

Abstract
Objective
To identify risk alleles relevant to the causal and biologic mechanisms of antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV).
Methods
A genome‐wide association study and subsequent replication study were conducted in a total cohort of 1,986 cases of AAV (patients with granulomatosis with polyangiitis [Wegener's] [GPA] or microscopic polyangiitis [MPA]) and 4,723 healthy controls. Meta‐analysis of these data sets and functional annotation of identified risk loci were performed, and candidate disease variants with unknown functional effects were investigated for their impact on gene expression and/or protein function.
Results
Among the genome‐wide significant associations identified, the largest effect on risk of AAV came from the single‐nucleotide polymorphism variants rs141530233 and rs1042169 at the HLA–DPB1 locus (odds ratio [OR] 2.99 and OR 2.82, respectively) which, together with a third variant, rs386699872, constitute a triallelic risk haplotype associated with reduced expression of the HLA–DPB1 gene and HLA–DP protein in B cells and monocytes and with increased frequency of complementary proteinase 3 (PR3)–reactive T cells relative to that in carriers of the protective haplotype. Significant associations were also observed at the SERPINA1 and PTPN22 loci, the peak signals arising from functionally relevant missense variants, and at PRTN3, in which the top‐scoring variant correlated with increased PRTN3 expression in neutrophils. Effects of individual loci on AAV risk differed between patients with GPA and those with MPA or between patients with PR3‐ANCAs and those with myeloperoxidase‐ANCAs, but the collective population attributable fraction for these variants was substantive, at 77%.
Conclusion
This study reveals the association of susceptibility to GPA and MPA with functional gene variants that explain much of the genetic etiology of AAV, could influence and possibly be predictors of the clinical presentation, and appear to alter immune cell proteins and responses likely to be key factors in the pathogenesis of AAV.
Granulomatosis with polyangiitis (Wegener's) (GPA) and microscopic polyangiitis (MPA) are life‐threatening necrotizing vasculitides that are strongly associated with the presence of antineutrophil cytoplasmic antibodies (ANCAs) reactive to proteinase 3 (PR3) or myeloperoxidase (MPO). Although often considered a single disease, GPA and MPA diverge in important respects, such as in the extent of their association with PR3‐reactive ANCAs compared to MPO‐reactive ANCAs, the risk of relapsing disease, and the association of GPA with granulomatous inflammation. The etiology of AAV remains unknown; however, genome‐wide association studies (GWAS) performed in a North American GPA cohort and a European GPA/MPA cohort confirmed the findings from candidate gene analyses identifying strong associations of these diseases with major histocompatibility complex (MHC) class II region alleles 1, 2. A genome‐wide significant association at the SERPINA1 locus was also identified in the European cohort study, with both this and several associations with MHC alleles being differentially detected between patient subsets defined by the presence of PR3‐ANCAs or MPO‐ANCAs 2.
These findings have not yet been replicated, and knowledge remains rudimentary regarding the non‐MHC loci and specific disease‐causal variants predisposing to GPA and/or to MPA. Therefore, we sought to further define the genetic variation underpinning the susceptibility to GPA and MPA by conducting a new GWAS and a validation study of a larger, independently ascertained North American–based cohort of GPA/MPA patients and healthy controls, involving functional annotation of the risk loci to identify candidate disease‐causal alleles.
PATIENTS AND METHODS
Subjects
All study subjects were of self‐reported European ancestry, with the diagnosis of AAV, and specifically of GPA or MPA, being based on the American College of Rheumatology modified criteria for the classification of vasculitis 3. The discovery cohort included the following subjects: 779 AAV cases recruited via 13 centers from the Vasculitis Clinical Research Consortium (VCRC), which conducts studies involving vasculitis patients in the US, Canada, and elsewhere; 438 AAV cases recruited via the Wegener's Granulomatosis Genetic Repository (WGGER), a study conducted at 8 centers in the US from 2001 to 2005; and 378 AAV cases from the University of North Carolina Kidney Center (key clinical and serologic features are provided in Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). The controls included 202 healthy subjects recruited from the WGGER, and 3,121 historic controls whose genotype data were obtained from the Resource for Genetic Epidemiology Research on Aging study 4, 5. The replication cohort included 505 AAV cases and 1,477 healthy controls recruited independently from Canada and the US via a Toronto‐based AAV study, and 114 independent cases recruited via the VCRC. Demographic data and samples of peripheral blood cells and/or saliva were obtained from all subjects after their provision of written informed consent. The local institutional review boards approved the study.
Genotyping methods
For the GWAS, 1,615 AAV cases and 202 healthy controls were genotyped at the Mount Sinai Hospital Clinical Genomics Centre, and 3,121 historic controls were genotyped at Affymetrix (Santa Cruz, CA) using the Axiom Biobank 1 Genotyping array. This array tests 628,679 single‐nucleotide polymorphisms (SNPs), including 246,000 genome‐wide association markers (36.5%), 265,000 nonsynonymous coding SNPs (39.3%), 70,000 loss‐of‐function SNPs (10.4%), 23,000 expression quantitative trait loci (eQTL) SNPs (3.4%), 2,000 pharmacogenetic markers (0.3%), and 27,679 “custom” markers. Genotypes were called and processed using Affymetrix Genotyping Console version 4.2 and SNPolisher software. Quality control filtering was performed using Golden Helix SVS software (version 8.3.4) and with a genotype call rate of >95%, individual sample call rate of >97%, and exclusion of SNPs with a Hardy‐Weinberg equilibrium (HWE) P value of <10−5. After filtering, genotypes derived from SNP markers common to both data sets were merged in a single file containing 1,528 cases, 3,309 controls, and 333,040 SNPs. A set of linkage disequilibrium (LD)–pruned SNPs with a minor allele frequency (MAF) of >5% was used to estimate identity by descent (ibd) and ancestry. For each pair of individuals with an estimated ibd of >0.25, the sample with the lower call rate was removed. Principal components analysis was used to exclude samples from subjects with non‐European ancestry 6. In total, 1,371 cases, 3,258 controls, and 333,035 SNPs passed quality control filters (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract).
For the replication study, 8 SNPs in 6 gene loci were genotyped in 619 AAV cases and 1,477 controls using Sequenom iPLEX assays. One other SNP (rs62132293) at the PRTN3 locus was genotyped using TaqMan (Applied Biosystems). After quality control filtering, 615 cases, 1,465 controls, and 9 SNPs were retained for analysis. For the replication study of the patients with MPO‐ANCAs/perinuclear ANCAs (designated herein as MPO‐ANCAs), 3 additional SNPs (rs3998159, rs7454108, and rs1049072) at the HLA–DQA2 and DQB1 loci were genotyped on the same platform. The GWAS and replication data sets were then combined for meta‐analysis.
Statistical analysis
Association tests and meta‐analyses
For the discovery data set, case–control association tests were conducted using logistic regression, with the principal components differing between cases and controls included as covariates, to adjust for population stratification. Calculation of the genomic control factor using EigenStrat showed a minimal inflation value of 1.012 and 0.991 before and after adjustment for the top 3 eigenvectors, respectively (see Supplementary Figure 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). An analysis of all of the nonzero eigenvalues established that 221,341 independent tests were conducted from 280,677 autosomal markers, which, with Bonferroni correction, established the P value for genome‐wide significance as <2.2 × 10−7.
Meta‐analysis of the data from the GWAS and replication logistic regression analyses was conducted using the basic meta‐analysis function in Plink version 1.9 (6,7). Differences between patients with GPA and patients with MPA and between patients with PR3‐ANCAs/cytoplasmic ANCAs (designated herein as PR3‐ANCAs) and those with MPO‐ANCAs were studied using this approach. Between‐study heterogeneity was tested by the chi‐square–based Cochrane's Q statistic. Heritability was estimated using genome‐wide complex trait analysis, as described by Lee et al 8, and assuming an AAV prevalence of 1/10,000. Prior to this analysis, we excluded sex chromosome data, SNPs with MAFs of <0.05 and missing rates of >0.01, individuals whose missing SNP rates were >0.01, SNPs with an HWE P value of <0.05, or markers with a significant difference in missingness between cases and controls.
Imputation
Genome‐wide imputation for the 4,629 samples in the discovery cohort was performed using 1000 Genomes Project Phase 3 data as the reference (release date October 2014) for the autosomes, and Phase 1 data (release date August 2012) as the reference for the X chromosome. Following removal of SNPs with call rates of <95%, MAFs of <0.001, and HWE P values of <10−5, SHAPEIT (http://shapeit.v2.r790.Ubuntu_12.04.4.static) was used to derive phased genotypes, and the phased data were imputed using IMPUTE version 2 (http://impute_v2.3.2_x86_64_static) to assess ∼5‐Mb nonoverlapping intervals 9. Imputation within defined regions was performed using IMPUTE version 2 without prephasing.
