

Abstract
Hepatic insulin resistance and hepatosteatosis in diet-induced obesity are associated with various metabolic diseases, yet the underlying mechanisms remain to be fully elucidated. Here we show that the expression levels of the disulfide-bond A oxidoreductase-like protein (DsbA-L) are significantly reduced in the liver of obese mice and humans. Liver-specific knockout or adenovirus-mediated overexpression of DsbA-L exacerbates or alleviates, respectively, high-fat diet–induced mitochondrial dysfunction, hepatosteatosis, and insulin resistance in mice. Mechanistically, we found that DsbA-L is localized in mitochondria and that its deficiency is associated with impairment of maximum respiratory capacity, elevated cellular oxidative stress, and increased JNK activity. Our results identify DsbA-L as a critical regulator of mitochondrial function, and its down-regulation in the liver may contribute to obesity-induced hepatosteatosis and whole body insulin resistance.—Chen, H., Bai, J., Dong, F., Fang, H., Zhang, Y., Meng, W., Liu, B., Luo, Y., Liu, M., Bai, Y., Abdul-Ghani, M. A., Li, R., Wu, J., Zeng, R., Zhou, Z., Dong, L. Q., Liu, F. Hepatic DsbA-L protects mice from diet-induced hepatosteatosis and insulin resistance.
Keywords: NAFLD, mitochondria, oxidative stress
Obesity, which is associated with various metabolic abnormalities, has become an epidemic in the industrialized and many rapidly developing countries (1). Obesity increases hepatic-free fatty acid uptake and de novo lipogenesis, which can lead to nonalcoholic fatty liver disease (NAFLD) and hepatic insulin resistance. Several mechanisms, including increased endoplasmic reticulum and mitochondrial stress, insufficient fatty acid β-oxidation, and/or increased leakage of reactive oxygen species (ROS), have been found to be relevant risk factors for NAFLD and hepatic insulin resistance (2–5). However, while these findings suggest that impaired mitochondrial function could be a key factor in obesity-induced liver dysfunction, the underlying mechanisms are yet to be further elucidated.
Dysregulated ROS levels are a hallmark of mitochondrial dysfunction. In obese status, the chronic uptake of excess energy substrates, especially lipids, leads to increased ROS production (6). Elevated oxidative stress and ROS levels are found in the liver of high-fat diet (HFD)-fed rats or mice (7, 8). Consistent with these findings, HFD feeding decreases hepatic mitochondrial activity (9). In addition, the activity of mitochondrial respiratory chain, the major site responsible for mitochondrial ROS generation, is dramatically impaired in patients with nonalcoholic steatohepatitis (10). These findings suggest a potential link between impaired mitochondrial function, oxidative stress, and obesity-induced metabolic diseases. However, how obesity scales up mitochondrial ROS production is incompletely understood.
Disulfide-bond A oxidoreductase-like protein (DsbA-L), originally termed glutathione S-transferase (GST)-κ, is a 25-kDa protein initially found in the matrix of rat liver mitochondria (11). Amino acid sequence and 3-dimensional structural analysis reveal that GST-κ is more closely related to the protein disulfide-bond A oxidoreductase from Escherichia coli than to other members of the canonical GST superfamily (12). Consistent with this, GST-κ lacks the SNAIL/TRAIL (Ser-Asn-Ala-Ile-Leu/Thr-Arg-Ala-Ile-Leu) motif found in all other classes of soluble GSTs and does not bind to agarose-coupled glutathione (13), suggesting that this protein is not a classic GST. We renamed the protein DsbA-L and show that it promotes adiponectin multimerization in adipocytes (14). In addition, overexpression of DsbA-L in adipocytes prevents endoplasmic reticulum stress-induced adiponectin down-regulation (15, 16) and protects mice against HFD-induced obesity and insulin resistance (17). However, while DsbA-L is highly expressed in the liver, the hepatic function of DsbA-L remains unknown.
In the present study, we found that expression levels of DsbA-L were significantly reduced in the liver of HFD-fed mice, db/db mice, and obese human subjects compared to their respective normal-weight controls. In addition, liver-specific knockout of the Dsba-l gene impaired hepatocyte mitochondrial functions and exacerbated HFD-induced hepatosteatosis and insulin resistance. On the other hand, adenovirus-mediated overexpression of DsbA-L protected mice from HFD-induced hepatosteatosis and insulin resistance. DsbA-L is localized in mitochondria, and its deficiency leads to impaired mitochondrial function. Our results identified DsbA-L as a key component of mitochondrial function in the liver and revealed a potential mechanism underlying obesity-induced hepatic mitochondrial dysfunction, hepatosteatosis, and insulin resistance.
MATERIALS AND METHODS
Human study
Twenty-seven male Chinese human liver specimens stored in paraffin wax were obtained from the Second Xiangya Hospital of Central South University. Total protein of the specimen in paraffin wax was extracted with Formalin-Fixed Paraffin-Embedded (FFPE) Total Protein Extraction Kit (Sango Biotech, Shanghai, China). Medical records, including age, height, body weight, fasting glucose levels, and ultrasound diagnosis, were obtained together with the specimen from the department of pathology. The protocol was approved by the ethics committee of the Second Xiangya Hospital.
DsbA-L-suppressed hepatocytes
The mouse hepatocyte cell line used in this study was provided by D. Accili (Columbia University, New York, NY, USA) (18). These cells were successfully used in our previous studies (19). The sense and antisense sequences of small interfering RNA (siRNA) were chemically synthesized and ligated into the pSIREN-RetroQ (BD Knockout RNAi system; BD Biosciences, San Jose, CA, USA). The sequences for siRNA and scramble control are GCATGGAGCAACCAGAGAT and GCCGTAGCACTGGAAGAAA, respectively. For generation of DsbA-L-suppressed siRNA hepatocyte stable cell lines, mouse hepatocytes were cotransfected with pSIREN-RetroQ/DsbA-L (or its scramble control) and pSV2-puromycin. Positive colonies were selected by puromycin 4 µg/ml and verified by Western blot analysis against DsbA-L. DsbA-L-suppressed mouse hepatocytes and their scramble control cells were cultured in minimum essential medium, α modification, with 10% fetal bovine serum and 0.8 µg/ml dexamethasone in the cell culture hood with carbon dioxide set at 5% and temperature set at 30°C. Multiple colonies of scramble and RNA interference (RNAi) stable cells have been selected in the study with similar results achieved. Cells were incubated with 5 μM MitoSox (Molecular Probes, Eugene, OR, USA) for 20 min before mitochondrial ROS detection by confocal microscopy. MitoTempo (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and Tempol (Sigma-Aldrich, St. Louis, MO, USA) were applied in the study as mitochondria-specific and nonspecific antioxidants, respectively.
