

Abstract
Current assessment of biomaterial biocompatibility is typically implemented in wild type rodent models. Unfortunately, different characteristics of the immune systems in rodents versus humans limit the capability of these models to mimic the human immune response to naturally derived biomaterials. Here we investigated the utility of humanized mice as an improved model for testing naturally derived biomaterials. Two injectable hydrogels derived from decellularized porcine or human cadaveric myocardium were compared. Three days and one week after subcutaneous injection, the hydrogels were analyzed for early and mid-phase immune responses, respectively. Immune cells in the humanized mouse model, particularly T-helper cells, responded distinctly between the xenogeneic and allogeneic biomaterials. The allogeneic extracellular matrix derived hydrogels elicited significantly reduced total, human specific, and CD4+ T-helper cell infiltration in humanized mice compared to xenogeneic extracellular matrix hydrogels, which was not recapitulated in wild type mice. T-helper cells, in response to the allogeneic hydrogel material, were also less polarized towards a pro-remodeling Th2 phenotype compared to xenogeneic extracellular matrix hydrogels in humanized mice. In both models, both biomaterials induced the infiltration of macrophages polarized towards a M2 phenotype and T-helper cells polarized towards a Th-2 phenotype. In conclusion, these studies showed the importance of testing naturally derived biomaterials in immune competent animals and the potential of utilizing this humanized mouse model for further studying human immune cell responses in an in vivo environment.
Keywords: Immune Response, Extracellular Matrix (ECM), Cardiac Tissue Engineering, Biocompatibility, Scaffold, Macrophage
1. Introduction
The field of decellularized extracellular matrix (ECM) based biomaterials is rapidly growing and has developed therapies for numerous applications including wound healing, hernia repair, skeletal muscle defect repair, and myocardial infarction [1–4]. Decellularized ECM biomaterials are an attractive platform for biomaterial therapies since tissue derived ECM can promote tissue remodeling by influencing cellular metabolism, proliferation, migration, maturation, and differentiation [5]. In fact, these biomaterials, derived from xenogeneic and allogeneic tissue sources [6, 7], have been successfully implanted into millions of patients [8]. Xenogeneic materials, from porcine tissue for example, are readily available and can be produced from younger tissue sources, which is desirable for regenerative medicine therapies [9]. However, xenogeneic materials can have potential immunogenic issues, regulatory hurdles and xenogeneic disease transfer. Allogeneic materials avoid some concerns associated with xenogeneic materials, but are typically from older and more limited cadaveric sources, and can have larger batch variability.
While xenogeneic and allogeneic sources for decellularized ECM have been widely used to date, preclinical understanding of these scaffolds is mostly based off immune responses to these matrices in rodents and a few large animals [1, 10, 11]. Given difficulties with obtaining sequential patient biopsies, no one has thoroughly monitored or understood the human immune response to these materials. Although connected evolutionarily, rodents typically used for biocompatibility testing provide limited representation of the human immune response. Differences in immune cell receptors, cytokine expression and response to various stimuli highlight how responses in rodents might not correlate with outcomes in humans [12]. Even non-human hominids have various biomedical differences from humans [13]. This combined with our incomplete understanding of the human immune system has led to the removal of several well characterized materials from the market [14, 15].
One method to address these shortcomings is the use of a humanized mouse (Hu-mice) model for preclinical assessment of the human immune response. Over the last 20 years, significant improvements have transformed Hu-mice into a valuable model for mimicking the human immune response [16–18]. In particular, Hu-mice developed by implantation of human fetal thymus tissue and injection of human CD34+ fetal liver cells into immune compromised NSG mice have been shown to be robust and contain human T-cells, B-cells, and dendritic cells, allowing the ability to reject xenogeneic tissue [19]. This model has been used extensively for studying autoimmune disease, virus infections, xenogeneic transplantation, and more recently allogeneic stem cell transplantation [20]. However, it has yet to be exploited in the biomaterials field. In this study, we utilized this Hu- mouse model to assess the human immune response to decellularized ECM biomaterials, specifically injectable hydrogels derived from porcine or human myocardium, which were initially developed to treat the heart post-myocardial infarction [11, 21–23]. Our goal with this study was to evaluate the utility of the Hu-mice for evaluating biocompatibility and studying the human immune response to biomaterials prior to clinical translation. We hypothesized that this model would demonstrate different immune responses to human versus xenogeneic ECM, unlike a wild type rodent model.
2. Methods and Materials
All experiments in this study were performed in accordance with the guidelines established by the committee on Animal Research at the University of California, San Diego, and the American Association for Accreditation of Laboratory Animal Care.
2.1. Fabrication of PMM, HMM, and NDM
Both the porcine myocardial matrix (PMM) and human myocardial matrix (HMM) were developed and characterized according to established protocols [21, 23]. Human hearts were obtained from donor patients whose hearts could not be used for transplantation under an institutionally approved protocol. In brief, left ventricular tissue (porcine or human) was isolated and chopped into small pieces. The tissue was spun in phosphate buffered saline (PBS) containing 1% (wt/vol) sodium dodecyl sulfate (SDS) (Fischer Scientific, Fair Lawn, NJ) with 0.5% penicillin streptomycin (PS) (Gibco, Life Technologies, Grand Island, NY) of 10,000 U/mL until fully decellularized. The human tissue was treated with additional lipid and DNA/RNA removal steps that were needed to fully decellularize the tissue [23]. Once decellularized, the remaining ECM was lyophilized, milled, and partially digested with pepsin into a liquid form as previously described [21, 23]. Non-decellularized myocardial matrix (NDM) was also produced from porcine ventricular tissue as a control. The tissue was simply rinsed in the PBS and PS solution with no SDS for one day. Then, the non-decellularized porcine tissue was processed into an injectable form using the same methods as the decellularized myocardial matrix. Finally, the materials were lyophilized and stored at −80°C until re-suspending with sterile water prior to injection.
2.2. Hydrogel Characterization
Porcine and human myocardial matrix hydrogels were imaged for nano-scale topography and fiber formation with scanning electron microscopy (SEM) as previously described [23, 24]. In brief, samples were gelled for 24 hours at 37°C and then fixed in a solution of 4% paraformald ehyde and 4% glutaraldehyde for 24 hours. Next, the gels were dehydrated with a series of gradated ethanol rinses. Then, fixed and dehydrated hydrogels were processed in an automated critical point drier (Leica EM CPD300, Leica, Vienna). Mounted samples were subsequently sputter coated (Leica SCD500, Leica, Vienna) with platinum while being rotated. The samples were then imaged on a FE-SEM (Sigma VP, Zeiss Ltd, Cambridge, UK) at 0.6 kV using the in-lens SE1 detector.
Immunohistochemistry was performed on the porcine derived material to assess removal of the alpha-gal epitope. Freshly isolated porcine left ventricular tissue, decellularized porcine myocardium, and porcine myocardial matrix hydrogels were fresh frozen in OTC for cyrosectioning. Sections (20 μm) were mounted onto glass slides, fixed in 4% paraformaldehyde for 10 minutes and permeabilized in acetone for 1.5 min. Slide samples were either stained with hematoxylin and eosin (H&E) or prepared for immunohistochemistry. Samples were blocked with a buffered solution containing bovine serum albumin (BSA) and stained for at least 12 hours at 4°C with M86 anti-alpha-gal (1:10, Enzo Life Sciences, Framingdale, NY) followed by incubation for 30 minutes with secondary anti-mouse Alexa Fluor 488 antibody (1:100, Life technologies, Carlsbad, CA) [25]. Hoechst 33342 was used to stain nuclei. Slides were imaged with a Carl Zeiss Observer D1 and Zeiss AxioVision SE64 software (Carl Zeiss, Jena, Germany).
2.3. Humanized Mouse Model
NOD.Cg-PrkdcscidII2rgtm1wjl/SzJ (NSG) (The Jackson Laboratory) mice of 6–10 weeks of age after conditioning with sublethal (2.25 Gy) total body irradiation underwent the following procedure, as previously described, to create the humanized mouse model (Hu-mice) [17]. First, the mice were transplanted under the kidney capsule with a piece of human fetal thymic tissue of about 1 mm3 that had been previously frozen. Next, the animals were transfused intravenously with 1−5 × 105 human CD34+ fetal liver cells from the same patient donor. Human fetal tissue, from Advanced Bioscience Resource, with gestational ages of 17–20 weeks was utilized.
2.4. Biomaterial Injection and Harvesting
Animals were briefly put under anesthesia using either 2.5% isoflurane or via injection with ketamine and xylazine. Each mouse was injected with only one type of biomaterial and received four 250 μL evenly spaced subcutaneous injections in the dorsal region. Each injection was premixed with 0.5 μL of sterile india ink to visually label the matrices for ease of identification upon harvesting. The injections, along with neighboring dermal tissue and spleens, were harvested three days and 1-week later for analysis with histology and immunohistochemistry (n=8−16), flow cytometry (n=4), or qRT-PCR (n=8−12). Along with the Hu-mice, both male NSG and male Balb/c (Jackson Laboratories and Harlan Laboratories, respectively) of the same ages were used for immune compromised and wild type immune system controls, respectively.
