

Abstract
Major depressive disorder (MDD) and substance use disorders (SUDs) are prevalent, disabling, and challenging illnesses for which new treatment options are needed, particularly in comorbid cases. Neuroimaging studies of the functional architecture of the brain suggest common neural substrates underlying MDD and SUDs. Intrinsic brain activity is organized into a set of functional networks, of which two are particularly relevant to psychiatry. The salience network (SN) is crucial for cognitive control and response inhibition, and deficits in SN function are implicated across a wide variety of psychiatric disorders, including MDD and SUDs. The ventromedial network (VMN) corresponds to the classic reward circuit, and pathological VMN activity for drug cues/negative stimuli is seen in SUDs/MDD. Noninvasive brain stimulation (NIBS) techniques, including rTMS and tDCS, have been used to enhance cortico–striatal–thalamic activity through the core SN nodes in the dorsal anterior cingulate cortex, dorsolateral prefrontal cortex, and anterior insula. Improvements in both MDD and SUD symptoms ensue, including in comorbid cases, via enhanced cognitive control. Inhibition of the VMN also appears promising in preclinical studies for quenching the pathological incentive salience underlying SUDs and MDD. Evolving techniques may further enhance the efficacy of NIBS for MDD and SUD cases that are unresponsive to conventional treatments.
Keywords: addiction, depression, fMRI, rTMS, tDCS
Introduction
Major depressive disorder (MDD) and substance use disorders (SUDs) are challenging illnesses that produce significant burdens on patients and the healthcare system. Mental illness and SUDs are the leading worldwide cause of years lived with a disability (YLD),1 and MDD is the second leading psychiatric cause of YLD.2 The societal burden of MDD and SUDs have also dramatically increased over the last 20 years,2 emphasizing the importance of access to care and effective treatment options.
For MDD, the mainstays of conventional treatment are pharmacotherapy and psychotherapy. Studies of real‐world effectiveness suggest that about one‐third of patients will remit on an initial trial of antidepressant medication, while another one‐third will remit after 1–3 additional medication trials.3, 4 The remaining one‐third of patients are labeled as having treatment‐resistant depression (TRD), with a low likelihood of remission (10–15%) on further trials.4 TRD affects approximately 2% of the general population.5 To address the challenge of treating TRD, combination therapies (antidepressant + antipsychotic, or antidepressant + anticonvulsant)6, 7, 8, 9 and electroconvulsive therapy (ECT)10 have yielded promising clinical results. Even for TRD, however, these intensive interventions achieve varied remission rates between approximately 30%11 and 50%,6, 7, 8 and the relapse rate following ECT is 50% by 2 years.12
As with MDD, in SUD patients the mainstays of conventional treatment are pharmacotherapy and psychotherapy. Lifetime rates of drug use are 92% for alcohol, 74% for tobacco, 42% for cannabis, and 16% for cocaine, in the United States.13 The lifetime prevalence of alcohol dependence is 13%, and up to 3% for illicit substances.14, 15, 16 The most common psychotherapeutic approach to substance dependence is cognitive behavioral therapy.17, 18 A meta‐analysis of 34 randomized controlled trials on cognitive behavioral therapy for SUDs demonstrated that the average effect size was moderate (d = 0.45), with the highest effects for cannabis, cocaine, and opioid treatment.19 A form of behavioral therapy known as contingency management appears to be a particularly potent tool for multiple classes of SUD patients.20, 21 Contingency management, however, requires that an individual is able to regulate/control their drug intake in order to get an alternative non‐drug reinforcer. This may be difficult for many patients, as disruptions in response inhibition and in the neural circuitry required for response inhibition are hallmarks of addiction.22
From a pharmacotherapy perspective, the therapeutic approach varies with the substance being abused. Pharmacotherapy may complement behavioral approaches by replacing the abused substance with a less harmful substitute23, 24 and thereby reduce the social and personal harm associated with the drug. However, dependences on several classes of drugs, including cocaine, have no approved pharmacotherapy, and relapse rates in the first 6 months after an outpatient treatment program are often higher than 75%.25 Some evidence also suggests that pharmacotherapy that interacts with neurotransmitter systems related to reward learning may enhance impulse control itself.26
High rates of treatment resistance and relapse represent a common challenge for MDD or SUDs. However, effective MDD and SUD treatment response is frequently hampered even further by the comorbidity of the two conditions. Among individuals with MDD, approximately 25–40% have a comorbid SUD. Conversely, MDD is among the common psychiatric disorders that have high comorbidity with all types of SUDs.27, 28 These patients report poorer response rates compared to their singly diagnosed counterparts from a 12‐step program,29 from single‐medication trials,30, 31, 32 and from cognitive behavioral therapy.33 Likewise, MDD patients with nicotine dependence also have increased difficulty with smoking cessation, with antidepressants having little influence on abstinence, and these patients are more likely to develop an episode of depression post–smoking cessation.34, 35, 36, 37 Hence, treatment strategies should ideally accommodate the frequent comorbidity of these illnesses.
Treatment strategies for MDD and SUDs are traditionally approached separately and sequentially. Such an approach implicitly assumes that if the primary diagnosis is addressed, secondary diagnoses may resolve on their own.38 However, limited success rates for conventional approaches to MDD and SUDs raise several fundamental questions. Is it helpful to identify one of the disorders—MDD or SUD—as the primary disorder? Does the underlying pathophysiology of MDD and SUDs support separate treatment strategies in comorbid cases? Finally, can the neurobiology of SUDs and MDD suggest new treatment strategies that might address the problem of comorbidity, while improving remission and relapse rates for both disorders?
To answer these questions, we can take advantage of recent advances described in the neuroimaging literature on human brain function. These include an increasingly robust model of the functional neuroanatomy and network architecture of the brain, a vast body of neuroimaging literature on the pathophysiology of SUDs and MDD in humans, and, finally, a set of automated, quantitative meta‐analytic software that renders this enormous literature more tractable. Such tools allow us to detect consistent and statistically robust patterns by combining scans in thousands of patients and healthy individuals.
Of course, advances in the functional neuroanatomy of SUDs and MDD are of limited immediate clinical interest unless they can translate into anatomically specific therapeutic interventions. While it is difficult to aim conventional treatments at specific brain circuits, noninvasive brain stimulation (NIBS) is an emerging treatment modality that exerts neuroanatomically specific effects. There are two particular NIBS technologies that are quickly translating from research into clinical practice: repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS). Repetitive TMS has held regulatory approval for MDD in many jurisdictions for several years and is being explored for SUDs in clinical trials. Transcranial DCS is a few years behind rTMS in translational progress but is likewise being explored with encouraging results in both MDD and SUDs. Unlike medications or therapy, the success of NIBS depends critically on the choice of target circuit and the type of stimulation (inhibitory or excitatory) applied to that circuit.39, 40, 41
This review is intended to serve four purposes. First, it will summarize key advances in our understanding of the functional architecture of the brain. Second, it will review how this emerging model of brain function relates to the pathophysiology behind SUDs and MDD. Third, it will review NIBS for the treatment of SUDs and MDD, particularly in cases where conventional treatments fail. Finally, it will review a number of promising areas for future research on NIBS in SUDs and MDD.
Functional architecture of the human brain
Functional networks of intrinsic brain activity
One of the key advances in the neuroimaging literature over the last 20 years is the demonstration that brain regions organize their activity into coherent functional networks.42 On functional magnetic resonance imaging (fMRI), these networks appear as correlations of the low‐frequency fluctuations in blood oxygenation level–dependent (BOLD) signal between brain regions.43 Many networks were originally identified through data‐driven methods for analyzing brain activity at rest and are termed resting‐state networks. However, these networks reliably appear in ongoing brain activity during tasks as well,44 and meta‐analyses of task‐based activation also reveal consistent functional networks similar to those identified at rest.45
The precise number of functional networks and the functional role of each network are still being studied. An emerging consensus has been identified between 7 and 20 distinct functional networks.43 One recent resting‐state fMRI analysis in 1000 healthy individuals found a stable seven‐network parcellation; these networks were further subdivided into a stable 17‐network parcellation (Fig. 1).46 Many of these cortical networks also have correlated counterparts in the striatum47 and cerebellum,48 thus hinting that they may represent integrated, whole‐brain functional circuits.
Figure 1.
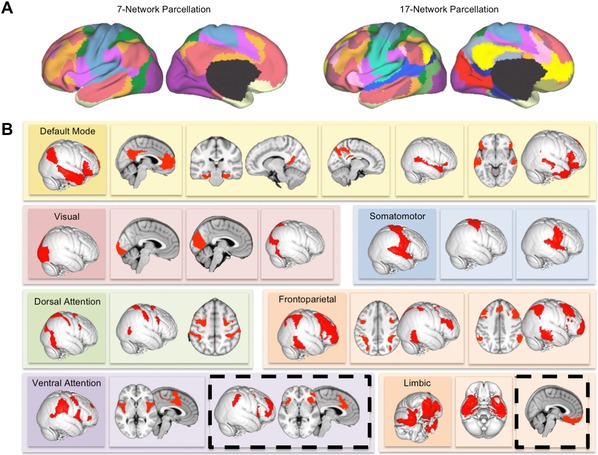
Functional networks in the intrinsic activity of the brain. (A) The intrinsic ongoing activity of the brain at rest or during task performance can be decomposed into sets or networks of brain regions that show correlated patterns of activation and deactivation over time. A set of at least seven distinct functional networks has been identified as consistently appearing in large datasets of up to 1000 individuals. However, these seven networks contain smaller sub‐networks. A 17‐network parcellation has been identified as stable across individuals. (B) The 17 resting‐state networks identified by Yeo et al.46 include low‐level visual and somatosensory cortical areas, higher‐level networks involving premotor and sensory association areas, and larger frontoparietal networks involved in attention, cognition, and executive control. However, two networks (highlighted in dashed black lines) are of particular interest in MDD and SUDs: the more anterior of the two subnetworks of the ventral attention network and the ventromedial subnetwork of the limbic network. Adapted from Yeo et al.46
The default‐mode network (DMN)49 is the best known and most studied of these functional networks. The DMN contains subcomponents that serve various introspective functions ranging from mind wandering,50 recollection and prospection,51 rumination,52 and self‐reflection.51 Other networks act in opposition to the DMN and activate during behaviorally regulated task performance and externally focused cognition. These networks include frontoparietal networks (FPNs) and related areas sometimes known as the dorsal (DAN) and ventral attention networks (VAN).46 Other networks include lower‐level sensory and motor cortices and ventromedial limbic networks that encompass the temporal pole and the orbitofrontal cortex (OFC)46 (Fig. 1).
Relevance of functional networks to MDD and SUDs
Several functional networks have been studied extensively in MDD and SUDs, particularly the DMN. However, there are two functional networks that are of particular interest in both SUDs and MDD, and it is worthwhile to briefly characterize these networks.
The first network of interest is the anterior cinguloinsular network (aCIN), or salience network (SN).46 Figure 2 visualizes the SN on the basis of a quantitative meta‐analysis of SN neuroimaging studies analyzed using Neurosynth.45 The SN activates for salient sensory events,53, 54 transitions from introspection to task performance,55 and during task initiation and switching.56 A remarkable recent study highlighted the central importance of the SN as a common neural substrate across psychiatric illness categories.57 The authors performed a meta‐analysis of structural abnormalities across six psychiatric disorder categories, including MDD and SUDs, and found that all of them showed gray matter volume reductions in the dorsal anterior cingulate cortex (dACC) and anterior insula. Further analyses demonstrated that these areas belonged to a common functional network in healthy controls, both at rest and during task performance.57 This network corresponded almost exactly to the SN.45, 46 Thus, of the 7–17 networks previously discussed, the SN merits particular attention for its role in MDD, SUDs, and other psychiatric pathophysiology.
Figure 2.
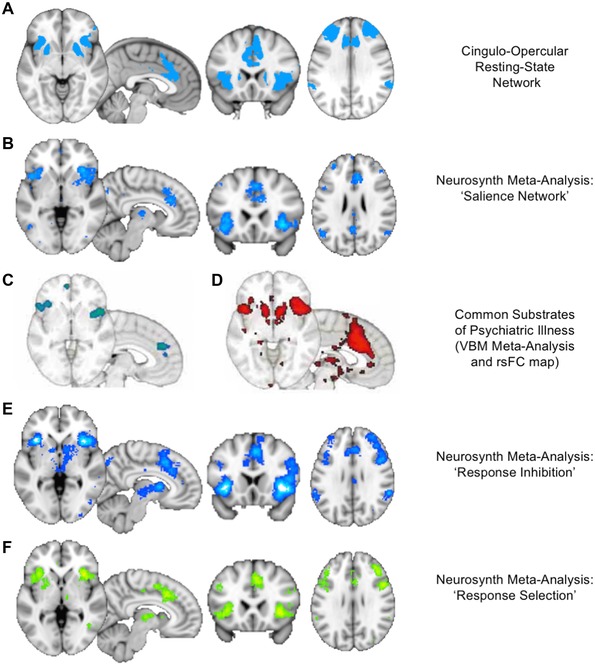
The salience network (SN). The reader is encouraged to replicate and explore the depicted networks in the Neurosynth tool.45 (A) The cingulo–opercular network from the parcellation of Figure 1 includes prominent nodes in the dorsal anterior cingulate cortex (dACC), anterior insula, dorsolateral prefrontal cortex (DLPFC), and inferior parietal lobule, as well as the dorsal anterior caudate nucleus. (B) A Neurosynth meta‐analysis45 using the term “salience network” reveals the close correspondence of this network to the cingulo–opercular network identified above. Note that the mediodorsal thalamus can also be seen in the network in this analysis. (C) The areas identified as common sites of gray matter loss across MDD, SUDs, and several other categories of psychiatric disorders in a meta‐analysis of 193 voxel‐based morphometry studies57 correspond closely to SN nodes in the dACC and anterior insula. (D) Resting‐state functional connectivity maps seeding from the nodes in C reveal a network that corresponds closely to the rest of the SN, as seen in A and B.45 (E, F) Neurosynth meta‐analyses using the terms “response inhibition” and “response selection” yield maps of activation that correspond closely to the SN, thus highlighting the role of the SN as a neural substrate for cognitive control.57
The second network of interest is the ventro‐medial network (VMN), encompassing the nucleus accumbens (NAcc), medial OFC (mOFC), and ventromedial prefrontal cortex (VMPFC) (Fig. 3, created using Neurosynth33). This circuit is best known as the reward pathway and also includes components of the mediodorsal thalamus and midbrain dopaminergic structures. The VMN activates not only for rewards, but also for stimuli of other incentive value, including losses.58 The role of the VMN in SUDs is well documented,59, 60 particularly for mediating abnormal incentive salience of drug cues, which is proposed to drive craving and relapse.61 VMN dysfunction is linked to anhedonia in MDD62 and activates paradoxically for negative stimuli, suggesting that negative cues have abnormal incentive salience in MDD, analogous to drug cues in SUDs.63
Figure 3.
