

Abstract
For many decades it has been known that tumor DNA is shed into the blood. As a consequence of technological limitations, researchers were unable to comprehensively characterize circulating DNA. The advent of ultrasensitive and highly specific molecular assays has provided a comprehensive profile of the molecular characteristics and dynamics of circulating DNA in healthy subjects and cancer patients. With these new tools in hand, significant interest has been provoked for an innovative type of tumor biopsy termed a “liquid biopsy”. Liquid biopsies are obtained by minimal invasive blood draws from cancer patients. Circulating cancer cells, exosomes and a variety of molecules contained within the liquid biopsy including cell‐free circulating tumor DNA (ctDNA) can serve as promising tools to track cancer evolution. Attractive features of ctDNA are that ctDNA isolation is straightforward, ctDNA levels increase or decrease in response to the degree of tumor burden and ctDNA contains DNA mutations found in both primary and metastatic lesions. Consequently, the analysis of circulating DNA for cancer‐specific mutations might prove to be a valuable tool for cancer detection. Moreover, the capacity to screen for ctDNA in serial liquid biopsies offers the possibility to monitor tumor progression and responses to therapy and to influence treatment decisions that ultimately may improve patient survival. Here we focus on mutation detection in ctDNA and provide an overview of the characteristics of ctDNA, detection methods for ctDNA and the feasibility of ctDNA to monitor tumor dynamics. Current challenges associate with ctDNA will also be discussed.
Keywords: ctDNA, liquid biopsy, minimal‐invasive
Abbreviations
- cfDNA
cell free DNA
- CTC
circulating tumor cell
- ctDNA
circulating tumor DNA
- dPCR
digital PCR
- NGS
next‐generation sequencing
- NSCLC
Non‐small cell lung cancers
- PCR
polymerase chain reaction.
The diagnosis, staging and therapeutic management of tumors relies upon cancer imaging systems and histologic or cytologic examination of tumor specimens.1, 2 The widespread integration of biomedical imaging systems with other diagnostic and screening tools has contributed to decreased mortality rates for certain cancer types.1, 2 Although a tumor biopsy is a key diagnostic tool that also guides therapy, tumor biopsies have limitations. For instance, tumor location and size may limit the utility of biopsies. With regard to the molecular diagnosis of DNA mutations, a tumor biopsy may not fully capture the molecular heterogeneity within a tumor.2, 3, 4 Consequently, a tumor may respond poorly when only a portion of tumor cells within a tumor mass harbor genetic mutations that render them susceptible to targeted molecular therapies.4 Moreover, the molecular differences between primary and metastatic tumor lesions not captured by tumor biopsies may negatively impact overall tumor burden in patients.3, 5 Despite these limitations, tumor biopsies remain a primary diagnostic tool as histopathologic examination of tumor specimens is required to render an accurate medical diagnosis.
Recent technological advancements in next‐generation sequencing and quantitative polymerase chain reaction (PCR) have contributed to the growing interest amongst clinicians and researchers in an innovative type of biopsy termed a liquid biopsy.2, 5, 6 In a liquid biopsy, blood is drawn, processed and then analyzed for the presence of circulating tumor cells (CTCs),7 cancer‐derived exosomes8 and cell‐free tumor DNA (ctDNA).4 The major obstacle for liquid biopsies to be effective as diagnostic and screening tools is sensitivity since the majority of cells in the blood are normal cells and blood also contains exosomes and cell‐free DNA released by normal cells.4, 9 For example the frequency of cell‐free DNA containing tumor‐associated mutations might be as low as 0.01%4, 6, 9 and between 0 and 6 CTCs have been detected per 7.5 mL of peripheral blood.7, 10 To meet this challenge, researchers have devised a variety of highly sensitive and specific molecular diagnostic techniques.6, 10 An attractive feature of liquid biopsies is the potential to monitor tumor progression and response to therapy in a minimal‐invasive manner.9 In addition, the analysis of serial liquid biopsies may lead to the identification of newly acquired DNA mutations in therapy resistant tumors that make them vulnerable to other cancer drugs.11
Circulating tumor cells (CTCs)10 and tumor‐derived proteins,12 RNAs.8 and ctDNA4 are considered as potential cancer biomarkers contained within a liquid biopsy. The isolation, characterization and analysis of each of them have their unique challenges. The advent of highly sensitivity PCR methods and refinements to next‐generation sequencing (NGS) has sparked much interest in ctDNA as a viable tool to provide diagnostic and prognostic information of cancers monitoring.2 These include DNA mutations, epigenetic alterations and other forms of tumor‐specific abnormalities. Here, we provide a discussion on the functionality of ctDNA as a clinically feasible tool to monitor tumor development by detecting tumor specific mutations.