2.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
After harvesting, tissue was immediately flash frozen in liquid nitrogen and stored at −80°C until processed. Tissue was homogenized and then run through an RNEasy kit (Qiagen, Germantown, MD) along with an on-column DNase digestion step (Qiagen) to extract RNA with minimal genomic DNA contamination. Superscript III Reverse Transcriptase kit (Applied Biosystems, Foster City, MA) was used to synthesize cDNA. Then, SYBR Green PCR Master Mix (Applied Biosystems) was used with forward and reverse primers at a final concentration of 1 μM. Primers for T-helper cells were designed as mouse or human specific for analyzing Balb/c and Hu-mice samples, respectively, with lack of amplification with human primers confirmed in mouse samples. Primers included: mouse ARG1 (F: 5′-GAACACGGCAGTGGCTTTAAC-3′, R: 5′-TGCTTAGTTCTGTCTGCTTTGC-3′), mouse NOS2 (F: 5′-CAGCTGGGCTGTACAAACCTT-3′, R: 5′-CATTGGAAGTGAAGCGTTTCG-3′), mouse GATA3 (F: 5′-CTCGGCCATTCGTACATGGAA-3′, R: 5′-GGATACCTCTGCACCGTAGC-3′), mouse TBX21 (F: 5′-AGCAAGGACGGCGAATCTT-3′, R: 5′-GGGTGGACATATAAGCGGTTC-3′), human GATA3 (F: 5′-CGGCATCTGTCTTGTCCCTA-3′, R: 5′-ATGCACGCTGGTAGCTCATA-3′), human TBX21 (F: 5′-ACAGCTATGAGGCTGAGTTTCGA-3′, R: 5′-GGCCTCGGTAGTAGGACATGGT-3′), human CRTH2 (F: 5′-CCCTCTGGGCACTGGTAATC-3′, R: 5′-CAGGTGGAGGAATGAGACGG-3′), human CCR5 (F: 5′-CAAAAAGAAGGTCTTCATTACACC-3′, R′: 5′-CCTGTGCCTCTTCTTCTCATTTCG-3′), and GAPDH (F: 5′-CATCAAGAAGGTGGTGAAGC-3′, R: 5′-GTTGTCATACCAGGAAATGAGC-3′). Samples were run in technical duplicates along with negative controls without template cDNA to confirm lack of contamination in PCR reagents. PCR reactions were run on a CFX95™ Real-Time System (Biorad, Hercules, CA) with the following thermal cycler settings: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15s at 95°C, and 1 min at 60–65°C based on pre-determined optimal primer efficiency amplification temperature. After completing 40 cycles of PCR amplification, automated melting curve analysis, consisting of increasing the thermal cycler temperature from 50°C to 95°C at 5°C increments lasting 5s each, was used to confirm formation of a singular PCR amplicon for each primer set. Bio-Rad CFX Manager™ 3.0 (Biorad) was used for determining cycle threshold (ct) values from recorded SYBR green signal.
Gene expression ratios for assessing immune cell response polarization were calculated by modification of methods by Livak et al. for relative gene expression analysis [26]. The Δct was calculated between representative genes for either macrophage or T-helper cell polarized phenotypes. Fold change was then determined by 2−(Gene 1 — Gene 2) and normalized to fold change of corresponding NDM ratios for comparison. Ratios of ARG1/NOS2 and GATA3/TBX21 were selected to quantify macrophage and T-helper cell polarization, respectively, with CRTH2/CCR5 specifically for human T-helper cells. GAPDH values were used to confirm consistent loading between PCR reactions.
2.6. Histology and Immunohistochemistry
Each injection was harvested and divided in half for either paraffin embedding or fresh frozen in OCT for cryosectioning. Samples were sectioned to obtain a transverse section of the biomaterial and neighboring dermal tissue. Paraffin embedded samples were de-paraffinized and then stained with H&E from slides taken from five evenly spaced locations. Cryosections from three different evenly spaced locations were used for all immunohistochemistry. Slides were fixed with acetone or 4% paraformaldehyde and blocked with a buffered solution containing bovine serum albumin, goat serum and/or donkey serum based on the antibody. The following primary antibodies were incubated for 1 hour at room temperature or for 12–18 hours at 4°C: anti- F4/80 (1:200 dilution, eBiosciences, San Diego, CA), anti-iNOS (1:50 dilution, Abcam, Cambridge, MA), anti-CD206 (1:50 dilution, Santa Cruz, Dallas, TX), anti-mCD3 (1:100 dilution, Abcam), anti-mCD4 (1:200 dilution, Bioss, Woburn, MA), anti-mCD8 (1:200 dilution, Bioss), anti-human nuclei (1:250 dilution, Millipore), anti-hCD3 (1:50 dilution, Abcam), anti-hCD4 (1:50 dilution, Becton Dickinson, Franklin Lakes, NJ), anti-hCD8 (1:500 dilution, Becton Dickinson). The following secondary antibodies were incubated for 30–45 minutes: anti-rat Alexa Fluor 568 (1:250 dilution), anti-rabbit Alexa Fluor 488 (1:500 dilution), anti-mouse Alexa Fluor 488 (1:500 dilution), anti-rat Alexa Fluor 488 (1:400 dilution), anti-rabbit Alexa Fluor 568 (1:200 dilution), and anti-rabbit Alexa Fluor 647 (1:200 dilution). Bright field images were taken with Leica Aperio ScanScope® CS2 and fluorescent images with the Leica Ariol® system (Leica). Cellular density and human nuclei quantification were done via an automated counting program as part of the Leica Ariol® system. Macrophage and T-cell co-staining quantification was done via custom MATLAB scripts (Mathworks, Natick, MA). An individual blinded to the study groups identified the biomaterial outline, and the core of the biomaterial was defined as 200 μm interior to the designated biomaterial outline.
2.7. Flow Cytometry
Cells were isolated from injected tissue and spleens by first chopping the tissue into small pieces and digesting with 4 Wunsch Units of Liberase Blendzyme TM (Roche, Indianapolis, IN), 4 Wunsch Units of Liberase Blendzyme TH (Roche), 100 Kunitz Units of DNase 1 (Stem Cell), and 200 μL of 1 M HEPES (Gibco) in 10 mL of 199 Media (Gibco) at 37°C. The reaction was quenched and then filtered through a 40 μm cell filter. Cells were rinsed and suspended in a concentration of 1 × 106 cells/mL or less in FACS buffer. Cells were then divided and stained for 45 minutes on ice with the following conjugated primary antibodies or their corresponding isotope control antibody to remove non-specific binding: APC-anti-mCD3 (1:40 dilution, Biolegend, San Diego, CA), PE-anti-mCD4 (1:80 dilution, Biolegend), FITC-anti-mCD8 (1:50 dilution, Biolegend), APC-anti-mCD19 (1:20 dilution, Biolegend), PE-anti-mCD68 (1:80 dilution, Biolegend). Cells were either analyzed immediately or fixed and then analyzed on a FACSCanto Flow Cytometer machine using FACS Diva software (Becton Dickinson).
2.8. Statistics
All data and plots are presented as mean ± SEM. Significance was determined with a one-way ANOVA using a Tukey post-hoc test for histology, flow cytometry and qRT-PCR data, and an unpaired student’s t-test for total and human nuclei data with a p<0.05.
3. Results
3.1. Study Design
We assessed two hydrogels, a porcine myocardial matrix (PMM) and a human myocardial matrix (HMM), in the humanized model (Hu-mice) versus age-matched wild type Balb/c mice. Both biomaterials were derived from decellularized myocardium and processed into injectable liquids that self-assemble into hydrogels with nanofibrous architecture (Figure 1). These hydrogels were previously shown to have similar structure and mechanical properties (~2.5–6 Pa storage modulus) [23]. We confirmed this processing substantially removes the alpha-gal epitope in the porcine derived material (Figure S1). As a control, we examined these materials in age matched NSG mice, which lack T-cells, B-cells, and natural killer cells, and are used to create the Hu-mice (Figure S2). Each material was injected subcutaneously, a common delivery route for ISO standard biocompatibility tests, and at least eight different injections (n=8−16) were analyzed histologically three days and one week post injection. These time points were selected for assessing the early and mid-phase immune response since previous studies have shown macrophage response and polarization during this timeframe to be representative of long term biocompatibility and tissue repair [27–29]. Longer time points were not investigated since these biomaterials degrade completely by 2–3 weeks in vivo.
Figure 1. Injectable myocardial matrix hydrogels derived from porcine or human tissue.

The biomaterials were derived from either fresh porcine or human cadaveric left ventricular myocardium. The tissues were decellularized and then processed into an injectable form by enzymatic digestion. Once in the liquid form, the materials self-assembled into myocardial matrix hydrogels at physiological conditions. The hydrogels contained a similar nano-scale architecture as shown via SEM images (scale bars are 5 μm).
3.2. Greater Cellular Infiltration into Biomaterial Core of Xenogeneic Versus Allogeneic ECM
Samples were sectioned and stained with hematoxylin and eosin (H&E) for histological analysis (Figure 2A). Both decellularized biomaterials, PMM and HMM, showed moderate to minimal infiltration of mononuclear cells in both animal models (Figure S2). No foreign body giant cells or other signs of material rejection were observed for these decellularized materials. Minimal to no fibrous capsule formation was observed. Cellular infiltration was observed to be unevenly distributed in the injected material, particularly towards the center in the Hu-mice samples (Figure 2A). To distinguish cells clustered at the material border versus cells that infiltrated deeper into the biomaterial, cell density was quantified as two measures: cells throughout the whole biomaterial and cells in only the core region as defined as 200 μm inward from the biomaterial border [29].