Animal experiments and procedures
DsbA-LloxP/loxP mice were generated in the 129J background and crossed with C57BL/6 mice for 6 generations before experiments. DsbA-L liver-specific knockout mice were generated by crossing DsbA-LloxP/loxP mice with albumin-Cre transgenic mice (The Jackson Laboratory, Bar Harbor, ME, USA). DsbA-L liver-specific knockout mice and their wild-type littermates were fed with a 45% HFD (Research Diet, New Brunswick, NJ, USA) at 1 mo of age for 8 wk to induce hyperglycemia and insulin resistance. For the adenovirus-mediated DsbA-L overexpression experiment, 8-wk-old C57/BL6 mice were fed with a normal diet or a 60% HFD (Research Diet) for 12 wk. A total of 2 × 109 pfu of green fluorescent protein (GFP) or myc-tagged DsbA-L adenovirus was injected via tail vein. Glucose tolerance test was performed on day 6 after injection and insulin tolerance test on day 8 d after injection, respectively. On d 10 after injection, mice were unfed overnight and then humanely killed to obtain liver tissues for Western blot analysis. All animal care and treatments were in accordance with guidelines of the University of Texas Health Science Center at San Antonio for Animal Studies; the protocol was approved by the Ethics Committee of Animal Care and Use (12085x).
Oxygen consumption assays
Mouse primary hepatocytes were isolated and cultured in Seahorse microplates (Seahorse Bioscience, North Billerica, MA, USA). A comprehensive assay of hepatocytes mitochondrial bioenergetic function was performed using the Mito-Stress Test Kit (Seahorse Bioscience). In brief, primary hepatocytes were isolated by collagenase perfusion technique (20) and plated in Seahorse microplates 1 d before measurement. The shift of the bioenergetic profile of the cell induced by sequential injections of 4 μM oligomycin, 2 μM FCCP (carbonylcyanide-p-trifluoromethoxyphenylhydrazone), 1 μM rotenone, and antimycin were recorded by an XF24 Extracellular Flux Analyzer (Seahorse Bioscience).
Hyperinsulinemic–euglycemic clamp studies
Hyperinsulinemic–euglycemic clamp studies were performed as described (21). Briefly, the right internal jugular vein was cannulated, and the indwelling catheters were extended to the left atrium by surgery under an anesthetized condition. After a minimum 5 d recovery period, 3H-glucose (NET331C; PerkinElmer, Waltham, MA, USA) was infused, and blood glucose were kept at ∼110 mg/dl by monitoring glucose infusion rate during the clamp. Blood samples were collected via tail-tip bleeds for determination of peripheral tissue glucose disposal and hepatic glucose production (HGP).
Tissue triglyceride content measurement
Tissue triglyceride (TG) levels were determined as previously described (22). Briefly, liver tissues (∼ 50 mg) were weighed and homogenized in PBS. 150 μl of homogenate was mixed with 1 ml of 2:1 chloroform–methanol and 0.1 ml of 0.1% sulfuric acid to extract the lipids. A total of 0.5 ml of 0.6% NaCl was added, followed by centrifugation at 800 g for 20 min at 4°C. The organic phase was transferred to a new tube, and then the samples were dried under nitrogen gas and reconstituted with 100 μl 1% Triton X-100/PBS. TG concentrations were detected using a Triglyceride Assay Kit (Cayman Chemicals, Ann Arbor, MI, USA).
ATP level assays
ATP levels in tissue homogenate were measured by a chemiluminescent assay with an ATP determination kit (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instruction. To determine the ATP production rate, aliquots of mitochondria freshly isolated were incubated with a luciferin–luciferase ATP-monitoring reagent in the reaction buffer (ATP Bioluminescence Assay Kit; Roche, Berlin, Germany), following the manufacturer’s instructions (23). The reaction was performed in an opaque 96-well plate, and the kinetic of luciferase signal were detected at 560 nm using the Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Winooski, VT, USA).
Mitochondrial isolation and ROS measurement
Mitochondria from liver tissue were isolated on the basis of a published protocol (24). All the mitochondrial isolation steps were performed on ice or at 4°C. Amplex Red assays were performed in assay buffer (5 mM pyruvate and malate, 2 mM ADP, 100 mM KCl, 2 mM MgCl2, 1 mM EDTA, 4 mM KH2PO4, 20 mM TES, pH7.4) with 30 μg of mitochondrial protein after addition of horseradish peroxidase. Hydrogen peroxide release kinetics from the mitochondria were recorded at excitation 560 nm and emission 590 nm using a fluorescence plate reader. Mitochondrial ROS level was quantified according to a hydrogen peroxide standard curve.
Data analysis
Quantification of Western blot results were analyzed by ImageJ software (Image Processing and Analysis in Java; U.S. National Institutes of Health, Bethesda, MD, USA). Quantitative data are presented as means ± sem. Statistic analysis was performed by SPSS 18.0 software (IBM SPSS, Chicago, IL, USA). Significance of 2 groups was determined by unpaired, 2-tailed Student’s t test. One-way ANOVA was applied for multiple group comparison. The Student’s t test is used to determine the significance of correlation coefficient. All statistical tests with a value of P < 0.05 were considered significant.
RESULTS
Hepatic DsbA-L expression levels are negatively correlated with obesity
DsbA-L is highly expressed in mouse liver (14), but its physiologic role in the liver is unknown. To address this question, we asked whether hepatic expression of DsbA-L is affected by obesity. Real-time quantitative PCR (qPCR) and Western blot analysis revealed that both the mRNA (Fig. 1A) and protein (Fig. 1B) levels of DsbA-L were significantly reduced in the liver of HFD-induced obese mice compared to chow-fed mice. A significant reduction in DsbA-L expression was also observed in the liver of db/db mice compared to control mice (Fig. 1C). To determine whether DsbA-L deficiency is associated with obesity in humans, we examined the protein levels of DsbA-L in liver specimens from male human subjects (Supplemental Table 1). We found that DsbA-L expression levels are negatively correlated with body mass index (Fig. 1D). Taken together with the finding that most of the obese human subjects who were enrolled onto the current study were diagnosed with fatty liver disease (Supplemental Fig. 1), these results suggest that down-regulation of hepatic DsbA-L may play an important role in obesity-induced fatty liver and metabolic dysfunction.
Figure 1.