Origins and Molecular Characteristics of ctDNA
In healthy individuals, cell free DNA (cfDNA) is released into the circulation by cells undergoing apoptosis.13, 14 Apoptotic cells shed DNA fragments approximately 185 to 200 bp in length.14, 15 In contrast, cfDNA of cancer patients is derived from both non‐malignant and malignant cells. Interestingly, the percentage of circulating DNA derived from cancer cells ranges from 3% to as much as 93%.16 Since cancer cells die through multiple mechanisms that ultimately lead to DNA cleavage such as apoptosis, necrosis and autophagy,17 ctDNA displays less uniformity in size and integrity relative to cfDNA in healthy individuals.14, 15, 17 Some researcher groups report that ctDNA has less integrity and is smaller relative to cfDNA13, 14, 15 while others report the opposite.18, 19 These conflicting results may be a result of the different DNA isolation methods used and the sample sources tested. More recently, one study showed that decreased cfDNA integrity and increased cfDNA concentrations distinguish normal individuals from patients with primary and metastatic breast cancer.20 A second study used single‐base pair resolution sequencing to demonstrate that the shorter DNA fragments in cancer patient plasma preferentially contained tumor‐associated copy number alterations.21 Collectively, these two reports suggest that the isolation and analysis of short DNA fragments may improve the sensitivity of detection of ctDNA. Irrespective of DNA fragment size, the ability to detect and quantify ctDNA fragments carrying tumor‐specific mutations has maintained interest in ctDNA as a useful tool to monitor tumor growth.
Detection of ctDNA
Given that the frequency of mutant DNA alleles in cfDNA is as low as 0.01%,4 highly sensitive and specific detection methods are required for ctDNA to serve as a clinically feasible approach. Here we provide a brief description of the ctDNA detection methods capable of such high specificity and sensitivity. Initial studies on ctDNA relied upon real‐time allele‐specific PCR to detect mutations of interest.22 However, primarily patients with high tumor burden, and thus higher ctDNA levels in the plasma,23 were chosen for these studies as the sensitivity and specificity of real‐time PCR approaches is limited.22 Digital PCR (dPCR) has increased sensitivity and specificity relative to real‐time PCR.22 With dPCR, a DNA sample is segregated using limiting dilution such that individual DNA molecules are captured within water‐oil emulsion droplets or chambers.24 A portion of the droplets or chambers will contain no DNA while others will contain individual DNA molecules. The segregated individual DNA molecules may be wild type or mutant in nucleotide sequence. Using unique sets of primers and probes, mutant and normal DNA sequences are amplified, quantified and the percent mutant allele frequency determined. For liquid biopsy samples, dPCR is capable of detecting mutant alleles with a fractional abundance of 0.005%25 to 0.04%.26 There are several advantageous features to dPCR including precision, sensitivity and increased resistance to PCR inhibitors.24, 26, 27 In addition, dPCR is capable of absolute measurements of rare DNA alleles without the need for a standard curve.24, 28, 29 Recently novel modifications to dPCR have been introduced with the intention of detecting highly infrequent mutant DNA molecules in plasma.9, 30, 31, 32 The ability to detect these extremely rare mutant DNA molecules might allow for efficient screening and diagnosis of cancers at earlier stages with liquid biopsies.