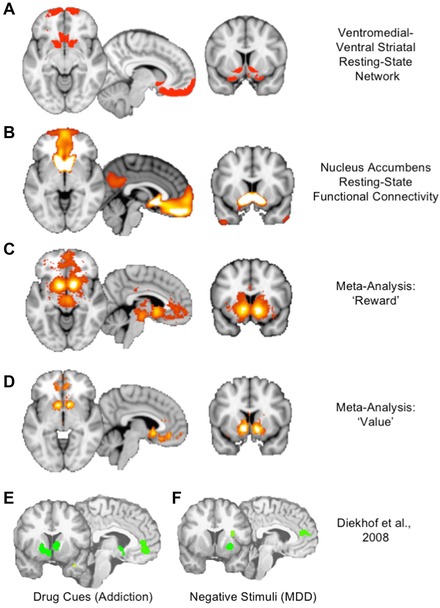
The ventromedial network (VMN). The reader is encouraged to replicate and explore the depicted networks in the Neurosynth tool.45 (A) The VMN consists of the cortical and striatal nodes of the ventral striatal–ventromedial prefrontal network from the parcellation of Figure 1. (B) A resting‐state functional connectivity map seeded from the nucleus accumbens illustrates the strong connection to the ventromedial prefrontal cortex (VMPFC) and frontal pole.45 (C) A Neurosynth meta‐analysis using the term “reward” reveals the classic reward circuit, including mesolimbic dopaminergic structures in the ventral tegmental area (VTA) and substantia nigra (SN), the ventral striatum, and a specific subregion of the VMPFC slightly posterior to the medial frontal pole.45 (D) A Neurosynth meta‐analysis using the term “value” reveals the striatal and cortical components of the VMN, suggesting a broader role beyond reward to include valuation of incentives.45 (E) A meta‐analysis of regions activated by drug cues in patients with addiction reveals a circuit corresponding closely to the VMN, illustrating the pathological distortion of reward value for drug cues in addiction.63 (F) A meta‐analysis of regions activated by negative emotional stimuli in MDD reveals a similar signature of pathological activation of reward‐related areas for negative rather than positive cues, illustrating a common pathophysiology of distorted incentive salience in the VMN across SUDs and MDD.63
The opposed functions of the SN and VMN
The SN is critical for “switching” brain activity between the introspective DMN and the externally focused FPNs.64 It is also active during performance‐related errors and task initiation.56, 65 The functions of the SN map rather well on to the Research Domain Criteria (RDoC) construct of cognitive control: specifically, the subdomain of response inhibition/response selection. A quantitative fMRI meta‐analysis using Neurosynth45 revealed a network closely matching the SN (Fig. 2). This function stands in opposition to the functions of the VMN in mediating reward, incentive salience, and value assignment in similar meta‐analyses using Neurosynth45 (Fig. 3). These two circuits have been proposed to play opposing roles in behavioral regulation: the VMN as a “drive network” mediating craving and urge, and the SN as a “gatekeeper network” mediating response selection and inhibition.66
If the roles of these networks are indeed opposed, we might predict one to be active when the other is not, and vice versa.67 In fact, this is precisely the case. For many of the functional networks previously discussed, there also exists a map of corresponding negatively correlated regions: the anti‐network.68 The anti‐network for the VMN, shown by qualitative meta‐analysis, consists of the SN and corresponding striatothalamic partners. Conversely, the anti‐network for the SN is the VMN, as evident in functional connectivity data45 (Fig. 4). Thus, the SN and VMN may play reciprocal roles as each other's anti‐networks, with opposing patterns of activity even in the resting brain. This functional architecture might explain the tendency for highly salient stimuli to reduce inhibitory control or, conversely, for cognitively demanding activities to attenuate urges, cravings, or emotions.
Figure 4.
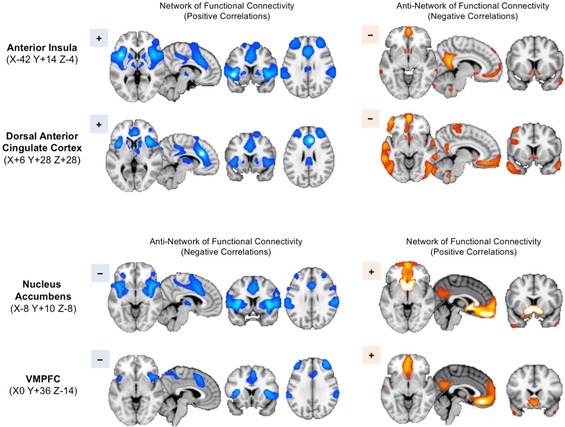
Reciprocal relationship of SN and VMN activity. The reader is encouraged to replicate and explore the depicted networks in the Neurosynth tool. Resting‐state functional connectivity maps are generated using the Neurosynth tool45 from the seed coordinates indicated at left. Seeds in the anterior insula and dACC reveal a network of positively correlated regions throughout the other nodes of the SN, as expected (upper left). Notably, the anti‐network of these SN seeds (i.e., regions showing negative rather than positive correlations) includes the key VMN nodes in the ventral striatum, VMPFC, and temporal poles (upper right), as may be seen by comparison with Figure 3. Conversely, seeds in the nucleus accumbens and VMPFC reveal a network of positively correlated regions corresponding to the VMN (lower right). The anti‐network of these seeds corresponds well to the SN (lower left). In order to highlight the correspondence of the networks and anti‐networks, blue colors are used for the SN networks and the VMN anti‐networks, while orange colors are used for the VMN networks and SN anti‐networks.
Coordination among the functional networks
Modeling the brain as a network can allow us to map information flow among its regions. Approaches to the study of the brain using graph theory, a set of mathematical techniques allowing formal analysis of network structures, consider individual brain regions as “nodes” and functional connections between correlated regions/nodes as “edges.” Such analyses identify clique‐like “communities” of nodes and “hubs” that bridge together different communities within a larger network.69 This means that graph theoretical approaches can characterize the structural and functional interactions between networks and nodes.70
Reassuringly, graph theoretical analyses of the brain extract 10–12 communities that correspond to the same data‐driven functional networks70 and also show how these communities connect to one another (Fig. 5A). Generally, sensory and motor networks lie on the functional outskirts of the brain, as isolated functional units with few connections outside of their network. This makes sense given their roles as pathways for processing elementary sensory input and motor output.
Figure 5.
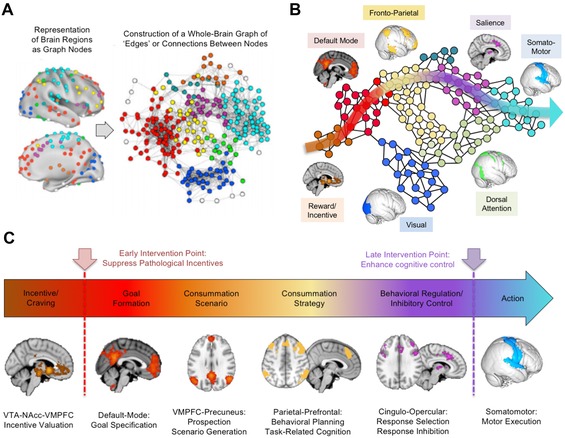
Network architecture of the brain from incentive formation to behavioral execution. (A) The network architecture of the brain can be derived by placing nodes to cover the entire cerebrum and then extracting the intrinsic activity of these nodes over time. By connecting the well‐correlated nodes with “edges,” a graph of the network architecture of the brain can be constructed. Within this architecture, the functional networks illustrated in Figure 1 appear as clusters of cliques, and the larger relationship between the networks can be seen.70 (B) A schematic derived from A illustrates a trajectory of information flow for behavioral control. This trajectory begins in the reward circuitry of the VMN, passing through the default‐mode and frontoparietal networks and then the SN, before exiting the cerebrum via the nodes of the sensorimotor cortex to direct bodily movements. (C) This pathway of connections passes from one brain region to the next and allows the mapping of basic drives into specific incentives or cravings via the VMPFC and then the elaboration of these incentives into specific goals and scenarios via the default‐mode network, followed by the refinement of these scenarios into specific strategies or plans for consummation of the goal. However, before these strategies can be executed as motor actions in the sensorimotor cortex, they must pass through the nodes of the SN, which thus sits in a gatekeeper position for response selection and behavioral inhibition. This functional architecture suggests that two points of intervention may be possible in SUDs and MDD: suppressing the pathological incentives early in this pathway at the VMPFC and/or strengthening the gatekeeper functions of response selection late in the pathway, via excitatory stimulation of the SN.
What about other prominent networks, such as the DMN itself? The intuitive expectation is that the DMN should be situated centrally, in a crossroads position linking the other networks together. Instead, the DMN appears as a peripheral, close‐knit module much like the visual cortex, but taking feedback from the VMN rather than the retina. The DMN's position is not that of an integrative controller, but rather another specialized “think tank,” linking the incentive functions of the VMN to the high‐level executive and cognitive functions of the FPNs, but not in close communication with most other networks (Fig. 5B).
Does another functional network hold the crossroads position? One recent study using graph theoretical methods identified nodes standing in critical hub positions between many networks. These nodes lay not in the DMN but in the SN itself.71 Close inspection of the whole‐brain graph showed that the SN is in a gatekeeper position between the deliberative functions of the DMN and FPN and the behavioral output of the somatomotor cortex (Fig. 5B).
This overall functional architecture provides a framework for understanding the roles of the VMN, DMN, FPN, SN, and motor cortex in behavioral control (Fig. 5C). The trajectory of information flow originates within the basic drives of midbrain dopaminergic regions, which are elaborated into specific incentives or urges via the VMN and elaborated further into goals and mapped onto relevant scenarios via the DMN. The FPNs then develop these goals and scenarios into consummatory strategies, which are broken down into cognitions and behaviors that can be either inhibited or selected via the gatekeeper‐like SN. If selected, responses finally map to the action‐control systems of the somatomotor cortex to initiate overt behavior.
This urges‐to‐actions trajectory through the brain offers early and late points for pathology to arise in MDD or SUDs. Early pathology at the VMN–DMN interface would drive priorities away from core survival needs, assigning inappropriately high incentive salience to drug cues (in the setting of SUDs) or negative emotional cues (in the setting of MDD). Late pathology near the SN would interfere with response selection and inhibitory control, producing a pattern of chronic emotional liability, intrusive thinking, and impulsive behavior. These more elementary dysfunctions may cut across the diagnostic entities of MDD and SUDs, and may also shed light on MDD and SUD comorbidity as arriving from more fundamental disruptions in network architecture. Importantly, the neural overlap between MDD and SUDs may be an opportunity, rather than an obstacle, for interventions that can address both illnesses concurrently by targeting the underlying circuitry of the SN and VMN using NIBS.
Functional architecture in psychiatric disorders
Neural circuit disruptions in SUDs
It is challenging to concisely summarize the neural circuits that are involved in all SUDs, since each substance has a unique pharmacologic profile and addiction involves multiple temporal phases, including regular controlled use, regular uncontrolled/habitual use, abstinence, and relapse. However, there appear to be common neurobiological pathways operating across substances of abuse, involving dysfunction of both the VMN and its anti‐network, the SN.
Although patients with SUDs can become dependent on a variety of drugs that have diverse initial pharmacologic actions, nearly all drugs of abuse have a common final pathway in which they modulate reward circuitry in the ventral striatum–ventral tegmental area (VTA) pathway.72 For example, cocaine modulates this pathway directly by increasing dopamine transmission, while nicotine also has direct actions on the VTA via nicotinic receptors. Opiates and alcohol, however, modulate the ventral striatum and VTA indirectly via GABAergic disinhibition. While the majority of basic science research in addiction has focused on this subcortical reward pathway, recent work has demonstrated that many cortical and subcortical regions interact with this mesolimbic dopamine pathway, especially the VMN and its anti‐network, the SN, as described below.