Hepatic DsbA-L levels are negatively correlated with obesity. A, B) mRNA (A) or protein (B) levels of DsbA-L in liver of male C57BL/6 mice (n = 8 per group) fed normal chow diet (NC) or HFD for 8 wk were detected by real-time qPCR or Western blot analysis. Data are normalized to actin for loading control. C) DsbA-L protein levels in liver of 2 mo old male db/db mice and control littermates. All values represent means ± sem. *P < 0.05, **P < 0.01 by Student’s t test. D) Expression levels of DsbA-L in human liver were determined by Western blot analysis. Pearson correlation coefficient was applied to show correlation between DsbA-L protein level and body mass index in human subjects.
Liver-specific DsbA-L-knockout mice show increased susceptibility to diet-induced insulin resistance
To investigate the in vivo role of DsbA-L in the regulation of metabolic function, we produced mice in which exons 1–3 of the DsbA-L gene were floxed using the Cre/loxP system (Fig. 2A). Liver-specific DsbA-L-knockout mice (hereafter referred to as DsbA-LLKO mice) were generated by crossing the DsbA-L gene floxed mice with albumin-Cre transgenic mice, as demonstrated by both PCR and Western blot experiments (Fig. 2B–E). DsbA-LLKO mice were born at mendelian ratios and displayed normal appearance under normal chow feeding conditions (data not shown). In addition, little difference in either body weight or food intake was observed between DsbA-LLKO mice and floxed control littermates under either normal diet or HFD feeding conditions (Supplemental Fig. 2). However, under HFD feeding conditions, the DsbA-LLKO mice displayed increased glucose and insulin intolerance compared to the floxed control mice (Fig. 3A, B). Hyperinsulinemic–euglycemic clamp studies showed that the glucose infusion rate of the DsbA-LLKO mice was significantly lower than that of the control mice (Fig. 3C), suggesting that DsbA-L deficiency in the liver exacerbated insulin resistance. Consistent with this, insulin greatly suppressed HGP of the control mice but had much less effect on HGP rate of the DsbA-LLKO mice (58 and 29% for the control mice and the DsbA-LLKO mice, respectively) (Fig. 3D). In line with these results, the DsbA-LLKO mice showed reduced insulin signaling in the liver, as demonstrated by decreased phosphorylation of PKB and its downstream target, Foxo1, in response to insulin stimulation (Fig. 3E). The DsbA-LLKO mice also displayed a tendency to decrease peripheral glucose disposal rate, though the difference between DsbA-LLKO mice and the control mice did not reach statistical significance (Fig. 3F).
Figure 2.

Generation of liver-specific DsbA-L knockout mice. A) Schematic representation showing construction of DsbA-L targeting vector. B) Genotyping PCR showing 5′ (top) and 3′ (bottom) loxP site insertion with primers indicated. Primer sequences are as follows: primer a, 5′-CTGGATGGCTTCTGTTAGAG-3′; primer b, 5′-AAGACTGCCACTCTGCAATG-3′; primer c, 5′-AGGGATGGGAGAAGCAAGAAC-3′; primer d, 5′-GGATGGAGGACCGTGTCATC-3′). C) Genotyping PCR for albumin-Cre transgenic mice. D) DNA were extracted from primary hepatocytes, and PCR was performed to confirm successful loxP site recombination mediated by Cre with primers a and d, as indicated in A. E) Western blots confirmed generation of DsbA-LLKO (LKO) mice.
Figure 3.
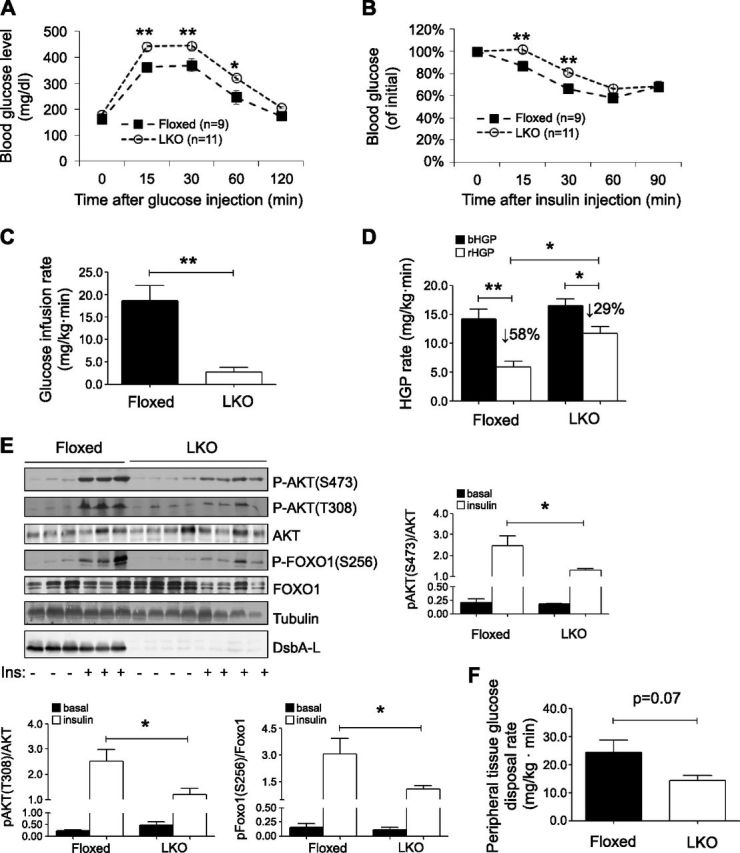
Liver-specific knockout of DsbA-L exacerbated diet-induced insulin resistance. A, B) Glucose tolerance test (A) and insulin tolerance test (B) were performed on DsbA-LLKO (LKO) and floxed control mice fed with HFD for 8 wk. C) Glucose infusion rate of DsbA-LLKO and floxed control mice fed with HFD for 8 wk was measured by hyperinsulinemic–euglycemic clamp studies (n = 4 per group). D) HGP under basal (bHGP) and during hyperinsulinemic–euglycemic clamp (rHGP). E) HFD-fed DsbA-LLKO and floxed control mice were unfed overnight before intraperitoneal injection of insulin (1 U/kg). Mice were humanely killed 15 min after insulin injection, and tissues were dissected on ice and frozen in liquid nitrogen until homogenization. Insulin-stimulated phosphorylation of PKB and Foxo1 in liver of DsbA-LLKO and floxed control mice was determined by Western blot analysis. F) Peripheral tissue glucose disposal during hyperinsulinemic–euglycemic clamp. All values represent means ± sem. *P < 0.05, **P < 0.01 (1-way ANOVA).