The analytical sensitivity of dPCR is one advantageous feature that is counterbalanced by the limited capacity of dPCR to interrogate multiple genomic alterations simultaneously.33 Without prior and precise knowledge of the DNA rearrangements, translocations and mutations of interest, dPCR is compromised in its utility to detect cancer and monitor tumor growth. Massive parallel sequencing, also known as next‐generation sequencing (NGS), offers to circumvent this limitation as NGS has the ability of detect multiple somatic DNA alterations simultaneously.33, 34, 35 Early ctDNA studies with NGS demonstrated the ability of NGS to detect chromosomal rearrangements and chromosomal copy number changes.35, 36 In the setting of metastatic prostate cancer, Heitzer and colleagues36 devised a genome‐wide NGS approach capable of identifying novel genomic alterations in ctDNA associated with the emergence of metastatic tumor clones. Impressively, this approach provided genomic cancer profiles in 2 days thus making it a more feasible test in a clinical setting.36 A shortcoming of most NGS‐based approaches is the low sensitivity that renders them unfit to be reliable molecular diagnostic tests.34, 35, 37 More recently, Newman and colleagues devised an ultrasensitive NGS‐based approached termed CAncer Personalized Profiling by deep Sequencing (CAPP‐Seq) with the ability to detect mutant allele fractions as low as 0.02% and with 96% specificity.38 Importantly, CAPP‐Seq reduces cost to approximately $200–300 US dollars per test.33 Further refinements to both dPCR and NGS may ultimately make ctDNA cancer screening a reality.
Cancer Surveillance With ctDNA
A small number of studies have examined the feasibility of ctDNA in detecting early stage cancers and fewer still have considered ctDNA for screening pre‐symptomatic cancer patients.39, 40, 41, 42, 43 Since effective screening of asymptomatic patients requires prior knowledge of mutations in genes commonly altered in low grade cancers, ctDNA is most suited for the detection of advanced cancers with well‐known mutations. Complicating matters further is the observation that healthy subjects have detectable mutations in their circulating DNA. For instance, during minimal‐invasive prenatal testing, Amant and coworkers detected genomic alterations in 3 of 4,000 asymptomatic pregnant women that were later confirmed to have malignancies by magnetic resonance imaging and biopsy.40 In addition, the percentage of individuals with mutations in the blood increases with age44, 45 at a rate that exceeds the clinical incidence of hematological malignancies in the general population.44 Moreover, independent studies reported that mutations in KRAS and TP53 in the circulating DNA of non‐cancer control subjects range from 1 to 3.6%.46 Collectively these studies suggest that cfDNA of healthy subjects contains mutations that might pose a significant challenge in accurately diagnosing and screening patients for cancer.47 On the other hand, pre‐symptomatic testing for cancer might be more feasible with ctDNA than originally thought. Interestingly, Sausen and coworkers demonstrated that pancreatic cancer recurrence was predicted 6.5 months earlier with ctDNA detection relative to CT scanning48; thus supporting the notion that ctDNA might be useful in screening and diagnosing asymptomatic patients. Additional studies combining ctDNA analysis with biomedical imaging will ultimately determine the benefits of ctDNA in cancer surveillance as an adjunct diagnostic test.
Monitoring tumor burden and responses to therapy using ctDNA
Studies in multiple cancers have shown a pattern where ctDNA levels buildup as tumors progress and then decline following surgery or drug treatment.23, 32, 37, 49, 50 Thus it seems logical that ctDNA levels might be used as a surrogate marker of tumor burden and therapeutic responses. Conceptually, ctDNA has clear advantages to monitor tumor burden and therapeutic response compared with protein cancer markers and biomedical imaging. For instance, ctDNA has a half‐live of approximately 2 h9 while proteins persist in the blood for weeks to months,51, 52 thus analysis of ctDNA should permit a more rapid assessment of tumor changes within hours rather than weeks to months.53 Tumor specific protein markers have the limitation of being elevated in circumstances not related to tumor growth54 while elevated ctDNA levels are associated with tumor changes. Moreover, a direct comparison of ctDNA, CTC and cancer antigen 15–3 levels in the same patients revealed that ctDNA levels showed a greater correlation with tumor burden and the earliest measure of response to therapy,23 suggesting that ctDNA levels more accurately predict tumor changes relative to protein markers55 (Fig. 1). With regard to imaging techniques, increases in ctDNA levels predicted tumor recurrence much earlier than conventional tumor imaging techniques.38, 48, 56
Figure 1.