First, the SN is hypoactive in SUDs. Specifically, in SUD patients, the dACC and insula display hypoactivity to a variety of executive tasks, including the Stroop task,73, 74 response inhibition75, 76 (although this finding is mixed77), and emotion regulation.78 The dACC also displays abnormal connectivity on resting‐state and graph theoretical measures in SUDs.79 The abnormal network activation in the dACC may relate to the aberrant salience of substance‐specific cues.80 SUD patients display lowered structural and functional integrity in both the dACC and insula.81, 82 Although the role of the insula is still under debate, insular lesions have been reported to be associated with improved abstinence, possibly because disease‐relevant cues are less salient.83, 84 There is also an increase in dACC activation and an improvement in self‐control behavior following abstinence, suggesting a causal role in SUD psychopathology.85
The anti‐network of the SN, the VMN, is involved in limbic arousal in SUDs. Functional MRI studies have demonstrated that cue reactivity and craving are associated with elevated activity in the VMN, particularly in the ventral striatum, vmPFC, and OFC.86, 87, 88 Hyperactivation from the OFC is also seen during reward evaluation,89, 90, 91 risky decision making, personal relevance,92, 93, 94, 95 and goal‐driven processes associated with the ventral striatum. This abnormal VMN activity normalizes in smokers and cocaine users following treatment with varenicline/buproprion96 – 98 and methylphenidate,99 respectively. As in MDD, SUD patients also show VMN hypoactivation for natural rewards, compared to healthy controls.100
Connectivity between the VMN and SN is also impaired in SUDs, with SUD patients showing reduced corticostriatal VMN and SN resting‐state functional connectivity.101, 102 Furthermore, altered ACC corticocortical and corticostriatal connectivity between these circuits are related to SUD severity,101 high‐risk behavior, and genetic polymorphisms associated with SUDs.103
Neural circuit disruptions in MDD
As with SUDs, MDD has been linked to a pattern of SN hypoactivity and VMN hyperactivity104 in a wide variety of network‐based descriptions of the illness over the last decade.105, 106 Ventral prefrontal activation in MDD predicted dorsal inactivation, in keeping with the framework of opposed SN and VMN activity.107 Inhibition of SN control mechanisms by pathological salience signals from the VMN may create a self‐perpetuating imbalance, resulting in the persistent low mood of MDD rather than the transient low mood of normal sadness.
MDD patients display a similar disruption of incentive salience for primary rewards108, 109 and instead become attuned to disease‐specific stimuli. For example, the rostral ACC and ventral striatum are hypoactive during reward feedback110, 111, 112 and hyperactive during self‐referential negative processing.113, 114 MDD patients also show an absence of ventral striatal and VMPFC/OFC inactivation for pleasant sights and tastes but heightened activation in the caudate in response to viewing aversive images.115 Altered responses from the subgenual cingulate and OFC are also found in MDD during rumination and negative self‐focus.116
As in SUDs, MDD patients show reduced activation in SN regions at rest. Reflecting this deficiency, MDD patients exhibit task‐related hyperactivation from the dACC on many cognitive control tasks, including the Stroop117, 118 and n‐back tasks.119 The DMPFC is also inappropriately activated during positive affect processing in MDD, which normalizes after successful treatment.120 Additionally, the anterior insula and DMPFC are inappropriately active during negative affect.121 Increased activation after treatment also results in improved resting‐state activity in the cingulate in MDD.122 On structural imaging, TRD patients tend to show volumetric alterations in regions of the SN and cognitive deficits relative to their treated counterparts.123
Neural similarities in MDD/SUDs
As noted earlier, voxel‐based morphometry studies of MDD and SUDs reveal a common neural substrate of decreased gray matter affecting the dACC and anterior insula—both key nodes of the SN.57 One interpretation of this neural overlap is that both SUDs and MDD feature deficits in self‐regulation of impulses, cognition, emotion, and behavior—or in RDoC terms, response inhibition/selection. SUDs and MDD also share a common feature of inappropriate VMN activation as shown by fMRI. In SUDs, the VMN's reward circuit activates in response to drug cues, despite their lack of primary survival value. In MDD, the VMN activates in response to negative rather than positive affective stimuli. One interpretation is that, for MDD, negative emotional stimuli acquire aberrant incentive salience, as with drug cues in SUDs. If so, both SUD and MDD patients also share an underlying pathology of distorted incentive salience.63
The shared neural substrates of SUDs and MDD may help to account for the high prevalence of MDD/SUD comorbidity. From a therapeutic standpoint, the shared neural circuitry between SUDs and MDD may also present an opportunity to address both disorders concurrently, using NIBS interventions. On this view, NIBS treatments should not be regarded as antidepressant or anti‐substance use. Rather, they should be considered to offer two more nuanced approaches to treatment, cutting across diagnostic categories: enhancing cognitive control by targeting the nodes of the SN or relieving the distorted incentive salience of substance/negative cues by inhibiting the VMN.
One prediction of this framework is that any NIBS interventions that target SN nodes (i.e., DLPFC, DMPFC, or anterior insula) with excitatory stimulation should exert a therapeutic effect across both MDD and SUDs, by enhancing cortico–striatal–thalamic connectivity through the SN on fMRI and by enhancing capacity for response selection/inhibition. Another prediction is that any NIBS interventions that target the VMN (by stimulating the frontal pole or VMPFC) should also exert a therapeutic effect across both MDD and SUDs by reduced cortico–striatal–thalamic connectivity through the VMN on fMRI, and by reducing incentive salience of drug cues/negative affective stimuli.
NIBS as a treatment for MDD/SUDs
Repetitive TMS overview
Repetitive TMS applies powerful, focused, magnetic field pulses to target regions of the brain via a handheld induction coil placed against the scalp. By applying trains of pulses over several minutes, rTMS increases or decreases target brain region activity. While experimental applications of rTMS usually involve 1–3 sessions on a single day, therapeutic applications of rTMS typically require 20–30 daily sessions.124, 125
Repetitive TMS appears to act through the mechanisms of synaptic plasticity: long‐term potentiation and depression, although there is conflict among the effects of genetic polymorphisms, such as brain‐derived neurotrophic factor, on these functions.126, 127 Generally, the direction of the effect depends on the intensity, duration, and stimulation pattern.128, 129 High‐frequency stimulation (5–20 Hz) is classically considered excitatory, while low‐frequency stimulation (1 Hz) is inhibitory.130, 131 Some recent rTMS studies use stimulation patterns that mimic theta electroencephalography (EEG) rhythms, thought to be especially efficient for inducing plasticity.132, 133 For example, two advantages of theta‐burst stimulation (TBS)—classified as intermittent TBS (iTBS), which is generally excitatory, or continuous TBS (cTBS), which is generally inhibitory—are that the time required for a session of stimulation is rather brief and that it is as potent as conventional stimulation, both in model systems such as the motor cortex133 and in clinical applications.134, 135
One current drawback to all forms of rTMS concerns the variability of effect: a certain percentage of individuals show neutral or inhibitory effects from excitatory stimulation and, conversely, some individuals show excitatory effects from inhibitory stimulation.128, 136 This variability is observed both in model systems such as the motor cortex during single sessions and in therapeutic applications of rTMS over multiple sessions.137, 138
Therapeutic effects of rTMS in neuropsychiatric disease may ensue through changes in cortico–striatal–thalamic circuits stemming from SN and VMN nodes. Positron emission tomography (PET) studies of rTMS reveal changes in striatal dopamine receptor occupancy following rTMS, with the changes localized to the specific region of the striatum that serves the cortical target (DMPFC and DLPFC) of stimulation.139, 140 In keeping with this observation, dopamine agonists and antagonists can potentiate or block the effects of rTMS.141 Baseline cortico–striatal–thalamic functional connectivity on resting‐state fMRI predicts treatment outcome across multiple conditions, including MDD,142 eating disorders,138 OCD,143 and movement disorders.144 Pre–post treatment changes in this measure also track outcomes across all of these disorders. Thus, rTMS is posited to exert therapeutic effects by enhancing or suppressing cortico–striatal–thalamic circuit integrity from the SN and/or VMN.
Transcranial DCS overview
Transcranial DCS applies lower‐energy stimulation to the brain, using a montage of over two scalp electrodes, each typically 3–7 cm across. Constant‐current stimulation is applied through the electrodes at intensities of just 1–2 mA. A tDCS session typically lasts 5–30 min, and a therapeutic course typically involves daily stimulation over 10–30 days.145
Transcranial DCS seeks to modulate the synaptic activity of target brain regions, rather than directly eliciting action potentials, as does rTMS.146 Anodal stimulation is classically considered to increase cortical excitability in the underlying brain region, while cathodal stimulation is considered to be inhibitory. However, as with rTMS, some individuals show the opposite pattern of effect, and variability remains a problematic issue.147 Newer patterns of stimulation include transcranial random noise stimulation (tRNS), as well as transcranial alternating current stimulation (tACS). The latter approach is considered promising, since stimulation can be tuned to match specific EEG frequency bands for more potent or more selective effects.148 In one notable recent example, investigators were able to induce lucid dreaming in healthy subjects during rapid eye movement (REM) sleep by applying tACS at 20–40 Hz, but not at 2–12 Hz.149
The mechanisms of tDCS/tACS are still under investigation.150 However, the technique does appear to modulate the activity of resting‐state networks on fMRI151 and may also modulate cortico–striatal functional connectivity.152 In these respects, its mechanisms may resemble those of rTMS to some degree, and, for therapeutic purposes, both techniques have been used to target similar brain regions in similar illnesses. However, at the physiological level, there are likely to be important mechanistic differences between the two approaches, the details of which require further study.146
Transcranial DCS is likely capable of stimulating many of the same regions as rTMS, although, once again, focal stimulation becomes more difficult for deeper structures. Electrical field simulations suggest that the direct effects of tDCS on neural activity are most prominent near superficial brain regions directly under the electrode, although deeper effects may be possible.153 Preclinical studies indicate that tDCS can modulate the excitability of the motor cortex and DLPFC,154 as well as deeper structures of the medial wall, such as the motor cortex of the lower limb,155 supplementary motor area,156 and DMPFC.157 There are also suggestions that tDCS may be capable of modulating the reward value of stimuli during task performance, suggesting engagement of deeper VMN nodes, such as the VMPFC and even the midbrain.158 Thus, many of the areas of interest for NIBS in MDD and SUDs are likely to be accessible to both tDCS and rTMS.
NIBS as a treatment for MDD
MDD is the original and best‐studied therapeutic indication for rTMS. To date, dozens of large‐scale randomized controlled trials have confirmed efficacy for rTMS in MDD, as summarized in recent meta‐analyses.159, 160 Response and remission rates in the most recent large studies are approximately 50–55% and 30–35%, respectively;161, 162 real‐world effectiveness studies report similar outcomes.159, 160 Course lengths of 26–28 sessions are required for maximum effect;124 the poorer outcomes of early trials may be attributable in part to inadequate course lengths of 5–10 sessions.160
The most widely used rTMS protocol in MDD is high‐frequency left DLPFC stimulation. Some studies alternatively used low‐frequency right DLPFC rTMS,161 or both.162 Meta‐analyses have not found marked outcome differences between these approaches,160 and no one stimulation pattern appears markedly superior.162 Few NIBS studies have sought targeted non‐DLPFC nodes of the SN or VMN in MDD.163 One exception is the DMPFC,164 which was recently targeted with rTMS in a sham‐controlled study and in several open‐label case series,142, 165, 166 with promising results. However, no rTMS trials have targeted the anterior insula or the VMPFC in MDD; these remain theoretically promising targets for intervention.
Mechanistic studies show that DLPFC or DMPFC stimulation may enhance cognitive control in healthy subjects and MDD patients. DLPFC or DMPFC excitatory rTMS in healthy subjects can enhance impulse control via delay‐discounting tasks;167 likewise, rTMS of the presupplementary motor area, just posterior to the DMPFC, improves stop‐signal task performance.168 Neurally, rTMS may enhance impulse control via strengthened DLPFC and DMPFC frontal–striatal–cortical circuit integrity on fMRI,138 and, on PET, rTMS changes dopamine receptor occupancy in these same striatal regions.169 In MDD, cortico–striatal–thalamic connectivity on fMRI predicts and correlates with treatment outcomes.142 These findings suggest that rTMS could be relieving MDD by enhancing cortico–striatal–thalamic circuit integrity in the nodes of the SN, thereby enhancing cognitive control over negative cognition and affect.
The literature on tDCS in MDD is more nascent. To date, 10 randomized controlled trials have been completed, with a recent meta‐analysis supporting tDCS efficacy in MDD.145 Outcomes appeared comparable to antidepressant medication in one comparative trial.170 Again, 20–30 sessions may be required for maximal efficacy;171 the shorter, 10‐session courses and relatively small sample sizes used in many tDCS trials may imply an underestimate of the true efficacy of the technique.145
To date, all tDCS trials in MDD have targeted the left DLPFC, using anodal, excitatory stimulation over the F3 EEG site; the inhibitory cathode has been variously applied to either the right DLPFC via the F4 EEG site or to the neighboring right supraorbital area near Fp2/F8. Mechanistically, anodal tDCS over the left DLPFC enhances cognitive control in healthy participants on a working memory task with negative emotional distracters.172 In a more direct demonstration, anodal DLPFC tDCS abolished the effect of negative emotional distractors on a working memory task in MDD patients,173 suggesting that the mechanisms of effect may likewise involve enhanced cognitive control. The functional anatomy of the SN suggests that similar effects might also be achieved via excitatory tDCS of the right DLPFC, the DMPFC, and the anterior insula. Frontopolar tDCS is also of interest for the anhedonic symptoms of MDD, since anodal stimulation of this site engages the VMPFC and VTA on fMRI, and enhances perceived attractiveness of faces.158 These regions are therefore important candidate targets for future tDCS trials in MDD.
NIBS as a treatment for SUDs and comorbid SUD/MDD
NIBS is attracting increasing interest as a novel therapeutic intervention in SUDs. To date, approximately 25 original research reports have been published on the efficacy of rTMS as a tool to decrease craving, along with at least six reviews on rTMS in addiction.174 The types of SUDs, the targets of stimulation, and the patterns of stimulation have varied across these studies. However, as in MDD, most so far have sought to enhance cognitive control mechanisms by targeting the nodes of the SN.174 The alternative approach, of attenuating craving and incentive salience via the VMN, is also beginning to be explored.175
For alcohol cravings, most studies have targeted the left or right DLPFC. In sham‐controlled studies of rTMS, low‐frequency right DLPFC stimulation reduced alcohol craving in some reports176 but not others,177, 178 and high‐frequency left DLPFC stimulation reduced attention to alcohol cues but did not reduce cravings.179 In open‐label case reports, bilateral high‐frequency rTMS reduced alcohol cravings in three patients,180 and low‐frequency dACC rTMS likewise reduced refractory cravings in one patient; however, this patient eventually relapsed following rTMS.181 For DLPFC tDCS, left anodal/right cathodal stimulation reduced alcohol cravings in initial studies;182, 183 however, relapse rates improved with this montage.184 Reversing the polarity to left cathodal/right anodal DLPFC tDCS yielded superior abstinence rates at 6 months, despite no differences in craving.185 Such findings are consistent with a model in which SN stimulation exerts therapeutic effects by enhancing cognitive control rather than reducing cravings.
