

Abstract
Background
Protein flexibility, which has been referred as a dynamic behavior has various roles in proteins’ functions. Furthermore, for some developed tools in bioinformatics, such as protein-protein docking software, considering the protein flexibility, causes a higher degree of accuracy. Through undertaking the present work, we have accomplished the quantification plus analysis of the variations in the human Cyclin Dependent Kinase 2 (hCDK2) protein flexibility without affecting a significant change in its initial environment or the protein per se.
Objectives
The main goal of the present research was to calculate variations in the flexibility for each residue of the hCDK2, analysis of their flexibility variations through clustering, and to investigate the functional aspects of the residues with high flexibility variations.
Materials and Methods
Using Gromacs package (version 4.5.4), three independent molecular dynamics (MD) simulations of the hCDK2 protein (PDB ID: 1HCL) was accomplished with no significant changes in their initial environments, structures, or conformations, followed by Root Mean Square Fluctuations (RMSF) calculation of these MD trajectories. The amount of variations in these three curves of RMSF was calculated using two formulas.
Results
More than 50% of the variation in the flexibility (the distance between the maximum and the minimum amount of the RMSF) was found at the region of Val-154. As well, there are other major flexibility fluctuations in other residues. These residues were mostly positioned in the vicinity of the functional residues. The subsequent works were done, as followed by clustering all hCDK2 residues into four groups considering the amount of their variability with respect to flexibility and their position in the RMSF curves.
Conclusions
This work has introduced a new class of flexibility aspect of the proteins’ residues. It could also help designing and engineering proteins, with introducing a new dynamic aspect of hCDK2, and accordingly, for the other similar globular proteins. In addition, it could provide a better computational calculation of the protein flexibility, which is, especially important in the comparative studies of the proteins’ flexibility.
Keywords: Flexibility fluctuation, Human CDK2 (hCDK2) protein, Molecular Dynamics-Root Mean Square Fluctuation (MD-RMSF), Molecular dynamics simulation, Protein flexibility, RMSF Standard Deviation (RMSF-SD)
1. Background
Protein flexibility probably is a phenomenon that can make the existence of the many protein conformations possible in contrast to a limited number. Many conformations in a protein are needed for doing many functions. For instance, based on searching IntAct PPI database with the UniprotKB accession number of hCDK2: P24941, hCDK2 has at least 200 different interactions with other proteins in addition to interactions by itself (1). As a structural characteristic, protein flexibility plays many functional roles with respect to different aspects of the proteins as well. Examples in this regard are enzyme catalysis and ligand-receptor interactions, substrate and ligand orientation, the turnover rate of the substrates, and reduction of the free energy barrier in the active sites (2).
In structural bioinformatics tools, there are many instances regarding protein flexibility applications and usefulness; as an example, improvement of the small ligand-receptor docking (3, 4). Additionally, new algorithms in the field of protein designing, as well as modeling, have been improved by applying the flexibility information (5, 6).
Also, there is no indication for introducing the effect of permanent environmental parameters in the protein flexibility representatives such as X-ray crystallographic B-factor. In X-ray crystal structures, B-factor is calculated as a representative of the flexibility in absence of the existing ions and enough water molecules; the two permanent agents in a normal physiological environment of the proteins. Furthermore, in the present method of calculating X-ray B-factor, the final quantities have not amplitude of variation for residues, whereas, based on a number of evidences (7) and results obtained in this work, these amplitude of variations for a considerable number of residues is almost unavoidable. Due to this fact, efforts regarding prediction of the protein flexibility seem to be also in a wrong direction to some extent, as, X-ray crystallographic B-factor has mostly been chosen as their templates (8-11).
Since 2005 and even before, fluctuation in the protein flexibility has been observed in several researches, such as study conducted by Lange et al. (12). In their study, in which the two long MD simulations have been accomplished for B1 domain of the G protein, considering or quantifying this fluctuation and causes for the observed phenomenon could hardly be targeted or investigated. The objective of their research was to compare NMR driven flexibility with MD simulation ones (12). Also, there were a number of studies which have used a method called as the Multiple Molecular Dynamics Simulation (MMDS) (13-17). However, MD simulations will be repeated with exactly the same initial snapshots in this method, while the initial snapshots of the repeating MD simulations have some differences with each other in our method.
There are several reports that have studied the flexibility of the hCDK2 protein, applying a comparative approach (18-22), that is studying the flexibility of this protein in significantly different conditions of the protein structure or environment, such as phosphorylation, ATP binding, protein binding etc. However, in our work we have studied variations and changes in the flexibility of hCDK2 protein with no significant changes in its initial environment, structure or even its conformation. In a deeper insight, the flexibility variation, while it doesn’t accompany with a significant change in the protein environment, structure, or conformation has rarely been studied. It is a different concept related to the low or high frequency fluctuations in the protein structures. We believe this line of research needs furthermore scrutiny.
2. Objectives
Our main goal, and findings through undertaking the present study could briefly be explained and consisted as summarized below:
1-Throughout the entire length of the hCDK2, there are domains with flexibility fluctuations (RMSF as representative of the flexibility). As well, there are several other domains with no significant fluctuations in the independent MD simulations as calculated with no significant variations in the environment, structure, or conformation of the hCDK2 protein (PDB ID: 1HCL). Most areas of RMSF with variable forms are located on the surface of the protein, but, there are some variable parts out of hCDK2’s surface. In hCDK2 MD simulations, this fluctuation in Val-154 could reach more than 50% of the distance between maximum and minimum RMSF points in all these three MD simulations.
2-The second goal was to investigate the probable functional incorporation of the residues with highly variable flexibilities in hCDK2.
3-The third goal was to emphasize on, and introduce a better approach for calculation of a computational parameter in order to measure protein flexibility, mostly supporting the comparative study of the protein flexibility.
3. Materials and Methods
3.1. Protein Models
The crystal structural model for hCDK2 protein was downloaded as a PDB file with 1HCL PDB ID (23) from protein data bank (24). In this crystal structure model, residues from 37 to 40 are missed. To repair this missing part, we used version 9.11 of Modeller package (25, 26). Therefore, in this homology modeling, 1HCL PDB structure was used as the template of the protein by itself. We generated 100 structures and based on DOPE score the best structure was selected. Also, DOPE score profile for the protein’s residues in all points was in an acceptable region.