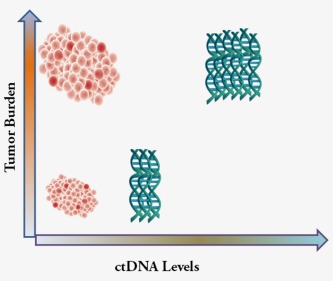
Tumor burden and plasma ctDNA levels show a direct correlation. As tumor burden increases, ctDNA accumulates in the plasma. With therapeutic intervention, tumor burden and ctDNA levels decrease. Thus, ctDNA can serve as a surrogate marker of tumor progression and regression. [Color figure can be viewed at wileyonlinelibrary.com]
The genetic basis of resistance by tumors to targeted molecular therapies has recently been determined.57, 58, 59, 60 Non‐small cell lung cancers (NSCLC) bearing activating mutations in the EGFR gene are extremely sensitive to the small molecule inhibitors erlotinib and gefitinib.59 In approximately 50% of NSCLC patients resistance to EGFR inhibition is driven by a gatekeeper mutation in the EGFR kinase domain that changes threonine 790 to a methionine (T790M). The T790M mutation increases the affinity of the EGFR kinase domain for ATP while decreasing its affinity to erlotinib and gefitinib. In the case of colorectal cancers, mutations in KRAS (G12R and G13D) activate survival pathways that drive resistance to EGFR monoclonal antibodies.56 With such precise knowledge of the molecular basis of response to targeted therapies, the possibility of detecting resistance in a minimal‐invasive manner in the blood has been examined.61, 62 As might be predicted, detection of the T790M gatekeeper mutation was shown to correlate with therapy resistance and disease progression for NSCLC patients treated with EGFR inhibitors.11, 63 Impressively, in some cases, T790M‐driven resistance was detected 16 weeks before tumor progression was detectable with radiographic imaging.63 In colon cancer, ctDNA analysis revealed that the presence of mutant KRAS was associated either primary or acquired resistance to EGFR inhibition.62, 64 Upon withdraw of anti‐EGFR antibodies, mutant Ras levels dropped and relatively novel mutations associated with either primary or acquired resistance were found in NRAS, MET, ERBB2, FLT3, EGFR and MAPK21.62
Thus far, several studies show that the analysis of ctDNA levels is a powerful tool to monitor tumor dynamics and response to therapy. In analyzing ctDNA, novel mutations associated with resistance to conventional and targeted therapy have been identified.62, 65 These newly identified mutations might offer new avenues of treatment for cancer patients with advanced disease.
Current Challenges and Limitations
Although ctDNA represents a promising “real time” tool for tumor characterization and is now being extensively studied, there are still many challenges that need to be overcome before it's routinely used by clinicians. One major challenge with analysis of ctDNA is sensitivity and specificity. ctDNA is not always detectable in peripheral blood due to the extremely low level presented in the blood. In addition, current digital PCR method preferably measures relatively longer fragments, i.e., >120 bp, while smaller fragments derived from tumors are not detected efficiently.66 Therefore, the results from ctDNA analysis might be false negative. Many studies have shown an average of 70–80% concordance between tumor somatic mutations and the presence of ctDNA.67, 68 This could be improved by the advances in genomic approaches that have higher sensitivity to identify all ctDNA in the blood. Additionally, we do not have a standard and validated procedure for samples handling and liquid biopsy analysis, both aspects would affect the final results. A standard operation procedure should be established and be strictly followed to ensure we get the right information from ctDNA analysis.
In addition to ctDNA, CTCs, cell free RNAs and exosomes also attract extensive attention for their potential clinical application. Compared to these measurements, which can address tumor status at DNA, RNA and protein levels, ctDNA results are restricted to genomic alterations. Some drug resistance can only be reflected by mRNA or protein expression analysis, such as AR‐V7.69 In this situation, ctDNA analysis may not be a good choice to monitor tumor development. In contrast to tumor tissues, ctDNA represents mainly the genome of dying tumor cells, or the cells that respond to therapy, one cannot ruled the exist of resistant cells or subpopulations that contain few or no dying cells based on ctDNA analysis. Selection of the appropriate time points for ctDNA screening will be very important in some situation. Also, results from ctDNA analysis cannot determine the exact origin of a mutation, i.e., whether it is somatic or germline. The discovery of germline mutations might be an important indication for genetic screening for the patients' family members as well.