For stimulants (cocaine and methamphetamine), one open‐label study reported reduced spontaneous craving with rTMS of the left DLPFC at high frequencies,186 while another found effects for right low‐frequency, but not left high‐frequency, DLPFC rTMS.187 Of note, rTMS of the left DLPFC at low frequencies has increased cue‐induced craving,188 and anodal tDCS of the left DLPFC has increased risky decision making in cocaine users,189 once again suggesting that the stimulation parameters may be important in determining whether cognitive control is enhanced or diminished. A recent study of tDCS in crack cocaine dependence accordingly reversed the polarity of stimulation to left cathodal/right anodal DLPFC tDCS, reporting a reduction in craving with five sessions of active but not sham stimulation.190
For nicotine, a slightly wider variety of targets have been studied across the SN, including the DLPFC and anterior insula. Left DLPFC rTMS, using low‐ or high‐frequency stimulation, has reduced cravings in experimental studies.191, 192 One clinical trial of high‐frequency DLPFC rTMS reported reductions in the number of cigarettes smoked, even in the absence of changes in craving.193 A more recent trial targeted the anterior insula, as well as the DLPFC, bilaterally with a helmet‐shaped deep rTMS coil, in a large sample of 115 patients.194 Thirteen sessions of high‐ but not low‐frequency or sham rTMS reduced cigarette consumption and increased abstinence rates, with stronger effects when the patients were exposed to smoking cues during stimulation. However, there were no significant effects on craving despite the reduction in use, once again suggesting a mechanism of cognitive control enhancement rather than craving reduction per se.
With respect to tDCS in nicotine dependence, a preliminary study found that one session of active but not sham anodal bilateral DLPFC tDCS reduced cigarette cravings.195 Another preliminary study196 found a significant reduction in cigarettes smoked for active but not sham DLPFC stimulation using the left anodal/right cathodal polarity that had proven useful in alcohol and crack cocaine use, as above. A more recent study using the same type of tDCS197 found a reduction in cigarettes smoked after 5 days of active but not sham stimulation. Furthermore, the active group showed signs of enhanced cognitive control, in the form of a greater propensity to reject offers of cigarettes (but not money) in a decision‐making task known as the Ultimatum game. On an important side note, at least one report has noted that nicotine patches abolish the effects of both anodal and cathodal tDCS in healthy volunteers,198 potentially posing a challenge to the therapeutic use of tDCS in tobacco cessation.
One final point concerns the potential of rTMS for treating comorbid MDD/SUDs in tandem, by addressing their common deficits in cognitive control. A recent study199 used high‐frequency bilateral DLPFC rTMS via a deep helmet coil, and compared outcomes in patients with MDD versus MDD and alcohol dependence. One major difference between deep TMS and conventional rTMS is the coil geometry; deep TMS coils employ complex helmet‐shaped windings that are able to reach deeper cortical structures.200 Depression scores improved 55% and 62% in the two groups, while scores on the Clinical Global Impression scale improved 67% and 78%. Improvement was actually significantly greater in the comorbid MDD/alcohol dependence patients than in the patients with MDD alone. It is also worth noting that a recent meta‐analysis found that deep TMS appears to achieve antidepressant effects over multiple sessions.201 The suggestion is that the presence of SUDs may not interfere with the outcomes of rTMS for MDD and indeed that the presence of comorbid SUDs might be an indicator of broader underlying deficits in cognitive control that render DLPFC rTMS more likely to be successful. This possibility warrants future investigation.
Future directions for NIBS in MDD and SUDs
Enhancing cognitive control
Cognitive control, and specifically response selection/inhibition, is presented in this review as one of the key transdiagnostic deficits across MDD and SUDs. Its associated functional network, the SN, maps closely onto a set of areas affected across multiple categories of psychiatric illness. Of its core cortical nodes, investigations have relied heavily on the DLPFC for NIBS in MDD and SUDs. The dACC and anterior insula, however, actually appear more prominently and consistently in the network than the DLPFC.57 Hence, it may be productive to pursue studies of dACC or anterior insula excitatory NIBS, under the hypothesis that these targets will surpass the DLPFC for enhancing cognitive control and thus achieve superior clinical outcomes in MDD and SUDs.
For the dACC, early evidence supports this hypothesis. In healthy controls, excitatory dorsomedial rTMS can reduce impulsivity on a delay‐discounting task,167 and there is a growing literature supporting clinical efficacy for DMPFC rTMS across multiple disorders.142, 165, 166 DMPFC rTMS also shows initial promise for treating acute and chronic craving.202 Dorsal ACC activity is higher in response to addiction cues,93, 203 especially when personally relevant,92, 93, 95 and during other tasks involving cognitive control and response inhibition.73, 75, 204
Reducing incentive salience
Reward circuitry pathology, and specifically distorted incentive salience, is also presented in this review as a common feature of MDD and SUDs. The associated network, VMN, is a candidate target for NIBS treatments. At present, rTMS and tDCS are unlikely to be able to stimulate the ventral striatum directly because of the depth of this structure. However, the cortical nodes of the VMN are not markedly deeper than the dACC or anterior insula. At least one deep rTMS coil has been designed to target the VMPFC,205 although it has not yet been used in MDD or SUDs. Likewise, at least one tDCS study has successfully modulated the VMPFC, along with the VMN circuit into the midbrain, enhancing perceived attractiveness of faces.158 In SUD patients, one study recently targeted the frontopolar cortex206 and found that high‐frequency rTMS increased cue‐induced cigarette craving, as would be expected for excitatory stimulation of this incentive pathway. Thus, stimulation of the VMN appears feasible and may exert effects on disease‐relevant reward function.
Could inhibitory TMS to the VMPFC be beneficial for SUD patients?
While the majority of rTMS studies to date have focused on the DLPFC and its downstream targets in the dorsal striatum, a recent study demonstrated that it is also possible to activate the VMPFC and its targets in the ventral striatum with TMS. Using integrated TMS/MRI scanning, they demonstrated that it is possible to differentially activate the frontal–striatal systems that govern executive control from those that govern limbic arousal by applying single‐pulse TMS to the DLPFC and the VMPFC/frontal pole, respectively.66 TMS pulses applied to the frontal pole (EEG coordinate: FP1) led to elevated BOLD signal in the VMPFC, ventral striatum, and OFC—core regions of the VMN involved in limbic arousal and craving. TMS pulses applied to the DLPFC in healthy individuals at rest (EEG coordinate: F3) led to elevated BOLD signal in the DLPFC and dorsal striatum—core regions of the SN involved in executive control (Fig. 6A). Additionally, elevated DLPFC activity was accompanied by a reciprocal decrease in VMPFC activity, highlighting the reciprocal activity of these two networks.
Figure 6.

Preclinical evidence for targeting the VMN in SUDs. (A) TMS–fMRI studies reveal that stimulation of the DLPFC elicits activation in the corresponding corticostriatal circuit through the dorsal caudate nucleus, as well as other nodes of the SN. Stimulation over the frontal pole, in contrast, elicits activation in VMN nodes, including the VMPFC and ventral striatum.66 (B) Applying an inhibitory pattern of rTMS to the frontal pole (two trains of 1800 pulses of cTBS, 60 s apart) causes a reduction in TMS‐evoked activation in the VMN and other limbic‐network regions, including the ventral striatum and orbitofrontal cortex (OFC). The degree of inhibition is proportional to the intensity of stimulation. This evidence suggests that inhibitory stimulation of the frontal pole may successfully reduce activation in the cortical and striatal nodes of the VMN, which could have therapeutic value for reducing cravings in SUDs and the incentive salience of negative emotional cues in MDD.
Having demonstrated that it was possible to modulate activity in the VMPFC and ventral striatum, the authors performed a study in cocaine users175 in which they applied an inhibitory form of TMS (continuous theta burst stimulation) to the medial PFC (FP1) while the participants were engaged in a craving‐induction task. Specifically, patients underwent a functional MRI scan immediately before and after a single session of real or sham cTBS. Immediately before the cTBS session, participants were asked to describe the last time that they used cocaine, using standardized techniques from exposure therapy. They were then primed and asked to think about this event while the cTBS was administered. It was demonstrated that, relative to sham, active stimulation significantly decreased stimulus‐evoked BOLD signal in the VMPFC and the ventral striatum—critical brain regions for craving (Fig. 6B). Additionally, these data revealed a significant correlation between the TMS pulse intensity and the effect on these neural circuits (Fig. 6C). Thus, inhibitory rTMS to the frontal pole appears to be a successful strategy for engaging and attenuating the VMN target, especially when an inhibitory dose of rTMS is applied to a neural circuit that is in a primed state (e.g., thinking about drug cues).
There are hints that this strategy may be more useful than SN excitatory stimulation in at least some patients with SUDs. Although the SN and VMN show reciprocal activity in healthy subjects, this may not be true in all individuals. In cocaine users, a recent MRI–TMS study showed that DLPFC rTMS did not elicit the usual pattern of reciprocal deactivation of the VMN207 (Fig. 7). Thus, although some individuals may be able to achieve the desired suppression of the VMN indirectly during excitatory stimulation of the SN, other patients may require the VMN to be targeted directly. Investigating this hypothesis in MDD and SUD patients with neuroimaging methods will be an important area for future study.
Figure 7.

Aberrant functional connectivity of the SN and VMN in cocaine users versus MDD patients.207 TMS of the DLPFC (F3 EEG site) during fMRI elicits local activation of the DLPFC itself, and reciprocal deactivation of the striatum and VMPFC, highlighting the reciprocal relationship of the SN and VMN in healthy controls. In cocaine users, however, TMS of the DLPFC elicited only local activation, suggesting a possible absence of the usual reciprocal relationship between SN and VMN may contribute to the pathophysiology of SUDs. If so, SN excitation alone may fail to inhibit the VMN and therefore may not exert the same degree of therapeutic effect in such cases. Instead, direct intervention to inhibit the VMN may be required.
Controlling the cognitive state during treatment
Few NIBS trials to date have controlled patients’ cognitive state during treatment. Brain activity during stimulation is known to have an important influence on the effects of rTMS,208, 209, 210 as evident even in the difference between resting and active motor threshold during motor cortex stimulation.
Repetitive TMS of the SN for nicotine addiction is more effective in the presence of smoking cues.194 Conversely, in MDD, negative stimuli exposure during rTMS may disrupt the beneficial effects of treatment.209 With frontopolar stimulation, the effects of rTMS on cigarette craving were different, depending on whether the patient was presented with smoking or neutral cues during stimulation.206 Thus, future efforts to improve outcomes for NIBS in MDD/SUDs will likely benefit from more rigorous manipulation of the cognitive state during treatment.
Optimizing the treatment protocol
Available evidence concurs with clinical experience in suggesting that maximal effects of rTMS in MDD require 20–30 sessions of treatment, regardless of site or protocol.161, 162, 166 The limited evidence for tDCS likewise suggests that benefits continue to accumulate over 30 sessions in MDD.171 For this reason, early literature may underestimate the therapeutic potential of NIBS, and longer courses are required for future studies to establish the true effect size.
Long courses of treatment may negatively affect treatment adherence, as well as the scalability of NIBS as a treatment in the larger population. SUD patients respond better to briefer treatment interventions than to extended ones.211 However, more sessions need not require more days of treatment. Recent rTMS studies have begun to explore the use of multiple daily sessions separated by short intervals. These protocols are safe and tolerable, and have reduced the treatment course to 10 days with 2x daily stimulation,212 5 days with 4x daily stimulation,213 and, in one small case series, 2 days with 15 sessions.214 The optimal session number and interval have not been systematically investigated under randomized conditions, and this is an important area for future study. Accelerated regimens may be facilitated by cTBS and iTBS, which achieve similar effects as longer conventional protocols despite requiring only 1–3 min for delivery.134, 166 Other products that have the potential to be clinically impactful include deep TMS201 and external trigeminal nerve stimulation.215
Reducing the variability of effect for NIBS
Although different NIBS protocols are classically considered to be excitatory or inhibitory, many individuals display effects opposite to the usual direction, or no effect at all. For 1‐Hz rTMS, approximately 50% of individuals show excitation rather than inhibition;128, 136 likewise, for 10‐Hz rTMS, a similarly large proportion show inhibition rather than excitation.128 Variability is also problematic for cTBS and iTBS.216 Likewise, for tDCS, one study found that only 36% showed the classic pattern of excitatory effects for anodal and inhibitory for cathodal stimulation, and the opposite was true in 21%.147 The variability is not confined to the motor cortex: fMRI studies reveal similarly inconsistent effects of 1‐Hz parietal rTMS on resting‐state functional connectivity.136
This variability of effect likely impedes successful treatment outcome. Currently, there is no known reliable biological or neuroimaging marker to predict a priori which individuals will respond best to a particular stimulation target or stimulation protocol. However, group‐level differences between responders and non‐responders to treatment may help to elucidate these markers. For example, recent studies have found that 30 sessions of 10‐Hz DMPFC rTMS strengthens cortico–striatal–thalamic resting‐state connectivity in patients (responders) with low baseline connectivity, but weakens it in those with high baseline connectivity (non‐responders).138 Thus, variability of NIBS effects must be addressed in future studies to improve treatment outcomes.