Liver-specific knockout of DsbA-L exacerbates diet-induced hepatosteatosis
DsbA-LLKO mice exhibit normal liver morphology compared to their wild-type littermates under normal chow feeding conditions (Fig. 4A). However, DsbA-LLKO mice display more severe hepatosteatosis, as well as higher liver weight (Fig. 4A–C), compared to the floxed control mice under HFD feeding condition, which is associated with a significant increase of TG levels in DsbA-LLKO mouse liver (Fig. 4D, E). Consistent with this, the expression levels of lipogenic genes such as Fasn and Srebp1 were significantly higher and the levels of fatty acid oxidation genes such as Cpt1 and Acadm were significantly lower in the liver of DsbA-LLKO mice compared to control mice under HFD feeding condition (Fig. 4F). However, liver-specific knockout of DsbA-L had no significant effect on the expression of CD36, Mttp, and Mapob under either normal chow or HFD feeding conditions (Fig. 4F). Taken together, these findings suggest that increased lipogenesis and/or insufficient fatty acid oxidation, rather than lipid update and secretion, may contribute to the exacerbated hepatosteatosis in the DsbA-LLKO mice.
Figure 4.
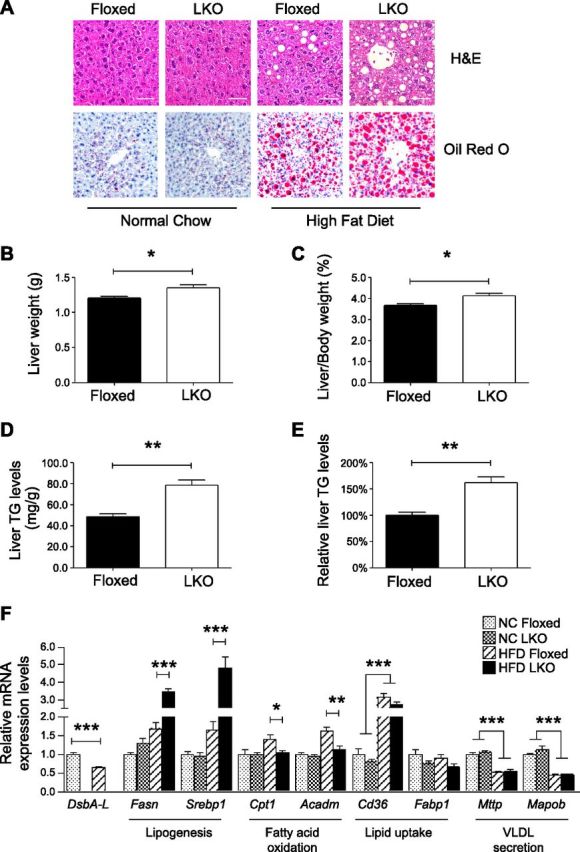
Liver-specific knockout of DsbA-L exacerbates diet-induced hepatosteatosis. A) Representative images of hematoxylin and eosin staining (top) and Oil Red O staining (bottom) of liver tissues from normal chow and HFD-fed DsbA-LLKO mice and control littermates. Scale bars, 50 μm. B, C) Absolute liver weight (B) and percentage to body weight (C) of HFD-fed DsbA-LLKO mice and control littermates (n = 5 per group). D, E) Absolute (D) and relative (E) TG levels were measured and presented (n = 5 per group). F) Expression levels of genes involved in lipogenesis, fatty acid oxidation, lipid uptake, and very low-density lipoprotein secretion in liver of DsbA-LLKO mice and their floxed control littermates were determined by real-time qPCR and normalized to actin (n = 4 per group). All values represent means ± sem. *P < 0.05, **P < 0.01, ***P < 0.001 (1-way ANOVA).
Adenovirus-mediated overexpression of DsbA-L improves diet-induced insulin resistance and hepatosteatosis in mice
To determine whether enhancing hepatic DsbA-L expression could protect against diet-induced metabolic dysfunction, we transiently overexpressed GFP or GFP plus myc-tagged DsbA-L in mice via tail vein injection of adenoviruses encoding these proteins (Fig. 5A). Overexpressing DsbA-L did not cause liver morphology change in mice fed with normal chow as indicated by hematoxylin and eosin staining (Fig. 5B). However, overexpression of DsbA-L alleviated HFD-induced glucose intolerance, insulin resistance, and hepatosteatosis (Fig. 5B–D). Liver TG levels were significantly reduced in the DsbA-L-overexpressing mice compared to the GFP-expressing control mice fed with HFD (Fig. 5E, F). Real-time qPCR experiments demonstrated that hepatic overexpression of DsbA-L significantly suppressed HFD feeding-induced hepatic expression of lipogenic genes such as Fasn and Srebp1 (Fig. 5G). HFD increased the expression of Acc1 but down-regulated the expression of Acc2, the mitochondrial isoform of acetyl-coA carboxylase (25). The liver of HFD-fed mice exhibit higher expression levels of PPARα (Fig. 5G), probably due to a compensatory effect resulted from enhanced free fatty acid loading (26). Interestingly, the expression levels of Cpt1 and Acadm are significantly higher in DsbA-L-overexpressing mouse liver (Fig. 5G), suggesting that DsbA-L may enhance the adaptive metabolic capability to alleviate HFD-induced metabolic dysfunction. HFD feeding also up-regulated hepatic expression of Lpl and CD36 in mice, which was not significantly affected by DsbA-L overexpression (Fig. 5G). Taken together, these results provide further evidence of a role for DsbA-L in regulating lipogenesis and fatty acid oxidation.
Figure 5.