Conclusion and perspectives
The shedding of ctDNA into the circulation offers researchers and clinicians a novel target that carries the same DNA mutations found in primary and metastatic cancer lesions. Access to ctDNA is minimal‐invasive and simple as it involves a standard blood draw. With newly developed ultrasensitive and specific methods to detect and characterize genomic alteration present in ctDNA the opportunity exists to monitor tumor progression and recurrence. Tumor burden and recurrence have been shown to directly correlate to ctDNA levels in patients and the frequency of ctDNA molecules carrying mutations that confer resistance to cancer therapies. In some studies, detection of ctDNA is superior in predicting tumor recurrence relative to conventional tumor imaging techniques. With further refinements to the methods to detect and quantify ctDNA, it may be possible to screen asymptomatic patients with ctDNA to detect cancers at their earliest stages (Fig. 2). As discussed above, many limitations associated with ctDNA still exist. Further studies should be launched to validate the potential of ctDNA for routine clinical use. However, based on the large amounts of promising data and with the rapid advances and the decreased cost of next‐generation sequencing, we anticipate there is much promise in influencing cancer patient treatment and survival with ctDNA. In addition to ctDNA, cfRNA, CTCs, circulating exosomes or blood platelets are also promising as novel liquid biopsy candidates.70, 71, 72 The advantages and disadvantages of these technologies are worth to be further explored.
Figure 2.
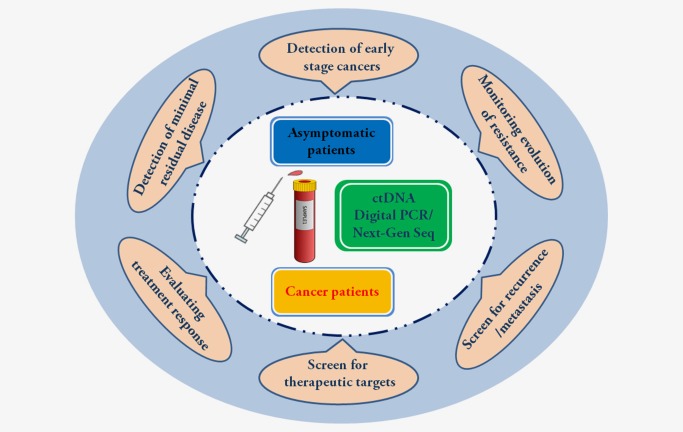
Clinical application of ctDNA as a tool for cancer monitoring. ctDNA can be obtained from plasma and in combination with digital PCR and next‐generation sequencing (Next‐Gen Seq) might allow for detection of cancers at their earliest stages. In some reports, ctDNA detection predicts cancer recurrence or progression months earlier than conventional cancer imaging methods. ctDNA could be applied in cancer diagnosis, prognosis and therapy monitoring. [Color figure can be viewed at wileyonlinelibrary.com]
Contributor Information
Xin Yi, Email: yix@geneplus.org.cn.
Xuefeng Xia, Email: xuefengx@gmail.com.
References
- 1. Fass L. Imaging and cancer: a review. Mol Oncol 2008;2:115–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Francis G, Stein S. Circulating cell‐free tumour DNA in the management of cancer. Int J Mol Sci 2015;16:14122–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allison KH, Sledge GW. Heterogeneity and cancer. Oncology (Williston Park) 2014;28:772–8. [PubMed] [Google Scholar]
- 4. Yong E. Cancer biomarkers: Written in blood. Nature 2014;511:524–6. [DOI] [PubMed] [Google Scholar]
- 5. De Mattos‐Arruda L, Weigelt B, Cortes J, et al. Capturing intra‐tumor genetic heterogeneity by de novo mutation profiling of circulating cell‐free tumor DNA: a proof‐of‐principle. Ann Oncol 2014;25:1729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 2005;102:16368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lalmahomed ZS, Kraan J, Gratama JW, et al. Circulating tumor cells and sample size: the more, the better. J Clin Oncol 2010;28:e288–9; author reply e90. [DOI] [PubMed] [Google Scholar]
- 8. Sohn W, Kim J, Kang SH, et al. Serum exosomal microRNAs as novel biomarkers for hepatocellular carcinoma. Exp Mol Med 2015;47:e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayes DF, Cristofanilli M, Budd GT, et al. Circulating tumor cells at each follow‐up time point during therapy of metastatic breast cancer patients predict progression‐free and overall survival. Clin Cancer Res 2006;12:4218–24. [DOI] [PubMed] [Google Scholar]
- 11. Uchida J, Imamura F, Kukita Y, et al. Dynamics of circulating tumor DNA represented by the activating and resistant mutations in epidermal growth factor receptor tyrosine kinase inhibitor treatment. Cancer Sci 2016;107:353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawakami K, Fujita Y, Kato T, et al. Integrin beta4 and vinculin contained in exosomes are potential markers for progression of prostate cancer associated with taxane‐resistance. Int J Oncol 2015;47:384–90. [DOI] [PubMed] [Google Scholar]
- 13. Mouliere F, Rosenfeld N. Circulating tumor‐derived DNA is shorter than somatic DNA in plasma. Proc Natl Acad Sci U S A 2015;112:3178–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giacona MB, Ruben GC, Iczkowski KA, et al. Cell‐free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas 1998;17:89–97. [DOI] [PubMed] [Google Scholar]
