKE mutant was unaffected by LiCl versus NaCl treatments (Fig. 2b). Similar to the SVKE mutant, the AVKI mutant exhibited no significant difference between groups (Fig. 2c). Thus, for Li+ to exert its influence on surface levels of GluA2, binding to the GRIP scaffold is required, while binding to PICK1 is not sufficient.
Figure 2. GRIP is required for a Li+ treatment-induced increase in surface GluA2.
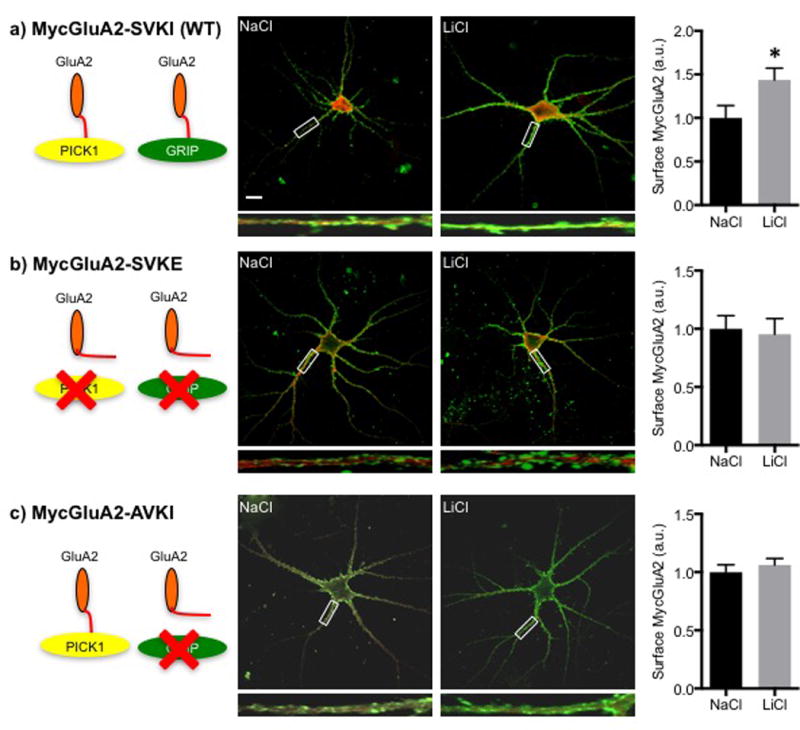
Schematic diagrams show the binding properties of MycGluA2 and its mutants (Greger et al, 2002) a) GluA2-SVKI (WT) binds to PICK1 and GRIP; b) GluA2-SVKE is unable to bind to either PICK1 or GRIP, and c) GluA2-AVKI is able to bind to PICK1 but not GRIP. Also given are representative confocal images of cultured hippocampal neurons expressing the various mutants of MycGluA2 from Sindbis virus vectors and treated with NaCl or LiCl showing surface GluA2 (green) and total GluA2 (red). Scale bar indicates 10μm. Higher magnification images of highlighted dendrites are shown below. Summary graphs show that: a) LiCl treatment was able to increase surface GluA2-SVKI (WT) levels in cultured hippocampal neurons (n=12 neurons, *p<0.05, unpaired two-tailed Student’s t tests), but b–c) LiCl treatment was unable to alter surface levels of mutant GluA2 (n=30 neurons in b) and 50 neurons in c)), suggesting that the ability of MycGluA2 to bind to GRIP is required for a Li+ treatment-induced increase in surface GluA2 in cultured hippocampal neurons.