3.2. MD Simulation Procedure
To accomplish molecular dynamics simulation studies of the hCDK2 protein, three independent MD simulations were run using Gromacs package 4.5.4 (27, 28). Amber99sb was implemented as the force field with SPC water as selected water model (29-31). The distance between protein molecule and the walls of the box was nine angstroms. 20219 water molecules filled each simulation box and 4 Cl ions to neutralize system’s charge positioned energetically favorable. Then the system was minimized by the steepest descent algorithm into 1000 steps with implementing 0.002 ps as the time step. In the next step, MD simulation was run in the position-restrained condition as an attempt to reach equilibration and relaxation of the system with 20 ps time scale. 10 ns MD simulations were run with 2 fs time step under NPT condition and using of Berendsen’s coupling method to keep a constant pressure and temperature. For coulombtype, Particle Mesh-Ewald (PME) method was used.
For results depicted in Figure 2, normalization was done using the following equation (32):
Figure 2 .
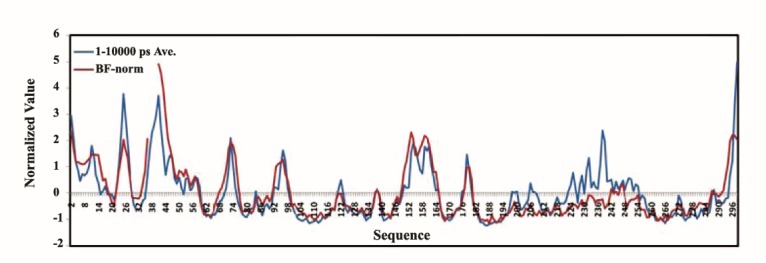
Normalized Crystallographic Cα-B-factor of the hCDK2 protein (PDB ID = 1HCL) synchronized with the normalized average of RMSFs of MD1, 2, and 3 of the hCDK2 for 10 ns time scale; red curve plotted normalized crystallographic B-factor of the hCDK2 protein and the blue curve represent normalized average RMSF of hCDK2 MD 1, 2, and 3. In the red curve from residue 37 till 40 are missed as these residues are missed in the crystallographic structure file
B-factorNorm= (B-factorNorm-μ)/σ
In this equation, μ is the average of the raw B-factors for backbone atoms in each residue and s is the standard deviation obtained from the raw B-factors.
3.3. Measuring Flexibility Variation
The percent of RMSF variation for each selected residue was calculated as follows:
PRF= (LMax-LMin)/(GMax-GMin)×100
Where, LMax and LMin are the local maximum and the local minimum of the RMSF in the three repeats of MD trajectories for each selected residue respectively, GMax and GMin are the global maximum and the global minimum in all points of the three MD simulations’ RMSFs. In our calculations, GMax (Global Max) of RMSF curves was 0.263 for Leu-25 from MD3 and GMin (Global Min) of RMSF curves was 0.034 for Trp-187 from MD2. PRF was the percent of the RMSF fluctuation for each selected residue. To select global maximum and minimum we disregarded the first and the last residues of the hCDK2 sequence due to their extra flexibility.
As another parameter for estimating the RMSF variations, we calculated the RMSF standard deviation (RMSF-SD) for each residue through MD simulations as repeated three times.
3.4. ASA Calculation and Normalization
To calculate relative ASA of the hCDK2 protein (PDB ID: 1HCL), we have used GetArea server (33) and the raw data were converted with the sliding window algorithm, with a window size of 9 to the final form and then the data were normalized implementing the following equation in order to be compatible with RMSF and RMSF-SD data (32):
ASANorm= (ASARaw-μ)/σ
In the above equation, μ is the average of raw ASA data (i.e. relative form for each residue and after implementing sliding window algorithm) and s is their standard deviation.
3.5. 3D Structural Alignment, Structural Preparation and Visualization
Deep view from Swiss Institute of Bioinformatics (SIB) (34) and MUSTANG 3.2.1 software (35) was used for 3D structural alignment and VMD 1.9 (36) and ViewerLite 4.2 were used to visualize and prepare the structural files.
4. Results
Human CDK2 is a typical globular protein regarding with many aspects, i.e. the protein has a range of secondary structures and also it has a highly flexible fragment (37-48) based on its inactive free form of the X-ray crystallographic B-factor (PDB ID = 1HCL) (37). Functionally, this protein plays an important role in the regulation of many cellular events in eukaryotic (human in this context) cell cycle. Because of various functions that this protein is involved in, there are several levels of the regulatory mechanisms for this protein’s functions, such as activation and inactivation through protein-protein interactions (e.g. cyclins as its positive regulators, in addition to the interaction with the inhibitory proteins that are involved in its negative regulation), phosphorylation, ATP binding and sub-cellular localization (19, 21). Each of these regulatory mechanisms has an especial influence on its flexibility. Therefore, we have chosen this protein in our study on the flexibility fluctuations as a representative of a typical globular protein with many function.
4.1. Comparison Crystallographic B-factor with RMSF of hCDK2 along with MD Trajectories
For all the three independent hCDK2 MD simulations, Cα-RMSFs were calculated and for the hCDK2 X-ray crystallographic data (PDB ID = 1HCL), Cα-B-factor was extracted. The average correlation coefficients (CC) between each one of the Cα-RMSFs and Cα-B-factor are displayed in Figure 1. The average CC for RMSFs of the three independent MD trajectories along with 10 ns is also presented in this figure. As could be seen the CC between the averages of Cα-RMSFs and crystallographic Cα-B-factor shows a very mild elevated inclination during 10 ns time scale. This parameter reaches to 0.82 in the 1-10000 ps point. However, this CC is much higher than average CC between these two parameters in previous reports. For instance, a study in 2012 has reported the average for CC of 0.68 between Cα-B-factor and Cα-MD-RMSF for almost 43 proteins (32). The average CC between each couple of Cα-MD-RMSFs of the three independent MD trajectories during 10 ns displays a peak in 1-8000 ps point. The amount of CC in this peak has reached to 0.89. Although, this quantity is relatively low for 1-500 ps and 1-1000 ps. Another characteristic of this curve is that it is not continually increasing. Also, CC displays a reduction in the start and final points. Therefore, it is acceptable that similarity between RMSFs of the independent repeats for some MD trajectories is not perpetually additive along with the time of the simulation, but until 2000 ps, it could be intensively additive. Figure 1 also illustrates that an RMSF of a single MD trajectory such as MD1 could significantly be away from the best CC with X-ray B-factor (i.e. the green curve). However, the best way to reach abetter CC with X-ray B-factor is to calculate the average RMSF from at least three independent MD trajectories and from the point of 2000 ps, as the CC could reach to more than 0.80, following to which it could rise very gentle with increasing the simulation time. Also from the timepoint of 2000 ps, similarities between RMSFs of the independent repeats of MD trajectories could be acceptable in the scale of all 10000 ps. These data nevertheless could be considered for the most other globular proteins which are relatively similar to hCDK2. Furthermore, these results are important since hCDK2 is an important and popular protein in the protein science and engineering. For instance, in protein-protein and small ligand binding studies, in silico site directed mutations, protein modifications and other studies of hCDK2, these data could help researchers.