Conditional analysis
To test for multiple independent effects within the HLA region, a logistic regression framework was used to assess individual HLA alleles for association, including the top 3 principal components as covariates to account for population stratification. After we had identified the most significant marker, we tested for additional independent effects by including the dose of the top markers in a joint model. Conditional analysis was performed using the proc logistic module of SAS (version 9.2) to obtain odds ratios (ORs) when all top markers were jointly analyzed.
Population attributable fraction (PAF)
The PAF was estimated using ORs from a multivariate logistic regression model incorporating SNPs from multiple loci, so that each OR is adjusted for the effects of the other SNPs. The PAF for effects from an allele at a single locus was determined as follows:
where OR is the odds ratio associated with the allele genotype, and RAF is the allele frequency of the risk variant. For computation over multiple loci, the following formula was used:
Random forest analysis
The potential role of combinations of alleles in the risk of AAV was evaluated by random forest analysis using classification and regression tree (CART) methodology 10. Data from 11 risk‐associated variants were subjected to analyses in which CARTs were repetitively built using two‐thirds of the samples and variables. The CARTs were then used to classify the remaining one‐third of the data. Eight variants that improved the model fit by ≥3% were retained to build a CART. The rpart program in R and the Gini index measure were used to identify optimal splits of the data, with the complexity parameter set to 0.001 and the data then pruned to include only those nodes containing at least 20 observations. This final model had a classification accuracy of 73%. ORs were calculated with the following formula:
The Woolf approximation was used to compute standard errors and confidence intervals.
Functional annotation
The online Probabilistic Identification of Causal SNPs (PICS) algorithm (http://www.broadinstitute.org/pubs/finemapping/?q=pics) was used to identify variants at each risk locus with a PICS probability of >0.0275, consistent with that used by Farh et al 11. We then used the Ensembl Variant Effect Predictor web tool to annotate these variants for predicted functional consequences (http://www.ensembl.org/info/docs/tools/vep/index.html) and used Genevar 12, seeQTL (http://www.bios.unc.edu/research/genomic_software/seeQTL/) 13, and the University of Chicago eQTL browser (http://eqtl.uchicago.edu/cgi‐bin/gbrowse/eqtl/) to identify eQTLs.
Cellular assays
Peripheral blood mononuclear cells (PBMCs) and polymorphonuclear leukocytes (obtained from patients at Mount Sinai Hospital) were isolated over Ficoll‐Hypaque. For quantitative polymerase chain reaction (qPCR), RNA (500 ng) was reverse transcribed using random hexamer primers and SuperScript III reverse transcriptase (Invitrogen), and qPCR was performed using SYBR Green and the gene‐appropriate primer pairs (listed in Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). Samples were run on an ABI Prism 7900HT system (Applied Biosystems), and the fold change in expression of the specific gene relative to the internal control gene (COX5B for PRTN3; GAPDH for HLA–DPB1) was calculated using the method 14.
For flow cytometry, PMBCs were stained with phycoerythrin‐conjugated anti‐CD19 antibodies, allophycocyanin–Cy7–conjugated anti‐CD14 antibodies (BD Biosciences), and/or fluorescein isothiocyanate (FITC)–conjugated HLA–DP antibodies (Leinco) or FITC‐conjugated murine IgG (BD Biosciences). The cells were then analyzed using a FACSCanto cytometer (BD Biosciences) and FlowJo software.
For ELISpot experiments, 20‐mer peptides, which were selected using published data or the NetMHCII version 2.2 prediction algorithm (http://www.cbs.dtu.dk), were synthesized and purified by the manufacturer (Genscript) (for details, see Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract).
PBMCs (2 × 10−5) from PR3‐ANCA–positive vasculitis patients and healthy controls were suspended in 20% fetal bovine serum–supplemented RPMI and incubated in 96‐well ELISpot plates (Millipore) precoated with an anti‐human interferon‐γ (IFNγ) monoclonal antibody (eBioscience). Cells were stimulated for 24 hours at 37°C with 10 μg/ml peptide or 1 μg/ml concanavalin A (Sigma) and incubated with a biotinylated mouse anti‐human IFNγ antibody (eBioscience), avidin–horseradish peroxidase (eBioscience), and aminoethylcarbazole solution (BD ELISpot), and an ImmunoSpot reader and software (Cellular Technology) were used to detect IFNγ‐releasing cells.
RESULTS
GPA/MPA susceptibility loci
After filtering and correction for population substructure, our GWAS discovery data set included 333,035 SNPs genotyped in 1,371 subject with AAV (GPA or MPA) and 3,258 healthy controls, with no evidence of inflation of the test statistic (λGC = 0.991) (see Supplementary Figures 1 and 2 and Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract).
Association analysis of this cohort identified 120 SNPs across the MHC class II locus achieving genome‐wide significance levels, with the strongest signals emanating from the DPB1, DPA1, DQA1, and DQB1 genes (Table 1 and Supplementary Table 4 [http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract]). Four other SNPs across 3 non‐MHC gene loci also achieved genome‐wide significance levels and were taken forward, together with 5 top‐scoring SNPs from the MHC region, for a replication study in an independent cohort of 615 cases and 1,465 controls (Table 1; details also shown in Supplementary Figure 1 and Supplementary Table 1 [http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract]). Because all of the associations tested were replicated (at a threshold of P ≤ 0.05), the GWAS and replication data sets were combined for a meta‐analysis, and the 9 associations were also explored in patient subgroups defined by GPA or MPA phenotypes or by ANCA specificities and/or immunofluorescence patterns (PR3‐ANCAs or MPO‐ANCAs).
Results of the meta‐analysis confirmed the MHC class II region as the locus most strongly associated with AAV susceptibility (Table 1). The peak association signals arose from 2 HLA–DPB1 gene variants, rs141530233 (P = 1.13 × 10−89) and rs1042169 (P = 1.12 × 10−84), with other significant associations observed in the HLA–DPA1¸ DQA1, and DQB1 genes (Table 1; see also Supplementary Figures 3 and 4 [http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract]). Associations with DPA1 and DPB1 remained strong in the GPA and PR3‐ANCA subgroups, but not in the MPA or MPO‐ANCA subgroups. Conversely, the DQB1 association was much stronger in patients with MPO‐ANCAs compared to those with PR3‐ANCAs (Table 2). In view of this divergence, a GWAS, replication analysis, and meta‐analysis were also performed de novo to compare patients with either PR3‐ANCAs or MPO‐ANCAs to healthy controls. These analyses revealed significant associations of the MPO‐ANCA phenotype with the variants rs3998159 (P = 5.24 × 10−25) and rs7454108 (P = 5.03 × 10−25) at the HLA–DQA2 locus (Table 3). Neither this association nor any other significant associations beyond those detected in the AAV total cohort GWAS were detected in the subset GWAS analysis of PR3‐ANCA–positive AAV cases and healthy controls (results available in Supplementary Tables 5 and 6, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract).
Table 1.