Better methods to control patient brain activity during stimulation may ameliorate some of the interindividual variability. For example, 20‐Hz rTMS and newer forms of cTBS more consistently show effects both in the motor cortex128 and on resting‐state functional connectivity.136 Quadripulse stimulation (QPS) uses 4‐pulse bursts of stimulation that can be excitatory or inhibitory depending on the inter‐pulse interval,217 and effects are longer lasting and appear consistent across greater than 85–90% of individuals. Unfortunately, most rTMS devices cannot perform QPS without significant hardware modification, and QPS protocols may require 30 min of stimulation for optimal effects.218 Nonetheless, studies of novel rTMS protocols should be pursued to achieve better consistency in the effects.
Selecting and phenotyping patients
Important considerations in SUD trials are the type of substance dependence and the selection of SUD patients that are seeking treatment. Studies often do not disclose patient treatment‐seeking status or whether patients are active substance users during NIBS treatments. Generally, patients are not abstinent for tobacco‐ or alcohol‐related NIBS trials, while for illicit drug trials patients have completed detoxification. The active use of certain substances may influence the effect of NIBS. For example, tobacco use in healthy controls can reduce NIBS‐induced inhibition of the motor cortex.198 In cigarette smokers, NIBS‐induced facilitation was found to be abolished during nicotine withdrawal.219 Future studies should take into account these sources of heterogeneity, as they may alter clinical outcomes.
Diagnostic criteria are another important source of heterogeneity. MDD and SUDs are increasingly recognized to encompass a wide variety of subtypes, or endophenotypes. In MDD, for example, distinct endophenotypes have recently been proposed for patients with prominent memory impairment, neuroticism, cognitive control, and anhedonia.220 The latter three have recently been linked to VMN disconnectivity and, consequently, poor response to DMPFC rTMS.165 The presence of neutrally distinctive endophenotypes suggests that treatment targeted to individual pathology may improve success rates. For example, patients without cognitive control deficits, but with poor reward sensitivity, may be poor candidates for standard SN rTMS and might instead be better suited to stimulation of the VMN. Conversely, patients with mood liability, cognitive‐control deficits, and multiple comorbidities, such as SUDs, might be identified as particularly good responders to SN NIBS.
A common practice in clinical trials of NIBS is to attempt to limit patient heterogeneity by excluding MDD patients with comorbid SUDs or active substance dependence. However, given that SN pathology may give rise to multiple comorbidities, this practice may be counterproductive in that it excludes good treatment candidates. A more productive approach may be to adopt generous inclusion criteria and to carefully characterize each patient before treatment.
Conclusions
Neuroimaging has revealed two functional networks playing key roles in MDD and SUD pathophysiology. The SN mediates cognitive control, and its hypofunction is a common feature across multiple psychiatric illnesses. The SN's anti‐network, the VMN, plays a key role in reward and incentive salience. Distorted incentive salience is a common feature of both MDD and SUDs. MDD/SUD comorbidity, and the tendency for symptoms of one disorder to exacerbate the other, can therefore be understood as reflecting a shared set of reciprocal neural substrates: inappropriate VMN activation, resulting in pathological incentives, and/or insufficient SN activation, resulting in an inability to exert cognitive control over those incentives. NIBS targeting the SN may enhance cognitive control, thereby relieving a deficit common to MDD and SUDs. NIBS targeting the VMN is less well explored, but may attenuate pathological incentive salience, thereby reducing craving in SUDs and negative affect in MDD.
In conclusion, NIBS offers a promising new approach to treat MDD and SUDs. By strengthening cognitive control and quelling pathological incentive salience, NIBS may address underlying deficits common to both disorders, and may be particularly well suited to comorbid cases. Given the high prevalence and social impact of MDD and SUDs and high nonresponse rates, new treatment options will be a welcome development for clinicians and patients alike.
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1. Whiteford, H.A. , Degenhardt L., Rehm J., et al 2013. Global burden of disease attributable to mental and substance use disorders: findings from the global burden of disease study 2010. Lancet 382: 1575–1586. [DOI] [PubMed] [Google Scholar]
- 2. Ferrari, A.J. , Charlson F.J., Norman R.E., et al 2013. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 10: e1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rush, A.J. 2007. Limitations in efficacy of antidepressant monotherapy. J. Clin. Psychiatry 68(Suppl.): 8–10. [PubMed] [Google Scholar]
- 4. Rush, A.J. , Trivedi M.H., Wisniewski S.R., et al 2006. Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am. J. Psychiatry 163: 1905–1917. [DOI] [PubMed] [Google Scholar]
- 5. Nemeroff, C.B. 2007. Prevalence and management of treatment‐resistant depression. J. Clin. Psychiatry 68: 17–25. [PubMed] [Google Scholar]
- 6. Rocha, F.L. , Fuzikawa C., Riera R. & Hara C.. 2012. Combination of antidepressants in the treatment of major depressive disorder: a systematic review and meta‐analysis. J. Clin. Psychopharmacol. 32: 278–281. [DOI] [PubMed] [Google Scholar]
- 7. Papakostas, G.I. , Shelton R.C., Smith J. & Fava M.. 2007. Augmentation of antidepressants with atypical antipsychotic medications for treatment‐resistant major depressive disorder: a meta‐analysis. J. Clin. Psychiatry 68: 826–831. [DOI] [PubMed] [Google Scholar]
- 8. Blier, P. , Ward H.E., Tremblay P., et al 2010. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double‐blind randomized study. Am. J. Psychiatry 167: 281–288. [DOI] [PubMed] [Google Scholar]
- 9. Turner, P. & Kantaria R.A.. 2014. Systematic review and meta‐analysis of the evidence base for add‐on treatment for patients with major depressive disorder who have not responded to antidepressant treatment: a European perspective. J. Psychopharmacol. 28: 85–98. [DOI] [PubMed] [Google Scholar]
- 10. Dierckx, B. , Heijnen W.T., van den Broek W.W. & Birkenhäger T.K.. 2012. Efficacy of electroconvulsive therapy in bipolar versus unipolar major depression: a meta‐analysis. Bipolar Disord. 14: 146–150. [DOI] [PubMed] [Google Scholar]
- 11. Swan, J.S. , Macvicar R., Christmas D., et al 2014. Cognitive Behavioural Analysis System of Psychotherapy (CBASP) for chronic depression: clinical characteristics and six month clinical outcomes in an open case series. J. Affect. Disord. 152–154: 268–276. [DOI] [PubMed] [Google Scholar]
- 12. Jelovac, A. , Kolshus E. & McLoughlin D.M.. 2013. Relapse following successful electroconvulsive therapy for major depression: a meta‐analysis. Neuropsychopharmacology 38: 2467–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Degenhardt, L. , Chiu W.T., Sampson N., et al 2008. Toward a global view of alcohol, tobacco, cannabis, and cocaine use: findings from the WHO world mental health surveys. PLoS Med. 5: 1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.U.S. Department of Health and Human Services (HHS). Substance Abuse and Mental Health Services Administration. 2010. Results from the 2010 National Survey on Drug Use and Health: Summary of National Findings. Vol. I 1–143. Rockville, MD: Substance Abuse and Mental Health Services Administration. [Google Scholar]
- 15. Grant, B.F. , Chou S.P., Goldstein R.B., et al 2008. Prevalence, correlates, disability, and comorbidity of DSM‐IV borderline personality disorder: results from the Wave 2 National Epidemiologic Survey on Alcohol and Related Conditions. J. Clin. Psychiatry 69: 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merikangas, K.R. & McClair V.L.. 2012. Epidemiology of substance use disorders. Hum. Genet. 131: 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dutra, L. , Stathopoulou G., Basden S.L., et al 2008. A meta‐analytic review of psychosocial interventions for substance use disorders. Am. J. Psychiatry 165: 179–187. [DOI] [PubMed] [Google Scholar]
- 18. Magill, M. & Ray La.. 2009. Cognitive‐behavioral treatment with adult alcohol and illicit drug users: a meta‐analysis of randomized controlled trials. J. Stud. Alcohol Drugs 70: 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McHugh, R.K. , Hearon B.A. & Otto M.W.. 2010. Cognitive behavioral therapy for substance use disorders. Psychiatr. Clin. North Am. 33: 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prendergast, M. , Podus D., Finney J., et al 2006. Contingency management for treatment of substance use disorders: a meta‐analysis. Addiction 101: 1546–1560. [DOI] [PubMed] [Google Scholar]
- 21. Stitzer, M. & Petry N.. 2006. Contingency management for treatment of substance abuse. Annu. Rev. Clin. Psychol. 2: 411–434. [DOI] [PubMed] [Google Scholar]
- 22. Morein‐Zamir, S. & Robbins T.W.. 2014. Fronto‐striatal circuits in response‐inhibition: relevance to addiction. Brain Res 1628: 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anton, R.F. , O'Malley S.S., Ciraulo D.A., et al 2006. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA 295: 2003–2017. [DOI] [PubMed] [Google Scholar]
- 24. Johnson, R.E. , Jaffe J.H. & Fudala P.J.. 1992. A controlled trial of buprenorphine treatment for opioid dependence. JAMA 267: 2750–2755. [PubMed] [Google Scholar]
- 25. Sinha, R. 2011. New findings on biological factors predicting addiction relapse vulnerability. Curr. Psychiatry Rep. 13: 398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blum, K. , Braverman E.R., Holder J.M., et al 2000. Reward deficiency syndrome: a biogenetic model for the diagnosis and treatment of impulsive, addictive, and compulsive behaviors. J. Psychoactive Drugs 32(Suppl.): 1–112. [DOI] [PubMed] [Google Scholar]
- 27. Hasin, D.S. , Goodwin R.D., Stinson F.S. & Grant B.F.. 2005. Epidemiology of major depressive disorder: results from the National Epidemiologic Survey on Alcoholism and Related Conditions. Arch. Gen. Psychiatry 62: 1097–1106. [DOI] [PubMed] [Google Scholar]
- 28. Kessler, R.C. , Chiu W.T., Demler O., et al 2005. Prevalence, severity, and comorbidity of 12‐month DSM‐IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 62: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelly, J. , Mckellar J.D. & Moos R.. 2003. Major depression in patients with substance use disorders: relationship to 12‐step self‐help involvement and substance use outcomes. Addiction 98: 499–508. [DOI] [PubMed] [Google Scholar]
- 30. Nunes, E.V. & Levin F.R.. 2004. Treatment of depression in patients with alcohol or other drug dependence: a meta‐analysis. JAMA 291: 1887–1896. [DOI] [PubMed] [Google Scholar]
- 31. Nunes, E.V. , Sullivan M.A. & Levin F.R.. 2004. Treatment of depression in patients with opiate dependence. Biol. Psychiatry 56: 793–802. [DOI] [PubMed] [Google Scholar]
- 32. Raby Wilfrid, N. , Rubin Eric A., Garawi F., et al 2014. A randomized, double‐blind, placebo‐controlled trial of venlafaxine for the treatment of depressed cocaine‐dependent patients. Am. J. Addict. 23: 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riper, H. , Andersson G., Hunter S.B., et al 2014. Treatment of comorbid alcohol use disorders and depression with cognitive‐behavioural therapy and motivational interviewing: a meta‐analysis. Addiction 109: 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glassman, A.H. , Covey L.S., Stetner F. & Rivelli S.. 2001. Smoking cessation and the course of major depression: a follow‐up study. Lancet 357: 1929–1932. [DOI] [PubMed] [Google Scholar]
- 35. Covey, L.S. , Glassman A.H. & Stetner F.. 1998. Cigarette smoking and major depression. J. Addict. Dis. 17: 35–46. [DOI] [PubMed] [Google Scholar]
- 36. Covey, L.S. , Glassman A.H., Stetner F., et al 2002. A randomized trial of sertraline as a cessation aid for smokers with a history of major depression. Am. J. Psychiatry 159: 1731–1737. [DOI] [PubMed] [Google Scholar]
- 37. Gierisch, J.M. , Bastian L.A., Calhoun P.S., et al 2012. Smoking cessation interventions for patients with depression: a systematic review and meta‐analysis. J. Gen. Intern. Med. 27: 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pettinati, H.M. , O'Brien C.P. & Dundon W.D.. 2013. Current status of co‐occurring mood and substance use disorders: a new therapeutic target. Am. J. Psychiatry 170: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fox, M.D. , Liu H. & Pascual‐Leone A.. 2013. Identification of reproducible individualized targets for treatment of depression with TMS based on intrinsic connectivity. Neuroimage 66: 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimbrell, T.A. , Little J.T. & Dunn R.T., et al 1999. Frequency dependence of antidepressant response to left prefrontal repetitive transcranial magnetic stimulation (rTMS) as a function of baseline cerebral glucose metabolism. Biol. Psychiatry 46: 1603–1613. [DOI] [PubMed] [Google Scholar]
- 41. Speer, A.M. , Kimbrell T.A., Wassermann E.M., et al 2000. Opposite effects of high and low frequency rTMS on regional brain activity in depressed patients. Biol. Psychiatry 48: 1133–1141. [DOI] [PubMed] [Google Scholar]
- 42. Sporns, O. 2011. The human connectome: a complex network. Ann. N. Y. Acad. Sci. 1224: 109–125. [DOI] [PubMed] [Google Scholar]