Figure 1 .
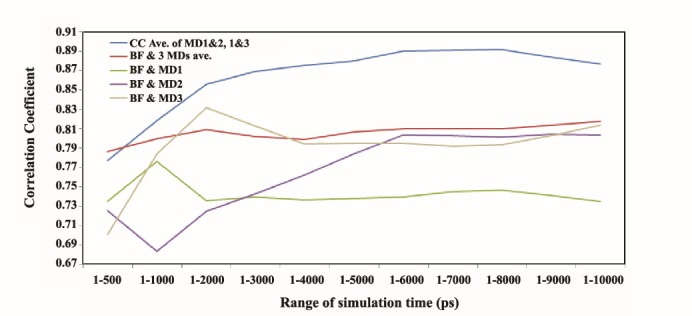
Correlation Coefficient (CC) between Cα-MD-RMSFs among themselves and with crystallographic Cα-B-factor (BF) of hCDK2 during 10 ns time scale of the MD simulations. Blue curve depicts the plot for the average CC between Cα-RMSFs of MD1 and 2, 1 and 3, and 2 and 3 during 10 ns of the MD trajectories time scale. The red curve is for CC between the average of three Cα-RMSFs and Cα-B-factor during the same time for the MD trajectories. The green curve shows the CC plot between Cα-B-factor and Cα-RMSF of the MD1. Purple curve depicts CC between Cα-B-factor and Cα-RMSF of the MD2 the gray curve indicates CC between Cα-B-factor and Cα-RMSF of the MD3
The normalized hCDK2 crystallographic Cα-B-factor and average of Cα-MD-RMSFs are plotted in Figure 2. A highlevel of correlation between these two representatives of the protein flexibility could be seen in this figure. Interestingly, this feature illustrates the robustness of a computational technique i.e. Molecular Dynamics Simulation as well.
4.2. Cα-MD-RMSFs of hCDK2, Variable Versus Non-variable Residues
We found that RMSF (which has been used as a representative for protein flexibility) of the hCDK2 protein has variable and non-variable regions within these three independent repeats of MD simulations (Figure 3). There are more than 10 peaks and dips in each three RMSFs curves within all through the sequence length of hCDK2. The differences between these three independent MD trajectories of the hCDK2 are included of the initial positions of the explicit water molecules and 4 Cl- as counter ions, very low differences of the initial hCDK2 conformations of the three simulations (as much as RMSDs = 0.30 and 0.39 angstrom of the 3D alignment of the all pairs of the three initial conformations of hCDK2 for backbone and the whole atoms respectively), as well as the differences in the initial velocities within MD simulations. We have explained more about these different factors in the next section of the article.
Figure 3 .
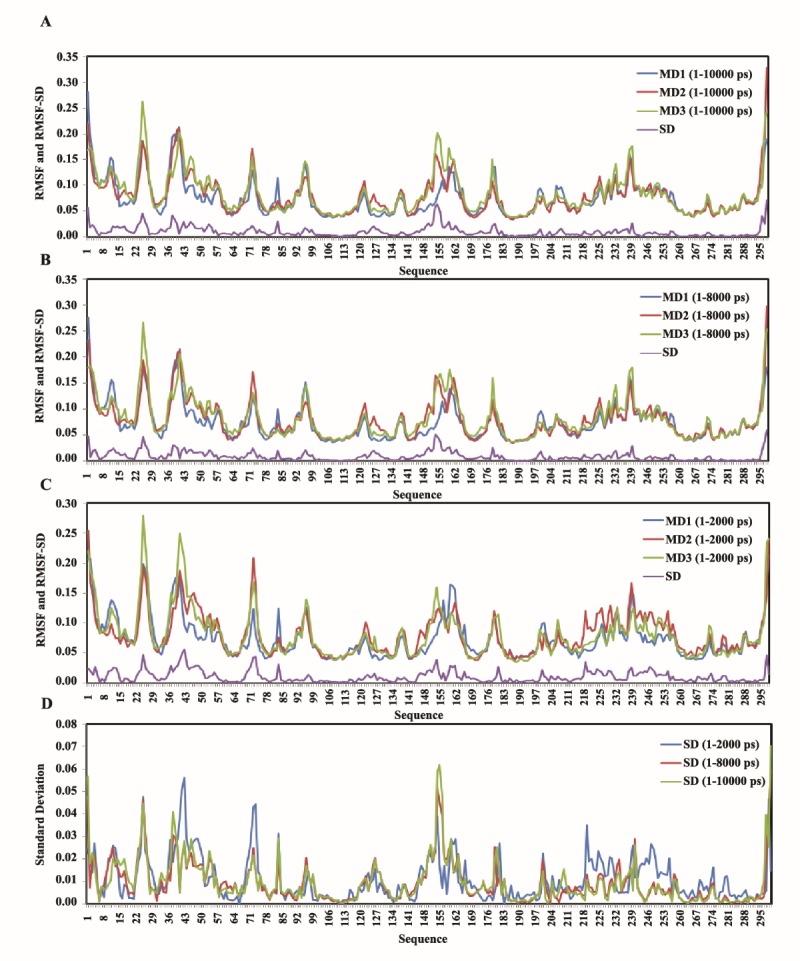
RMSF and RMSF-SDs of 1-2000, 1-8000 and 1-10000 ps time scale of the three independent MD trajectories; A: depicts the curves for 1-10000 ps, B: for 1-8000 ps, C: for 1-2000 ps, and D: is RMSF-SDs for the three MD trajectories in 1-10000, 1-8000 and 1-2000 ps time scale
Nevertheless, the illustration of these variations in the flexibility of hCDK2 protein, as we mentioned, is presented in Figure 3. In this figure, comparative forms of RMSFs of the three amplitude of the time scales are captured. It is clear in these panels, that there are considerable variations in different regions of the plots as well as regions with the variable RMSF (or flexibility) have a degree of alterations even within a change in the time scale amplitude. These changes are much larger when comparison is done between Figure 3C (1-2000 ps time scale) and Figure 3B (1-8000 ps time scale) versus comparison between Figure 3A and Figure 3B (1-10000 ps time scale). In the other words, the RMSF variations within three independent MD trajectories are very similar in 1-10000 ps time scale (Figure 3A) with 1-8000 ps time scale (Figure 3B). However, they are very different with 1-2000 (Figure 3C). Anyhow, the amplitude of this RMSF variation even in 1-10000 and 1-8000 time scales could reach to 53.63% of possible distance between the maximum and minimum of the RMSF for all the three RMSF plots in the point of Val-154 (Table 1). Figure 3D demonstrates a comparative form of the RMSF-SDs for the three different time scales in panel A, B, and C. In this panel, a larger difference between RMSF-SD curves for the time scales 1-10000 ps and 1-8000 ps with 1-2000 ps is clear.