Results of GWAS, replication, and combined analyses of associations with antineutrophil cytoplasmic autoantibody–associated vasculitisa
|
GWAS (n = 1,371 cases, n = 3,258 controls) |
Replication analysis (n = 615 cases, n = 1,465 controls) |
Combined analysis (n = 1,986 cases, n = 4,723 controls) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Risk | RAF | OR | RAF | OR | OR | |||||||||||
| SNP | Locus | Position | Gene | allele | Cases | Controls | P b | (95% CI)c | Cases | Controls | P d | (95% CI) | P c | (95% CI) | ||
| rs141530233 | 6p21.32 | 33048688 | HLA–DPB1 | A dele | 0.86 | 0.70 | 5.93 × 10−56 | 2.76 (2.44–3.13) | 0.90 | 0.69 | 2.45 × 10−39 | 4.00 (3.23–5.00) | 1.13 × 10−89 | 2.99 (2.69–3.33) | ||
| rs1042169 | 6p21.32 | 33048686 | HLA–DPB1 | G | 0.86 | 0.70 | 4.41 × 10−52 | 2.57 (2.27–2.94) | 0.90 | 0.68 | 1.94 × 10−39 | 4.00 (3.23–5.00) | 1.12 × 10−84 | 2.82 (2.54–3.13) | ||
| rs9277341 | 6p21.32 | 33039625 | HLA–DPA1 | T | 0.84 | 0.70 | 1.62 × 10−40 | 2.21 (1.96–2.50) | 0.87 | 0.66 | 3.58 × 10−34 | 3.13 (2.63–3.70) | 6.09 × 10−71 | 2.44 (2.21–2.69) | ||
| rs35242582 | 6p21.32 | 32600057 | HLA–DQA1 | A | 0.82 | 0.74 | 3.34 × 10−16 | 1.61 (1.43–1.79) | 0.82 | 0.74 | 3.59 × 10−8 | 1.59 (1.35–1.89) | 6.34 × 10−23 | 1.60 (1.46–1.76) | ||
| rs1049072 | 6p21.32 | 32634355 | HLA–DQB1 | A | 0.23 | 0.17 | 4.23 × 10−10 | 1.43 (1.28–1.59) | 0.21 | 0.17 | 1.69 × 10−3 | 1.30 (1.10–1.54) | 6.46 × 10−13 | 1.40 (1.28–1.53) | ||
| rs6679677 | 1p13.2 | 114303808 | PTPN22 | A | 0.13 | 0.09 | 2.40 × 10−8 | 1.49 (1.30–1.72) | 0.11 | 0.09 | 4.57 × 10−2 | 1.25 (1.00–1.55) | 1.88 × 10−8 | 1.40 (1.25–1.57) | ||
| rs62132293 | 19p13.3 | 838178 | PRTN3 | G | 0.37 | 0.31 | 5.55 × 10−8 | 1.30 (1.18–1.43) | 0.37 | 0.31 | 6.81 × 10−5 | 1.33 (1.15–1.52) | 8.60 × 10−11 | 1.29 (1.19–1.39) | ||
| rs28929474 | 14q32.13 | 94844947 | SERPINA1 | T | 0.04 | 0.02 | 8.26 × 10−8 | 2.09 (1.59–2.73) | 0.04 | 0.02 | 6.72 × 10−5 | 2.18 (1.49–3.20) | 3.09 × 10−12 | 2.18 (1.75–2.71) | ||
| rs2476601 | 1p13.2 | 114377568 | PTPN22 (R620W) | A | 0.13 | 0.10 | 3.03 × 10−7 | 1.45 (1.26–1.66) | 0.11 | 0.09 | 5.38 × 10−2 | 1.24 (1.00–1.53) | 1.86 × 10−7 | 1.36 (1.21–1.53) | ||
RAF = risk allele frequency; OR = odds ratio; 95% CI = 95% confidence interval.
EigenStrat P value.
P values for the replication analysis and combined genome‐wide association study (GWAS) data sets were calculated using the Cochran‐Mantel‐Haenszel method, which combines allele frequency counts.
Plink P value.
The single‐nucleotide polymorphism (SNP) rs141530233 is an insertion/deletion polymorphism, with the risk genotype lacking an adenosine residue at nucleotide position 33048688 (adenosine deletion [A del]) and the nonrisk genotype containing this adenosine residue.
Table 2.
MHC and non‐MHC associations with ANCA‐associated vasculitis according to clinically and serologically defined subgroups of patientsa
| Clinical syndrome | ANCA specificity | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Risk | Overall analysis of combined cohort (n = 1,986 cases, n = 4,723 controls) |
GPA (n = 1,556) vs. controls (n = 4,723) |
MPA (n = 236) vs. controls (n = 4,723) |
GPA (n = 1,556) vs. MPA (n = 236) |
PR3‐cANCA (n = 1,361) vs. controls (n = 4,723) |
MPO‐pANCA (n = 378) vs. controls (n = 4,723) |
PR3‐cANCA (n = 1,361) vs. MPO‐pANCA (n = 378) |
||||||||||||||||
| SNP | Locus | Gene | allele | P b | OR | P b | OR | P b | OR | P b | OR | P b | OR | P b | OR | P b | OR | ||||||
| rs141530233 | 6p21.32 | HLA–DPB1 | A del | 1.13 × 10−89 | 2.99 | 3.80 × 10−93 | 3.82 | 9.45 × 10−5 | 1.58 | 1.45 × 10−9 | 2.04 | 1.33 × 10−106 | 6.19 | 1.50 × 10−2 | 1.24 | 3.53 × 10−32 | 3.93 | ||||||
| rs1042169 | 6p21.32 | HLA–DPB1 | G | 1.12 × 10−84 | 2.82 | 1.09 × 10−90 | 3.66 | 2.22 × 10−3 | 1.40 | 9.50 × 10−12 | 2.18 | 6.53 × 10−106 | 6.09 | 1.27 × 10−1 | 1.14 | 3.44 × 10−36 | 4.27 | ||||||
| rs9277341 | 6p21.32 | HLA–DPA1 | T | 6.09 × 10−71 | 2.44 | 2.78 × 10−73 | 2.86 | 9.40 × 10−4 | 1.45 | 4.96 × 10−7 | 1.79 | 4.52 × 10−84 | 3.69 | 3.61 × 10−3 | 1.29 | 4.55 × 10−20 | 2.61 | ||||||
| rs35242582 | 6p21.32 | HLA–DQA1 | A | 6.34 × 10−23 | 1.60 | 1.60 × 10−20 | 1.63 | 8.91 × 10−3 | 1.36 | 1.36 × 10−1 | 1.20 | 5.78 × 10−18 | 1.62 | 2.34 × 10−7 | 1.68 | 7.67 × 10−1 | 1.03 | ||||||
| rs1049072 | 6p21.32 | HLA–DQB1 | A | 6.46 × 10−13 | 1.40 | 1.40 × 10−7 | 1.31 | 4.16 × 10−9 | 1.89 | 2.99 × 10−3 | 1.39 | 3.82 × 10−3 | 1.17 | 2.13 × 10−24 | 2.37 | 7.53 × 10−13 | 1.94 | ||||||
| rs6679677 | 1p13.2 | PTPN22 | A | 1.88 × 10−8 | 1.40 | 2.38 × 10−7 | 1.40 | 8.96 × 10−4 | 1.58 | 5.48 × 10−1 | 1.09 | 7.89 × 10−6 | 1.36 | 8.83 × 10−7 | 1.71 | 1.08 × 10−1 | 1.21 | ||||||
| rs62132293 | 19p13.3 | PRTN3 | G | 8.60 × 10−11 | 1.29 | 7.06 × 10−11 | 1.32 | 1.12 × 10−1 | 1.17 | 2.70 × 10−1 | 1.12 | 3.59 × 10−13 | 1.39 | 5.66 × 10−1 | 1.05 | 3.22 × 10−5 | 1.45 | ||||||
| rs28929474 | 14q32.13 | SERPINA1 | T | 3.09 × 10−12 | 2.18 | 3.53 × 10−13 | 2.35 | 2.06 × 10−2 | 1.88 | 3.86 × 10−1 | 1.26 | 1.29 × 10−13 | 2.43 | 4.96 × 10−3 | 1.87 | 1.92 × 10−1 | 1.34 | ||||||
| rs2476601 | 1p13.2 | PTPN22 (R620W) | A | 1.86 × 10−7 | 1.36 | 1.77 × 10−6 | 1.36 | 1.31 × 10−3 | 1.56 | 4.95 × 10−1 | 1.10 | 3.19 × 10−5 | 1.33 | 5.85 × 10−6 | 1.64 | 1.40 × 10−1 | 1.19 | ||||||
MHC = major histocompatibility complex; ANCA = antineutrophil cytoplasmic autoantibody; GPA = granulomatosis with polyangiitis (Wegener's); MPA = microscopic polyangiitis; PR3 = proteinase 3; cANCA = cytoplasmic ANCA; MPO = myeloperoxidase; pANCA = perinuclear ANCA; SNP = single‐nucleotide polymorphism; OR = odds ratio; A del = adenosine deletion.
EigenStrat P value.
Table 3.