Binding of PICK1 is not required for synaptic GluA2 elevation by Li+
PICK1 is a second GluA2-interacting protein that contributes to synaptic plasticity by promoting the trafficking of GluA2 (Chung et al, 2000; Hanley, 2008; Perez et al, 2001; Srivastava et al, 1998; Terashima et al, 2008; Xia et al, 1999). Although our findings using mutant GluA2 suggest that PICK1 may not be required for a Li+-induced increase in GluA2 surface expression (Fig. 2), we decided to investigate further a possible role for PICK1 in the mechanism by which Li+ regulates synapses. To directly address this question, we employed PICK1 knockdown via shRNA. In a control for knockdown, we infected cultured hippocampal neurons with a lentivirus expressing shRNA that targets PICK1 mRNA for degradation or with a control lentivirus not expressing shRNA (vector). PICK shRNA was sufficient to reduce PICK1 protein levels in infected neurons (Fig. 3a). To determine surface GluA2 levels, we first infected PICK1 shRNA to reduce PICK1 levels. We expressed WT MycGluA2 by sindbis virus infection 10–12 days after shRNA silencing and treated with either NaCl or LiCl as was done previously. We then assayed for surface MycGluA2 by live staining and quantitative immune cytochemistry. A significant increase in surface MycGluA2 was seen in the control neurons treated with LiCl relative to cells treated with NaCl (Fig. 3b), consistent with the previous data (Fig. 1d and 2a). In PICK1 knockdown neurons, LiCl also elevated MycGluA2 surface levels relative to the NaCl controls (Fig. 3b). This confirms that PICK1 is not required for the Li+-induced increase of surface MycGluA2.
Figure 3. PICK1 is not necessary for a Li+ treatment-induced increase in surface GluA2.
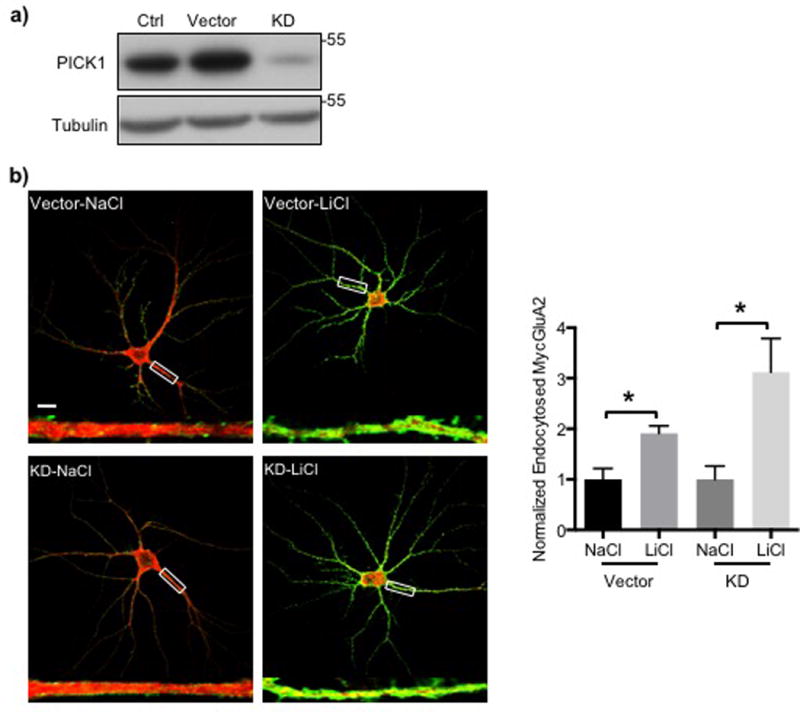
a) Representative immunoblots of cell extracts from PICK1 knockdown (KD) cultured hippocampal neurons showing that PICK1 shRNA significantly reduced PICK1 protein levels in these neurons. b) Representative confocal images of hippocampal neurons treated with NaCl or LiCl in the presence of absence of PICK1 shRNA showing surface GluA2 (green) and total GluA2 (red). Scale bar indicates 10μm. Higher magnification images of highlighted dendrites are shown below. Summary graphs show that reduction of PICK1 levels had no effect of LiCl treatment on surface GluA2 levels (n=4 neurons in each condition, *p<0.05 and ***p<0.001, one-way ANOVA with Fisher’s LSD test).