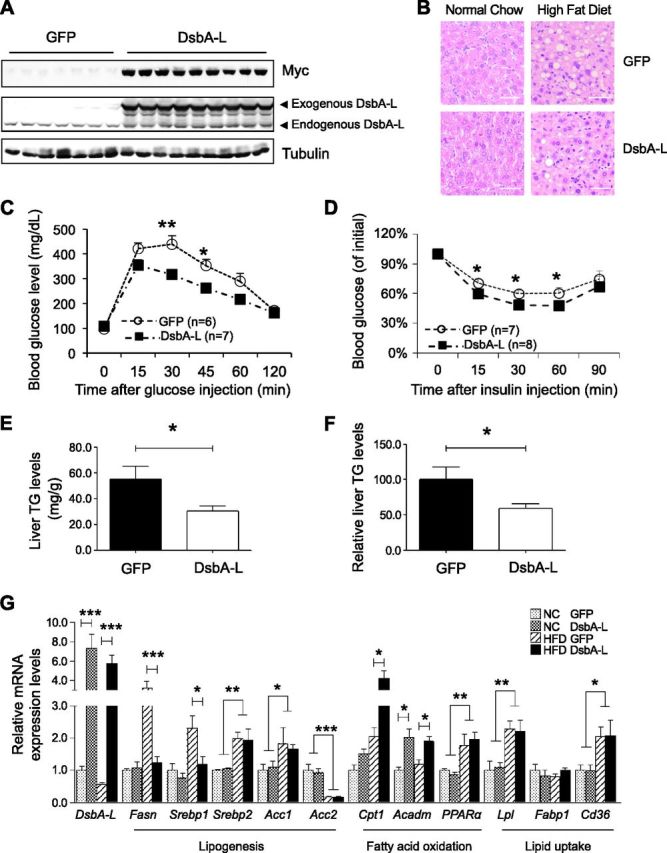
Overexpression of DsbA-L protects mice against diet-induced insulin resistance and hepatosteatosis. Normal diet- or HFD-fed C57BL/6 male mice were injected with adenovirus encoding GFP or myc-tagged DsbA-L via tail vein (2 × 109 pfu per mouse). A) Expression of myc-tagged DsbA-L was detected by Western blot analysis at d 10 after adenovirus injection. B) Mice were humanely killed 10 d after adenovirus injection; hematoxylin and eosin staining was performed in liver tissues of mice. Scale bars, 50 μm. C, D) Glucose tolerance (C) and insulin tolerance (D) tests (D) were performed on d 6 and 8 after injection, respectively. E, F) Absolute (E) and relative (F) TG levels in liver of DsbA-L-overexpressed mice and control mice (n = 7 per group). G) Expression levels of genes involved in lipogenesis, fatty acid oxidation, and lipid uptake were determined in DsbA-L-overexpressed and control mice by real-time qPCR and normalized to actin (n = 7 per group). All values represent means ± sem. *P < 0.05, **P < 0.01, ***P < 0.001 (1-way ANOVA).
DsbA-L deficiency leads to mitochondrial dysfunction in hepatocytes
DsbA-L was previously found to localize in the matrix of mitochondria (11, 27). In agreement with this finding, we expressed an exogenous myc-tagged DsbA-L in mouse hepatocytes and found a noticeable colocalization of DsbA-L with MitoTracker by confocal immunofluorescence study (Fig. 6A). To determine whether disrupting DsbA-L expression affects mitochondrial function in hepatocytes, we measured the oxygen consumption rate in primary hepatocytes isolated from HFD-fed DsbA-LLKO mice and floxed control littermates using a Seahorse XF24 analyzer. Hepatocytes from DsbA-L-deficient mice exhibited a similar oxygen consumption rate compared to that of hepatocytes isolated from control mice at the basal condition or when the cells were treated with the ATP synthase inhibitor oligomycin (Fig. 6B). However, treatment of the electron transport chain accelerator FCCP results in a much less stimulatory effect in oxygen consumption in DsbA-L-deficient hepatocytes, suggesting an impaired mitochondrial respiratory reservoir (Fig. 6B). Oxygen consumption rates returned to similar levels in both the DsbA-L-deficient and control hepatocytes when the cells were treated with the mitochondrial respiration inhibitors rotenone and antimycin (Fig. 6B). In addition, no difference was observed in extracellular acidification rate (Fig. 6C). To further determine the role of DsbA-L in mitochondrial function, we examined ATP levels in the liver of HFD-fed DsbA-LLKO mice and control mice. Both the levels (Fig. 6D) and the production rate (Fig. 6E) of ATP were significantly reduced in the liver of the DsbA-LLKO mice compared to control mice. On the other hand, the DsbA-LLKO mice showed a significant increase in mitochondrial ROS production (Fig. 6F).
Figure 6.

DsbA-L regulates mitochondrial function in hepatocytes. A) Localization of DsbA-L in mitochondria of mouse hepatocytes as determined by confocal immunofluorescence experiments. Scale bars, 20 μm. B, C) Primary hepatocytes isolated from HFD-fed DsbA-LLKO mice and their floxed control littermates were treated with compounds as indicated. Oxygen consumption rate (B) and extracellular acidification rate (C) were measured (n = 4 per group). D) ATP levels in liver tissue homogenates of HFD-fed DsbA-LLKO mice and their floxed control littermates were determined and normalized to protein content (n = 4 per group). E) ATP production rates of mitochondria isolated from liver of HFD-fed DsbA-LLKO mice and their floxed control littermates were determined (n = 4 per group). F) H2O2 production rate in mitochondria isolated from liver of HFD-fed DsbA-LLKO mice and their floxed control littermates was determined (n = 6 group). Results are means ± sem. *P < 0.05 (Student’s t test).
Suppression of DsbA-L promoted oxidative stress, increased JNK activity, and impaired insulin signaling
To better understand the mechanisms that underlie the phenotype observed in DsbA-LLKO mice, we generated stable cell lines in which the expression of DsbA-L is suppressed by RNAi (Fig. 7A). Consistent with in vivo findings, DsbA-L RNAi stable hepatocytes display decreased intracellular ATP levels (Fig. 7B) but increased ROS levels (Fig. 7C). Interestingly, a much stronger signal derived from DsbA-L suppressed stable hepatocytes is observed after incubation with MitoSox (Fig. 7D), suggesting that the elevated ROS levels were mainly produced from mitochondria. Increased ROS level has been shown to activate a number of serine/threonine kinase signaling cascades that act on critical components of the insulin signaling pathway (28–30). In agreement with this, we found that suppressing DsbA-L expression led to JNK activation both in cultured hepatocytes (Fig. 7E) and in vivo (Fig. 7F). In addition, insulin-stimulated PKB phosphorylation was largely diminished in DsbA-L-suppressed RNAi stable hepatocytes compared to scramble control cells (Fig. 7G).
Figure 7.