Table 1. The local maximums and minimums as well as the globals in RMSF-SD curves of the MD simulation repeats for the hCDK2. In the first column, superscripted numbers indicate the group of each selected residue. Group 1 is included of residues with the high RMSF-SD in or near the RMSF peaks, group 2 is for residues with high RMSF-SDs but in or near the dips of the RMSF plot, group 3 indicates residues with a low RMSF-SDs and in or near the peaks of RMSF plot, and group 4 includes residues with low RMSF-SDs in or near the dips of RMSF plot. In the last column, the negatively charged residues are shown as “Bold” and for positively charged residues as “Underline”. 2nd column displays percent of the RMSF for each selected residue in comparison with the distance between between the Max and the Min of the RMSFs for all points in the three RMSF curves. 3nd column indicates the Percent of RMSF Fluctuation (PRF) for each selected residue in comparison with all distance between the Max and the Min of RMSF curves at 1-10000 ps time scale. The formula for calculating PRF has been mentioned in the methods section .
| hCDK2 selected Residues | Max of 3 RMSFs (%) | PRF (%) | Selected points sequence |
| Asn-031 | 63.42% | 17.31% | MENFQ |
| Glu-131 | 45.83% | 15.25% | GEGTY |
| Leu-251 | 100% | 34.18% | NKLTG |
| Asp-381 | 73.77% | 34.61% | RLDTE |
| Gly-432 | 58.09% | 21.59% | TEGVP |
| Ser-461 | 59.50% | 25.13% | VPSTA |
| Leu-542 | 45.03% | 20.02% | ISLLK |
| Glu-731 | 65.40% | 18.53% | HTENK |
| His-842 | 43.55% | 23.78% | FLHQD |
| Arg-1262 | 31.59% | 16.96% | LHRDL |
| Arg-1501 | 42.29% | 22.25% | LARAF |
| Val-1541 | 76.86% | 53.63% | FGVPV |
| Tyr-1591 | 65.63% | 24.91% | RTYTH |
| Glu-1621 | 48.99% | 17.00% | THEVV |
| Tyr-1791 | 52.00% | 21.98% | CKYYS |
| Ser-2391 | 67.00% | 22.55% | KPSFP |
| Leu-2961 | 63.57% | 31.56% | PHLRL |
| Lys-064 | 36.47% | 0.92% | FQKVE |
| Ala-2144 | 28.78% | 5.11% | YKARN |
| Glu-283 | 42.33% | 3.10% | TGEVV |
| Val-304 | 25.12% | 4.24% | EVVAL |
| Ile-354 | 30.07% | 8.30% | KKIRL |
| The-413 | 81.00% | 4.41% | TETEG |
| Ile-524 | 32.43% | 8.13% | REISL |
| Leu-583 | 38.64% | 2.49% | KELNH |
| Leu-674 | 21.66% | 2.32% | KLLDV |
| Lys-753 | 34.41% | 4.76% | ENKLY |
| Phe-804 | 18.31% | 1.79% | LVFEF |
| Asp-864 | 22.69% | 3.80% | HQDLK |
| Met-914 | 25.31% | 2.80% | KFMDA |
| Ser-943 | 32.66% | 1.60% | DASLA |
| Leu-1014 | 21.28% | 1.92% | IPLPL |
| Gln-1104 | 16.18% | 0.96% | LFQLL |
| Cys-1184 | 22.42% | 1.35% | AFCHS |
| Val-1233 | 30.91% | 4.41% | HRVLH |
| Leu-1334 | 16.82% | 1.92% | QNLLI |
| Ile-1354 | 18.20% | 1.09% | LLINT |
| Ala-1404 | 21.09% | 2.53% | EGAIK |
| Trp-1674 | 20.97% | 3.80% | TLWYR |
| Leu-1754 | 25.24% | 4.50% | ILLGC |
| Ser-1814 | 24.78% | 3.06% | YYSTA |
| Trp-1874 | 14.35% | 1.66% | DIWSL |
| Ser-1884 | 14.50% | 0.52% | IWSLG |
| Arg-2004 | 31.67% | 1.70% | TRRAL |
| Pro-2044 | 25.81% | 2.62% | LFPGD |
| Arg-2174 | 24.70% | 1.35% | IFRTL |
| Val-2264 | 33.73% | 2.18% | EVVWP |
| Trp-2434 | 30.57% | 0.48% | PKWAR |
| Leu-2554 | 26.76% | 0.83% | PPLDE |
| Leu-2624 | 16.14% | 0.35% | RSLLS |
| Ala-2804 | 17.28% | 0.52% | KAALA |
| Phe-2854 | 20.06% | 0.66% | HPFFQ |
| Asp-2883 | 31.78% | 0.44% | FQDVT |
For a deeper analysis of these variations, we tried to cluster the variable and non-variable regions of RMSF plot of hCDK2 in two ways. In the first approach, we peeked residues with RMSF-SDs higher than 0.02 (Figure 4B) and bolded them in the 3D structure of the hCDK2 (Figure 4A) along with the most considered functional residues of this protein which have been indexed in UniProtKB database within the hCDK2 entry (AC = P24941). As it is clear in Figure 4A, residues with highly variable RMSF (RMSF-SD > 0.02) which have been illustrated with red CPK model are mostly located around the most important functional residues of the hCDK2 protein. However, except for two residues, the other 28 functional residues were not located in the high RMSF-SD regions. Therefore, most residues with a high RMSF-SDs have intended to be located around the functional residues much more than to be the functional residues by themselves.
Figure 4 .