Collectively, these data suggest that: 1) Li+ regulates the synaptic levels of GRIP through control of the levels of δ-catenin, and, 2) the ability of GluA2 interaction with GRIP to stabilize MycGluA2 on the plasma membrane (Osten et al, 2000). It is also consistent with the interaction with PICK1 promoting the trafficking of these receptors between different membranes rather than stabilizing the receptor at the plasma membrane (Lu et al, 2005; Perez et al, 2001).
Li+ increases synaptic GluA2 in vivo
Our studies with cultured neurons revealed that Li+ is sufficient to increase surface GluA2, providing a potential mechanism for its pro-cognitive effects. As an additional test of whether this mechanism is relevant to medical applications of Li+, such as treatment of BD (Geddes et al, 2004), we tested the effects of Li+ on synaptic GluA2 expression in vivo. To determine if Li+ increased endogenous GluA2 in vivo, we fed mice Li+ chow for one week. Mice were the sacrificed for synaptic protein analysis. To determine serum Li+ levels, blood was collected at the time of sacrifice. The average Li+ level in serum in Li+ chow fed animals (0.5mEq/l) was significantly higher than for standard chow fed animals (<0.2 mEq/L, p<0.05). Because Li+ crosses the blood brain barrier (Forester et al, 2009), this protocol enabled us to assay its effects on synaptic proteins in Li+ fed animals. We isolated the PSD from the hippocampus of Li+-chow or normal chow fed animals and found through Western blotting that GSK3β phosphorylation was significantly higher in Li+-chow fed animals than control mice, indicating that GSK3β kinase activity was successfully inhibited (Fig. 4), as seen in cultured neurons (Fig. 2b–c). Furthermore, the levels of β and δ-catenin, which are substrates for GSK3β-mediated protein degradation, were significantly elevated by Li+ treatment (Fig. 4), consistent with findings in cultured neurons (Fig. 1b–c). Moreover, the δ-catenin-interacting synaptic scaffolding protein, GRIP, but not a second, structurally unrelated scaffold, PSD95, was also increased, providing a basis for an elevation of synaptic GluA2 in the hippocampus of Li+ fed animals (Fig. 5), and recapitulating what was found in cultured neurons (Fig. 1b–c). As seen in cultured neurons (Fig. 1c), synaptic GluA1 levels were also significantly elevated in the hippocampus of Li+-chow fed animals (Fig. 4), suggesting that increased AMPARs are GluA1/2 heteromers. Therefore, this suggests that changes of synaptic scaffolding and AMPARs induced by Li+ in cultured neurons also take place in vivo.
Figure 4. Li+ increases synaptic GluA2 and its associated scaffolding protein in vivo.
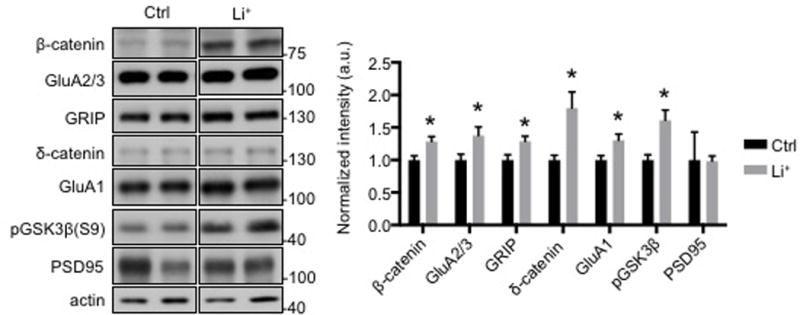
a) Representative immunoblots and b) quantitative analysis of hippocampal PSD from mice fed with normal mouse chow and Li+ chow for 7 days showing that Li+ increased GluA2/3, GRIP, and δ-catenin levels (n=8 animals, *p<0.05, unpaired two-tailed Student’s t tests). In vivo GSK3β inhibition by Li+ was confirmed by an increase in its phosphorylation and increased β -catenin levels.
Fig. 5. Li+ increased the peak amplitude but not the frequency of miniature ESPCs of hippocampal neurons in culture.