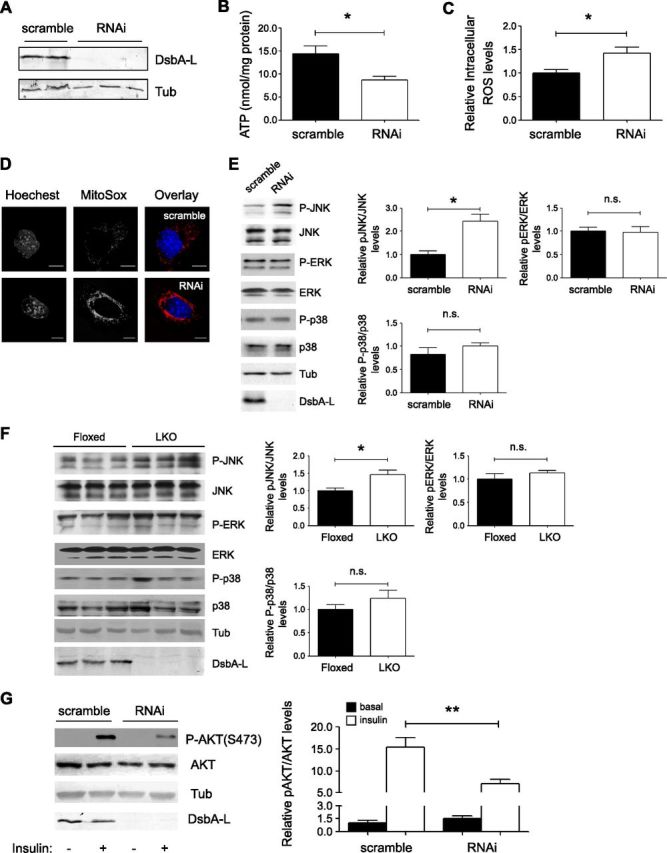
Deficiency of DsbA-L results in elevated oxidative stress, JNK activity, and impaired insulin signaling. A) DsbA-L expression in scramble and RNAi stable mouse hepatocytes. B) Cellular ATP levels were determined and normalized to protein content. C) Cells were incubated with H2DCFDA, and intracellular ROS levels were measured and are presented as relative levels. D) Cells were incubated with MitoSox, and mitochondrial ROS level was determined by confocal microscopy. E, F) Western blot results of signaling molecules in vitro (E) and in vivo (F). G) Western blot results of insulin-stimulated PKB phosphorylation in scramble and DsbA-L RNAi stable mouse hepatocytes. Data are representative of 3 independent experiments, and all values represent means ± sem. *P < 0.05, **P < 0.01 (1-way ANOVA).
Relieving mitochondrial oxidative stress decreases DsbA-L deficiency–induced JNK activity and improves insulin signaling
To determine whether activation of JNK mediates DsbA-L deficiency–induced inhibition of insulin signaling, we treated the cells with N-acetylcysteine (NAC), a widely used antioxidant, and MitoTempo, a ROS scavenger that preferably accumulates in the mitochondria. After this treatment, we found that JNK activity was greatly reduced in DsbA-L-suppressed hepatocytes (Fig. 8A, B). Interestingly, we did not observe a noticeable effect from the cytosolic antioxidant Tempol, a structural mimic of MitoTempo. A recovery of insulin-stimulated PKB phosphorylation was also observed after NAC or MitoTempo treatment (Fig. 8C, D). This experimental result indicates that quenching free radicals generated in the mitochondria could successfully relieve JNK activity in DsbA-L-suppressed hepatocytes and improve insulin signaling.
Figure 8.
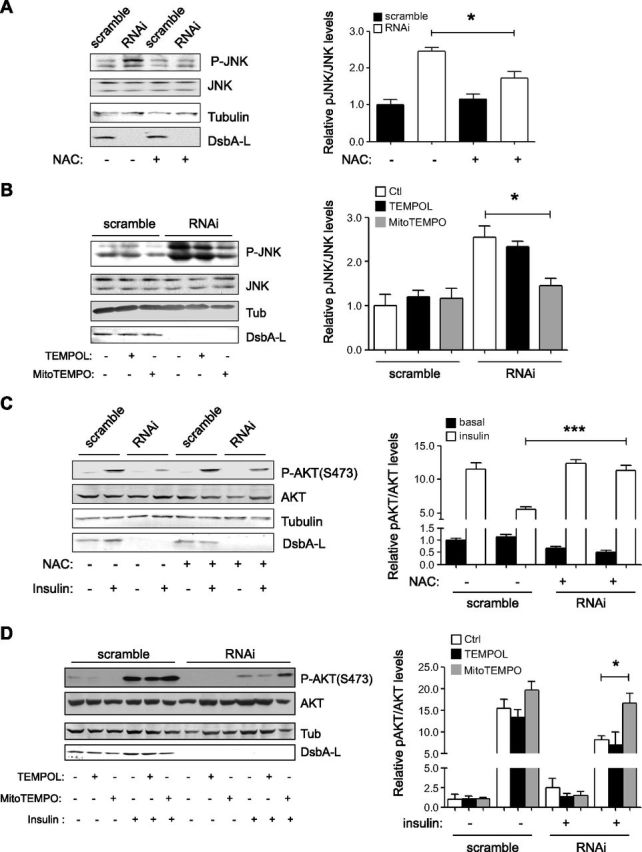
Mitochondria-specific antioxidants suppress JNK activity and improve insulin signaling. A, B) Scramble and DsbA-L RNAi stable mouse hepatocytes were incubated with NAC (A) or MitoTempo (B) for 20 h, and JNK phosphorylation was determined by Western blot analysis. B, C) Cells were incubated with NAC (C) or MitoTempo (D) for 20 h. Then cells were washed and incubated with serum-free medium for 2 h before treatment with insulin (10 nM) for 5 min. Cells in culture dish were lysed, and insulin-stimulated PKB phosphorylation was determined by Western blot analysis. Data are representative of 3 independent experiments, and all values represent means ± sem. *P < 0.05, **P < 0.01, ***P < 0.001 (1-way ANOVA).
DISCUSSION
Hepatic insulin resistance and mitochondrial dysfunction are frequently observed in metabolic diseases such as NAFLD, obesity, and type 2 diabetes (31, 32). However, the mechanism underlying diet-induced hepatic insulin resistance and mitochondrial dysfunction remains incompletely understood.
DsbA-L was discovered in rat liver mitochondrial matrix (11). However, the functional role of DsbA-L in mitochondria remains largely unknown. In this study, we showed that liver-specific disrupting DsbA-L expression impaired mitochondrial function (Fig. 6), demonstrating that DsbA-L plays an important role in the maintenance of mitochondrial respiratory capacity and oxidative homeostasis. In addition, DsbA-L deficiency in the liver exacerbated diet-induced insulin resistance (Fig. 3) and hepatosteatosis (Fig. 4). On the other hand, increasing hepatic DsbA-L levels protected mice against HFD-induced hepatosteatosis and insulin resistance (Fig. 5). Taken together with the finding that DsbA-L levels were significantly reduced in the liver of obese human subjects and mice (Fig. 1), these findings suggest that DsbA-L down-regulation and consequent mitochondrial dysfunction could be a potential mechanism underlying obesity-induced hepatosteatosis and hepatic insulin resistance. The mechanism by which DsbA-L deficiency led to loss of maximum respiratory capacity and insulin resistance in hepatocytes remains unclear. We found an induction of ROS production in DsbA-L-suppressed hepatocytes (Figs. 6F and 7C, D), which may impair mitochondrial respiratory complexes activity and trigger a vicious cycle that generates more ROS, resulting in inadequate mitochondrial adaption under HFD condition. Elevated ROS may activate JNK as reported (33), which adversely act on insulin signaling.