The first form of clustering variable and non-variable regions in RMSF plot of the hCDK2 within three independent MD trajectories. A: Residues with red CPK model compose those residues with a high RMSF-SD more than 0.02. Residues with blue color in CPK model are nucleotide-binding regions of hCDK2 and the green ones are the other functional regions such as regions which undergo modifications and a number of other binding sites. B: Illustrates RMSF-SD plot for RMSFs of three independent MD trajectories at 1-10000 ps time scale
In the second method of clustering of the residues with either variable or non-variable RMSFs, we considered the most local minimum and maximum values, in addition to the globals of RMSF-SD curves of 1-10000 ps time scale (Figure 4B) and the residues in or near the peaks and dips as summarized in Table 1. These selected residues, which are located in or near the top or bottom of the peaks and dips were clustered in four groups. Group 1 includes residues in or near the peaks with high RMSF-SDs, group 2 includes of residues in or near the dips with high RMSF-SDs, group 3 is composed of residues in or near the peaks with low RMSF-SDs, and finaly the group 4 is composed of the residues in or near the dips with low RMSF-SDs. These selected residues in the four groups reveal that RMSF fluctuation (as a representative of the flexibility fluctuation) occurs in various regions of RMSF plot. Therefore, this phenomenon has not a simple description. Considering Table 1, we included penta-peptide sequence around each selected residue in this table. It is clear that residues with a high RMSF-SD are mostly included of the group 1 and they mostly have at least one charged residue in the middle or in the immediately adjacent position of the middle of the penta-peptide. However, less number of selected residues with high RMSF-SD are included in group 2. In half of group 2 residues, there are no charged residues in the middle or beside of the middle position. Also in the other half, there is, at least, one charged residue in the middle or beside it. In fact, there are some exceptions in the group 1, such as Ser-46 and Val-154, which in these positions all residues of penta-peptides unexpectedly are uncharged and the segments are highly hydrophobic. Based on table 1 from the selected residues with low RMSF-SD, most of them are located in or near the dips of RMSF plot (Figure 3) named as group 4 and lower number of them are located in or near the peaks of RMSF plot, named as group 3. Most of the selected residues in the group 4 are not included of the charged residues in the middle or adjacent to the middle position of their penta-peptides. In a reversed condition all members of the group 3 have at least one charged residue in the middle or adjacent to the middle position of their penta-peptides except for Ser-94.
4.3. Different Initial Conditions between Three MD Trajectories of hCDK2
Based on the multiple structural alignments between zero frames of the MD1, MD2, and MD3, RMSD for each couple of these conformations was equally just 0.30 angstrom for backbones and 0.39 for all atoms alignments. It means that there are very small structural differences between each couple of these three initial conformations of our present MD simulations. However, there is a significant difference in one of the ions positions from all the four ions (that exist in the environment of each MD’s protein) of the initial snapshots (Figure 5). In addition, the initial positions of water molecules and the initial velocities of the whole systems in the beginning of these three independent repeats of MD simulations of the hCDK2 are different. From the previous literature in this context, there are some evidences that could tell us each of these differences could cause a variation in RMSF of the introduced points of hCDK2. The literature review for these evidences are incorporated in the discussion section. Despite the causes of these variations for some points of RMSF of this protein, there are other aspects related to this phenomenon, which should be considered. Firstly, what would be the nature of this phenomenon? Is it a technical issue or an intrinsic characteristic of this protein, and even other proteins? If it is intrinsic then it implies that protein flexibility is a very sensitive feature of the proteins and it is not as rigid as B-factor parameter, which can be extracted from a crystal structure of a protein. Secondly, if these variations in the flexibilities are intrinsic, then, for studying flexibility changes which are caused by any local structural alterations, such as protein mutations, modifications, and ligand binding, these variations must be considered in designing of the method. For example, regarding these type of studies which have been done using MD simulation, there is a method called as Multiple Molecular Dynamics (MMD) Simulation (14-16). In this method of MD simulation for one beginning snapshot, multiple repeats will be run with just different initial velocities and an average of the results will be considered or, at least, all the repeats will be notified. However, there are many researches in the field of protein flexibility investigation, even in recent years, which have done with MD simulations without using this method, or any other procedures, considering this possibility of flexibility variations in proteins. Actually, the use of MMD simulation is nearly rare in comparison with the number of investigations in the field of MD simulation studies of the protein flexibility. As it is clear, our method of calculating the MD-RMSF is not MMD simulation, but, additionally it considers all the possible variable factors including the initial velocities in repeating MD simulations for calculating the RMSF as a representative for protein flexibility. There are a number of evidence for these flexibility variations which have been obtained from NMR technique for crambin protein (7). Therefore, it is not only just as a computational technique, which could imply to these kinds of variations, but also, there is an experimental technique that could confirm this phenomenon for a protein other than the hCDK2.
Figure 5 .
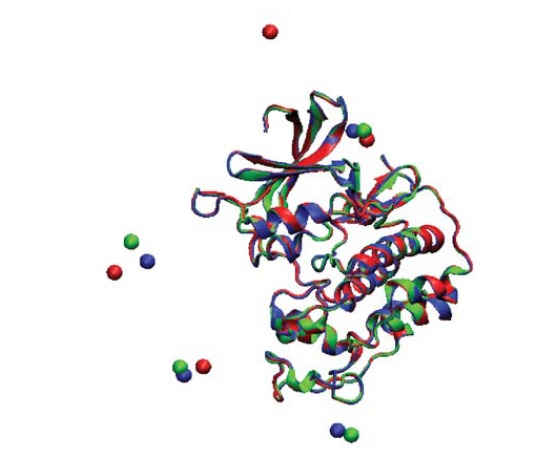
Structural alignment of the zero snapshots of three MD simulations in the present study; Cl ions positions are considered in this alignment. Zero snapshots of MD1, 2 and 3 are represented by Green, red, and blue colors, respectively
Regarding the probable effects of counter ions in our MD simulation environments, in addition to the literature reviews, we have investigated the existed lysozyme X-ray crystal structures with and without ions. Also, it was monitored that their B-factors are included of significant fluctuations in some regions. However, in X-ray crystal structures because of other variant conditions such as temperature, it is not possible to do a satisfactory comparison.
4.4. Correlation of RMSF and RMSF-SD with the ASA in hCDK2
The calculated correlation coefficient of RMSFs average with the relative ASA (Figure 6A) and RMSF-SD (Figure 6B) were 0.63 and 0.24 respectively, where, the sliding window method was used for relative ASA with 9 residues as window size. RMSF-SD had been considered as a representative of the flexibility fluctuation in our repeats of the hCDK2 MD simulations. As it is illustrated in Figure 6B, this fluctuation is placed nearly everywhere of ASA curve obtained for hCDK2. The correlation coefficients also express that there are some flexible regions in the partly buried areas of the protein but most of the flexible areas are located in near the surface of the protein. However, the flexibility fluctuation could be located everywhere of the ASA plot of the hCDK2 protein. Therefore, flexibility is a complicated phenomenon, which its description, modeling, or finding of its driving force is not a simple task. Distribution of the flexibility and its fluctuation in both exposed and buried areas of the protein does mainly reveal the role of both the buried and the exposed residues in these two phenomena. However, based on correlation coefficients and what is seen in Figure 6, the exposed residues are mostly located at flexible parts. Assuming that the polar and charged residues are mostly located in the exposed areas, the above results could imply to a more incorporated role for these types of residues than apolar residues in the protein flexibility and probably its fluctuations. However, based on the presented results in Table 1 there are considerable exceptions about this conclusion.