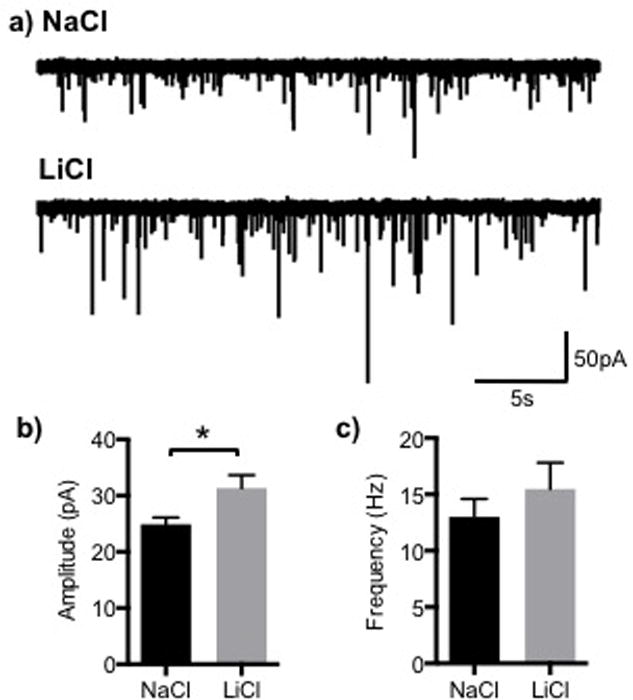
a) Representative mEPSC traces from cultured hippocampal neurons treated overnight with NaCl (control) or LiCl. Graphs are shown of: b) the peak amplitude and c) frequency of mEPSCs. (n= 13 and 18 cells with Na+ and Li+ treatment from 3 different cultures, respectively, *p<0.05)
Li+ increases mEPSC amplitude but not frequency
To determine whether Li+ treatment had functional consequences on AMPAR currents, we compared mEPSCs in cultured hippocampal neurons, treated overnight with Li+, with control cultures treated with Na+. Li+ treatment overnight increased mEPSC amplitude but did not alter mEPSC frequency (Fig. 5). The increase in mEPSC amplitude is consistent with the trafficking of new GluA2 containing AMPARs to synapses, stabilized by elevated levels of the scaffolding proteins, δ-catenin and GRIP following Li+ treatment.
Discussion
We have investigated the effect of Li+ on the trafficking of AMPAR subunit, GluA2, and on its scaffolding proteins in cultured hippocampal neurons and in mouse brain. In both systems, Li+ elevated GluA2 synaptic levels relative to the control. Furthermore, the elevation in cultured neurons depended on the ability of GluA2 to interact with the GRIP scaffold, which is stabilized at synapses via interaction with δ-catenin (Silverman et al, 2007). Our work supports a model in which Li+, acting through its ability to inhibit GSK3β, stabilizes δ-catenin, which interacts with the juxtamembrane region of the cadherin synaptic cell adhesion molecules (Lu et al, 1999; Oh et al, 2009; Paffenholz et al, 1997). Phosphorylation of δ-catenin by GSK3β increases the rate of δ-catenin proteolysis (Bareiss et al, 2010; Oh et al, 2009). In our model, inhibition of GSK3β by Li+ is able to increase δ-catenin at synapses, and δ-catenin in turn binds to the scaffolding protein, GRIP, which anchors GluA2 at synapses. The model holds that by stabilizing synaptic δ-catenin, Li+ elevates the scaffolds and in turn, synaptic GluA2 (Silverman et al, 2007). The finding that a chemical inhibitor of GSK3β elevated δ-catenin in heterologous cells (Oh et al, 2009) is consistent with δ-catenin being the relevant target of Li+ in the current study. We cannot exclude that other activities of Li+ contribute to the synaptic increase of GluA2. Li+ also increased the synaptic levels of GluA1, both in vitro and in vivo. This suggests that GluA1/2 heteromers are stabilized at the synapse by Li+. Li+ did not alter the synaptic levels of PSD95, a major scaffold that is structurally distinct from GRIP, indicating selectivity of action of Li+ on scaffold levels. Because ABP, a scaffold closely related to GRIP (Srivastava et al, 1998) also binds δ-catenin and GluA2, it may also be elevated by Li+ and contribute to the increases in GluA2 synaptic levels. Because the activity of PICK1, a protein that functions in GluA2 trafficking to and from the plasma membrane (Hanley, 2008), does not seem to play a large role in the mechanism of action of Li+, GluA2 stabilization by Li+ is not mediated by PICK1-dependent trafficking.