MHC and non‐MHC associations with ANCA‐associated vasculitis in the MPO‐pANCA subgroup (assessed in GWAS, replication, and combined analyses) compared to the PR3‐cANCA subgroup (in combined analysis)a
| Patients with MPO‐pANCAs | Patients with PR3‐cANCAs in | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
GWAS (n = 324 patients, n = 3,258 controls) |
Replication analysis (n = 54 patients, n = 1,465 controls) |
Combined association analysis (n = 378 patients, n = 4,723 controls) |
combined association analysis (n = 1,361 patients, n = 4,723 controls) |
|||||||||||||
| Risk | RAF | OR | RAF | OR | OR | OR | ||||||||||
| SNP | Locus | Position | Gene | allele | Cases | Controls | P b | (95% CI) | Cases | Controls | P b | (95% CI) | P b | (95% CI) | P b | (95% CI) |
| rs3998159 | 6p21.32 | 32682019 | HLA–DQA2 | C | 0.23 | 0.10 | 3.47 × 10−19 | 2.61 (2.12–3.22) | 0.25 | 0.09 | 7.11 × 10−7 | 3.25 (2.04–5.18) | 5.24 × 10−25 | 2.72 (2.24–3.22) | 5.18 × 10−1 | 1.05 (0.91–1.20) |
| rs7454108 | 6p21.32 | 32681483 | HLA–DQA2 | C | 0.23 | 0.10 | 3.90 × 10−19 | 2.61 (2.12–3.23) | 0.25 | 0.09 | 4.78 × 10−7 | 3.34 (2.09–5.33) | 5.03 × 10−25 | 2.73 (2.25–3.24) | 5.48 × 10−1 | 1.04 (0.90–1.20) |
| rs1049072 | 6p21.32 | 32634355 | HLA–DQB1 | A | 0.32 | 0.17 | 1.63 × 10−18 | 2.27 (1.89–2.72) | 0.35 | 0.17 | 3.16 × 10−6 | 2.60 (1.74–3.88) | 2.13 × 10−24 | 2.37 (2.01–2.78) | 3.82 × 10−3 | 1.17 (1.06–1.31) |
MHC = major histocompatibility complex; ANCA = antineutrophil cytoplasmic autoantibody; MPO = myeloperoxidase; pANCA = perinuclear ANCA; GWAS = genome‐wide association study; PR3 = proteinase 3; cANCA = cytoplasmic ANCA; RAF = risk allele frequency; SNP = single‐nucleotide polymorphism; OR = odds ratio; 95% CI = 95% confidence interval.
EigenStrat P value.
Among the non‐MHC associations identified in the total AAV GWAS, the strongest signal arose from a SNP (rs28929474) in the SERPINA1 gene (P = 3.09 × 10−12) encoding an α1 anti‐trypsin null (“Z”) allele that was previously implicated in GPA by candidate gene analysis (Table 1; see also Supplementary Figures 3 and 4 [http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract]) 15. This association was limited to the GPA and PR3‐ANCA subsets (Table 2) and is consistent with prior GWAS data showing that a significant association with another SERPINA1 SNP, rs7151526, depended entirely on the concomitant presence of the Z allele 2.
A significant association of AAV with rs62132293, a SNP located 2.6 kb upstream of the PRTN3 transcription start site, was also observed (P = 8.60 × 10−11); this finding is in keeping with previously observed associations at this locus (albeit with different SNPs) in AAV candidate gene and GWAS analyses (Table 1 and Supplementary Figures 3 and 4 [http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract]) 2, 16. This association was limited to the GPA and PR3‐ANCA subsets (Table 2), consistent with the pathophysiologic relevance of the PRTN3‐encoded PR3 serine protease to these phenotypes 17.
Significant associations with AAV were observed at the PTPN22 rs6679677 and rs2476601 loci (P = 1.88 × 10−8 and P = 1.86 × 10−7, respectively) (Table 1 and Supplementary Figures 3 and 4 [http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract]). Consistent with their equivalent effect sizes, these variants are in almost complete LD (r2 = 0.99). However, the rs2476601 variant encodes an Arg620Trp substitution in the Lyp phosphatase associated with risk of multiple autoimmune diseases, including giant cell arteritis 18, 19. Although not consistently observed in candidate gene studies 20, 21, the association of rs2476601 with GPA/MPA is strongly supported by our data, and unlike most of the other significant associations observed, this allele's strength of association did not differ between the subgroups (Table 2).
The discovery GWAS of either the entire cohort or the PR3‐ANCA subgroup revealed no reliable associations at the SEMA6A gene locus, possibly because those analyses involved a different case–control cohort. In the current discovery GWAS, there were 189 SNPs identified in the 1‐MB region around SEMA6A, of which the most significant variant, rs12521259, was located ∼100 kb upstream of SEMA6A (P for association = 9.11 × 10−3 in the full cohort and P = 4.87 × 10−3 in the PR3‐ANCA subgroup).
Analyses of the associations in patient subgroups defined by the presence or absence of lung or kidney disease revealed a modest association with AAV risk at the HLA–DPA1 locus in patients with kidney involvement (P = 8.15 × 10−3), whereas no significant subgroup differences were apparent at the other risk loci (results in Supplementary Table 7, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). Modest associations with HLA–DPB1 and HLA–DPA1 were observed but restricted to the patients with ANCA‐positive AAV; this finding may be a reflection of the relatively low numbers of ANCA‐negative cases analyzed.
Contribution of risk alleles jointly to disease susceptibility
As the strongest associations in all subgroups were with MHC gene SNPs, independence of the individual allele associations was explored by forward logistic regression selection analysis. Beginning with the most significant SNP identified in the total cohort or in the PR3‐ANCA or MPO‐ANCA subgroups, additional significant variants were incorporated into the analysis until no variants significant at the P < 5 × 10−8 level remained. This analysis revealed that variants in several of the class II genes studied in each group were jointly significantly associated with AAV risk (results in Supplementary Table 8, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract).
These analyses showed that the array heritability of AAV was a mean ± SD 0.2197 ± 0.0204, while in analyses with the HLA region removed, the array heritability was 0.138 ± 0.022. The PAF for the risk loci to disease was also assessed, and the collective contribution of these loci to risk of AAV was found to be substantive (PAF of 77%), albeit variable (PAFs between 30% and 87%) across different the subgroups (Table 4).
Table 4.
Population risk estimates for disease‐associated SNPs at the MHC and non‐MHC loci in all patients with AAV and in clinically and serologically defined subgroups of patientsa
| Clinical syndrome | ANCA specificity | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Combined AAV cohort | GPA | MPA | PR3‐cANCAs | MPO‐pANCAs | ||||||||||||
| Gene | SNP | RAF | OR | PAF | OR | PAF | OR | PAF | OR | PAF | OR | PAF | ||||
| HLA–DPB1 | rs141530233 | 0.70 | 2.36 | 0.49 | 3.01 | 0.58 | 1.64 | 0.31 | 3.98 | 0.68 | 1.01 | 0.00 | ||||
| HLA–DPA1 | rs9277341 | 0.70 | 1.62 | 0.30 | 1.81 | 0.36 | 1.26 | 0.00 | 1.84 | 0.37 | 1.03 | 0.00 | ||||
| HLA–DQA1 | rs35242582 | 0.74 | 1.39 | 0.22 | 1.46 | 0.26 | 1.06 | 0.00 | 1.27 | 0.17 | 1.02 | 0.00 | ||||
| HLA–DQB1 | rs1049072 | 0.17 | 1.33 | 0.05 | 1.19 | 0.00 | 1.91 | 0.13 | 1.16 | 0.00 | 2.64 | 0.22 | ||||
| PRTN3 | rs62132293 | 0.31 | 1.27 | 0.08 | 1.30 | 0.09 | 1.18 | 0.00 | 1.59 | 0.16 | 1.10 | 0.00 | ||||
| SERPINA1 | rs28929474 | 0.02 | 2.13 | 0.02 | 2.43 | 0.02 | 1.98 | 0.00 | 3.64 | 0.04 | 2.98 | 0.00 | ||||
| PTPN22 (R620W) | rs2476601 | 0.10 | 1.45 | 0.04 | 1.47 | 0.04 | 1.62 | 0.06 | 1.71 | 0.06 | 2.18 | 0.10 | ||||
| Total | – | – | – | 0.77 | – | 0.83 | – | 0.43 | – | 0.87 | – | 0.30 | ||||
Population risk estimates included the risk allele frequency (RAF), odds ratio (OR), and population attributable fraction (PAF) calculated in the overall combined cohort of patients with antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis (AAV), in the clinical subgroups of patients with granulomatosis with polyangiitis (Wegener's) (GPA) and those with microscopic polyangiitis (MPA), and in the serologically defined subgroups of patients with proteinase 3 (PR3)–cytoplasmic ANCAs (cANCAs) and those with myeloperoxidase (MPO) perinuclear ANCAs. SNPs = single‐nucleotide polymorphisms; MHC = major histocompatibility complex.
The extent to which the risk variants/variant combinations might be predictive of disease was also evaluated using random forest and CART methods. These analyses confirmed the strong association of HLA class II alleles with AAV risk (details in Supplementary Figure 5, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). Furthermore, homozygosity for the relatively common DPB1 risk allele rs141530233 and DPA1 risk allele rs9277341, together with homozygosity or heterozygosity for the rare SERPINA1 risk variant rs2829474, identified a subgroup of individuals with an OR of >10 for developing AAV.