- 43. Damoiseaux, J.S. , Rombouts S.A.R.B., Barkhof F., et al 2006. Consistent resting‐state networks across healthy subjects. Proc. Natl. Acad. Sci. U.S.A. 103: 13848–13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith, S.M. , Fox P.T., Miller K.L., et al 2009. Correspondence of the brain's functional architecture during activation and rest. Proc. Natl. Acad. Sci. U.S.A. 106: 13040–13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yarkoni, T. , Poldrack R.A., Nichols T.E., et al 2011. Large‐scale automated synthesis of human functional neuroimaging data. Nat. Methods 8: 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomas Yeo, B.T. , Krienen F.M., Sepulcre J., et al 2011. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 106: 1125–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choi, E.Y. , Yeo B.T.T. & Buckner R.L.. 2012. The organization of the human striatum estimated by intrinsic functional connectivity. J. Neurophysiol. 108: 2242–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buckner, R.L. , Krienen F.M., Castellanos A., et al 2011. The organization of the human cerebellum estimated by intrinsic functional connectivity. J. Neurophysiol. 106: 2322–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Greicius, M.D. , Krasnow B., Reiss A.L. & Menon V.. 2003. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc. Natl. Acad. Sci. U.S.A. 100: 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kucyi, A. & Davis K.D.. 2014. Dynamic functional connectivity of the default mode network tracks daydreaming. Neuroimage 100: 471–480. [DOI] [PubMed] [Google Scholar]
- 51. Spreng, R.N. & Grady C.L.. 2010. Patterns of brain activity supporting autobiographical memory, prospection, and theory of mind, and their relationship to the default mode network. J. Cogn. Neurosci. 22: 1112–1123. [DOI] [PubMed] [Google Scholar]
- 52. Hamilton, J.P. , Furman D.J., Chang C., et al 2011. Default‐mode and task‐positive network activity in major depressive disorder: implications for adaptive and maladaptive rumination. Biol. Psychiatry 70: 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Downar, J. , Crawley A.P., Mikulis D.J. & Davis K.D.. 2000. A multimodal cortical network for the detection of changes in the sensory environment. Nat. Neurosci. 3: 277–283. [DOI] [PubMed] [Google Scholar]
- 54. Downar, J. , Crawley A.P., Mikulis D.J. & Davis K.D.. 2001. The effect of task relevance on the cortical response to changes in visual and auditory stimuli: an event‐related fMRI study. Neuroimage 14: 1256–1267. [DOI] [PubMed] [Google Scholar]
- 55. Sridharan, D. , Levitin D.J. & Menon V.. 2008. A critical role for the right fronto‐insular cortex in switching between central‐executive and default‐mode networks. Proc. Natl. Acad. Sci. U.S.A. 105: 12569–12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dosenbach, N.U.F. , Visscher K.M., Palmer E.D., et al 2006. A core system for the implementation of task sets. Neuron 50: 799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goodkind, M. , Eickhoff S.B., Oathes D.J., et al 2015. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry 5797: 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cooper, J.C. & Knutson B.. 2008. Valence and salience contribute to nucleus accumbens activation. Neuroimage 39: 538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Volkow, N.D. , Wang G.‐J., Fowler J.S., et al 2011. Addiction: beyond dopamine reward circuitry. Proc. Natl. Acad. Sci. U.S.A. 108: 15037–15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koob, G.F. 2009. Dynamics of neuronal circuits in addiction: reward, antireward, and emotional memory. Pharmacopsychiatry 42 (Suppl.): S32–S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Berridge, K.C. 2012. From prediction error to incentive salience: mesolimbic computation of reward motivation. Eur. J. Neurosci. 35: 1124–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tremblay, L.K. , Naranjo C.A., Graham S.J., et al 2005. Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Arch. Gen. Psychiatry 62: 1228–1236. [DOI] [PubMed] [Google Scholar]
- 63. Diekhof, E.K. , Falkai P. & Gruber O.. 2008. Functional neuroimaging of reward processing and decision‐making: a review of aberrant motivational and affective processing in addiction and mood disorders. Brain Res. Rev. 59: 164–184. [DOI] [PubMed] [Google Scholar]
- 64. Menon, V. 2011. Large‐scale brain networks and psychopathology: a unifying triple network model. Trends Cogn. Sci. 15: 483–506. [DOI] [PubMed] [Google Scholar]
- 65. Dosenbach, N.U.F. , Fair D.A., Cohen A.L., et al 2008. A dual‐networks architecture of top‐down control . Trends Cogn. Sci. 12: 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hanlon, C.A. , Canterberry M., Taylor J.J., et al 2013. Probing the frontostriatal loops involved in executive and limbic processing via interleaved TMS and functional MRI at two prefrontal locations: a pilot study. PLoS One 8: e67917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fox, M.D. , Snyder A.Z., Vincent J.L., et al 2005. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc. Natl. Acad. Sci. U.S.A. 102: 9673–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Uddin, L.Q. , Kelly A.M., Biswal B.B., et al 2009. Functional connectivity of default mode network components: correlation, anticorrelation, and causality. Hum. Brain Mapp. 30: 625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bullmore, E. & Sporns O.. 2009. Complex brain networks: graph theoretical analysis of structural and functional systems. Nat. Rev. Neurosci. 10: 186–198. [DOI] [PubMed] [Google Scholar]
- 70. Power, J.D. , Cohen A.L., Nelson S.M., et al 2011. Functional network organization of the human brain. Neuron 72: 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Power, J.D. , Schlaggar B.L., Lessov‐Schlaggar C.N. & Petersen S.E.. 2013. Evidence for hubs in human functional brain networks. Neuron 79: 798–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nestler, E.J. 2005. Is there a common molecular pathway for addiction? Nat. Neurosci. 8: 1445–1449. [DOI] [PubMed] [Google Scholar]
- 73. Moeller, S.J. , Hajcak G., Parvaz M.A., et al 2012. Psychophysiological prediction of choice: relevance to insight and drug addiction. Brain 135: 3481–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bolla, K. , Ernst M., Kiehl K., et al 2004. Prefrontal cortical dysfunction in abstinent cocaine abusers. J. Neuropsychiatry Clin. Neurosci. 16: 456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lee, T.M.C. , Zhou W.‐H., Luo X.‐J., et al 2005. Neural activity associated with cognitive regulation in heroin users: a fMRI study. Neurosci. Lett. 382: 211–216. [DOI] [PubMed] [Google Scholar]
- 76. Hester, R. , Nestor L. & Garavan H.. 2009. Impaired error awareness and anterior cingulate cortex hypoactivity in chronic cannabis users. Neuropsychopharmacology 34: 2450–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Connolly, C.G. , Foxe J.J., Nierenberg J., et al 2012. The neurobiology of cognitive control in successful cocaine abstinence. Drug Alcohol Depend. 121: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Denier, N. , Gerber H., Vogel M., et al 2013. Reduction in cerebral perfusion after heroin administration: a resting state arterial spin labeling study. PLoS One 8: e71461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu, J. , Liang J., Qin W., et al 2009. Dysfunctional connectivity patterns in chronic heroin users: an fMRI study. Neurosci. Lett. 460: 72–77. [DOI] [PubMed] [Google Scholar]
- 80. Zhang, X. , Salmeron B.J., Ross T.J., et al 2011. Anatomical differences and network characteristics underlying smoking cue reactivity. Neuroimage 54: 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Radeloff, D. , Willmann K., Otto L., et al 2014. High‐fat taste challenge reveals altered striatal response in women recovered from bulimia nervosa: a pilot study. World J. Biol. Psychiatry 15: 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rando, K. , Hong K.I., Bhagwagar Z., et al 2011. Association of frontal and posterior cortical gray matter volume with time to alcohol relapse: a prospective study. Am. J. Psychiatry 168: 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Naqvi, N.H. & Bechara A.. 2010. The insula and drug addiction: an interoceptive view of pleasure, urges, and decision‐making. Brain Struct. Funct. 214: 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Naqvi, N.H. , Rudrauf D., Damasio H. & Bechara A.. 2007. Damage to the insula disrupts addiction to cigarette smoking. Science 315: 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Moeller, S.J. , Tomasi D., Woicik P.A., et al 2012. Enhanced midbrain response at 6‐month follow‐up in cocaine addiction, association with reduced drug‐related choice. Addict. Biol. 17: 1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kilts, C.D. , Gross R.E., Ely T.D. & Drexler K.P.G.. 2004. The neural correlates of cue‐induced craving in cocaine‐dependent women. Am. J. Psychiatry 161: 233–241. [DOI] [PubMed] [Google Scholar]
- 87. Risinger, R.C. , Salmeron B.J., Ross T.J., et al 2005. Neural correlates of high and craving during cocaine self‐administration using BOLD fMRI. Neuroimage 26: 1097–1108. [DOI] [PubMed] [Google Scholar]
- 88. Li, Q. , Wang Y., Zhang Y., et al 2012. Craving correlates with mesolimbic responses to heroin‐related cues in short‐term abstinence from heroin: an event‐related fMRI study. Brain Res. 1469: 63–72. [DOI] [PubMed] [Google Scholar]
- 89. Rosenberg, D.R. , MacMaster F.P., Mirza Y., et al 2005. Reduced anterior cingulate glutamate in pediatric major depression: a magnetic resonance spectroscopy study. Biol. Psychiatry 58: 700–704. [DOI] [PubMed] [Google Scholar]
- 90. Franklin, T.R. , Wang Z., Wang J., et al 2007. Limbic activation to cigarette smoking cues independent of nicotine withdrawal: a perfusion fMRI study. Neuropsychopharmacology 32: 2301–2309. [DOI] [PubMed] [Google Scholar]
- 91. McClernon, F.J. , Hiott F.B., Huettel S.A. & Rose J.E.. 2005. Abstinence‐induced changes in self‐report craving correlate with event‐related FMRI responses to smoking cues. Neuropsychopharmacology 30: 1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Engelmann, J.M. , Versace F., Robinson J.D., et al 2012. Neural substrates of smoking cue reactivity: a meta‐analysis of fMRI studies. Neuroimage 60: 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kühn, S. & Gallinat J.. 2011. Common biology of craving across legal and illegal drugs: a quantitative meta‐analysis of cue‐reactivity brain response. Eur. J. Neurosci. 33: 1318–1326. [DOI] [PubMed] [Google Scholar]
- 94. Schacht, J.P. , Anton R.F. & Myrick H.. 2013. Functional neuroimaging studies of alcohol cue reactivity: a quantitative meta‐analysis and systematic review. Addict. Biol. 18: 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Moeller, S.J. & Goldstein R.Z.. 2014. Impaired self‐awareness in human addiction: deficient attribution of personal relevance. Trends Cogn. Sci. 18: 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Franklin, T. , Wang Z., Suh J.J., et al 2011. Effects of varenicline on smoking cue–triggered neural and craving responses. Arch. Gen. Psychiatry 68: 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Menossi, H.S. , Goudriaan A.E., De Azevedo‐Marques Périco C., et al 2013. Neural bases of pharmacological treatment of nicotine dependence – insights from functional brain imaging: a systematic review. CNS Drugs 27: 921–941. [DOI] [PubMed] [Google Scholar]
- 98. Culbertson, C.S. , Bramen J., Cohen M.S., et al 2011. Effect of bupropion treatment on brain activation induced by cigarette‐related cues in smokers. Arch. Gen. Psychiatry 68: 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Goldstein, R.Z. , Woicik P.A., Maloney T., et al 2010. Oral methylphenidate normalizes cingulate activity in cocaine addiction during a salient cognitive task. Proc. Natl. Acad. Sci. U.S.A. 107: 16667–16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lubman, D.I. , Yücel M., Kettle J.W.L., et al 2009. Responsiveness to drug cues and natural rewards in opiate addiction: associations with later heroin use. Arch. Gen. Psychiatry 66: 205–212. [DOI] [PubMed] [Google Scholar]
- 101. Zhang, Y. , Gong J., Xie C., et al 2015. Alterations in brain connectivity in three sub‐regions of the anterior cingulate cortex in heroin‐dependent individuals: evidence from resting state fMRI. Neuroscience 284: 998–1010. [DOI] [PubMed] [Google Scholar]
- 102. Hu, Y. , Salmeron B.J., Gu H., et al 2015. Impaired functional connectivity within and between frontostriatal circuits and its association with compulsive drug use and trait impulsivity in cocaine addiction. JAMA Psychiatry 72: 584–592. [DOI] [PubMed] [Google Scholar]
- 103. Hong, L.E. , Hodgkinson C.A., Yang Y., et al 2010. A genetically modulated, intrinsic cingulate circuit supports human nicotine addiction. Proc. Natl. Acad. Sci. U.S.A. 107: 13509–13514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Koenigs, M. & Grafman J.. 2009. The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behav. Brain Res. 201: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mayberg, H.S. , Lozano A.M., Voon V., et al 2005. Deep brain stimulation for treatment‐resistant depression. Neuron 45: 651–660. [DOI] [PubMed] [Google Scholar]
- 106. Brody, A.L. , Saxena S., Mandelkern M.A., et al 2001. Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol. Psychiatry 50: 171–178. [DOI] [PubMed] [Google Scholar]