In the current study, we found that liver-specific knockout of DsbA-L impaired mitochondrial function, leading to increased ROS production. Elevated ROS levels have been shown to activate JNK, which is known to inhibit insulin signaling and mediate TNF-α- and palmitate-induced insulin resistance (34, 35). Consistent with these results, we found that DsbA-L deficiency increased ROS levels and JNK phosphorylation in mouse hepatocytes. In addition, reducing mitochondrial ROS levels in the DsbA-L-suppressed hepatocytes decreased JNK phosphorylation and increased insulin-stimulated PKB phosphorylation. These findings suggest a potential mechanism by which DsbA-L deficiency leads to JNK activation and impaired insulin signaling. However, we cannot exclude the possibility that additional mechanisms, such as accumulated fatty acid metabolites due to impaired mitochondrial function (36) and/or increased mitochondrial stress, may account for the increased insulin resistance in the liver of the DsbA-LLKO mice. Future studies will be needed to explicitly examine these possibilities.
In this study, we demonstrated that DsbA-L is localized in mouse liver mitochondria and plays an important role in regulating mitochondrial function. An important question waiting to be answered is how DsbA-L mitochondrial localization is regulated. It has been reported that the first 6 residues at the N terminus of DsbA-L may function as the mitochondrial targeting signal (11, 37). However, deleting these residues did not affect DsbA-L mitochondrial localization in adipocytes (16) and in hepatocytes (data not shown), suggesting that localization of DsbA-L to mitochondria may be mediated by a nonconventional mechanism. Future studies will be needed to characterize the mechanisms regulating DsbA-L localization in mitochondria, which could help us better understand the mechanism by which DsbA-L regulates mitochondrial functions.
In summary, we identified DsbA-L as a key regulator of mitochondrial integrity and function. In addition, we found that down-regulation of hepatic DsbA-L is associated with obesity in both mice and humans. Our findings indicate that DsbA-L as a target molecule in response to diet-induced mitochondrial dysfunction, which provides a newly described mechanism underlying obesity-induced hepatosteatosis and insulin resistance.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors appreciate helpful discussion with B. Fromenty (Université de Rennes, Rennes, France) and acknowledge the help of H. Zhang and P. Zhou (Second Xiangya Hospital) for technical support. This work was supported, in part, by the U.S. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (Grants DK76902 to F.L. and DK102965 to L.Q.D.), the National Basic Research Program of China (Grant 2014CB910501 to F.L.), and the National Nature Science Foundation of China (Grant 81500621 to H.Z.C.). H.Z.C. was supported by a predoctoral fellowship from the American Heart Association (11PRE7970021) and a fellowship from the Translational Science Training Across Disciplines program at the University of Texas Health Science Center at San Antonio, with funding provided by the University of Texas System Graduate Programs Initiative. Y.D.B. was supported by the Owens Medical Research Foundation.
Glossary
- Cpt1
carnitine palmitoyl transferase 1
- DsbA-L
disulfide-bond A oxidoreductase-like protein
- DsbA-LLKO
liver-specific DsbA-L knockout
- FCCP
carbonylcyanide-p-trifluoromethoxyphenylhydrazone
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HFD
high-fat diet
- HGP
hepatic glucose production
- NAC
N-acetylcysteine
- NAFLD
nonalcoholic fatty liver disease
- qPCR
quantitative PCR
- RNAi
RNA interference
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- TG
triglyceride
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
H. Chen, J. Bai, F. Dong, H. Fang, Y. Zhang, B. Meng, W., Liu, Y. Luo, and J. Wu performed experimental work and data analysis; H. Chen, J. Bai, Y. Bai, M. Liu, M. A. Abdul-Ghani, R. Li, R. Zeng, Z. Zhou, L. Q. Dong, and F. Liu were responsible for investigation, experimental design, and methods; H. Chen wrote the first draft of the article; H. Chen, J. Bai, L. Q. Dong, and F. Liu reviewed and edited the article; L. Q. Dong and F. Liu supervised the project; and F. Liu designed, rewrote, and reviewed the entire project and the article.