For HLA alleles, no subsets were defined by heterozygotes, and further modeling comparing the goodness‐of‐fit of additive, dominant, and recessive models showed that the risk imbued by the HLA–DPB1, DPA1, and DQA1 disease‐associated variants is recessively inherited, i.e., conferred by carriage of the common homozygous genotypes (see Supplementary Table 9, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). Associations of the homozygous risk genotypes with AAV were as follows: OR of 3.58 for rs141530233, OR of 2.69 for rs9277341, and OR of 1.80 for rs352425282. Similar results were found for the subgroups defined by carriage of PR3‐ANCAs or MPO‐ANCAs, with recessive models fitting best. These observations suggest a potential for genetic data to inform the distinction of patient subsets within the AAV population and identify an unusual recessive effect for the HLA region loci studied.
Disease‐associated PRTN3 polymorphism identified as a novel eQTL
To identify candidate causal variants, additional genotypes were imputed and the PICS algorithm was applied across each risk locus 11. Although peak association signals at a few loci were stronger for imputed SNPs than for observed SNPs (details in Supplementary Table 10, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract), among all variants with a PICS probability of >0.0275, the index SNPs derived from direct genotyping were consistently associated with the highest PICS values (see Supplementary Table 11, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). PICS values were particularly high for the index SNPs at the HLA–DPB1, SERPINA1, and PTPN22 loci, all of which are functional missense variants 15, 22, 23. The candidate causal variants at the other HLA gene loci were either synonymous, intronic, or upstream gene variants, but the majority of these noncoding variants, and even several HLA–DPB1 coding variants, have been annotated as eQTLs that influence gene expression in immune cell lineages 24.
None of the candidate variants at the PRTN3 locus were coding or reported eQTL SNPs. Increases in PRTN3 expression levels have, however, been observed in neutrophils from patients with AAV and implicated in the pathogenicity of AAV 17, 25. Because knowledge of neutrophil‐specific eQTLs remains limited, we evaluated the lead SNP at this locus (rs62132293) for allelic effects on PRTN3 expression in neutrophils. Results of qPCR analyses revealed cellular PRTN3 transcript levels to be significantly higher in those homozygous for the risk (G) allele than in donors with CC or CG genotypes (Figure 1). These results identify the rs62132293 SNP as an eQTL for PRTN3 and suggest that the causal variant at this locus engenders risk by its association with increased PRTN3 expression.
Figure 1.
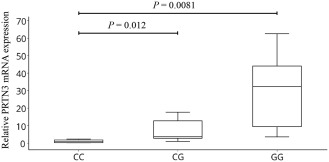
Association between the antineutrophil cytoplasmic autoantibody–associated vasculitis variant rs62132293 and increased expression of PRTN3. Levels of mRNA for PRTN3 were measured using quantititative polymerase chain reaction amplification of cDNA from peripheral blood polymorphonuclear leukocytes obtained from healthy donors with the rs62132293 CC genotype (n = 7), rs62132293 CG genotype (n = 9), or rs62132293 GG genotype (n = 6). PRTN3 expression levels, relative to the values for the calibrator reference gene COX5B, are presented as box plots, in which the boxes represent the 25th to 75th percentiles, the horizontal line within the boxes indicates the median, and the bars outside the boxes indicate the lowest and highest values. Data are representative of 3 independent experiments. P values were determined by unpaired t‐test.
Association of the rs141530233 risk variant with altered HLA–DPB1 expression and T cell responses
Among the candidate causal variants, the HLA–DPB1 rs141530233 and rs1042169 SNPs had the largest effects on risk, with respective ORs of 2.99 and 2.82 in the primary cohort and respective ORs of 6.19 and 6.09 in the PR3‐ANCA subset (Tables 1 and 2). These SNPs are, respectively, insertion/deletion (–/A) and missense (G/A) polymorphisms that map only 2 basepairs apart in exon 2 of the HLA–DPB1 gene, with their risk alleles in complete LD in the control cohort and the reference 1000 Genomes Project data sets. In the latter population, these alleles correlate perfectly with another insertion/deletion polymorphic variant, rs386699872 CA/G, 3 basepairs downstream of rs141530233, suggesting that these variants comprise a triallelic risk and nonrisk HLA–DPB1 haplotype (see Supplementary Figure 6, http://onlinelibrary.wiley.com/doi/10.1002/art.40034/abstract). To confirm the haplotypic relatedness of the 3 variants, we sequenced this region in 100 study subjects who were homozygous for the rs141530233 and rs1042169 markers. Our findings confirmed the organization of the 3 variants in 2 haplotype blocks (as shown in Supplementary Figure 6), which is consistent with the findings in prior studies of a dimorphic polymorphism (GGPM versus DEAV) at the corresponding amino acid positions 84–87 of the HLA–DPB chain 22, 26, 27.
Effects of the rs141530233 SNP on gene expression have not been reported, but the linked missense rs1042169 G/A SNP has been catalogued as an eQTL, with presence of the homozygous GG genotype being correlated with increased expression of HLA–DPB1 in PBMCs 24. These variants are in LD with a SNP variant (rs9277534) in the downstream HLA–DPB1 3 ′‐untranslated region, for which the homozygous genotype is associated with lower levels of HLA–DPB1 and HLA–DP expression in immune cells compared to those in subjects with the alternate homozygous genotype 28, 29. We therefore assessed the relationship between the triallelic AAV risk haplotype and HLA–DPB1/HLA–DP expression using PBMCs from healthy subjects carrying either risk or protective rs1042169 alleles. Results of qPCR analysis revealed HLA–DPB1 messenger RNA levels to be significantly lower in rs1042169 GG risk allele homozygotes than in subjects with the AA or GA genotypes (Figure 2A).
Figure 2.
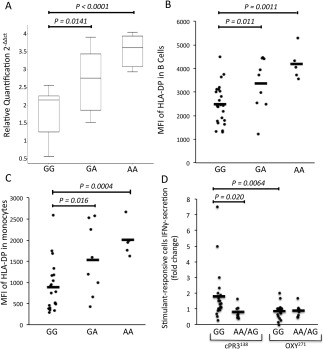
Association of the rs1042169 allele with differential HLA–DPB1 expression and T cell responses. A, HLA–DPB1 mRNA levels were detected by quantititative polymerase chain reaction amplification of cDNA from peripheral blood mononuclear cells (PBMCs) obtained from healthy donors with the rs1042169 GG genotype (n = 13), rs1042169 GA genotype (n = 8), or rs1042169 AA genotype (n = 7). HLA–DPB1 mRNA levels, relative to the values for the calibrator reference gene GAPDH, are shown as box plots, in which the boxes represent the 25th to 75th percentiles, the horizontal line within the boxes indicates the median, and the bars outside the boxes indicate the lowest and highest values. Data are representative of 3 independent experiments. B and C, Surface HLA–DP levels were evaluated by flow cytometric assay of B cells (B) and monocytes (C) in anti‐DP and anti‐CD19 or anti‐CD14 antibody–stained PBMCs obtained from donors with the rs1042169 GG genotype (n = 24), rs1042169 GA genotype (n = 9), or rs1042169 AA genotype (n = 5). D, PBMCs from patients positive for proteinase 3 (PR3)–specific antineutrophil cytoplasmic autoantibodies who were carriers of the rs10421699 GG genotype (n = 21) or the AA/AG genotype (n = 8) were stimulated with anti‐sense PR3 codons (cPR3) or an OXY control peptide and analyzed by ELISpot for interferon‐γ (IFNγ)–secreting T cells. Results are the mean fold change in stimulated cells relative to unstimulated cells. Symbols in B–D represent individual donors; horizontal bars indicate the mean. In A–C, P values were determined by unpaired t‐test. In D, P values were determined by Mann‐Whitney U test (GG versus AA/AG + cPR3138) or Wilcoxon's signed rank test (GG cPR3138 versus GG OXY271). MFI = mean fluorescence intensity.
Furthermore, flow cytometric analyses revealed significantly lower HLA–DP expression on CD19+ B cells and CD14+ monocytes from donors with the GG risk allele than on cells from donors with the GA or AA genotype (Figures 2B and C). Thus, the triallelic risk haplotype defined by the rs1042169 G variant is associated with reduced HLA–DPB1 transcript levels and HLA–DP surface expression in immune cells.
The finding that a triallelic haplotype was correlated with reduced expression of HLA–DPB1/HLA–DP and encoded a putative functionally important HLA–DP polymorphism that is highly associated with risk of AAV, and particularly PR3‐ANCA vasculitis, strongly suggests that this genetic variation influences HLA–DP–modulated immune responses that could be relevant to susceptibility.