Li+ is a therapeutic agent that is also an inhibitor of GSK3β (O’Brien et al, 2009). It has been established that inhibition of GSK3β has numerous protective effects in neurons, potentially providing a basis for the potent anti-manic activity of Li+ (Jope, 1999). Mutations in cellular inhibitors of GSK3β have been shown to promote Schizophrenia and BD (Freyberg et al, 2010). Other studies have indicated that Li+ regulates GSK3β not only by direct enzyme inhibition, but also blocking inhibitors of negative regulators of GSK3β to achieve the same regulatory effect (Beaulieu, 2012). Although Li+ interacts functionally with many upstream targets of GSK3β, the majority of the synaptic changes and neuronal regulation induced by Li+ is thought to occur downstream of GSK3β. For example, Li+ has been shown to increase the levels of β-catenin, a cell adhesion molecule important for cellular signaling (O’Brien et al, 2004). Indeed, studies of transgenic animal models suggest that increase in β-catenin is responsible for the behavioral mood-stabilizing effects of Li+ (Gould et al, 2007). The present study reinforces the importance of the role of downstream targets of GSK3β, specifically the synaptic proteins, δ-catenin and its associated scaffold proteins. Disruption of the interaction between these proteins has been shown to decrease the neuron’s ability to maintain AMPAR GluA2 at the surface (Restituito et al, 2011; Silverman et al, 2007). With Li+ inhibition of GSK3β, there is a potential for modifying synapse function by increasing δ-catenin plus the associated GRIP scaffold, which then can be utilized to increase GluA2 expression at the cellular surface in culture and in vivo.
Measurement of mEPSCs showed that Li+ treatment increases mEPSC amplitude, consistent with the delivery of new AMPA receptors to the synapse following Li+ treatment. mEPSC frequency, however, did not change, which rules out the possibility of presynaptic effects on AMPAR currents. A study employing GSK3β conditional knockout mice found that cell autonomous loss of GSK3β reduced hippocampal neuron spine density and mEPSC amplitude and frequency (Ochs et al, 2015). This result emphasizes the role of GSK3β in controlling spines and synapses. The different outcome for mEPSCs seen with GSK3β inhibition by Li+ in the current study may reflect the greater length of time of GSK3β inhibition in the knockout protocol or action by Li+ on other targets.
PICK1 is one of the primary proteins responsible for the trafficking of GluA2-containing AMPARs to and from the cellular surface and as such, it plays a crucial role in synaptic plasticity (Hanley, 2008). No clear relationship has been described between PICK1 and Li+, although much is known about Li+ on one hand and on the other about proteins that interact with PICK1, such as PKC (Chen et al, 2000). Our study showed that PICK1 did not play a major role in the mechanism by which Li+ regulated synaptic expression of GluA2. This would be consistent with the function of PICK1 in dissociating GluA2 from GRIP complexes and promoting surface receptor endocytosis rather than stabilization (Lu et al, 2005).
These studies raise the possibility that regulation of δ-catenin levels plays a significant role in brain function and is a therapeutic target. This possibility is supported by the role of δ-catenin mutation and knockout in Cri-du chat syndrome, synaptic regulation and behavior, and autism (Israely et al, 2004; Restituito et al, 2011; Silverman et al, 2007; Turner et al, 2015; Yuan et al, 2015).
Acknowledgments
We thank past and present members of the Ziff lab for helpful discussions. We extend special thanks to Joseph Pick for reviewing the paper and offering additional guidance. This work was supported by a grant from NIH, R37AG013620 (to EBZ) and R01 DA038616 (to MR).