- 107. Hamilton, J.P. , Chen G., Thomason M.E., et al 2011. Investigating neural primacy in Major Depressive Disorder: multivariate Granger causality analysis of resting‐state fMRI time‐series data. Mol. Psychiatry 16: 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nestler, E.J. & Carlezon W.A.. 2006. The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 59: 1151–1159. [DOI] [PubMed] [Google Scholar]
- 109. Tremblay, L.K. , Naranjo C.A., Graham S.J., et al 2005. Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Arch. Gen. Psychiatry 62: 1228–1236. [DOI] [PubMed] [Google Scholar]
- 110. Robinson, O.J. , Cools R., Carlisi C.O., et al 2012. Ventral striatum response during reward and punishment reversal learning in unmedicated major depressive disorder. Am. J. Psychiatry 169: 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pizzagalli, D.A. , Holmes A.J., Dillon D.G., et al 2009. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am. J. Psychiatry 166: 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Knutson, B. , Bhanji J.P., Cooney R.E., et al 2008. Neural responses to monetary incentives in major depression. Biol. Psychiatry 63: 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wagner, G. , Koch K., Schachtzabel C., et al 2013. Self‐referential processing influences functional activation during cognitive control: an fMRI study . Soc. Cogn. Affect. Neurosci. 8: 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wagner, G. , Schachtzabel C., Peikert G. & Bär K.‐J.. 2015. The neural basis of the abnormal self‐referential processing and its impact on cognitive control in depressed patients. Hum. Brain Mapp. 36: 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. McCabe, C. , Cowen P.J. & Harmer C.J.. 2009. Neural representation of reward in recovered depressed patients. Psychopharmacology (Berl.) 205: 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cooney, R.E. , Joormann J., Eugène F., et al 2010. Neural correlates of rumination in depression. Cogn. Affect. Behav. Neurosci. 10: 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wagner, G. , Sinsel E., Sobanski T., et al 2006. Cortical inefficiency in patients with unipolar depression: an event‐related fMRI study with the Stroop task. Biol. Psychiatry 59: 958–965. [DOI] [PubMed] [Google Scholar]
- 118. Holmes, A.J. & Pizzagalli D.A.. 2008. Response conflict and frontocingulate dysfunction in unmedicated participants with major depression. Neuropsychologia 46: 2904–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Harvey, P.O. , Fossati P., Pochon J.B., et al 2005. Cognitive control and brain resources in major depression: an fMRI study using the n‐back task. Neuroimage 26: 860–869. [DOI] [PubMed] [Google Scholar]
- 120. Bermpohl, F. , Walter M., Sajonz B., et al 2009. Attentional modulation of emotional stimulus processing in patients with major depression—alterations in prefrontal cortical regions. Neurosci. Lett. 463: 108–113. [DOI] [PubMed] [Google Scholar]
- 121. Mayberg, H.S. , Liotti M., Brannan S.K., et al 1999. Reciprocal limbic‐cortical function and negative mood: converging PET findings in depression and normal sadness. Am. J. Psychiatry 156: 675–682. [DOI] [PubMed] [Google Scholar]
- 122. Pizzagalli, D. , Pascual‐Marqui R.D., Nitschke J.B., et al 2001. Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am. J. Psychiatry 158: 405–415. [DOI] [PubMed] [Google Scholar]
- 123. Li, C.‐T. , Lin C.‐P., Chou K.‐H., et al 2010. Structural and cognitive deficits in remitting and non‐remitting recurrent depression: a voxel‐based morphometric study. Neuroimage 50: 347–56. [DOI] [PubMed] [Google Scholar]
- 124. Carpenter, L.L. , Janicak P.G., Aaronson S.T., et al 2012. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress. Anxiety 29: 587–596. [DOI] [PubMed] [Google Scholar]
- 125. Solvason, H.B. , Husain M., Fitzgerald P.B., et al 2014. Improvement in quality of life with left prefrontal transcranial magnetic stimulation in patients with pharmacoresistant major depression: acute and six month outcomes. Brain Stimul. 7: 219–225. [DOI] [PubMed] [Google Scholar]
- 126. Wang, H. , Wang X. & Scheich H.. 1996. LTD and LTP induced by transcranial magnetic stimulation in auditory cortex. Neuroreport 7: 521–525. [DOI] [PubMed] [Google Scholar]
- 127. Antal, A. , Chaieb L., Moliadze V., et al 2010. Brain‐derived neurotrophic factor (BDNF) gene polymorphisms shape cortical plasticity in humans. Brain Stimul. 3: 230–237. [DOI] [PubMed] [Google Scholar]
- 128. Maeda, F. , Keenan J.P., Tormos J.M., et al 2000. Interindividual variability of the modulatory effects of repetitive transcranial magnetic stimulation on cortical excitability. Exp. Brain Res. 133: 425–430. [DOI] [PubMed] [Google Scholar]
- 129. Pascual‐Leone, A. , Tormos J.M., Keenan J., et al 1998. Study and modulation of human cortical excitability with transcranial magnetic stimulation. J. Clin. Neurophysiol. 15: 333–343. [DOI] [PubMed] [Google Scholar]
- 130. Chen, R. , Classen J., Gerloff C., et al 1998. Depression of motor cortex excitability by low‐frequency transcranial magnetic stimulation. Neurology 48: 1398–1403. [DOI] [PubMed] [Google Scholar]
- 131. Pascual‐Leone, A. , Valls‐Solé J., Wassermann E.M. & Hallett M.. 1994. Responses to rapid‐rate transcranial magnetic stimulation of the human motor cortex. Brain 117(Pt 4): 847–858. [DOI] [PubMed] [Google Scholar]
- 132. Huang, Y.‐Z. , Edwards M.J., Rounis E., et al 2005. Theta burst stimulation of the human motor cortex. Neuron 45: 201–206. [DOI] [PubMed] [Google Scholar]
- 133. Huang, Y.‐Z. , Rothwell J.C., Lu C.‐S., et al 2009. The effect of continuous theta burst stimulation over premotor cortex on circuits in primary motor cortex and spinal cord. Clin. Neurophysiol. 120: 796–801. [DOI] [PubMed] [Google Scholar]
- 134. Li, C.‐T. , Chen M.‐H., Juan C.‐H., et al 2014. Efficacy of prefrontal theta‐burst stimulation in refractory depression: a randomized sham‐controlled study. Brain 137(Pt 7): 2088–2098. [DOI] [PubMed] [Google Scholar]
- 135. Bakker, N. , Shahab S., Giacobbe P., et al 2015. rTMS of the dorsomedial prefrontal cortex for major depression: safety, tolerability, effectiveness, and outcome predictors for 10 Hz versus intermittent theta‐burst stimulation. Brain Stimul. 8: 208–215. [DOI] [PubMed] [Google Scholar]
- 136. Eldaief, M.C. , Halko M.A., Buckner R.L. & Pascual‐Leone A.. 2011. Transcranial magnetic stimulation modulates the brain's intrinsic activity in a frequency‐dependent manner. Proc. Natl. Acad. Sci. U.S.A. 108: 21229–21234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Nettekoven, C. , Volz L.J., Leimbach M., et al 2015. Inter‐individual variability in cortical excitability and motor network connectivity following multiple blocks of rTMS. Neuroimage 118: 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Dunlop, J. , Woodside B., Lam E., et al 2015. Increases in frontostriatal connectivity are associated with response to dorsomedial repetitive transcranial magnetic stimulation in refractory binge/purge behaviors. Neuroimage Clin. 8: 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pogarell, O. , Koch W., Pöpperl G., et al 2007. Acute prefrontal rTMS increases striatal dopamine to a similar degree as d‐amphetamine. Psychiatry Res. 156: 251–255. [DOI] [PubMed] [Google Scholar]
- 140. Pogarell, O. , Koch W., Pöpperl G., et al 2006. Striatal dopamine release after prefrontal repetitive transcranial magnetic stimulation in major depression: preliminary results of a dynamic [123I] IBZM SPECT study. J. Psychiatr. Res. 40: 307–314. [DOI] [PubMed] [Google Scholar]
- 141. Monte‐Silva, K. , Ruge D., Teo J.T., et al 2011. D2 receptor block abolishes θ burst stimulation‐induced neuroplasticity in the human motor cortex. Neuropsychopharmacology 36: 2097–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Salomons, T.V. , Dunlop K., Kennedy S.H., et al 2014. Resting‐state cortico‐thalamic‐striatal connectivity predicts response to dorsomedial prefrontal rTMS in major depressive disorder. Neuropsychopharmacology 39: 488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dunlop, K. , Woodside B., Olmsted M., et al 2015. Reductions in cortico‐striatal hyperconnectivity accompany successful treatment of obsessive‐compulsive disorder with dorsomedial prefrontal rTMS. Neuropsychopharmacology doi:10.1038/npp.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Strafella, A.P. , Ko J.H., Grant J., et al 2005. Corticostriatal functional interactions in Parkinson's disease: a rTMS/[11C]raclopride PET study. Eur. J. Neurosci. 22: 2946–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Meron, D. , Hedger N., Garner M. & Baldwin D.S.. 2015. Transcranial direct current stimulation (tDCS) in the treatment of depression: systematic review and meta‐analysis of efficacy and tolerability. Neurosci. Biobehav. Rev. 57: 46–62. [DOI] [PubMed] [Google Scholar]
- 146. Brunoni, A.R. , Nitsche M.A., Bolognini N., et al 2012. Clinical research with transcranial direct current stimulation (tDCS): challenges and future directions. Brain Stimul. 5: 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wiethoff, S. , Hamada M. & Rothwell J.C.. 2014. Variability in response to transcranial direct current stimulation of the motor cortex. Brain Stimul. 7: 468–475. [DOI] [PubMed] [Google Scholar]
- 148. Reato, D. , Rahman A., Bikson M. & Parra L.C.. 2013. Effects of weak transcranial alternating current stimulation on brain activity‐a review of known mechanisms from animal studies. Front. Hum. Neurosci. 7: 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Voss, U. , Holzmann R., Hobson A., et al 2014. Induction of self awareness in dreams through frontal low current stimulation of gamma activity. Nat. Neurosci. 17: 810–812. [DOI] [PubMed] [Google Scholar]
- 150. Stagg, C.J. & Nitsche M.A.. 2011. Physiological basis of transcranial direct current stimulation. Neuroscientist 17: 37–53. [DOI] [PubMed] [Google Scholar]
- 151. Keeser, D. , Meindl T., Bor J., et al 2011. Prefrontal transcranial direct current stimulation changes connectivity of resting‐state networks during fMRI. J. Neurosci. 31: 15284–15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Polanía, R. , Paulus W. & Nitsche M.A.. 2012. Modulating cortico‐striatal and thalamo‐cortical functional connectivity with transcranial direct current stimulation. Hum. Brain Mapp. 33: 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Edwards, D. , Cortes M., Datta A., et al 2013. Physiological and modeling evidence for focal transcranial electrical brain stimulation in humans: a basis for high‐definition tDCS. Neuroimage 74: 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Fregni, F. , Boggio P.S., Nitsche M., et al 2005. Anodal transcranial direct current stimulation of prefrontal cortex enhances working memory. Exp. Brain Res. 166: 23–30. [DOI] [PubMed] [Google Scholar]
- 155. Bocci, T. , Marceglia S., Vergari M., et al 2015. Transcutaneous spinal direct current stimulation modulates human corticospinal system excitability. J. Neurophysiol. 114: 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Vollmann, H. , Conde V., Sewerin S., et al 2013. Anodal transcranial direct current stimulation (tDCS) over supplementary motor area (SMA) but not pre‐SMA promotes short‐term visuomotor learning. Brain Stimul. 6: 101–107. [DOI] [PubMed] [Google Scholar]
- 157. Hsu, T.‐Y. , Tseng L.‐Y., Yu J.‐X., et al 2011. Modulating inhibitory control with direct current stimulation of the superior medial frontal cortex. Neuroimage 56: 2249–2257. [DOI] [PubMed] [Google Scholar]
- 158. Chib, V.S. , Yun K., Takahashi H. & Shimojo S.. 2013. Noninvasive remote activation of the ventral midbrain by transcranial direct current stimulation of prefrontal cortex. Transl. Psychiatry 3: e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Kedzior, K. , Azorina V. & Reitz S.. 2014. More female patients and fewer stimuli per session are associated with the short‐term antidepressant properties of repetitive transcranial magnetic stimulation (rTMS): a meta‐analysis of 54 sham‐controlled studies published between 1997–2013. Neuropsychiatr. Dis. Treat. 10: 727–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Berlim, M.T. , van den Eynde F., Tovar‐Perdomo S. & Daskalakis Z.J.. 2014. Response, remission and drop‐out rates following high‐frequency repetitive transcranial magnetic stimulation (rTMS) for treating major depression: a systematic review and meta‐analysis of randomized, double‐blind and sham‐controlled trials. Psychol. Med. 44: 225–239. [DOI] [PubMed] [Google Scholar]
- 161. Brunelin, J. , Jalenques I., Trojak B., et al 2014. The efficacy and safety of low frequency repetitive transcranial magnetic stimulation for treatment‐resistant depression: the results from a large multicenter French RCT. Brain Stimul. 7: 855–863. [DOI] [PubMed] [Google Scholar]
- 162. Fitzgerald, P.B. , Hoy K., Gunewardene R., et al 2011. A randomized trial of unilateral and bilateral prefrontal cortex transcranial magnetic stimulation in treatment‐resistant major depression. Psychol. Med. 41: 1187–1196. [DOI] [PubMed] [Google Scholar]
- 163. Downar, J. & Daskalakis Z.J.. 2013. New targets for rTMS in depression: a review of convergent evidence. Brain Stimul. 6: 231–240. [DOI] [PubMed] [Google Scholar]
- 164. Kreuzer, P.M. , Schecklmann M., Lehner A., et al 2015. The ACDC pilot trial: targeting the anterior cingulate by double cone coil rTMS for the treatment of depression. Brain Stimul. 8: 240–246. [DOI] [PubMed] [Google Scholar]
- 165. Downar, J. , Geraci J., Salomons T.V., et al 2014. Anhedonia and reward‐circuit connectivity distinguish nonresponders from responders to dorsomedial prefrontal repetitive transcranial magnetic stimulation in major depression. Biol. Psychiatry 76: 176–185. [DOI] [PubMed] [Google Scholar]