Because T cells that respond to PR3 protein or peptides have been identified in patients with PR3‐ANCA–positive AAV, and because the frequency of T cells responding to a “complementary” peptide encoding anti‐sense PR3 codons (cPR3) has been correlated with the presence and activity of disease 30, 31, 32, 33, 34, 35, we stimulated PBMCs from patients carrying rs1042169 G and/or A alleles with putatively immunogenic cPR3 and PR3 peptides and used an IFNγ ELISpot assay to identify responding T cells. Whereas the PR3 peptides elicited either no response or minimal responses (data not shown), the cPR3 peptide, in most patients, evoked clear reactivity that was completely absent in cells stimulated with an irrelevant (OXY) peptide (Figure 2D) and in cells from healthy controls (data not shown). Frequencies of IFNγ‐producing cells differed strikingly among patients according to their rs1042169 allele status, with the numbers of responding T cells being significantly higher (P < 0.02) in risk allele homozygotes than in individuals having 1 or 2 copies of the protective A allele, and significantly higher in risk allele homozygotes following stimulation with cPR3 compared to stimulation with OXY (P < 0.0064). These findings are consistent with the presence of cPR3‐reactive T cells in PR3‐ANCA–positive vasculitis patients and suggest the possibility that the altered HLA–DP expression, and possibly function, associated with the HLA–DPB1 homozygous risk haplotype is correlated with increased numbers of autoreactive cells.
DISCUSSION
This study identifies MHC and non‐MHC gene variants that are associated with GPA/MPA susceptibility and with altered gene expression and/or function of proteins integral to immune responses. Our data reveal that the largest effect on risk emanates from a triallelic HLA–DPB1 haplotype underpinning a previously reported HLA–DPB amino acid polymorphism across positions 84–87 (22–27). Our data also support major roles for the PRTN3, SERPINA1, and PTPN22 genes in AAV susceptibility, providing the first evidence of a genome‐wide significant association with the PTPN22 rs2476601 functional variant, and identifying a correlation of the top‐scoring variant at SERPINA1 as a null allele and at PRTN3 as an eQTL allele with increased PRTN3 expression in neutrophils. Results of the CART analysis revealed the potential for these functional variants to be used to identify population subsets of individuals who would be at highly elevated risk of developing GPA or MPA, as indicated by the observed collective PAF of 77%. The estimated array heritability of 21% is comparable to previous estimates of the heritability of inflammatory bowel disease 36.
Among the MHC associations, the HLA–DPB1 risk haplotype alleles appear particularly significant, having a very strong effect on risk and underpinning a β‐chain polymorphism in the HLA–DP antigen‐binding pocket that modulates the protein's peptide‐binding properties and possibly its effects on T cell allorecognition 22. The physiologic significance of this haplotype can also be inferred on the basis of our data linking these risk alleles to decreased expression of HLA–DPB1 and HLA–DP and an increased frequency of cPR3 peptide–reactive T cells in patients with anti‐PR3 autoantibodies. Although understanding of the autoantigenic epitopes driving T cell responses in AAV is limited, our findings are consistent with prior data correlating alleles at linked HLA–DPB1 SNP loci to differential HLA–DPB1/HLA–DP expression and with the association of such expression changes, as well as the HLA–DP GGPM/DEAV variance, with differential outcomes of specific immune challenges 28, 29. Further investigation is required to define the extent to which the risk haplotype–associated increase in levels of autoreactive T cells reflects the failure to eliminate such cells during thymic selection and/or whether another mechanistic aberrancy may be involved.
Among the non‐MHC associations identified, direct causal effects of the PTPN22 risk variant are strongly suggested by previous data linking the associated Lyp variant to aberrant increases in lymphocyte antigen receptor signaling and dendritic cell activation 23. Direct contribution of the SERPINA1 rs28929474 risk variant to AAV pathogenesis has also been implied in a study that established a role for α1‐antitrypsin in inhibiting PR3 protease activity and, by extension, PR3‐induced inflammatory responses 37. Similarly, the most strongly associated PRTN3 variant has been found to increase the neutrophil expression of PRTN3, an aberrancy found often in PR3‐ANCA–positive patients, and this is correlated with pathogenic neutrophil activation, suggesting that altered PRTN3 expression mediated via this or another tightly linked variant functionally underpins the PRTN3 association with AAV 17, 38.
Our analyses revealed that the risk associated with the various AAV phenotypes was linked to joint effects of different genes across the HLA class II region. Consistent with a prior study that demonstrated genetic distinctions between PR3‐ANCA–positive vasculitis and MPO‐ANCA–positive vasculitis 2, we detected peak associations of the HLA–DPB1 and HLA–DPA1 variants with positivity for PR3‐ANCAs, whereas in those with MPO‐ANCAs, the HLA–DQA2 and HLA–DQB1 variants showed the strongest associations. Differential effects of these variants also distinguished patients with GPA from those with MPA, suggesting that GPA and PR3‐ANCA–positive AAV share a composite set of MHC class II risk alleles that is largely distinct from those conferring risk of MPA and MPO‐ANCA–positive AAV. Stronger associations at the PRTN3 and SERPINA1 loci appear to distinguish the GPA and PR3‐ANCA subsets from their counterpart subgroups. In contrast, effects of the PTPN22 locus on AAV risk seem equivalent across the different subsets, suggesting that the genetic disparities between subgroups do not reflect insufficient statistical power and are important determinants of phenotypic heterogeneity in AAV.
In summary, our study has illuminated MHC and non‐MHC gene variants that are strongly associated with AAV and that are differentially associated with key clinical and serologic disease subsets. The identified variants could potentially directly influence the pathogenesis of AAV. The extent to which, and the mechanisms whereby, these variants directly cause disease requires more investigation, and our data do not directly preclude the potential biologic relevance of other alleles in LD with these variants, particularly at the HLA–DPB1 and PRTN3 loci. Whether the sample size constrained our analysis of important subsets (such as patients with IgG‐ANCAs versus those with IgA‐ANCAs) or confounded detection of some important associations remains to be determined 39. Nonetheless, our findings identify a set of risk variants that explain much of the genetic risk of GPA/MPA, that appear to influence the clinical presentation of the disease, and that represent biologically important alleles with high potential to drive the aberrant immune responses contributing to the development of AAV.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Siminovitch had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Merkel, Xie, Monach, Ciavatta, Pinder, Zhang, Hirano, Edberg, Falk, Amos, Siminovitch.
Acquisition of data
Merkel, Xie, Monach, Ji, Ciavatta, Byun, Pinder, Zhao, Zhang, Tadesse, Qian, Weirauch, Nair, Tsoi, Pagnoux, Carette, Chung, Cuthbertson, Davis Jr., Dellaripa, Forbess, Gewurz‐Singer, Hoffman, Khalidi, Koening, Langford, Mahr, McAlear, Moreland, Seo, Specks, Spiera, Sreih, St.Clair, Stone, Ytterberg, Elder, Qu, Ochi, Hirano, Edberg, Falk, Amos, Siminovitch.
Analysis and interpretation of data
Merkel, Xie, Monach, Ji, Byun, Pinder, Zhao, Zhang, Tadesse, Weirauch, Chung, Elder, Qu, Ochi, Hirano, Edberg, Amos, Siminovitch.
ADDITIONAL DISCLOSURE
Dr. Davis is currently an employee of Pfizer Inc.
Supporting information
Supplementary Figure 1. Quality Control and Study Design. AAV = ANCA‐associated vasculitis; GERA = Genetic Epidemiology Research on Aging; VCRC = Vasculitis Clinical Research Consortium; WGGER = Wegener's Granulomatosis Genetic Repository; UNC = University of North Carolina. Panel A shows the outcomes of genotyping quality control for single nucleotide polymorphisms (SNPs) and genomic DNA from individual subjects. The requirements for SNPs to meet quality control standards were call rates of greater than 95%, p < 1 x 10−5 for test of Hardy‐Weinberg equilibrium and >0.01 for test of minor allele frequency. Panel B shows the numbers of cases and controls used in the discovery (GWAS) and replication cohorts and combined to generate a meta‐analysis data set.
Supplementary Figure 2. Quantile‐Quantile plot of test statistics for the genome‐wide association study. The –log10(p) values from EIGENSTRAT analysis are plotted on the Y axis against the expected –log10(p) values on the X axis after removing all individuals and SNPs that failed quality control. After genomic control correction, the inflation factor was λ = 0.991 with and 1.012 without eigenvector adjustment. (A) –log10(p) values for all GWAS SNPs. (B) –log10(p) values after removal of the HLA region SNPs.
Supplementary Figure 3. Results of the ANCA‐associated vasculitis genome‐wide association screen. The Y axis shows the –log10 P values (from EIGENSTRAT) for each single nucleotide polymorphism on each chromosome along the X axis. The dashed line indicates the genome‐wide significance threshold (P = 5.0 x 10−7).