References
- Bareiss S, Kim K, Lu Q. Delta-catenin/NPRAP: A new member of the glycogen synthase kinase-3beta signaling complex that promotes beta-catenin turnover in neurons. J Neurosci Res. 2010;88(11):2350–2363. doi: 10.1002/jnr.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J Psychiatry Neurosci. 2012;37(1):7–16. doi: 10.1503/jpn.110011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, et al. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96(24):14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Masana MI, Manji HK. Lithium regulates PKC-mediated intracellular cross-talk and gene expression in the CNS in vivo. Bipolar Disord. 2000;2(3 Pt 2):217–236. doi: 10.1034/j.1399-5618.2000.20303.x. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Manji HK. In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol Psychiatry. 2007;62(1):4–6. doi: 10.1016/j.biopsych.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20(19):7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev KK, Nishimune A, Henley JM, Nakanishi S. The protein kinase C alpha binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology. 1999;38(5):635–644. doi: 10.1016/s0028-3908(98)00230-5. [DOI] [PubMed] [Google Scholar]
- Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386(6622):279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Forester BP, Streeter CC, Berlow YA, Tian H, Wardrop M, Finn CT, et al. Brain lithium levels and effects on cognition and mood in geriatric bipolar disorder: a lithium-7 magnetic resonance spectroscopy study. The American journal of geriatric psychiatry: official journal of the American Association for Geriatric Psychiatry. 2009;17(1):13–23. doi: 10.1097/JGP.0b013e318172b3d0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg Z, Ferrando SJ, Javitch JA. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiatry. 2010;167(4):388–396. doi: 10.1176/appi.ajp.2009.08121873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JR, Burgess S, Hawton K, Jamison K, Goodwin GM. Long-term lithium therapy for bipolar disorder: systematic review and meta-analysis of randomized controlled trials. Am J Psychiatry. 2004;161(2):217–222. doi: 10.1176/appi.ajp.161.2.217. [DOI] [PubMed] [Google Scholar]
- Gould TD, Einat H, O’Donnell KC, Picchini AM, Schloesser RJ, Manji HK. Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology. 2007;32(10):2173–2183. doi: 10.1038/sj.npp.1301338. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34(5):759–772. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacology & therapeutics. 2008;118(1):152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Developmental biology. 1997;185(1):82–91. doi: 10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annual review of neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, Liu X. Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr Biol. 2004;14(18):1657–1663. doi: 10.1016/j.cub.2004.08.065. [DOI] [PubMed] [Google Scholar]
- Jope RS. A bimodal model of the mechanism of action of lithium. Mol Psychiatry. 1999;4(1):21–25. doi: 10.1038/sj.mp.4000444. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29(2):95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kim H, Han JR, Park J, Oh M, James SE, Chang S, et al. Delta-catenin-induced dendritic morphogenesis. An essential role of p190RhoGEF interaction through Akt1-mediated phosphorylation. The Journal of biological chemistry. 2008;283(2):977–987. doi: 10.1074/jbc.M707158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Violette CJ, Ziff EB. Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant. Neurobiol Aging. 2015;36(12):3239–3246. doi: 10.1016/j.neurobiolaging.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Donahue CP, Israely I, Liu X, Ochiishi T. Delta-catenin at the synaptic-adherens junction. Trends Cell Biol. 2005;15(3):172–178. doi: 10.1016/j.tcb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098) Neuropharmacology. 2001;40(8):1028–1033. doi: 10.1016/s0028-3908(00)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu Q, Paredes M, Medina M, Zhou J, Cavallo R, Peifer M, et al. delta-catenin, an adhesive junction-associated protein which promotes cell scattering. J Cell Biol. 1999;144(3):519–532. doi: 10.1083/jcb.144.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47(3):407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Marmol F. Lithium: bipolar disorder and neurodegenerative diseases Possible cellular mechanisms of the therapeutic effects of lithium. Progress in neuro-psychopharmacology & biological psychiatry. 2008;32(8):1761–1771. doi: 10.1016/j.pnpbp.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Medina M, Marinescu RC, Overhauser J, Kosik KS. Hemizygosity of delta-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome. Genomics. 2000;63(2):157–164. doi: 10.1006/geno.1999.6090. [DOI] [PubMed] [Google Scholar]