- 15. Mouliere F, Robert B, Arnau Peyrotte E, et al. High fragmentation characterizes tumour‐derived circulating DNA. PLoS One 2011;6:e23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001;61:1659–65. [PubMed] [Google Scholar]
- 17. Umetani N, Giuliano AE, Hiramatsu SH, et al. Prediction of breast tumor progression by integrity of free circulating DNA in serum. J Clin Oncol 2006;24:4270–6. [DOI] [PubMed] [Google Scholar]
- 18. Wang BG, Huang HY, Chen YC, et al. Increased plasma DNA integrity in cancer patients. Cancer Res 2003;63:3966–8. [PubMed] [Google Scholar]
- 19. Gao YJ, He YJ, Yang ZL, et al. Increased integrity of circulating cell‐free DNA in plasma of patients with acute leukemia. Clin Chem Lab Med 2010;48:1651–6. [DOI] [PubMed] [Google Scholar]
- 20. Madhavan D, Wallwiener M, Bents K, et al. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res Treat 2014;146:163–74. [DOI] [PubMed] [Google Scholar]
- 21. Jiang P, Chan CW, Chan KC, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A 2015;112:E1317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014;32:579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013;368:1199–209. [DOI] [PubMed] [Google Scholar]
- 24. Huggett JF, Whale A. Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin Chem 2013;59:1691–3. [DOI] [PubMed] [Google Scholar]
- 25. Sanmamed MF, Fernandez‐Landazuri S, Rodriguez C, et al. Quantitative cell‐free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow‐up of patients with melanoma being treated with BRAF inhibitors. Clin Chem 2015;61:297–304. [DOI] [PubMed] [Google Scholar]
- 26. Zhu G, Ye X, Dong Z, et al. Highly sensitive droplet digital PCR method for detection of EGFR‐activating mutations in plasma cell‐free DNA from patients with advanced non‐small cell lung cancer. J Mol Diagn 2015;17:265–72. [DOI] [PubMed] [Google Scholar]
- 27. Sanders R, Huggett JF, Bushell CA, et al. Evaluation of digital PCR for absolute DNA quantification. Anal Chem 2011;83:6474–84. [DOI] [PubMed] [Google Scholar]
- 28. Hindson BJ, Ness KD, Masquelier DA, et al. High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011;83:8604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pinheiro LB, Coleman VA, Hindson CM, et al. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 2012;84:1003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Castellanos‐Rizaldos E, Paweletz C, Song C, et al. Enhanced ratio of signals enables digital mutation scanning for rare allele detection. J Mol Diagn 2015;17:284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Isobe K, Hata Y, Tochigi N, et al. Usefulness of nanofluidic digital PCR arrays to quantify T790M mutation in EGFR‐mutant lung adenocarcinoma. Cancer Genomics Proteomics 2015;12:31–7. [PubMed] [Google Scholar]
- 32. Diehl F, Li M, He Y, et al. BEAMing: single‐molecule PCR on microparticles in water‐in‐oil emulsions. Nat Methods 2006;3:551–9. [DOI] [PubMed] [Google Scholar]
- 33. Bratman SV, Newman AM, Alizadeh AA, et al. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP‐Seq. Expert Rev Mol Diagn 2015;15:715–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murtaza M, Dawson SJ, Tsui DW, et al. Non‐invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013;497:108–12. [DOI] [PubMed] [Google Scholar]
- 35. Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole‐genome sequencing. Sci Transl Med 2012;4:162ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heitzer E, Ulz P, Belic J, et al. Tumor‐associated copy number changes in the circulation of patients with prostate cancer identified through whole‐genome sequencing. Genome Med 2013;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012;4:136ra68. [DOI] [PubMed] [Google Scholar]
- 38. Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20:548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garcia‐Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 2015;7:302ra133. [DOI] [PubMed] [Google Scholar]
- 40. Amant F, Verheecke M, Wlodarska I, et al. Presymptomatic identification of cancers in pregnant women during noninvasive prenatal testing. JAMA Oncol 2015;1:814–9. [DOI] [PubMed] [Google Scholar]
- 41. Jamal‐Hanjani M, Wilson GA, Horswell S, et al. Detection of ubiquitous and heterogeneous mutations in cell‐free DNA from patients with early‐stage non‐small‐cell lung cancer. Ann Oncol 2016;27:862–7. [DOI] [PubMed] [Google Scholar]