Supplementary Figure 4. Locus Zoom plots. Showing regional associations across the MHC and non‐MHC loci. The –log10 P values of single‐nucleotide polymorphisms genotyped in the discovery (•) and replication (▲) cohorts and included in the meta‐analysis (♦) are plotted against their chromosomal position at each locus. SNPs are coloured depending on their degree of correlation (r2) with the top SNP (as estimated on the basis of 1000 Genome European haplotypes, 2012) shown in purple. Genes and expressed sequence tags within each region are shown in the lower panels. HLA‐DP and DQ regions; PTPN22; PRTN3 and SERPINA1.
Supplementary Figure 5. Classification And Regression Tree (CART) model for predicting risk of GPA/MPA vasculitis. The CART analysis incorporated the disease‐associated SNPs identified in the initial ANCA‐associated vasculitis cohort meta‐analysis. The HLA‐DQA2 rs7454108 and PTPN22 rs6679677 and rs2476601 variants that did not substantially improve classification of cases and controls were removed from further analyses. Eight other variants (HLA‐DPA1 rs9277341, HLA‐DPB1 rs141530233, HLA‐DPB1 rs1042169, PRTN3 rs62132293, HLA‐DQA1 rs35242582, SERPINA1 rs28929474, HLA‐DQB1 rs104902, and HLA‐DQA2 rs39981589) all improved the model fit by at least 3% and were retained to build a CART. The three symbols ++, +‐, or – on each split represent minor variant homozygote, heterozygote, or homozygote, respectively. Odds ratios (OR) and confidence intervals (bracketed numbers) are shown for each node with the effects of specific variants on risk shown for each sequentially subclassified patient subset.
Supplementary Figure 6. Confirmation of triallelic HLA‐DPB1 risk and non‐risk haplotypes by direct sequencing analysis. Sequence analysis showing the HLA‐DPB1 exon 2 region rs1042169, rs141530233, rs386699872 risk and non‐risk haplotypes. A 201 bp segment across HLA‐DPB1 nucleotide positions 3,048,604 to 33,048,804 (GRCh37/hg19) was PCR amplified using primer pairs 5’‐GAGTACTGGAACAGCCAGAA and 3’‐TAAGGTCCCTTAGGCCAACC and the amplification products then directly Sanger sequenced in individuals identified in the genome‐wide association study as having homozygous risk (n = 50) or homozygous non‐risk (n = 50) rs1042169 and rs141530233 genotypes. A representative example of the sequence read‐out from each subgroup is shown with the nucleotide sequence and corresponding amino acid sequence and position shown below. The polymorphic alleles within each haplotype are circled. The sequence analysis confirmed 100% correlation of rs386699872 CA with the risk and rs386699872 G with the non‐risk rs1042169/rs141530233 haplotype.
Supplementary Table 1. Sequences of primer pairs used in quantitative PCR analyses.
Supplementary Table 2. Sequences of 20mer peptides used to evaluate T cell responses.
Supplementary Table 3. Summary of patient demographics, clinical data and quality controls outcomes by cohort
Supplementary Table 4. Results of genome‐wide association analysis for ANCAassociated vasculitis.
Supplementary Table 5. Results of genome‐wide association analysis for PR3‐ANCA/cANCA‐associated vasculitis.
Supplementary Table 6. Results of genome‐wide association analysis for MPO‐ANCA/pANCAassociated Vasculitis
Supplementary Table 7. Effects of organ involvement and ANCA status on MHC and non‐MHC associations with ANCA‐associated vasculitis.
Supplementary Table 8. Conditional analyses of risk alleles across the HLA locus
Supplementary Table 9. Evaluation of genetic models for susceptibility for ANCA‐associated vasculitis.
Supplementary Table 10. Comparison of peak observed SNPs and peak imputed SNPs at risk loci for (a) ANCAassociated vasculitis, (b) MPO‐ANCA/pANCA associated vasculitis.
Supplementary Table 11. Most‐likely disease causal variants identified by functional annotation.
Supported by the Erna Baird Memorial Grant, the Vasculitis Foundation, the Ontario Research Fund (grant RE‐05075), the University of Toronto Department of Medicine Challenge Grant, and the National Natural Science Foundation of China (grant 31270930). Dr. Siminovitch holds a tier 1 Canada Research Chair and the Sherman Family Chair in Genomic Medicine. The Vasculitis Clinical Research Consortium (VCRC) has received support from the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases grants U54‐AR‐057319 and R01‐AR‐047799, National Center for Research Resources grant U54‐RR‐019497, the Office of Rare Diseases Research, and the National Center for Advancing Translational Sciences). The VCRC is part of the Rare Diseases Clinical Research Network.
REFERENCES
- 1. Xie G, Roshandel D, Sherva R, Monach PA, Lu EY, Kung T, et al. Association of granulomatosis with polyangiitis (Wegener's) with HLA–DPB1*04 and SEMA6A gene variants: evidence from genome‐wide analysis. Arthritis Rheum 2013;65:2457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, et al. Genetically distinct subsets within ANCA‐associated vasculitis. N Engl J Med 2012;367:214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fries JF, Hunder GG, Bloch DA, Michel BA, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: summary. Arthritis Rheum 1990;33:1135–6. [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann TJ, Zhan Y, Kvale MN, Hesselson SE, Gollub J, Iribarren C, et al. Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics 2011;98:422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 2015;200:1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics 2010;26:2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome‐wide association studies. Am J Hum Genet 2011;88:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Breiman L, Friedman J, Olshen RA, Stone CJ. Classification and Regression Trees. Belmont, California: Chapman and Hall/CRC; 1984. [Google Scholar]
- 11. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518:337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang TP, Beazley C, Montgomery SB, Dimas AS, Gutierrez‐Arcelus M, Stranger BE, et al. Genevar: a database and Java application for the analysis and visualization of SNP‐gene associations in eQTL studies. Bioinformatics 2010;26:2474–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xia K, Shabalin AA, Huang S, Madar V, Zhou YH, Wang W, et al. seeQTL: a searchable database for human eQTLs. Bioinformatics 2012;28:451–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 15. Mahr AD, Edberg JC, Stone JH, Hoffman GS, St.Clair EW, Specks U, et al. Alpha1‐antitrypsin deficiency–related alleles Z and S and the risk of Wegener's granulomatosis. Arthritis Rheum 2010;62:3760–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gencik M, Meller S, Borgmann S, Fricke H. Proteinase 3 gene polymorphisms and Wegener's granulomatosis. Kidney Int 2000;58:2473–7. [DOI] [PubMed] [Google Scholar]
- 17. Rarok AA, Stegeman CA, Limburg PC, Kallenberg CG. Neutrophil membrane expression of proteinase 3 (PR3) is related to relapse in PR3‐ANCA‐associated vasculitis. J Am Soc Nephrol 2002;13:2232–8. [DOI] [PubMed] [Google Scholar]
- 18. Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med 2015;21:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carmona FD, Mackie SL, Martin JE, Taylor JC, Vaglio A, Eyre S, et al. A large‐scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet 2015;96:565–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jagiello P, Aries P, Arning L, Wagenleiter SE, Csernok E, Hellmich B, et al. The PTPN22 620W allele is a risk factor for Wegener's granulomatosis. Arthritis Rheum 2005;52:4039–43. [DOI] [PubMed] [Google Scholar]
- 21. Chung SA, Xie G, Roshandel D, Sherva R, Edberg JC, Kravitz M, et al. Meta‐analysis of genetic polymorphisms in granulomatosis with polyangiitis (Wegener's) reveals shared susceptibility loci with rheumatoid arthritis. Arthritis Rheum 2012;64:3463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diaz G, Amicosante M, Jaraquemada D, Butler RH, Guillen MV, Sanchez M, et al. Functional analysis of HLA‐DP polymorphism: a crucial role for DPβ residues 9, 11, 35, 55, 56, 69 and 84‐87 in T cell allorecognition and peptide binding. Int Immunol 2003;15:565–76. [DOI] [PubMed] [Google Scholar]
- 23. Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, et al. The autoimmune disease‐associated PTPN22 variant promotes calpain‐mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet 2011;43:902–7. [DOI] [PubMed] [Google Scholar]
- 24. The GTEx Consortium . The Genotype‐Tissue Expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang JJ, Pendergraft WF, Alcorta DA, Nachman PH, Hogan SL, Thomas RP, et al. Circumvention of normal constraints on granule protein gene expression in peripheral blood neutrophils and monocytes of patients with antineutrophil cytoplasmic autoantibody‐associated glomerulonephritis. J Am Soc Nephrol 2004;15:2103–14. [DOI] [PubMed] [Google Scholar]