Figure 2. Total cell infiltration.
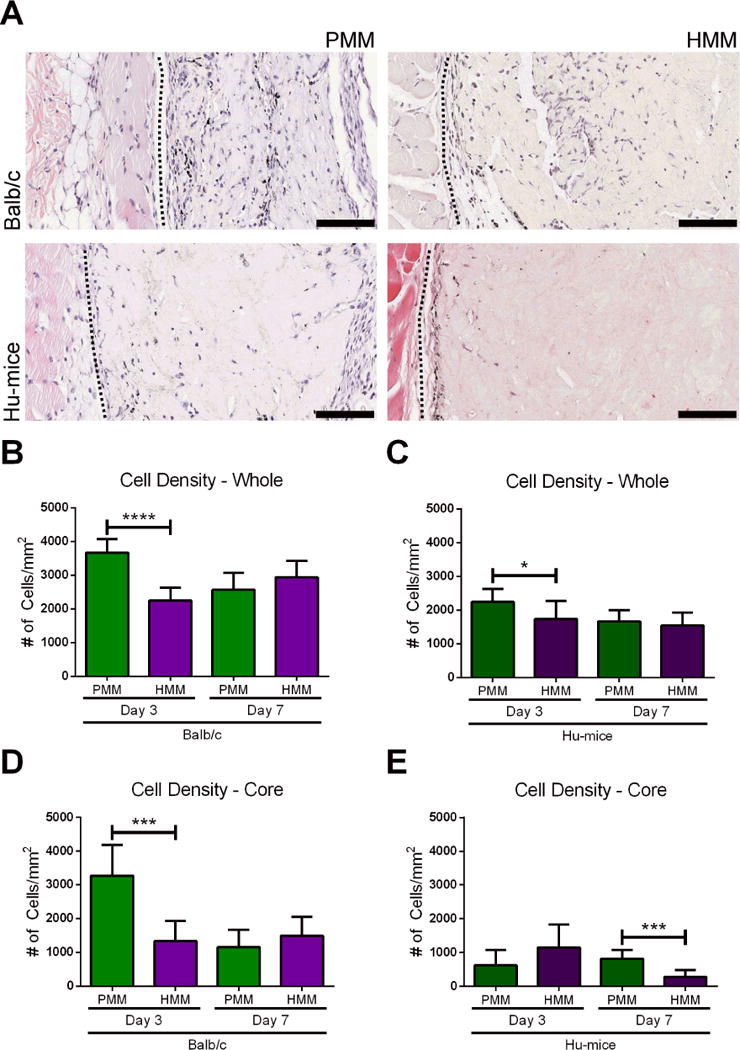
(A) Hematoxylin and eosin (H&E) images represent local tissue immune response one week after subcutaneous injections of porcine myocardial matrix (PMM) or human myocardial matrix (HMM) in Balb/c and a humanized mouse model (Hu-mice). Quantification of cellular density for the whole biomaterial (B, C) and infiltration into the core (D, E) of the biomaterial for each group is also shown (*p<0.05, ***p<0.001, ****p<0.001). On the left of each image is the panniculus carnosus muscle layer of the dermal tissue and on the right is the injected biomaterial. The biomaterial and dermal tissue are separated by a black dotted line. Scale bars are 100 μm.
Densities of the infiltrating cells into the whole biomaterial were significantly lower for both biomaterials in the Hu-mice when compared to wild type Balb/c animals (Figure S2A, B). This was expected since it is known that this model produces more moderate responses compared to a fully competent or wild type immune system [30]. Comparing differences in cell density throughput the whole biomaterial for PMM and HMM, significantly higher infiltration was observed in PMM in both Balb/c (Figure 2B) and Hu-mice (Figure 2C) at day 3, but no differences were observed between materials in either animal model at one week (Figure 2B, C). This same trend was observed when analyzing only the biomaterial core in Balb/c mice (Figure 2D); however, in Hu-mice, there was significantly decreased infiltration into the HMM core compared to the PMM core at one week (Figure 2E). This decreased infiltration notably occurred in HMM, which is an allogeneic material for the Hu-mice. In contrast, PMM and HMM both represent xenogeneic ECM in the Balb/c mice where they elicited similar responses. To confirm that the difference in cellular infiltration between the two materials at 1 week in the Hu-mice was due to the presence of human immune cells, we also evaluated immunocompromised NSG mice, which are used to generate the Hu-mice. Significantly less infiltration was observed for HMM in Hu-mice compared to NSG with both whole and core analysis (Figure S2B, D). Since the Hu-mice were made by implanting human tissue and cells into a NSG mouse, this further supported that these unique responses occurred from the allogeneic material interacting with the humanized component of the Hu-mouse model. Fewer cells also infiltrated into the core of both materials in the NSG mice compare to the Balb/c mice (Figure S2A, C), showing differences between immunocompromised and immunocompetent mice.
3.3. Minimal Human Cellular Infiltration for Allogeneic ECM
After investigation by histological analysis of the total cell density, we then investigated whether the observed differences resulted from varying human cell infiltration. Similar results were observed at day 3, but a notable difference in the prevalence and location of cells stained for human nuclei between PMM and HMM was observed at one week in the Hu-mice (Figure 3A). Both the percent and density of human cells trended higher in PMM compared to the HMM, with a significant difference in cell density observed within the core (Figure 3B–E). This demonstrated that human cells distinguished between the allogeneic versus xenogeneic biomaterial. There were also temporal differences between the two biomaterials, with the PMM eliciting increased human cell infiltration over time, unlike the HMM. (Figure 3 C, E).
Figure 3. Human nuclei infiltration.
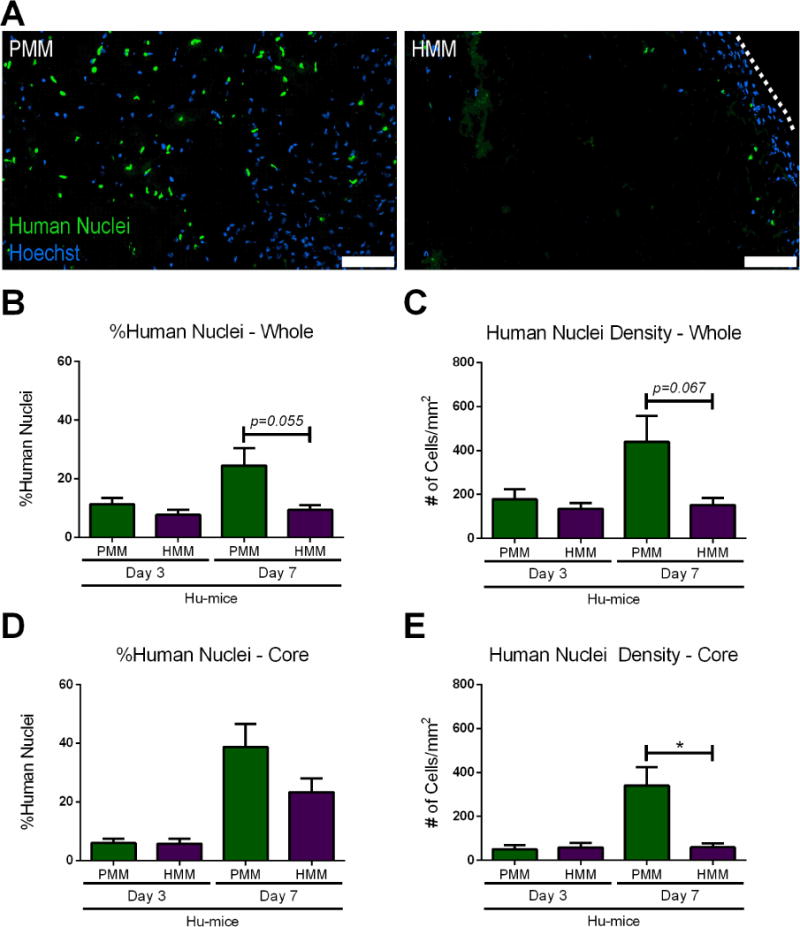
(A) Representative images of Hoechst stained nuclei (blue) and human nuclei staining (green) of humanized mice (Hu-mice) injected with either porcine myocardial matrix (PMM) or human myocardial matrix (HMM) at one week. The dotted white line indicates edge of HMM biomaterial. Quantification of percent and density of human nuclei in the whole biomaterial (B, C) and into the core (D, E) of the biomaterial (*p<0.05). Scale bars are 100 μm.