- 42. Beaver JA, Jelovac D, Balukrishna S, et al. Detection of cancer DNA in plasma of patients with early‐stage breast cancer. Clin Cancer Res 2014;20:2643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jaiswal S, Fontanillas P, Flannick J, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xie M, Lu C, Wang J, et al. Age‐related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gormally E, Vineis P, Matullo G, et al. TP53 and KRAS2 mutations in plasma DNA of healthy subjects and subsequent cancer occurrence: a prospective study. Cancer Res 2006;66:6871–6. [DOI] [PubMed] [Google Scholar]
- 47. Krimmel JD, Schmitt MW, Harrell MI, et al. Ultra‐deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc Natl Acad Sci U S A 2016;113:6005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sausen M, Phallen J, Adleff V, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun 2015;6:7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shinozaki M, O'Day SJ, Kitago M, et al. Utility of circulating B‐RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin Cancer Res 2007;13:2068–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bidard FC, Madic J, Mariani P, et al. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int J Cancer 2014;134:1207–13. [DOI] [PubMed] [Google Scholar]
- 51. Riedinger JM, Wafflart J, Ricolleau G, et al. CA 125 half‐life and CA 125 nadir during induction chemotherapy are independent predictors of epithelial ovarian cancer outcome: results of a French multicentric study. Ann Oncol 2006;17:1234‐ [DOI] [PubMed] [Google Scholar]
- 52. Ito K, Hibi K, Ando H, et al. Usefulness of analytical CEA doubling time and half‐life time for overlooked synchronous metastases in colorectal carcinoma. Jpn J Clin Oncol 2002;32:54–8. [DOI] [PubMed] [Google Scholar]
- 53. Kidess E, Heirich K, Wiggin M, et al. Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, high‐sensitivity multiplexed mutation detection platform. Oncotarget 2015;6:2549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate‐specific antigen level < or =4.0 ng per milliliter. N Engl J Med 2004;350:2239–46. [DOI] [PubMed] [Google Scholar]
- 55. Chen KZ, Lou F, Yang F, et al. Circulating Tumor DNA Detection in Early‐Stage Non‐Small Cell Lung Cancer Patients by Targeted Sequencing. Sci Rep 2016;6:31985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature 2012;486:532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005;2:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gomez GG, Wykosky J, Zanca C, et al. Therapeutic resistance in cancer: microRNA regulation of EGFR signaling networks. Cancer Biol Med 2013;10:192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005;11:4182–90. [DOI] [PubMed] [Google Scholar]
- 61. Taniguchi K, Uchida J, Nishino K, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res 2011;17:7808–15. [DOI] [PubMed] [Google Scholar]
- 62. Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015;21:827. [DOI] [PubMed] [Google Scholar]
- 63. Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR‐mutant lung cancer using quantitative next‐generation genotyping of cell‐free plasma DNA. Clin Cancer Res 2014;20:1698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 2016;7:11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Norton SE, Lechner JM, Williams T, Fernando MR. A stabilizing reagent prevents cell‐free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR. Clin Biochem 2013;46:1561–5. [DOI] [PubMed] [Google Scholar]
- 67. Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015;6:42008–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rapisuwon S, Vietsch EE, Wellstein A. Circulating biomarkers to monitor cancer progression and treatment. Comput Struct Biotechnol J 2016;14:211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Boudadi K, Antonarakis ES. Resistance to novel antiandrogen therapies in metastatic castration‐resistant prostate cancer. Clin Med Insights Oncol 2016;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Melo SA, Luecke LB, Kahlert C, et al. Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015;523:177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hoshino A, Costa‐Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527:329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Best MG, Sol N, Kooi I, et al. RNA‐Seq of tumor‐educated platelets enables blood‐based pan‐cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell 2015;28:666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]