- Metcalfe C, Bienz M. Inhibition of GSK3 by Wnt signalling–two contrasting models. Journal of cell science. 2011;124(Pt 21):3537–3544. doi: 10.1242/jcs.091991. [DOI] [PubMed] [Google Scholar]
- O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, et al. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24(30):6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien WT, Klein PS. Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans. 2009;37(Pt 5):1133–1138. doi: 10.1042/BST0371133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary O, Nolan Y. Glycogen synthase kinase-3 as a therapeutic target for cognitive dysfunction in neuropsychiatric disorders. CNS drugs. 2015;29(1):1–15. doi: 10.1007/s40263-014-0213-z. [DOI] [PubMed] [Google Scholar]
- O’Neill MJ, Bleakman D, Zimmerman DM, Nisenbaum ES. AMPA receptor potentiators for the treatment of CNS disorders. Curr Drug Targets CNS Neurol Disord. 2004;3(3):181–194. doi: 10.2174/1568007043337508. [DOI] [PubMed] [Google Scholar]
- Ochs SM, Dorostkar MM, Aramuni G, Schon C, Filser S, Poschl J, et al. Loss of neuronal GSK3beta reduces dendritic spine stability and attenuates excitatory synaptic transmission via beta-catenin. Molecular psychiatry. 2015;20(4):482–489. doi: 10.1038/mp.2014.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M, Kim H, Yang I, Park JH, Cong WT, Baek MC, et al. GSK-3 phosphorylates delta-catenin and negatively regulates its stability via ubiquitination/proteosome-mediated proteolysis. The Journal of biological chemistry. 2009;284(42):28579–28589. doi: 10.1074/jbc.M109.002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osten P, Khatri L, Perez JL, Kohr G, Giese G, Daly C, et al. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27(2):313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, et al. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21(1):99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- Paffenholz R, Franke WW. Identification and localization of a neurally expressed member of the plakoglobin/armadillo multigene family. Differentiation. 1997;61(5):293–304. doi: 10.1046/j.1432-0436.1997.6150293.x. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21(15):5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restituito S, Khatri L, Ninan I, Mathews PM, Liu X, Weinberg RJ, et al. Synaptic autoregulation by metalloproteases and gamma-secretase. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31(34):12083–12093. doi: 10.1523/JNEUROSCI.2513-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochemical and biophysical research communications. 2001;280(3):720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- Silverman JB, Restituito S, Lu W, Lee-Edwards L, Khatri L, Ziff EB. Synaptic anchorage of AMPA receptors by cadherins through neural plakophilin-related arm protein AMPA receptor-binding protein complexes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(32):8505–8516. doi: 10.1523/JNEUROSCI.1395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, et al. Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron. 1998;21(3):581–591. doi: 10.1016/s0896-6273(00)80568-1. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Cadherins: key molecules for selective cell-cell adhesion. IARC Sci Publ. 1988;(92):76–79. [PubMed] [Google Scholar]
- Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, et al. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57(6):872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Sharma K, Oh EC, Liu YP, Collins RL, Sosa MX, et al. Loss of delta-catenin function in severe autism. Nature. 2015;520(7545):51–56. doi: 10.1038/nature14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. The EMBO journal. 2012;31(12):2714–2736. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16(6):1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22(1):179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Xu C, Kim NG, Gumbiner BM. Regulation of protein stability by GSK3 mediated phosphorylation. Cell cycle. 2009;8(24):4032–4039. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Seong E, Beuscher JL, Arikkath J. delta-Catenin Regulates Spine Architecture via Cadherin and PDZ-dependent Interactions. The Journal of biological chemistry. 2015;290(17):10947–10957. doi: 10.1074/jbc.M114.632679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. The Journal of biological chemistry. 2003;278(35):33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]