3.4. Greater T-helper Cell Infiltration in Xenogeneic ECM Compared to Allogeneic ECM
Since the Hu-mouse model showed significant differences in the number of cells infiltrating between the allogeneic and xenogeneic materials, we then investigated differences in specific types of recruited immune cells. T-cells have been shown to be important for eliciting a pro-remodeling versus pro-inflammatory immune response to ECM biomaterials [31]. Given the larger numbers of infiltrating cells in the Balb/c model, presence of T-cells and subtypes in the biomaterial was confirmed with flow cytometry (Figure S3). In the Balb/c and Hu-mice, both T-helper cells (CD3+CD4+) and cytotoxic T-cells (CD3+CD8+) were detected with positive staining in spleen tissue (Figure S4), which in the Hu-mice, demonstrated successful reconstitution of mature human immune cells. Immunohistochemistry was then used to visualize cell infiltration and quantify spatial distribution of T-helper cells and cytotoxic T-cells in the PMM and HMM materials in the Balb/c and Hu-mice (Figure 4A). A nondecellularized porcine myocardial matrix (NDM) was also compared as a pro-inflammatory control in both mouse models.
Figure 4. T-helper cell and cytotoxic T-cell infiltration.
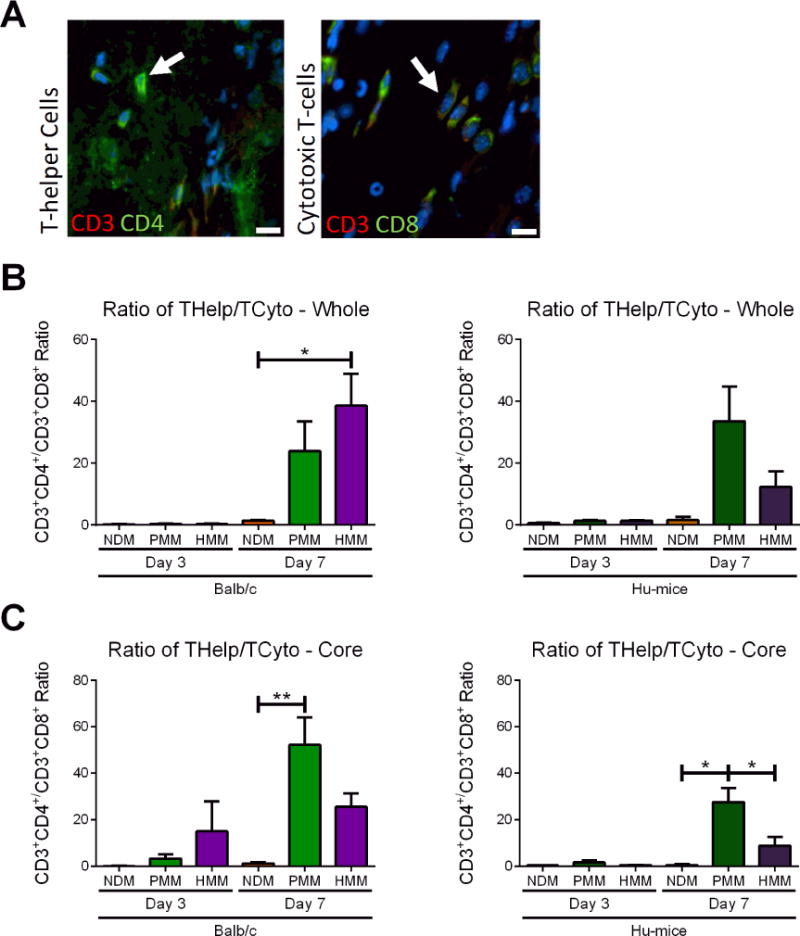
(A) Representative images of T-helper cells and cytotoxic T-cells with arrows indicating positive staining. Hoechst staining was used to label cellular nuclei (blue). T-helper cells were labeled by co-staining with CD3 (red) and CD4 (green). Cytotoxic T-cells were labeled by co-staining with CD3 (red) and CD8 (green). Ratios of cell density quantification of T-helper cells versus cytotoxic T-cells in whole biomaterial (B) and biomaterial core (C) in nondecellularized (NDM), porcine (PMM) and human myocardial matrix (HMM) at day 3 and 7 in Balb/c or Hu-mice. (*p<0.05, **p<0.01). Scale bars are shown at 10 μm.
Comparing the ratio of T-helper to cytotoxic T-cells, no significant differences were found among all materials at day 3 both in the whole and core biomaterial in both mouse models (Figure 4B, C). T-helper infiltration was generally low at this timepoint (Figure S5A, B) and NDM consistently recruited significantly greater cytotoxic T-cell densities compared to the decellularized materials for both Balb/c and Hu-mice (Figure S5C, D). By day 7, minimal numbers of cytotoxic T-cells were found in PMM and HMM groups, while cytotoxic T-cell infiltration continued to increase in NDM in both animal models (Figure S 5C, D). For all materials in both animal models, T-helper cell densities increased by day 7 with the decellularized materials becoming T-helper cell dominant compared to NDM (Figure 4B, C). This transition for the decellularized materials from a cytotoxic to a T-helper cell response has been previously observed [32] and suggests only NDM was being rejected as expected. Comparing the T-helper versus cytotoxic T-cell response between the decellularized materials, no significant differences were observed in the Balb/c mice and in the Hu-mice when the whole biomaterial was examined (Figure 4B). However, for the core analysis in the Hu-mice, PMM was significantly human T-helper cell dominant compared to both HMM and NDM (Figure 4C) with a significantly higher T-helper cell density than HMM (Figure S5B). This corresponded with previous human nuclei data (Figure 3E) indicating that PMM elicited greater human cell infiltration at one week. Collectively, these results showing minimal cytotoxic T-cell density and greater T-helper density in PMM in the Hu-mouse model suggested that the xenogeneic PMM material stimulated a greater pro-remodeling response compared to the allogeneic HMM material.
3.5. Similar Macrophage Infiltration and Polarization between Xenogeneic and Allogeneic ECM
Macrophage presence was also analyzed since macrophage phenotypic expression has been shown to be an important indicator of tissue remodeling outcomes for ECM based biomaterials [33, 34]. Although the presence of human macrophages has been previously demonstrated in this mouse model, minimal to no human macrophage infiltration into the biomaterial was found (data not shown); instead, mouse macrophages were highly abundant. Since the response was dictated by mouse macrophages, we evaluated the infiltration and polarization of these cells in both mouse models to determine if interaction with the human T cells lead to a unique immune response. Macrophage polarization is plastic and complex [35–37], but is often simplified into pro-inflammatory M1 and pro-remodeling M2 phenotypes [38]. Macrophages were identified with pan-macrophage marker F4/80, and polarization was evaluated with common markers for M2 (CD206) and M1 (iNOS) for pro-remodeling versus pro-inflammatory macrophages, respectively (Figure 5A) [27, 39]. In both mouse models at day 3, the ratio of M2/M1 macrophages for all three materials was below 1 in both whole and core biomaterial, suggesting that the short-term macrophage response was M1 dominant (Figure 5B, C). However, the total macrophage (Figure S6A) and M1 macrophage cell density (Figure S6C) was significantly greater in NDM compared to the decellularized materials in both animal models. At one week, NDM remained M1 dominant, while in the Balb/c and Hu-mice the decellularized materials switched to a M2 dominant response based on both whole and core biomaterial analysis (Figure 5B, C). Assessment of cell densities indicated that M2 macrophage cell density increased in all materials for both mouse models from day 3 to one week (Figure S6E, F). However, density of M1 macrophages remained greater or increased by one week in NDM while M1 density generally decreased in the decellularized materials in both mouse models (Figure S6C, D). This demonstrated a transition from a pro-inflammatory to pro-remodeling macrophage polarization for the decellularized materials, which is characteristic of wound healing [28]. This was also notably similar to the transition observed from our T-cell analysis (Figure 4B, C). Although, no distinct differences were observed in the response between PMM and HMM for the two animal models, these results showed that macrophages were dynamically responsive in the Hu-mouse model similar to a wild-type model. Interestingly, dual stained macrophages with both M1 and M2 markers were commonly observed, especially at the biomaterial border for all material groups (Figure S6G, H). This observation is not unique since previous studies have observed these results in vivo, and some studies have suggested that dual polarized macrophages are involved in angiogenesis [39]. As a result, these cells were separately grouped and not considered specifically M1 and M2 polarized in the ratio analysis.
Figure 5. Polarized macrophage infiltration.