Figure 6 .
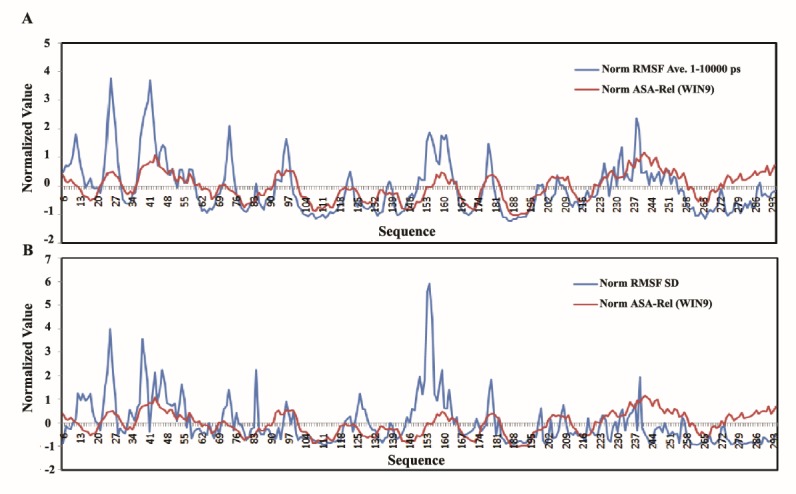
Correlation between RMSF-SD and ASA in hCDK2 in comparison with average RMSFs v.s. ASA; in panel A, Red curve is for win-9 normalized relative ASA and the blue one is for normalized RMSF average at 1-10000 ps. In panel B, red curve illustrates the normalized relative ASA calculated by using sliding window method as long as nine character and the blue curve is for the normalized RMSF-SD
5. Discussion
In this study, we have illustrated that a single MD simulation cannot be enough to calculate a good representative for protein flexibility even in 10 ns time scale. However, with our calculations for the RMSF in different amplitudes of the simulation time scale during our three independent repeats of 10 ns MD simulations, it was confirmed that similarities between MD simulation repeats will be remarkable from the 1-2000 ps and after that, it could get even better till 1-8000 ps. Also, CC between crystallographic Cα-B-factor and average Cα-MD-RMSF is increasing till 1-10000 ps, but smoothly. Our data from the first part of results section clearly illustrates that single MD simulations of a protein with almost same conditions have considerable differences with each other in RMSF. Thus, single MD simulations cannot be sufficient to analyze protein flexibility via MD simulation. Nevertheless, even in the recent studies, the number of published papers, which have used a single MD trajectory for RMSF calculation, are too many when compared to those that have applied any types of repeats. As we previously mentioned, a procedure which is called multiple molecular dynamics (MMD) simulation, has been introduced almost in 1998 through studies on crambin protein (17). This method is included of repeating MD trajectories for exactly same initial snapshot. In this method, just initial velocities are variable for repeating MD simulation method, however, in our presented method three additional variable factors have been incorporated. Based on the first part of our results RMSF average from at least three repeats of MD trajectories with at least 2000 ps time scale could be much better than single MD trajectory calculation even with 10000 ps. We achieved a good correlation between crystallographic Cα-B-factor and Cα-MD-RMSF using our method even better than previous reports.
In this work, we have introduced some details regarding flexibility fluctuations and bolding the non-constant intrinsic feature of the protein flexibility in a simple environment that just included of the water molecules and counter ions as one of our goals. We have calculated 10 ns MD simulations; then we proved that average RMSF of these simulations for hCDK2 protein has a CC much higher than previously reported average CC, which was calculated form single 5 ns MD simulations for more than 40 proteins between their RMSFs and X-ray B-factors (32). Interestingly, our results were better even in 1-1000 ps time scale.
A recent report about the effect of ions on DNA flexibility (38) has illustrated that these effects cannot be simply formulated. However, it is clear that the complexity of the protein structure is much larger than DNA. This increase in the complexity could be mostly because of the distribution of the charged residues in non-homogenous forms, whereas, DNA has relatively homogenous distribution of charges along its structure. However, there are not too many published research results regarding with direct study of the effect of the ions on protein flexibility, but, there are indirect studies in this context and some of which have been reviewed here. An in silico research has revealed the effect of counter ions on the stability of RMSD during the simulation (39). Based on their results counter ions could alter the protein structure during the simulation. In addition, some other studies have revealed the effect of ions on other protein structural properties, such as stability (40, 41). Recently, the role of interfacial waters was presented as the main driving force for the rotein flexibility (42). In this article, the main reason for this exposition has been referred to the correlation between properties of the interfacial water motions and protein motions. In another recently published research, it was pointed out that anchoring the surface charges of the proteins with these nearby water molecules have a significant effect on protein dynamics (43). In this paper there is more emphasis on the importance of electrostatic interactions in the formation of the flexibility, but with inserting role of anchoring of protein charges with the nearby water molecules. Also, there are evidence for the effect of different initial velocities in the MD simulations on the flexibility of a protein (17). For initial velocity, we are not sure whether it is a natural phenomenon or not. However, it seems that getting small differences in the initial velocities for the different copies of the same protein in different time and space conditions within a cell may not be an impossible event. Therefore, all factors, which are different in our three repeated MD simulations of hCDK2, could cause differences in their RMSFs. Consequently, variations in the protein flexibility of hCDK2 nearly in the same environment with no significant changes could be an intrinsic characteristic of this protein, and probably most of other proteins. As a further proof, it has been indicated from NMR studies of the crambin protein that NMR-RMSF has variations in the repeated experiments without significant changes in the environment of the protein (7).
In various algorithms designed to predict protein flexibility based on sequence, crystal structure B-factor has been used as a template, or, as a reference, and their predicted results have no amplitude of variation (11, 44). If we can accept that protein flexibility is intrinsically very sensitive and variable; without significant changes in the protein’s environment, therefore, it would be better to design algorithms for proteins’ flexibility prediction with the amplitude of variation. Also the diversity of the dynamics and structures in protein complexes in comparison with their free forms has been illustrated before (45); which could be another evidence for sensitivity of protein flexibility.