- 166. Bakker, N. , Shahab S., Giacobbe P., et al 2015. rTMS of the dorsomedial prefrontal cortex for major depression: safety, tolerability, effectiveness, and outcome predictors for 10 Hz versus intermittent theta‐burst stimulation. Brain Stimul. 8: 205–215. [DOI] [PubMed] [Google Scholar]
- 167. Cho, S.S. , Kosimori Y., Aminian K., et al 2015. Investing in the future: stimulation of the medial prefrontal cortex reduces discounting of delayed rewards. Neuropsychopharmacology 40: 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Watanabe, T. , Hanajima R., Shirota Y., et al 2015. Effects of rTMS of pre‐supplementary motor area on fronto basal ganglia network activity during stop‐signal task. J. Neurosci. 35: 4813–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Strafella, A.P. , Paus T., Barrett J. & Dagher A.. 2001. Repetitive transcranial magnetic stimulation of the human prefrontal cortex induces dopamine release in the caudate nucleus. J. Neurosci. 21: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Valiengo, L. , Benseñor I.M., Goulart A.C., et al 2013. The sertraline versus electrical current therapy for treating depression clinical study (select‐TDCS): results of the crossover and follow‐up phases. Depress. Anxiety 30: 646–653. [DOI] [PubMed] [Google Scholar]
- 171. Loo, C.K. , Alonzo A., Martin D., et al 2012. Transcranial direct current stimulation for depression: 3‐week, randomised, sham‐controlled trial. Br. J. Psychiatry 200: 52–59. [DOI] [PubMed] [Google Scholar]
- 172. Plewnia, C. , Schroeder P.A., Kunze R., et al 2015. Keep calm and carry on: improved frustration tolerance and processing speed by transcranial direct current stimulation (tDCS). PLoS One 10: e0122578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Wolkenstein, L. & Plewnia C.. 2013. Amelioration of cognitive control in depression by transcranial direct current stimulation. Biol. Psychiatry 73: 646–651. [DOI] [PubMed] [Google Scholar]
- 174. Gorelick, D.A. , Zangen A. & George M.S.. 2014. Transcranial magnetic stimulation in the treatment of substance addiction. Ann. N. Y. Acad. Sci. 1327: 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Hanlon, C.A. , Dowdle L.T., Austelle C.W., et al 2015. What goes up, can come down: novel brain stimulation paradigms may attenuate craving and craving‐related neural circuitry in substance dependent individuals. Brain Res 1628: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Mishra, B.R. , Nizamie S.H., Das B. & Praharaj S.K.. 2010. Efficacy of repetitive transcranial magnetic stimulation in alcohol dependence: a sham‐controlled study. Addiction 105: 49–55. [DOI] [PubMed] [Google Scholar]
- 177. Herremans, S.C. , Baeken C., Vanderbruggen N., et al 2012. No influence of one right‐sided prefrontal HF‐rTMS session on alcohol craving in recently detoxified alcohol‐dependent patients: results of a naturalistic study. Drug Alcohol Depend. 120: 209–213. [DOI] [PubMed] [Google Scholar]
- 178. Herremans, S.C. , Vanderhasselt M.‐A., De Raedt R. & Baeken C.. 2013. Reduced intra‐individual reaction time variability during a Go‐NoGo task in detoxified alcohol‐dependent patients after one right‐sided dorsolateral prefrontal HF‐rTMS session. Alcohol Alcohol. 48: 552–557. [DOI] [PubMed] [Google Scholar]
- 179. Höppner, J. , Broese T., Wendler L., et al 2011. Repetitive transcranial magnetic stimulation (rTMS) for treatment of alcohol dependence. World J. Biol. Psychiatry 12(Suppl.): 57–62. [DOI] [PubMed] [Google Scholar]
- 180. Rapinesi, C. , Kotzalidis G.D., Serata D., et al 2013. Efficacy of add‐on deep transcranial magnetic stimulation in comorbid alcohol dependence and dysthymic disorder: three case reports. Prim. Care Companion CNS Disord. 15: PCC.12m01438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. De Ridder, D. , Vanneste S., Kovacs S., et al 2011. Transient alcohol craving suppression by rTMS of dorsal anterior cingulate: an fMRI and LORETA EEG study. Neurosci. Lett. 496: 5–10. [DOI] [PubMed] [Google Scholar]
- 182. Boggio, P.S. , Sultani N., Fecteau S., et al 2008. Prefrontal cortex modulation using transcranial DC stimulation reduces alcohol craving: a double‐blind, sham‐controlled study. Drug Alcohol Depend. 92: 55–60. [DOI] [PubMed] [Google Scholar]
- 183. Nakamura‐Palacios, E.M. , de Almeida Benevides M.C., da Penha Zago‐Gomes M., et al 2012. Auditory event‐related potentials (P3) and cognitive changes induced by frontal direct current stimulation in alcoholics according to Lesch alcoholism typology. Int. J. Neuropsychopharmacol. 15: 601–616. [DOI] [PubMed] [Google Scholar]
- 184. da Silva, M.C. , Conti C.L., Klauss J., et al 2013. Behavioral effects of transcranial direct current stimulation (tDCS) induced dorsolateral prefrontal cortex plasticity in alcohol dependence. J. Physiol. (Paris) 107: 493–502. [DOI] [PubMed] [Google Scholar]
- 185. Klauss, J. , Penido Pinheiro L.C., Silva Merlo B.L., et al 2014. A randomized controlled trial of targeted prefrontal cortex modulation with tDCS in patients with alcohol dependence. Int. J. Neuropsychopharmacol. 17: 1793–1803. [DOI] [PubMed] [Google Scholar]
- 186. Politi, E. , Fauci E., Santoro A. & Smeraldi E.. 2008. Daily sessions of transcranial magnetic stimulation to the left prefrontal cortex gradually reduce cocaine craving. Am. J. Addict. 17: 345–346. [DOI] [PubMed] [Google Scholar]
- 187. Camprodon, J.A. , Martínez‐Raga J., Alonso‐Alonso M., et al 2007. One session of high frequency repetitive transcranial magnetic stimulation (rTMS) to the right prefrontal cortex transiently reduces cocaine craving. Drug Alcohol Depend. 86: 91–94. [DOI] [PubMed] [Google Scholar]
- 188. Li, X. , Malcolm R.J., Huebner K., et al 2013. Low frequency repetitive transcranial magnetic stimulation of the left dorsolateral prefrontal cortex transiently increases cue‐induced craving for methamphetamine: a preliminary study. Drug Alcohol Depend. 133: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Gorini, A. , Lucchiari C., Russell‐Edu W. & Pravettoni G.. 2014. Modulation of risky choices in recently abstinent dependent cocaine users: a transcranial direct‐current stimulation study. Front. Hum. Neurosci. 8: 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Batista, E.K. , Klauss J., Fregni F., et al 2015. A randomized placebo‐controlled trial of targeted prefrontal cortex modulation with bilateral tDCS in patients with crack‐cocaine dependence. Int. J. Neuropsychopharmacol. 18: doi:10.1093/ijnp/pyv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Hayashi, T. , Hyun J., Strafella A.P. & Dagher A.. 2013. Dorsolateral prefrontal and orbitofrontal cortex interactions during self‐control of cigarette craving. Proc. Natl. Acad. Sci. U.S.A. 110: 4422–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Li, X. , Hartwell K.J., Owens M., et al 2013. Repetitive transcranial magnetic stimulation of the dorsolateral prefrontal cortex reduces nicotine cue craving. Biol. Psychiatry 73: 714–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Eichhammer, P. 2003. High‐frequency repetitive transcranial magnetic stimulation decreases cigarette smoking. J. Clin. Psychiatry 64: 951–953. [DOI] [PubMed] [Google Scholar]
- 194. Dinur‐Klein, L. , Dannon P., Hadar A., et al 2014. Smoking cessation induced by deep repetitive transcranial magnetic stimulation of the prefrontal and insular cortices: a prospective, randomized controlled trial. Biol. Psychiatry 76: 742–749. [DOI] [PubMed] [Google Scholar]
- 195. Fregni, F. , Liguori P., Fecteau S., et al 2008. Cortical stimulation of the prefrontal cortex with transcranial direct current stimulation reduces cue‐provoked smoking craving: a randomized, sham‐controlled study. J. Clin. Psychiatry 69: 32–40. [DOI] [PubMed] [Google Scholar]
- 196. Boggio, P.S. , Liguori P., Sultani N., et al 2009. Cumulative priming effects of cortical stimulation on smoking cue‐induced craving. Neurosci. Lett. 463: 82–86. [DOI] [PubMed] [Google Scholar]
- 197. Fecteau, S. , Agosta S., Hone‐Blanchet A., et al 2014. Modulation of smoking and decision‐making behaviors with transcranial direct current stimulation in tobacco smokers: a preliminary study. Drug Alcohol Depend. 140: 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Thirugnanasambandam, N. , Grundey J., Adam K., et al 2011. Nicotinergic impact on focal and non‐focal neuroplasticity induced by non‐invasive brain stimulation in non‐smoking humans. Neuropsychopharmacology 36: 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Rapinesi, C. , Curto M., Kotzalidis G.D., et al 2015. Antidepressant effectiveness of deep Transcranial Magnetic Stimulation (dTMS) in patients with Major Depressive Disorder (MDD) with or without Alcohol Use Disorders (AUDs): a 6‐month, open label, follow‐up study. J. Affect. Disord. 174: 57–63. [DOI] [PubMed] [Google Scholar]
- 200. Roth, Y. , Zangen A. & Hallett M.. 2002. A coil design for transcranial magnetic stimulation of deep brain regions. J. Clin. Neurophysiol. 19: 361–370. [DOI] [PubMed] [Google Scholar]
- 201. Kedzior, K. , Gellersen H., Brachetti A. & Berlim M.. 2015. Deep transcranial magnetic stimulation (DTMS) in the treatment of major depression: an exploratory systematic review and meta‐analysis. J. Affect. Disord. 187: 73–83. [DOI] [PubMed] [Google Scholar]
- 202. Ceccanti, M. & Inghilleri M.. 2015. Deep TMS on alcoholics: effects on cortisolemia and dopamine pathway modulation. A pilot study. Can. J. Psychiatry 290: 283–290. [DOI] [PubMed] [Google Scholar]
- 203. Goudriaan, A.E. , De Ruiter M.B., Van Den Brink W., et al 2010. Brain activation patterns associated with cue reactivity and craving in abstinent problem gamblers, heavy smokers and healthy controls: an fMRI study. Addict. Biol. 15: 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Fu, L.P. , Bi G.H., Zou Z.T., et al 2008. Impaired response inhibition function in abstinent heroin dependents: an fMRI study. Neurosci. Lett. 438: 322–326. [DOI] [PubMed] [Google Scholar]
- 205. Lev‐Ran, S. , Shamay‐Tsoory S.G., Zangen A. & Levkovitz Y.. 2012. Transcranial magnetic stimulation of the ventromedial prefrontal cortex impairs theory of mind learning. Eur. Psychiatry 27: 285–289. [DOI] [PubMed] [Google Scholar]
- 206. Rose, J.E. , McClernon F.J., Froeliger B., et al 2011. Repetitive transcranial magnetic stimulation of the superior frontal gyrus modulates craving for cigarettes. Biol. Psychiatry 70: 794–799. [DOI] [PubMed] [Google Scholar]
- 207. Hanlon, C.A. , Canterberry M., Taylor J., et al 2013. Functional and structural integrity of frontal‐striatal circuits in cocaine users: an interleaved TMS‐BOLD & DTI study: Abstract Biol. Psych. 73: 94S. [Google Scholar]
- 208. Hallett, M. 2007. Transcranial magnetic stimulation: a primer. Neuron 55: 187–199. [DOI] [PubMed] [Google Scholar]
- 209. Isserles, M. , Rosenberg O., Dannon P., et al 2011. Cognitive‐emotional reactivation during deep transcranial magnetic stimulation over the prefrontal cortex of depressive patients affects antidepressant outcome . J. Affect. Disord. 128: 235–242. [DOI] [PubMed] [Google Scholar]
- 210. Amiaz, R. , Levy D., Vainiger D., et al 2009. Repeated high‐frequency transcranial magnetic stimulation over the dorsolateral prefrontal cortex reduces cigarette craving and consumption. Addiction 104: 653–660. [DOI] [PubMed] [Google Scholar]
- 211. Moyer, A. , Finney J.W., Swearingen C.E. & Vergun P.. 2002. Brief interventions for alcohol problems: a meta‐analytic review of controlled investigations in treatment‐seeking and non‐treatment‐seeking populations. Addiction 97: 279–292. [DOI] [PubMed] [Google Scholar]
- 212. McGirr, A. , Van den Eynde F., Tovar‐Perdomo S., et al 2015. Effectiveness and acceptability of accelerated repetitive transcranial magnetic stimulation (rTMS) for treatment‐resistant major depressive disorder: an open label trial. J. Affect. Disord. 173: 216–220. [DOI] [PubMed] [Google Scholar]
- 213. Baeken, C. , Marinazzo D., Wu G.‐R., et al 2014. Accelerated HF‐rTMS in treatment‐resistant unipolar depression: Insights from subgenual anterior cingulate functional connectivity. World J. Biol. Psychiatry 15: 286–297. [DOI] [PubMed] [Google Scholar]
- 214. Holtzheimer, P.E. , McDonald W.M., Mufti M., et al 2010. Accelerated repetitive transcranial magnetic stimulation for treatment‐resistant depression. Depress. Anxiety 27: 960–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Cook, I.A. , Schrader L.M., DeGiorgio C.M., et al 2013. Trigeminal nerve stimulation in major depressive disorder: acute outcomes in an open pilot study. Epilepsy Behav. 28: 221–226. [DOI] [PubMed] [Google Scholar]
- 216. Hamada, M. , Murase N., Hasan A., et al 2013. The role of interneuron networks in driving human motor cortical plasticity. Cereb. Cortex 23: 1593–1605. [DOI] [PubMed] [Google Scholar]
- 217. Hamada, M. & Ugawa Y.. 2010. Quadripulse stimulation–a new patterned rTMS. Restor. Neurol. Neurosci. 28: 419–424. [DOI] [PubMed] [Google Scholar]
- 218. Hamada, M. , Terao Y., Hanajima R., et al 2008. Bidirectional long‐term motor cortical plasticity and metaplasticity induced by quadripulse transcranial magnetic stimulation . J. Physiol. 586: 3927–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Grundey, J. , Thirugnanasambandam N., Kaminsky K., et al 2012. Neuroplasticity in cigarette smokers is altered under withdrawal and partially restituted by nicotine exposition. J. Neurosci. 32: 4156–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Goldstein, B.L. & Klein D.N.. 2014. A review of selected candidate endophenotypes for depression. Clin. Psychol. Rev. 34: 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]