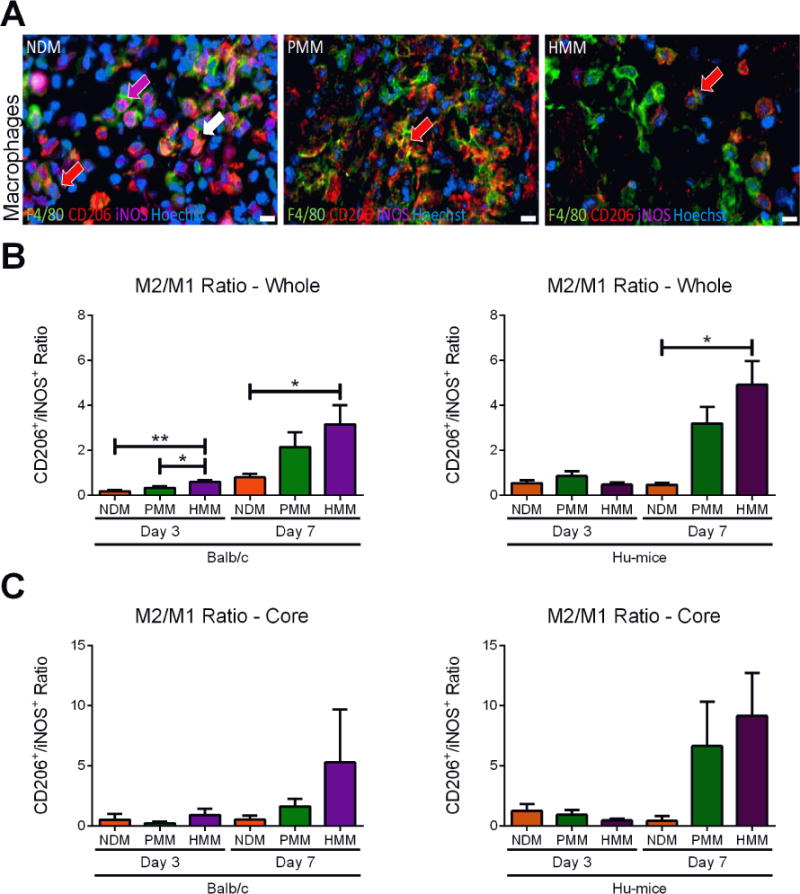
(A) Representative images at one week of polarized macrophage staining with a red arrow indicating a M2 macrophage, a magenta arrow indicating a M1 macrophage and white arrow indicating a macrophage stained with both M2 and M1 markers. Hoechst staining was used to label cellular nuclei (blue). Macrophages were labeled by co-staining with pan-macrophage marker F4/80 (green), M1 marker iNOS (magenta) and M2 marker CD206 (red). Ratios of cell density quantification of CD206+ to iNOS+ macrophages in whole biomaterial (B) and biomaterial core (C) in nondecellularized (NDM), porcine (PMM) and human myocardial matrix (HMM) at day 3 and 7 in Balb/c or Hu-mice. (*p<0.05, **p<0.01). Scale bars are shown at 10 μm.
3.6. Pro-Remodeling Immune Cell Polarization in ECM Biomaterials
Although we determined a T-helper dominant response to our decellularized materials as described above, T-helper cells are known to polarize and show dramatically different phenotypes depending on their local niche [40, 41]. For example, a T-helper cell’s dominant response is usually correlated with a pro-remodeling response, but T-helper cell infiltration can be connected with graft rejection as well [42]. Thus, we wanted to more fully characterize the T-helper cell polarization. T-helper cells can be divided into pro-inflammatory Th1 and pro-remodeling Th2 phenotypes [41, 43]. Polarization towards the Th2 phenotype was measured by comparing the ratio of expression of cell-specific master transcription factors GATA3 (Th2) to TBX21 (Th1) for NDM, PMM and HMM in both mouse models [43, 44]. An additional set of human specific T-helper cell markers for receptors CRTH2 (Th2) and CCR5 (Th1) was utilized to further confirm the results in Hu-mice [45–47]. At day 3, polarization was similar for all materials in both animal models (Figure 6A, B). This result likely corresponded with the previously determined low T-helper cell presence (Figure S5A) at this early time point. By one week, PMM was Th2 trending in Balb/c mice and significantly Th2 polarized in Hu-mice relative to NDM with both sets of T-helper cell markers. In contrast, HMM was only trending towards a Th2 polarization compared to NDM with these markers in the two mouse models (Figure 6A, B).
Figure 6. Gene expression ratios of T-helper cell and macrophage polarization.
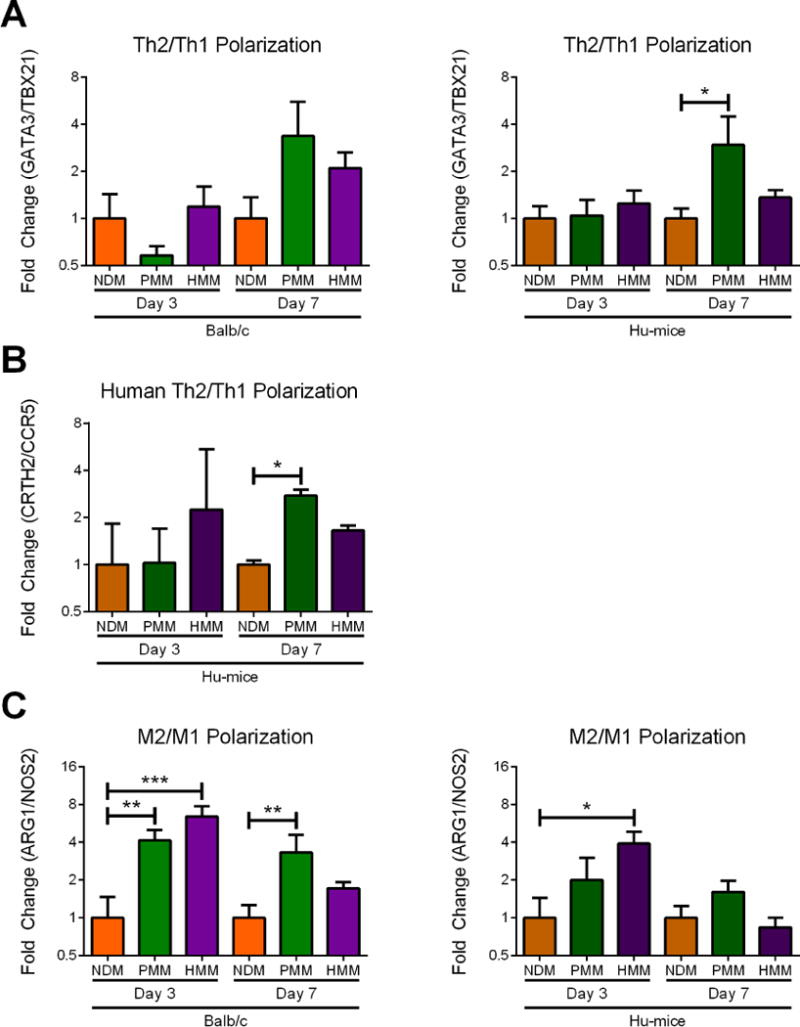
qRT-PCR was utilized to determine the degree of polarization of macrophages and T-helper cells at day 3 and one week in Balb/c and Hu-mice. (A) T-helper cell polarization of Th2/Th1 was measured as fold change ratio of GATA3/TBX21 gene expression. (B) A separate human T-helper cell polarization was measured as fold change ratio of CRTH2/CCR5 gene expression. (C) Macrophage polarization of M2/M1 was measured as a fold change ratio of ARG1/NOS2 gene expression. (*p<0.05, **p<0.01, ***p<0.001).
A ratio of related genes expressed in M1 (NOS2) and M2 (ARG1) was also used to measure macrophages polarization with qRT-PCR [27]. At day 3, HMM was significantly M2 polarized compared to NDM in both mouse models. PMM was significantly M2 polarized in Balb/c mice and M2 trending in Hu-mice (Figure 6C). Based on the cell staining, these results likely occurred from a greater M1 response in NDM compared to the decellularized materials considering the low M2 macrophage numbers previously shown at this timepoint (Figure 5, Figure S6). By one week, only PMM was significant in Balb/c mice and upward trending in the Hu-mice compared to NDM (Figure 6C). Therefore, notably only PMM elicited a significant Th2 polarization in the Hu-mice and a significant M2 polarization in the Balb/c mice compared to NDM while HMM was not significant compared to either PMM or NDM at one week.
4. Discussion
In this study, we investigated the ability of a humanized mouse (Hu-mice) model, which had previously been shown capable of rejecting xenogeneic tissue via a human T-cell mediated immune response [19], to differentiate between allogeneic and xenogeneic decellularized ECM. We tested a xenogeneic biomaterial sourced from porcine myocardial matrix (PMM), which was developed as a therapy for treating hearts post-myocardial infarction [21]. Similarly, a material derived from human cadaveric donor hearts (HMM) was produced as an allogeneic alternative [23]. The Hu-mouse model showed significant differences in the immune response to the allogeneic and xenogeneic materials that differentiated from a wild type animal model, suggesting that the Hu-mouse model could be an advantageous tool in studying the biocompatibility of tissue-derived materials and in vivo human immune cell responses.
Investigation of the infiltrating cells showed significant differences in cell densities found in the core of xenogeneic PMM compared to allogeneic HMM material in the Hu-mice. Greater human cell infiltration was observed at one week during the mid-phase immune response, while human cell interaction was similar at the earlier time point. Further evaluation of the infiltrating cells determined that the early day 3 response consisted mainly of M1 (iNOS+) macrophages and cytotoxic T-cells (CD3+, CD8+). These numbers decreased by one week with very few cytotoxic T-cells, and instead predominantly M2 macrophages (CD206+) and T-helper cells (CD3+, CD4+) were present. In contrast, the nondecellularized myocardial matrix (NDM) predominantly contained infiltration of M1 macrophages and cytotoxic T-cells throughout the study. This dynamic shift for the decellularized materials mimics the native wound healing response [28], suggesting that these materials could stimulate similar mechanisms when inducing tissue repair [11, 22]. Infiltration of T-helper cells was particularly distinct in PMM compared to HMM, which has a critical role in supporting a pro-regenerative response to biomaterial therapies [31]. This significantly different response was only observed in the Hu-mouse model potentially because both PMM and HMM are xenogeneic in the Balb/c, leading to similar levels of T-helper cells and macrophage infiltration.