As another goal in the present research, we have accomplished a deeper analysis of the flexibility fluctuation of hCDK2 protein with two different approaches for the clustering. Generally, these parts of our results could firstly imply a probable role of residues with variable flexibility in the functional aspects of the proteins, and secondly it demonstrates that the residues with variable and non-variable flexibilities are distributed all over the protein structure. Therefore, labeling a number of hCDK2 protein residues as the variable, or non-variable in their flexibilities is among our novel results achieved in this work, which could indicate a new structure-function relationship for a number of specific residues in the hCDK2 protein. However, the complexity of distribution of these two kinds of residues along with hCDK2 structure reveals that finding a detailed mechanism for this phenomenon (the flexibility fluctuation of the protein in an environment with almost no significant change) is a complicated issue.
Somehow in this work, it has been tried to introduce a new and possibly a better approach for investigating protein flexibility through MD simulations. There are several recent studies on new methods for analyzing conformational sampling and dynamics of proteins via MD simulations which imply the importance of this field (46-53), however, there is long pace remains to the attainment of the perfection (54).
As the final word, the new insight in to the structure-function relationship of residues which has been introduced in this work for hCDK2 could help protein designing and engineering. In addition, it could possibly help a better understanding of a number of proteins’ aspects in the mechanism of the protein-protein interaction, since, the specific interaction of a protein, such as hCDK2 with more than 200 different proteins could become possible by some residues with variable flexibilities. In addition, these results could help doing comparative flexibility studies via MD simulation more accurate than before.
Acknowledgments
Our appreciation to Mr Abdllah Shirazi, director of Soroush institute for supporting us with the hardware.
Funding/Support
The present work has been supported by Research Council of the Tehran University.
References
- 1.Marsh JA, Teichmann SA. Relative solvent accessible surface area predicts protein conformational changes upon binding. Structure. 2011;19:(6). doi: 10.1016/j.str.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kokkinidis M, Glykos NM, Fadouloglou VE. Protein flexibility and enzymatic catalysis. Adv Protein Chem Struct Biol. 2012;87:181–218. doi: 10.1016/b978-0-12-398312-1.00007-x. [DOI] [PubMed] [Google Scholar]
- 3.Lin JH. Accommodating protein flexibility for structure-based drug design. Curr Top Med Chem. 2011;11(2):171–178. doi: 10.2174/156802611794863580. [DOI] [PubMed] [Google Scholar]
- 4.Lexa KW, Carlson HA. Protein flexibility in docking and surface mapping. Q Rev Biophys. 2012;45(3):301–343. doi: 10.1017/s0033583512000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hallen MA, Keedy DA, Donald BR. Dead-end elimination with perturbations (DEEPer): a provable protein design algorithm with continuous sidechain and backbone flexibility. Proteins. 2013;81(1):18–39. doi: 10.1002/prot.24150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandell DJ, Kortemme T. Backbone flexibility in computational protein design. Curr Opin Biotechnol. 2009;20(4):420–428. doi: 10.1016/j.copbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Abaturov LV, Nosova NG. Protein conformational dynamics of crambin in crystal, solution and in the trajectories of molecular dynamics simulations. Biofizika. 2013;58(4):599–617. doi: 10.1134/s0006350913040027. [DOI] [PubMed] [Google Scholar]
- 8.Hwang H, Vreven T, Whitfield TW, Wiehe K, Weng Z. A machine learning approach for the prediction of protein surface loop flexibility. Proteins. 2011;79(8):2467–2474. doi: 10.1002/prot.23070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vihinen M, Torkkila E, Riikonen P. Accuracy of protein flexibility predictions. Proteins. 1994;19(2):141–149. doi: 10.1002/prot.340190207. [DOI] [PubMed] [Google Scholar]
- 10.Schlessinger A, Yachdav G, Rost B. PROFbval: predict flexible and rigid residues in proteins. Bioinformatics. 2006;22(7):891–893. doi: 10.1093/bioinformatics/btl032. [DOI] [PubMed] [Google Scholar]
- 11.Bornot A, Etchebest C, de Brevern AG. Predicting protein flexibility through the prediction of local structures. Proteins. 2011;79(3):839–852. doi: 10.1002/prot.22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lange OF, Grubmuller H, de Groot BL. Molecular dynamics simulations of protein G challenge NMR-derived correlated backbone motions. Angew Chem Int Ed Engl. 2005;44(22):3394–3399. doi: 10.1002/anie.200462957. [DOI] [PubMed] [Google Scholar]
- 13.Moonsamy S, Bhakat S, Walker RC, Soliman ME. Single Active Site Mutation Causes Serious Resistance of HIV Reverse Transcriptase to Lamivudine: Insight from Multiple Molecular Dynamics Simulations. Cell Biochem Biophys. 2015:1–14. doi: 10.1007/s12013-015-0709-2. [DOI] [PubMed] [Google Scholar]
- 14.Mancini G, Zazza C. F429 Regulation of Tunnels in Cytochrome P450 2B4: A Top Down Study of Multiple Molecular Dynamics Simulations. PloS ONE. 2015;10(9):e0137075. doi: 10.1371/journal.pone.0137075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bos F, Pleiss J. Multiple molecular dynamics simulations of TEM beta-lactamase: dynamics and water binding of the omega-loop. Biophys J. 2009;97(9):2550–2558. doi: 10.1016/j.bpj.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legge FS, Budi A, Treutlein H, Yarovsky I. Protein flexibility: multiple molecular dynamics simulations of insulin chain B. Biophys Chem. 2006;119(2):146–157. doi: 10.1016/j.bpc.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Caves LS, Evanseck JD, Karplus M. Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci. 1998;7(3):649–666. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR. et al. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem Biol. 2011;6(5):492–501. doi: 10.1021/cb100410m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartova I, Koca J, Otyepka M. Functional flexibility of human cyclin-dependent kinase-2 and its evolutionary conservation. Protein Sci. 2008;17(1):22–33. doi: 10.1110/ps.072951208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Vivo M, Cavalli A, Bottegoni G, Carloni P, Recanatini M. Role of phosphorylated Thr160 for the activation of the CDK2/Cyclin A complex. Proteins. 2006;62(1):89–98. doi: 10.1002/prot.20697. [DOI] [PubMed] [Google Scholar]