- 26. Doytchinova IA, Flower DR. In silico identification of supertypes for class II MHCs. J Immunol 2005;174:7085–95. [DOI] [PubMed] [Google Scholar]
- 27. Silveira LJ, McCanlies EC, Fingerlin TE, van Dyke MV, Mroz MM, Strand M, et al. Chronic beryllium disease, HLA‐DPB1, and the DP peptide binding groove. J Immunol 2012;189:4014–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thomas R, Thio CL, Apps R, Qi Y, Gao X, Marti D, et al. A novel variant marking HLA‐DP expression levels predicts recovery from hepatitis B virus infection. J Virol 2012;86:6979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petersdorf EW, Malkki M, O'hUigin C, Carrington M, Gooley T, Haagenson MD, et al. High HLA‐DP expression and graft‐versus‐host disease. N Engl J Med 2015;373:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van der Geld YM, Huitema MG, Franssen CF, van der Zee R, Limburg PC, Kallenberg CG. In vitro T lymphocyte responses to proteinase 3 (PR3) and linear peptides of PR3 in patients with Wegener's granulomatosis (WG). Clin Exp Immunol 2000;122:504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winek J, Mueller A, Csernok E, Gross WL, Lamprecht P. Frequency of proteinase 3 (PR3)‐specific autoreactive T cells determined by cytokine flow cytometry in Wegener's granulomatosis. J Autoimmun 2004;22:79–85. [DOI] [PubMed] [Google Scholar]
- 32. Popa ER, Franssen CF, Limburg PC, Huitema MG, Kallenberg CG, Tervaert JW. In vitro cytokine production and proliferation of T cells from patients with anti–proteinase 3– and antimyeloperoxidase‐associated vasculitis, in response to proteinase 3 and myeloperoxidase. Arthritis Rheum 2002;46:1894–904. [DOI] [PubMed] [Google Scholar]
- 33. Pendergraft WF III, Preston GA, Shah RR, Tropsha A, Carter CW Jr, Jennette JC, et al. Autoimmunity is triggered by cPR‐3(105–201), a protein complementary to human autoantigen proteinase‐3. Nat Med 2004;10:72–9. [DOI] [PubMed] [Google Scholar]
- 34. Csernok E, Ai M, Gross WL, Wicklein D, Petersen A, Lindner B, et al. Wegener autoantigen induces maturation of dendritic cells and licenses them for Th1 priming via the protease‐activated receptor‐2 pathway. Blood 2006;107:4440–8. [DOI] [PubMed] [Google Scholar]
- 35. Yang J, Bautz DJ, Lionaki S, Hogan SL, Chin H, Tisch RM, et al. ANCA patients have T cells responsive to complementary PR‐3 antigen. Kidney Int 2008;74:1159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen GB, Lee SH, Brion MJ, Montgomery GW, Wray NR, Radford‐Smith GL, et al. Estimation and partitioning of (co)heritability of inflammatory bowel disease from GWAS and immunochip data. Hum Mol Genet 2014;23:4710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duranton J, Bieth JG. Inhibition of proteinase 3 by α1‐antitrypsin in vitro predicts very fast inhibition in vivo. Am J Respir Cell Mol Biol 2003;29:57–61. [DOI] [PubMed] [Google Scholar]
- 38. Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest 2010;120:3209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kelley JM, Monach PA, Ji C, Zhou Y, Wu J, Tanaka S, et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci U S A 2011;108:20736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Quality Control and Study Design. AAV = ANCA‐associated vasculitis; GERA = Genetic Epidemiology Research on Aging; VCRC = Vasculitis Clinical Research Consortium; WGGER = Wegener's Granulomatosis Genetic Repository; UNC = University of North Carolina. Panel A shows the outcomes of genotyping quality control for single nucleotide polymorphisms (SNPs) and genomic DNA from individual subjects. The requirements for SNPs to meet quality control standards were call rates of greater than 95%, p < 1 x 10−5 for test of Hardy‐Weinberg equilibrium and >0.01 for test of minor allele frequency. Panel B shows the numbers of cases and controls used in the discovery (GWAS) and replication cohorts and combined to generate a meta‐analysis data set.
Supplementary Figure 2. Quantile‐Quantile plot of test statistics for the genome‐wide association study. The –log10(p) values from EIGENSTRAT analysis are plotted on the Y axis against the expected –log10(p) values on the X axis after removing all individuals and SNPs that failed quality control. After genomic control correction, the inflation factor was λ = 0.991 with and 1.012 without eigenvector adjustment. (A) –log10(p) values for all GWAS SNPs. (B) –log10(p) values after removal of the HLA region SNPs.
Supplementary Figure 3. Results of the ANCA‐associated vasculitis genome‐wide association screen. The Y axis shows the –log10 P values (from EIGENSTRAT) for each single nucleotide polymorphism on each chromosome along the X axis. The dashed line indicates the genome‐wide significance threshold (P = 5.0 x 10−7).
Supplementary Figure 4. Locus Zoom plots. Showing regional associations across the MHC and non‐MHC loci. The –log10 P values of single‐nucleotide polymorphisms genotyped in the discovery (•) and replication (▲) cohorts and included in the meta‐analysis (♦) are plotted against their chromosomal position at each locus. SNPs are coloured depending on their degree of correlation (r2) with the top SNP (as estimated on the basis of 1000 Genome European haplotypes, 2012) shown in purple. Genes and expressed sequence tags within each region are shown in the lower panels. HLA‐DP and DQ regions; PTPN22; PRTN3 and SERPINA1.
Supplementary Figure 5. Classification And Regression Tree (CART) model for predicting risk of GPA/MPA vasculitis. The CART analysis incorporated the disease‐associated SNPs identified in the initial ANCA‐associated vasculitis cohort meta‐analysis. The HLA‐DQA2 rs7454108 and PTPN22 rs6679677 and rs2476601 variants that did not substantially improve classification of cases and controls were removed from further analyses. Eight other variants (HLA‐DPA1 rs9277341, HLA‐DPB1 rs141530233, HLA‐DPB1 rs1042169, PRTN3 rs62132293, HLA‐DQA1 rs35242582, SERPINA1 rs28929474, HLA‐DQB1 rs104902, and HLA‐DQA2 rs39981589) all improved the model fit by at least 3% and were retained to build a CART. The three symbols ++, +‐, or – on each split represent minor variant homozygote, heterozygote, or homozygote, respectively. Odds ratios (OR) and confidence intervals (bracketed numbers) are shown for each node with the effects of specific variants on risk shown for each sequentially subclassified patient subset.
Supplementary Figure 6. Confirmation of triallelic HLA‐DPB1 risk and non‐risk haplotypes by direct sequencing analysis. Sequence analysis showing the HLA‐DPB1 exon 2 region rs1042169, rs141530233, rs386699872 risk and non‐risk haplotypes. A 201 bp segment across HLA‐DPB1 nucleotide positions 3,048,604 to 33,048,804 (GRCh37/hg19) was PCR amplified using primer pairs 5’‐GAGTACTGGAACAGCCAGAA and 3’‐TAAGGTCCCTTAGGCCAACC and the amplification products then directly Sanger sequenced in individuals identified in the genome‐wide association study as having homozygous risk (n = 50) or homozygous non‐risk (n = 50) rs1042169 and rs141530233 genotypes. A representative example of the sequence read‐out from each subgroup is shown with the nucleotide sequence and corresponding amino acid sequence and position shown below. The polymorphic alleles within each haplotype are circled. The sequence analysis confirmed 100% correlation of rs386699872 CA with the risk and rs386699872 G with the non‐risk rs1042169/rs141530233 haplotype.
Supplementary Table 1. Sequences of primer pairs used in quantitative PCR analyses.
Supplementary Table 2. Sequences of 20mer peptides used to evaluate T cell responses.
Supplementary Table 3. Summary of patient demographics, clinical data and quality controls outcomes by cohort
Supplementary Table 4. Results of genome‐wide association analysis for ANCAassociated vasculitis.
Supplementary Table 5. Results of genome‐wide association analysis for PR3‐ANCA/cANCA‐associated vasculitis.
Supplementary Table 6. Results of genome‐wide association analysis for MPO‐ANCA/pANCAassociated Vasculitis
Supplementary Table 7. Effects of organ involvement and ANCA status on MHC and non‐MHC associations with ANCA‐associated vasculitis.
Supplementary Table 8. Conditional analyses of risk alleles across the HLA locus
Supplementary Table 9. Evaluation of genetic models for susceptibility for ANCA‐associated vasculitis.
Supplementary Table 10. Comparison of peak observed SNPs and peak imputed SNPs at risk loci for (a) ANCAassociated vasculitis, (b) MPO‐ANCA/pANCA associated vasculitis.
Supplementary Table 11. Most‐likely disease causal variants identified by functional annotation.