Gene expression of cell specific markers were utilized to further characterization cell phenotypes towards pro-inflammatory and pro-remodeling subtypes. T-helper subtypes were assessed by cell-specific gene expression ratios towards pro-inflammatory Th1 or pro-remodeling Th2 phenotypes, respectively. This expression is directly correlated with separate phenotypes involving the production of IL-2 and interferon-γ (IFNγ) for Th1 and IL-4, IL-5, and IL-10 for Th2 [48]. Similarly, polarized macrophages were assessed towards the pro-remodeling M2 or pro-inflammatory M1 phenotype [27, 49]. M1 macrophages are known to produce inflammatory cytokines of TNF, IL-6, and IL-1β with high levels of IL-12 and IL-23, and low levels of IL-10. Whereas, the M2 polarized macrophages have low levels of IL-12 and IL-23 with high levels of IL-10 [38]. Early T-cell and macrophage response to the decellularized materials was Th1 and M2 polarized, respectively, which corresponded with low T-helper cell and lesser M1 macrophage densities from the cell staining analysis. At one week, only PMM was significantly shifted towards the Th2 phenotype and trending towards a M2 polarization compared to NDM in the Hu-mouse model. In contrast, both PMM and HMM were Th2 and M2 polarized in the Balb/c mouse at one week. However, analysis of macrophage cell densities for HMM in the Hu-mice supported that they were similarly more polarized towards a pro-remodeling state. This difference in the magnitude of M2 macrophage polarization measured by qRT-PCR could be due to the lesser presence of T-helper cells in HMM supporting the M2 macrophage phenotype [31].
Potentially, these results could suggest that allogeneic materials elicit lesser human T-helper involvement in the immune response, which reduces immunological concerns, but can also its limit pro-regenerative capability. However, it should be considered that limitations of the allogeneic tissue source might be responsible for these results. The efficacy of tissue decellularization, tissue source age, and cross-linking are crucial parameters that can significantly impact the host response [50]. For fabrication of the HMM material, older human cadaver hearts were utilized that required additional processing steps such as lipid removal and DNase/RNase treatment [23]. Older ECM is known to shift in composition [4], stimulate a lesser pro-remodeling macrophage response [51], and undergo increased cross-linking and fibrosis [52], which could create less ideal material properties for stimulating tissue repair. Likewise, additional processing steps were required to remove greater adipose tissue commonly found on older human myocardium and reduce nucleotide content to similar acceptable standards for therapeutic applications as the PMM material [23]. These steps could unintentionally strip important biological factors from the material. Previous assessment found that HMM did maintain a complex ECM protein composition with many similar material properties to PMM, though there was notably less sulfated glycosaminoglycan content [23, 53]. As the immune response is mediated by a multitude of cytokine signaling pathways, decreased sGAG content could reduce retention of endogenous chemoattract ant factors and limit the anti-inflammatory response elicited by HMM [54]. Although it could be hypothesized that young human hearts could provide an improved allogeneic scaffold, this is not a viable strategy considering the immediate need for young healthy human hearts for surgical transplantation, prevalence of cardiac diseases [55], and general shortages of healthy human organs [56]. Therefore, these shortcomings could be considered inherent restrictions of cardiac tissue derived allogeneic materials.
These results also demonstrate the insufficiency of immunocompromised NSG mice in modeling the response to biomaterials. There was significantly less cellular infiltration in NSG animals compared to immunocompetent Balb/c animals. Macrophages are present in both animal models, but T-cells are not present in the NSG mice. Therefore, macrophages alone are not sufficient to produce a full immune response to these materials. This is not surprising since the more severe xenogeneic tissue rejection has been shown to be largely a T-cell mediated response and involves the subsequent activation of macrophages [18, 19]. Thus, this raises concerns utilizing immune compromised animals in studies when a naturally derived material is used.
Although these results demonstrate the potential utility of this model, important limitations should be considered for further studies. One limitation of the Hu-mouse model that should be considered is the inability to measure the role of certain xenogeneic specific epitopes, including the anti-Gal epitope, which is involved in xenogeneic tissue rejection in humans [19]. For processed xenogeneic acellular materials, which have dramatically lower levels of the alpha-Gal epitopes similar to the materials used in this study, this has not been shown or been proven to cause adverse reactions in human patients [50]. Concerns regarding the potential influence of these epitopes could be addressed by evaluating epitope presence in the material, as done in this study, or perform additional tests in knockout mice of these epitopes [57]. The observed lack of human macrophage involvement compared to the larger population of mouse macrophages also limits the model’s capability to mimic this component of the human immune response.
Despite these limitation, human T-helper cells in the Hu-mouse model were able to distinguish between these biomaterials, which could indicate increased sensitivity to variations between xenogeneic versus allogeneic ECM or potentially other variables such as ECM age and material processing. The different responses suggested that the xenogeneic PMM is more favorable for eliciting a pro-remodeling immune response compared to the allogeneic HMM material, and supports its use in human trials. This study also demonstrates the utility of the Hu-mouse model for biomaterial testing, providing initial biocompatibility and evaluation of the immune response. With additional variables known to influence the immune response such as differences in the delivery method, location of implantation, and initial inflammatory state of the implantation site, there is much more potential utility with this model for evaluating the human immune cell response to biomaterial therapies. These results also warrant further mechanistic studies to improve our understanding of important human immune cell pathways to stimulate biomaterial induced tissue repair. Future improvements being tested in Hu-mice models such as earlier maturation of B-cell populations before xenogeneic graft-versus-host disease becomes a concern could allow for assessment of human humoral response to biomaterial implants [30, 58]. These results, therefore, convey that testing a xenogeneic biomaterial in preclinical studies in a wild type or immunocompetent animal could be sufficient for assessing biocompatibility. However, a wild type rodent model is likely to be insufficient to test a human derived, allogeneic material, and the Hu-mouse model provides a more thorough model of the human T-cell response to these materials. Collectively, our results with the Hu-mouse model demonstrate the importance of pursuing more representative preclinical models in the biomaterials field for confirming biocompatibility and improving our understanding the human cell response to biomaterial therapies.
5. Conclusion
In conclusion, we have utilized a xenogeneic material, PMM, and an allogeneic material, HMM, in a humanized mouse (Hu-mice) model, demonstrating its potential as a new tool for preclinical biomaterial testing. This work confirms that these naturally derived biomaterials elicit a T-cell mediated immune response with a pro-remodeling Th2 and M2 polarization. This was also accompanied by a notable pro-inflammatory to pro-remodeling shift for the early to mid-phase responses for both T-cells and macrophages, mimicking the native wound healing response. Both PMM and HMM in a wild type animal similarly favored a Th2 T-helper cell and M2 macrophage population with minimal cytotoxic T-cells present by one week after biomaterial injection. However, differences between the materials were then observed in the Hu-mouse model, namely a significantly reduced and minimal T-helper cell response to the allogeneic HMM. This suggests that the xenogeneic tissue derived biomaterial could be more suitable for eliciting pro-remodeling responses compared to similarly made materials from available allogeneic sources. Additionally, utilizing an immune compromised animal did not properly mimic the tissue response to ECM derived materials. Thus, the Hu-mouse model’s capability to better mimic the potential human immune response could be a useful tool to differentiate the biocompatibility and pro-remodeling qualities of biomaterials prior to clinical translation.
Supplementary Material
Figure S1: H&E and alpha-gal epitope staining of fresh porcine myocardium, decellularized tissue, and self-assembled hydrogel. Top row shows hematoxylin and eosin (H&E) staining with scale bar of 50 μm. Blue channel shows Hoechst staining for nuclei, and green channel shows alpha-gal epitope staining, demonstrating its substantial removal during material processing. Bottom scale bars are 100 μm.
Figure S2: Comparison of Total Cell Infiltration Between Animal Models. Quantification from H&E images of cellular density in the whole biomaterial (A, B) and infiltration into the core (C, D) in porcine (PMM) and human myocardial matrix (HMM) at three days and one week in Balb/c and Hu-mice. Results for NSG mice are also shown at one week. (*p<0.05, **p<0.01, ***p<0.001).
Figure S3: Flow cytometry cellular quantification in Balb/c animals for either PMM, HMM, or NDM. (A) Representative histograms or scatter plots for staining of macrophages (mCD68+), T-helper cells (mCD3+mCD4+), and cytotoxic T-cells (mCD3+mCD8+). B-cells (CD19+) were not detected in the biomaterials. (B) Averages and standard deviations for each treatment group and cell type analyzed with flow cytometry. (C) Ratio of T-helper cells to cytotoxic T-cells showing PMM and HMM favoring T-helper cells and NDM favoring cytotoxic T-cells. (***p<0.001).
Figure S4 Immunohistochemistry of T-helper cells and cytotoxic T-cells in either Balb/c or Hu-mice spleen. Hoechst staining (blue) was used to label nuclei. T-helper cells were labeled by co-staining with CD3 (red) and CD4 (green). Cytotoxic T-cells were labeled by co-staining with CD3 (red) and CD8 (green). Mouse specific antibodies were used for Balb/c spleens, and human specific antibodies were used for Hu-mice spleens. Scale bars are 10 μm.