REFERENCES
- 1.Milić S., Stimac D. (2012) Nonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatment. Dig. Dis. , 158–162 [DOI] [PubMed] [Google Scholar]
- 2.Hu F., Liu F. (2011) Mitochondrial stress: a bridge between mitochondrial dysfunction and metabolic diseases? Cell. Signal. , 1528–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nassir F., Ibdah J. A. (2014) Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. , 8713–8742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tarantino G., Savastano S., Colao A. (2010) Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World J. Gastroenterol. , 4773–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vernochet C., Kahn C. R. (2012) Mitochondria, obesity and aging. Aging (Albany NY) , 859–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher-Wellman K. H., Neufer P. D. (2012) Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. , 142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuzawa-Nagata N., Takamura T., Ando H., Nakamura S., Kurita S., Misu H., Ota T., Yokoyama M., Honda M., Miyamoto K., Kaneko S. (2008) Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism , 1071–1077 [DOI] [PubMed] [Google Scholar]
- 8.Vial G., Dubouchaud H., Couturier K., Cottet-Rousselle C., Taleux N., Athias A., Galinier A., Casteilla L., Leverve X. M. (2011) Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J. Hepatol. , 348–356 [DOI] [PubMed] [Google Scholar]
- 9.García-Ruiz I., Solís-Muñoz P., Fernández-Moreira D., Grau M., Colina F., Muñoz-Yagüe T., Solís-Herruzo J. A. (2014) High-fat diet decreases activity of the oxidative phosphorylation complexes and causes nonalcoholic steatohepatitis in mice. Dis. Model. Mech. , 1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pérez-Carreras M., Del Hoyo P., Martín M. A., Rubio J. C., Martín A., Castellano G., Colina F., Arenas J., Solis-Herruzo J. A. (2003) Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology , 999–1007 [DOI] [PubMed] [Google Scholar]
- 11.Harris J. M., Meyer D. J., Coles B., Ketterer B. (1991) A novel glutathione transferase (13-13) isolated from the matrix of rat liver mitochondria having structural similarity to class theta enzymes. Biochem. J. , 137–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ladner J. E., Parsons J. F., Rife C. L., Gilliland G. L., Armstrong R. N. (2004) Parallel evolutionary pathways for glutathione transferases: structure and mechanism of the mitochondrial class kappa enzyme rGSTK1-1. Biochemistry , 352–361 [DOI] [PubMed] [Google Scholar]
- 13.Pemble S. E., Wardle A. F., Taylor J. B. (1996) Glutathione S-transferase class kappa: characterization by the cloning of rat mitochondrial GST and identification of a human homologue. Biochem. J. , 749–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu M., Zhou L., Xu A., Lam K. S., Wetzel M. D., Xiang R., Zhang J., Xin X., Dong L. Q., Liu F. (2008) A disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc. Natl. Acad. Sci. USA , 18302–18307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou L., Liu M., Zhang J., Chen H., Dong L. Q., Liu F. (2010) DsbA-L alleviates endoplasmic reticulum stress-induced adiponectin downregulation. Diabetes , 2809–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M., Chen H., Wei L., Hu D., Dong K., Jia W., Dong L. Q., Liu F. (2015) Endoplasmic reticulum (ER) localization is critical for DsbA-L protein to suppress ER stress and adiponectin down-regulation in adipocytes. J. Biol. Chem. , 10143–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu M., Xiang R., Wilk S. A., Zhang N., Sloane L. B., Azarnoush K., Zhou L., Chen H., Xiang G., Walter C. A., Austad S. N., Musi N., DeFronzo R. A., Asmis R., Scherer P. E., Dong L. Q., Liu F. (2012) Fat-specific DsbA-L overexpression promotes adiponectin multimerization and protects mice from diet-induced obesity and insulin resistance. Diabetes , 2776–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rother K. I., Imai Y., Caruso M., Beguinot F., Formisano P., Accili D. (1998) Evidence that IRS-2 phosphorylation is required for insulin action in hepatocytes. J. Biol. Chem. , 17491–17497 [DOI] [PubMed] [Google Scholar]
- 19.Mao X., Kikani C. K., Riojas R. A., Langlais P., Wang L., Ramos F. J., Fang Q., Christ-Roberts C. Y., Hong J. Y., Kim R. Y., Liu F., Dong L. Q. (2006) APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. , 516–523 [DOI] [PubMed] [Google Scholar]
- 20.Glick D., Zhang W., Beaton M., Marsboom G., Gruber M., Simon M. C., Hart J., Dorn G. W. II, Brady M. J., Macleod K. F. (2012) BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol. Cell. Biol. , 2570–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryu J., Galan A. K., Xin X., Dong F., Abdul-Ghani M. A., Zhou L., Wang C., Li C., Holmes B. M., Sloane L. B., Austad S. N., Guo S., Musi N., DeFronzo R. A., Deng C., White M. F., Liu F., Dong L. Q. (2014) APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Reports , 1227–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newberry E. P., Xie Y., Kennedy S., Han X., Buhman K. K., Luo J., Gross R. W., Davidson N. O. (2003) Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid–binding protein gene. J. Biol. Chem. , 51664–51672 [DOI] [PubMed] [Google Scholar]
- 23.Abdul-Ghani M. A., Jani R., Chavez A., Molina-Carrion M., Tripathy D., Defronzo R. A. (2009) Mitochondrial reactive oxygen species generation in obese non-diabetic and type 2 diabetic participants. Diabetologia , 574–582 [DOI] [PubMed] [Google Scholar]
- 24.Fernández-Vizarra E., Ferrín G., Pérez-Martos A., Fernández-Silva P., Zeviani M., Enríquez J. A. (2010) Isolation of mitochondria for biogenetical studies: an update. Mitochondrion , 253–262 [DOI] [PubMed] [Google Scholar]
- 25.Abu-Elheiga L., Brinkley W. R., Zhong L., Chirala S. S., Woldegiorgis G., Wakil S. J. (2000) The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA , 1444–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benard O., Lim J., Apontes P., Jing X., Angeletti R. H., Chi Y. (2016) Impact of high-fat diet on the proteome of mouse liver. J. Nutr. Biochem. , 10–19 [DOI] [PubMed] [Google Scholar]
- 27.Thomson R. E., Bigley A. L., Foster J. R., Jowsey I. R., Elcombe C. R., Orton T. C., Hayes J. D. (2004) Tissue-specific expression and subcellular distribution of murine glutathione S-transferase class kappa. J. Histochem. Cytochem. , 653–662 [DOI] [PubMed] [Google Scholar]
- 28.Blair A. S., Hajduch E., Litherland G. J., Hundal H. S. (1999) Regulation of glucose transport and glycogen synthesis in L6 muscle cells during oxidative stress. Evidence for cross-talk between the insulin and SAPK2/p38 mitogen-activated protein kinase signaling pathways. J. Biol. Chem. , 36293–36299 [DOI] [PubMed] [Google Scholar]
- 29.Clerk A., Fuller S. J., Michael A., Sugden P. H. (1998) Stimulation of “stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J. Biol. Chem. , 7228–7234 [DOI] [PubMed] [Google Scholar]
- 30.Wang X., Martindale J. L., Liu Y., Holbrook N. J. (1998) The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J. , 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery M. K., Turner N. (2015) Mitochondrial dysfunction and insulin resistance: an update. Endocr. Connect. , R1–R15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shulman G. I. (2014) Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med. , 1131–1141 [DOI] [PubMed] [Google Scholar]
- 33.Seki E., Brenner D. A., Karin M. (2012) A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology , 307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishikawa T., Kukidome D., Sonoda K., Fujisawa K., Matsuhisa T., Motoshima H., Matsumura T., Araki E. (2007) Impact of mitochondrial ROS production in the pathogenesis of insulin resistance. Diabetes Res. Clin. Pract. (Suppl 1), S161–S164 [DOI] [PubMed] [Google Scholar]
- 35.Nakamura S., Takamura T., Matsuzawa-Nagata N., Takayama H., Misu H., Noda H., Nabemoto S., Kurita S., Ota T., Ando H., Miyamoto K., Kaneko S. (2009) Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. , 14809–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samuel V. T., Liu Z. X., Qu X., Elder B. D., Bilz S., Befroy D., Romanelli A. J., Shulman G. I. (2004) Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. , 32345–32353 [DOI] [PubMed] [Google Scholar]
- 37.Raza H. (2011) Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J. , 4243–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.