- 21.Barrett CP, Noble ME. Molecular motions of human cyclin-dependent kinase 2. J Biol Chem. 2005;280(14):13993–4005. doi: 10.1074/jbc.m407371200. [DOI] [PubMed] [Google Scholar]
- 22.Bartova I, Otyepka M, Kriz Z, Koca J. Activation and inhibition of cyclin-dependent kinase-2 by phosphorylation; a molecular dynamics study reveals the functional importance of the glycine-rich loop. Protein Sci. 2004;13(6):1449–1457. doi: 10.1110/ps.03578504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulze-Gahmen U, De Bondt HL, Kim S-H. High-resolution crystal structures of human cyclin-dependent kinase 2 with and without ATP: bound waters and natural ligand as guides for inhibitor design. J Med Chem. 1996;39(23):4540–4546. doi: 10.1021/jm960402a. [DOI] [PubMed] [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H. et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 26. Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci 2007;Chapter 2:Unit 2.9. DOI: 10.1002/0471140864.ps 0209s50. [DOI] [PubMed]
- 27.Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R. et al. GROMACS 45: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 29.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A. 2001;105(43):9954–9960. doi: 10.1021/jp003020w. [DOI] [Google Scholar]
- 31.Berendsen H, Postma J, Van Gunsteren W, Hermans J. Interaction models for water in relation to protein hydration. Intermolecular Forces. 1981;11(1):331–342. doi: 10.1007/978-94-015-7658-1_21. [DOI] [Google Scholar]
- 32.de Brevern AG, Bornot A, Craveur P, Etchebest C, Gelly JC. PredyFlexy: flexibility and local structure prediction from sequence. Nucleic Acids Res. 2012;40(Web Server issue):W317–322. doi: 10.1093/nar/gks482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19(3):319–333. doi: 10.1002/(sici)1096-987x(199802)19:3%3C319::aidjcc6%3E3.3.co;2-3. [DOI] [Google Scholar]
- 34.Kaplan W, Littlejohn TG. Swiss-PDB Viewer (Deep View) Brief Bioinforms. 2001;2(2):195–197. doi: 10.1093/bib/2.2.195. Epub 2001/07/24. [DOI] [PubMed] [Google Scholar]
- 35.Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. MUSTANG: a multiple structural alignment algorithm. Proteins. 2006;64(3):559–574. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–,38, 27. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 37.Schulze-Gahmen U, De Bondt HL, Kim SH. High-resolution crystal structures of human cyclin-dependent kinase 2 with and without ATP: bound waters and natural ligand as guides for inhibitor design. J Med Chem. 1996;39(23):4540–4546. doi: 10.1021/jm960402a. [DOI] [PubMed] [Google Scholar]
- 38.Savelyev A. Do monovalent mobile ions affect DNA’s flexibility at high salt content? Phys Chem Chem Phys: PCCP. 2012;14(7):2250–2254. doi: 10.1039/c2cp23499h. [DOI] [PubMed] [Google Scholar]
- 39.Drabik P, Liwo A, Czaplewski C, Ciarkowski J. The investigation of the effects of counterions in protein dynamics simulations. Protein Eng. 2001;14(10):747–752. doi: 10.1093/protein/14.10.747. [DOI] [PubMed] [Google Scholar]
- 40.Reyes-Alcaraz A, Martinez-Archundia M, Ramon E, Garriga P. Salt effects on the conformational stability of the visual G-protein-coupled receptor rhodopsin. Biophys J. 2011;101(11):2798–2806. doi: 10.1016/j.bpj.2011.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee YH, Won HS, Lee MH, Lee BJ. Effects of salt and nickel ion on the conformational stability of Bacillus pasteurii UreE. FEBS Lett. 2002;522(1-3):135–140. doi: 10.1016/s0014-5793(02)02919-8. [DOI] [PubMed] [Google Scholar]
- 42.Sophie Combet J-MZ. Further evidence that interfacial water is the main “driving force” of protein dynamics: a neutron scattering study on perdeuterated C-phycocyanin. Phys Chem Chem Phys. 2012;(14):4927–4934. doi: 10.1039/c2cp23725c. [DOI] [PubMed] [Google Scholar]
- 43.Pal S, Bandyopadhyay S. Importance of protein conformational motions and electrostatic anchoring sites on the dynamics and hydrogen bond properties of hydration water. Langmuir. 2013;29(4):1162–1173. doi: 10.1021/la303959m. [DOI] [PubMed] [Google Scholar]
- 44.Yuan Z, Bailey TL, Teasdale RD. Prediction of protein B-factor profiles. Proteins. 2005;58(4):905–912. doi: 10.1002/prot.20375. [DOI] [PubMed] [Google Scholar]
- 45.Marsh JA, Teichmann SA, Forman-Kay JD. Probing the diverse landscape of protein flexibility and binding. Curr Opin Struct Biol. 2012;22(5):643–650. doi: 10.1016/j.sbi.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Harada R, Takano Y, Baba T, Shigeta Y. Simple, yet powerful methodologies for conformational sampling of proteins. Phys Chem Chem Phys. 2015;17(9):6155–6173. doi: 10.1039/c4cp05262e. [DOI] [PubMed] [Google Scholar]
- 47.Eren D, Alakent B. Frequency response of a protein to local conformational perturbations. PLoS Comput Biol. 2013;9(9):e1003238. doi: 10.1371/journal.pcbi.1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harada R, Takano Y, Shigeta Y. Enhanced conformational sampling method for proteins based on the TaBoo SeArch algorithm: application to the folding of a mini-protein, chignolin. J Comput Chem. 2015;36(10):763–772. doi: 10.1002/jcc.23854. [DOI] [PubMed] [Google Scholar]
- 49.Markwick PR, McCammon JA. Studying functional dynamics in bio-molecules using accelerated molecular dynamics. Phys Chem Chem Phys. 2011;13(45):20053–20065. doi: 10.1039/c1cp22100k. [DOI] [PubMed] [Google Scholar]
- 50.Oblinsky DG, Vanschouwen BM, Gordon HL, Rothstein SM. Procrustean rotation in concert with principal component analysis of molecular dynamics trajectories: Quantifying global and local differences between conformational samples. J Chem Phys. 2009;131(22):225102. doi: 10.1063/1.3268625. [DOI] [PubMed] [Google Scholar]
- 51.Doshi U, Hamelberg D. Towards fast, rigorous and efficient conformational sampling of biomolecules: Advances in accelerated molecular dynamics. Biochim Biophys Acta. 2015;1850(5):878–888. doi: 10.1016/j.bbagen.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Ho BK, Agard DA. Probing the flexibility of large conformational changes in protein structures through local perturbations. PLoS Comput Biol. 2009;5(4):e1000343. doi: 10.1371/journal.pcbi.1000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang HW, Chu CH, Wang WC, Pai TW. A local average distance descriptor for flexible protein structure comparison. BMC Bioinformatics. 2014;15:95. doi: 10.1186/1471-2105-15-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vitalini F, Mey AS, Noe F, Keller BG. Dynamic properties of force fields. J Chem Phys. 2015;142(8):084101. doi: 10.1063/1.4909549. [DOI] [PubMed] [Google Scholar]