Figure S5: Quantified cell density for T-Helper cells and Cytotoxic T-cells. Cell density quantification of T-helper cells in whole biomaterial (A) and biomaterial core (B) in nondecellularized (NDM), porcine (PMM) and human myocardial matrix (HMM) at three days and one week in Balb/c and Hu-mice. Cytotoxic T-cell density in whole biomaterial (C) and biomaterial core (D) at three days and one week in Balb/c and Hu-mice. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Figure S6: Quantified cell density for total macrophages and polarized macrophage phenotypes. Cell density quantification of total macrophages in whole biomaterial (A) and biomaterial core (B) in nondecellularized (NDM), porcine (PMM) and human myocardial matrix (HMM) at three days and one week in Balb/c or Hu-mice. Cell density quantification of iNOS+ macrophages in whole biomaterial (C) and biomaterial core (D). Cell density quantification of CD206+ macrophages in whole biomaterial (E) and biomaterial core (F). Cell density quantification of dual stained iNOS+CD206+ macrophages in whole biomaterial (G) and biomaterial core (H). (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Acknowledgments
The authors would like to thank Matt Joens for his technical expertise in scanning electron microscopy. The authors would also like to thank Lifesharing for their assistance in obtaining the human cardiac tissue. Funding for this work was provided by CIRM (TR3-05559 and TR1-01277 to YX) and the NIH NHLBI (HL113468 to KLC). RMW received funding during the project from the NHLBI as a training grant recipient. TDJ received funding during the project as a Powell Fellow from the Powell Foundation, the NHLBI as a training grant recipient, and the NSF as a graduate student fellow. KLC is co-founder, consultant, board member, and holds equity interest in Ventrix, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pashuck ET, Stevens MM. Designing regenerative biomaterial therapies for the clinic. Science translational medicine. 2012;4(160):160sr4. doi: 10.1126/scitranslmed.3002717. [DOI] [PubMed] [Google Scholar]
- 2.Turner NJ, Badylak SF. Regeneration of skeletal muscle. Cell Tissue Res. 2012;347(3):759–774. doi: 10.1007/s00441-011-1185-7. [DOI] [PubMed] [Google Scholar]
- 3.Rane AA, Christman KL. Biomaterials for the treatment of myocardial infarction: a 5-year update. Journal of the American College of Cardiology. 2011;58(25):2615–2629. doi: 10.1016/j.jacc.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Tottey S, Johnson SA, Crapo PM, Reing JE, Zhang L, Jiang H, Medberry CJ, Reines B, Badylak SF. The effect of source animal age upon extracellular matrix scaffold properties. Biomaterials. 2011;32(1):128–136. doi: 10.1016/j.biomaterials.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowers SL, Banerjee I, Baudino TA. The extracellular matrix: at the center of it all. Journal of molecular and cellular cardiology. 2010;48(3):474–82. doi: 10.1016/j.yjmcc.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keane TJ, Londono R, Turner NJ, Badylak SF. Consequences of ineffective decellularization of biologic scaffolds on the host response. Biomaterials. 2012;33(6):1771–1781. doi: 10.1016/j.biomaterials.2011.10.054. [DOI] [PubMed] [Google Scholar]
- 7.Badylak SF. Xenogeneic extracellular matrix as a scaffold for tissue reconstruction. Transpl Immunol. 2004;12(3–4):367–377. doi: 10.1016/j.trim.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 8.Badylak SF, Gilbert TW. Immune response to biologic scaffold materials. Semin Immunol. 2008;20(2):109–116. doi: 10.1016/j.smim.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sicari BM, Johnson SA, Siu BF, Crapo PM, Daly KA, Jiang H, Medberry CJ, Tottey S, Turner NJ, Badylak SF. The effect of source animal age upon the in vivo remodeling characteristics of an extracellular matrix scaffold. Biomaterials. 2012;33(22):5524–5533. doi: 10.1016/j.biomaterials.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badylak SF. The extracellular matrix as a biologic scaffold material. Biomaterials. 2007;28(25):3587–3593. doi: 10.1016/j.biomaterials.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 11.Seif-Naraghi SB, Singelyn JM, Salvatore MA, Osborn KG, Wang JJ, Sampat U, Kwan OL, Strachan GM, Wong J, Schup-Magoffin PJ, Braden RL, Bartels K, DeQuach JA, Preul M, Kinsey AM, Demaria AN, Dib N, Christman KL. Safety and efficacy of an injectable extracellular matrix hydrogel for treating myocardial infarction. Science translational medicine. 2013;5(173):173ra25. doi: 10.1126/scitranslmed.3005503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. Journal of immunology. 2004;172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 13.Varki NM, Strobert E, Dick EJ, Benirschke K, Varki A. Biomedical differences between human and nonhuman hominids: potential roles for uniquely human aspects of sialic acid biology. Annu Rev Pathol. 2011;6:365–393. doi: 10.1146/annurev-pathol-011110-130315. [DOI] [PubMed] [Google Scholar]
- 14.Říhová B. Biocompatibility of biomaterials: hemocompatibility, immunocompatiblity and biocompatibility of solid polymeric materials and soluble targetable polymeric carriers. Advanced Drug Delivery Reviews. 1996;21(2):157–176. [Google Scholar]
- 15.Onuki Y, Bhardwaj U, Papadimitrakopoulos F. A review of the biocompatibility of implantable devices: current challenges to overcome foreign body response. J Diabetes Sci. 2008 doi: 10.1177/193229680800200610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legrand N, Weijer K. Experimental Models to Study Development and Function of the Human Immune System In Vivo. The Journal of Immunology. 2006 doi: 10.4049/jimmunol.176.4.2053. [DOI] [PubMed] [Google Scholar]
- 17.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108(2):487–492. doi: 10.1182/blood-2005-11-4388. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Duan Z, Zhao Y. Mouse models with human immunity and their application in biomedical research. Journal of Cellular and Molecular Medicine. 2009;13(6):1043–1058. doi: 10.1111/j.1582-4934.2008.00347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tonomura N, Shimizu A, Wang S, Yamada K, Tchipashvili V, Weir GC, Yang YG. Pig islet xenograft rejection in a mouse model with an established human immune system. Xenotransplantation. 2008;15(2):129–135. doi: 10.1111/j.1399-3089.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- 20.Rong Z, Wang M, Hu Z, Stradner M, Zhu S, Kong H, Yi H, Goldrath A, Yang YG, Xu Y, Fu X. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell. 2014;14(1):121–30. doi: 10.1016/j.stem.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singelyn JM, DeQuach JA, Seif-Naraghi SB, Littlefield RB, Schup-Magoffin PJ, Christman KL. Naturally derived myocardial matrix as an injectable scaffold for cardiac tissue engineering. Biomaterials. 2009;30(29):5409–5416. doi: 10.1016/j.biomaterials.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singelyn JM, Sundaramurthy P, Johnson TD, Schup-Magoffin PJ, Hu DP, Faulk DM, Wang J, Mayle KM, Bartels K, Salvatore M, Kinsey AM, Demaria AN, Dib N, Christman KL. Catheter-deliverable hydrogel derived from decellularized ventricular extracellular matrix increases endogenous cardiomyocytes and preserves cardiac function post-myocardial infarction. Journal of the American College of Cardiology. 2012;59(8):751–763. doi: 10.1016/j.jacc.2011.10.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson TD, DeQuach JA, Gaetani R, Ungerleider J, Elhag D, Nigam V, Behfar A, Christman KL. Human versus porcine tissue sourcing for an injectable myocardial matrix hydrogel. Biomater Sci. 2014;2(5):735–744. doi: 10.1039/C3BM60283D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson TD, Lin SY, Christman KL. Tailoring material properties of a nanofibrous extracellular matrix derived hydrogel. Nanotechnology. 2011;22(49):494015. doi: 10.1088/0957-4484/22/49/494015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galili U, LaTemple DC, Radic MZ. A sensitive assay for measuring alpha-Gal epitope expression on cells by a monoclonal anti-Gal antibody. Transplantation. 1998;65(8):1129–1132. doi: 10.1097/00007890-199804270-00020. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Brown BN, Valentin JE, Stewart-Akers AM, McCabe GP, Badylak SF. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials. 2009;30(8):1482–1491. doi: 10.1016/j.biomaterials.2008.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nassiri S, Zakeri I, Weingarten MS, Spiller KL. Relative Expression of Proinflammatory and Antiinflammatory Genes Reveals Differences between Healing and Nonhealing Human Chronic Diabetic Foot Ulcers. J Invest Dermatol. 2015;135(6):1700–3. doi: 10.1038/jid.2015.30. [DOI] [PubMed] [Google Scholar]
- 29.Yu T, Wang W, Nassiri S, Kwan T, Dang C, Liu W, Spiller KL. Temporal and spatial distribution of macrophage phenotype markers in the foreign body response to glutaraldehyde-crosslinked gelatin hydrogels. J Biomater Sci Polym Ed. 2016;27(8):721–42. doi: 10.1080/09205063.2016.1155881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vuyyuru R, Patton J, Manser T. Human immune system mice: current potential and limitations for translational research on human antibody responses. Immunologic research. 2011;51(2–3):257–66. doi: 10.1007/s12026-011-8243-9. [DOI] [PubMed] [Google Scholar]
- 31.Sadtler K, Estrellas K, Allen BW, Wolf MT, Fan H, Tam AJ, Patel CH, Luber BS, Wang H, Wagner KR, Powell JD, Housseau F, Pardoll DM, Elisseeff JH. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science. 2016;352(6283):366–70. doi: 10.1126/science.aad9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muhamed J, Revi D, Rajan A, Geetha S, Anilkumar TV. Biocompatibility and Immunophenotypic Characterization of a Porcine Cholecyst-derived Scaffold Implanted in Rats. Toxicol Pathol. 2015;43(4):536–45. doi: 10.1177/0192623314550722. [DOI] [PubMed] [Google Scholar]
- 33.Valentin JE, Stewart-Akers AM, Gilbert TW, Badylak SF. Macrophage participation in the degradation and remodeling of extracellular matrix scaffolds. Tissue Eng Part A. 2009;15(7):1687–1694. doi: 10.1089/ten.tea.2008.0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badylak SF, Valentin JE, Ravindra AK, McCabe GP, Stewart-Akers AM. Macrophage Phenotype as a Determinant of Biologic Scaffold Remodeling. Tissue Eng Part A. 2008;14(11):1835–1842. doi: 10.1089/ten.tea.2007.0264. [DOI] [PubMed] [Google Scholar]
- 35.Mosser D. The many faces of macrophage activation. J Leukocyte Biol. 2003 doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 36.Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N, Dormont D, Gras G. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142(3):481–489. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003 doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 38.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23(4):344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, Vunjak-Novakovic G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014;35(15):4477–88. doi: 10.1016/j.biomaterials.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown BN, Ratner BD, Goodman SB, Amar S, Badylak SF. Macrophage polarization: An opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials. 2012;33(15):3792–3802. doi: 10.1016/j.biomaterials.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reiner SL. Helper T cell differentiation, inside and out. Current opinion in immunology. 2001;13(3):351–5. doi: 10.1016/s0952-7915(00)00226-0. [DOI] [PubMed] [Google Scholar]
- 42.Pierson RN. Xenogeneic skin graft rejection is especially dependent on CD4+ T cells. Journal of Experimental Medicine. 1989;170(3):991–996. doi: 10.1084/jem.170.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakir H, Wang H, Lefebvre DE, Webb J, Scott FW. T-bet/GATA-3 ratio as a measure of the Th1/Th2 cytokine profile in mixed cell populations: predominant role of GATA-3. Journal of immunological methods. 2003;278(1–2):157–69. doi: 10.1016/s0022-1759(03)00200-x. [DOI] [PubMed] [Google Scholar]
- 44.Allman A, McPherson T, Badylak S, Merrill L, Kallakury B, Sheehan C, Raeder R, Metzger D. Xenogeneic extracellular matrix grafts elicit a Th2-restricted immune response. Transplantation. 2001;71(11):1631–1640. doi: 10.1097/00007890-200106150-00024. [DOI] [PubMed] [Google Scholar]
- 45.De Fanis U, Mori F, Kurnat RJ, Lee WK, Bova M, Adkinson NF, Casolaro V. GATA3 up-regulation associated with surface expression of CD294/CRTH2: a unique feature of human Th cells. Blood. 2007;109(10):4343–50. doi: 10.1182/blood-2006-05-025940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, Glimcher LH, Lord GM. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci U S A. 2009;106(42):17876–81. doi: 10.1073/pnas.0909357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boin F, De Fanis U, Bartlett SJ, Wigley FM, Rosen A, Casolaro V. T cell polarization identifies distinct clinical phenotypes in scleroderma lung disease. Arthritis Rheum. 2008;58(4):1165–74. doi: 10.1002/art.23406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes & development. 2000;14(14):1693–711. [PubMed] [Google Scholar]
- 49.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(43):13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Badylak SF. Decellularized Allogeneic and Xenogeneic Tissue as a Bioscaffold for Regenerative Medicine: Factors that Influence the Host Response. Ann Biomed Eng. 2014;42(7):1517–1527. doi: 10.1007/s10439-013-0963-7. [DOI] [PubMed] [Google Scholar]
- 51.Sicari BM, Johnson SA, Siu BF, Crapo PM, Daly KA, Jiang H, Medberry CJ, Tottey S, Turner NJ, Badylak SF. The effect of source animal age upon the in vivo remodeling characteristics of an extracellular matrix scaffold. Biomaterials. 2012;33(22):5524–33. doi: 10.1016/j.biomaterials.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Souza RR. Aging of myocardial collagen. Biogerontology. 2002;3(6):325–35. doi: 10.1023/a:1021312027486. [DOI] [PubMed] [Google Scholar]
- 53.Johnson TD, Hill RC, Dzieciatkowska M, Nigam V, Behfar A, Christman KL, Hansen KC. Quantification of decellularized human myocardial matrix: A comparison of six patients. Proteomics Clin Appl. 2016;10(1):75–83. doi: 10.1002/prca.201500048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hempel U, Matthaus C, Preissler C, Moller S, Hintze V, Dieter P. Artificial matrices with high-sulfated glycosaminoglycans and collagen are anti-inflammatory and pro-osteogenic for human mesenchymal stromal cells. J Cell Biochem. 2014;115(9):1561–71. doi: 10.1002/jcb.24814. [DOI] [PubMed] [Google Scholar]
- 55.M. Writing Group. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, C. American Heart Association Statistics, S. Stroke Statistics Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 56.Saidi RF, Kenari Hejazii SK. Challenges of organ shortage for transplantation: solutions and opportunities. Int J Organ Transplant Med. 2014;5(3):87–96. [PMC free article] [PubMed] [Google Scholar]
- 57.Galili U. The alpha-gal epitope and the anti-Gal antibody in xenotransplantation and in cancer immunotherapy. Immunol Cell Biol. 2005;83(6):674–86. doi: 10.1111/j.1440-1711.2005.01366.x. [DOI] [PubMed] [Google Scholar]
- 58.Seung E, Tager AM. Humoral immunity in humanized mice: a work in progress. J Infect Dis. 2013;208(2):S155–9. doi: 10.1093/infdis/jit448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: H&E and alpha-gal epitope staining of fresh porcine myocardium, decellularized tissue, and self-assembled hydrogel. Top row shows hematoxylin and eosin (H&E) staining with scale bar of 50 μm. Blue channel shows Hoechst staining for nuclei, and green channel shows alpha-gal epitope staining, demonstrating its substantial removal during material processing. Bottom scale bars are 100 μm.
Figure S2: Comparison of Total Cell Infiltration Between Animal Models. Quantification from H&E images of cellular density in the whole biomaterial (A, B) and infiltration into the core (C, D) in porcine (PMM) and human myocardial matrix (HMM) at three days and one week in Balb/c and Hu-mice. Results for NSG mice are also shown at one week. (*p<0.05, **p<0.01, ***p<0.001).
Figure S3: Flow cytometry cellular quantification in Balb/c animals for either PMM, HMM, or NDM. (A) Representative histograms or scatter plots for staining of macrophages (mCD68+), T-helper cells (mCD3+mCD4+), and cytotoxic T-cells (mCD3+mCD8+). B-cells (CD19+) were not detected in the biomaterials. (B) Averages and standard deviations for each treatment group and cell type analyzed with flow cytometry. (C) Ratio of T-helper cells to cytotoxic T-cells showing PMM and HMM favoring T-helper cells and NDM favoring cytotoxic T-cells. (***p<0.001).
Figure S4 Immunohistochemistry of T-helper cells and cytotoxic T-cells in either Balb/c or Hu-mice spleen. Hoechst staining (blue) was used to label nuclei. T-helper cells were labeled by co-staining with CD3 (red) and CD4 (green). Cytotoxic T-cells were labeled by co-staining with CD3 (red) and CD8 (green). Mouse specific antibodies were used for Balb/c spleens, and human specific antibodies were used for Hu-mice spleens. Scale bars are 10 μm.
Figure S5: Quantified cell density for T-Helper cells and Cytotoxic T-cells. Cell density quantification of T-helper cells in whole biomaterial (A) and biomaterial core (B) in nondecellularized (NDM), porcine (PMM) and human myocardial matrix (HMM) at three days and one week in Balb/c and Hu-mice. Cytotoxic T-cell density in whole biomaterial (C) and biomaterial core (D) at three days and one week in Balb/c and Hu-mice. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Figure S6: Quantified cell density for total macrophages and polarized macrophage phenotypes. Cell density quantification of total macrophages in whole biomaterial (A) and biomaterial core (B) in nondecellularized (NDM), porcine (PMM) and human myocardial matrix (HMM) at three days and one week in Balb/c or Hu-mice. Cell density quantification of iNOS+ macrophages in whole biomaterial (C) and biomaterial core (D). Cell density quantification of CD206+ macrophages in whole biomaterial (E) and biomaterial core (F). Cell density quantification of dual stained iNOS+CD206+ macrophages in whole biomaterial (G) and biomaterial core (H). (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).