

Abstract
2-Hydroxyglutarate (2HG) exists as two enantiomers, (R)-2HG and (S)-2HG, and both are implicated in tumor progression via their inhibitory effects on α-ketoglutarate (αKG)-dependent dioxygenases. The former is an oncometabolite that is induced by the neomorphic activity conferred by isocitrate dehydrogenase-1 and -2 (IDH1/2) mutations, whereas the latter is produced under pathologic processes such as hypoxia. Here, we report that IDH1/2 mutations induce a homologous recombination (HR) defect that renders tumor cells exquisitely sensitive to poly (ADP-ribose) polymerase (PARP) inhibitors. This “BRCAness” phenotype of IDH mutant cells can be completely reversed by treatment with small molecule inhibitors of the mutant IDH1 enzyme, and, conversely, it can be entirely recapitulated by treatment with either 2HG enantiomer alone in cells with intact IDH1/2 proteins. We demonstrate IDH1-dependent PARP inhibitor sensitivity in a range of clinically relevant models, including primary patient-derived glioma cells in culture and genetically matched tumor xenografts in vivo. These findings provide the basis for a possible therapeutic strategy exploiting the biological consequences of mutant IDH, rather than attempting to block 2HG production, by targeting the 2HG-dependent HR-deficiency with PARP inhibition. Furthermore, our results uncover an unexpected link between oncometabolites, altered DNA repair, and genetic instability.
Introduction
The normal function of isocitrate dehydrogenase (IDH) enzymes is to catalyze the conversion of isocitrate to α-ketoglutarate (αKG) in the citric acid cycle. Recurring IDH1 mutations were identified in two independent cancer genome sequencing projects focused on gliomas and acute myeloid leukemia (AML; (1, 2)). Subsequent studies revealed that IDH1 mutations occur in more than 70% of low grade gliomas and up to 20% of higher grade tumors (secondary glioblastoma multiforme; GBM), and approximately 10% of AML cases (3), 10% of cholangiocarcinoma (4), as well as in melanomas (5) and chondrosarcomas (6). Additionally, mutations were also identified in IDH2, the mitochondrial homolog of IDH1, in about 4% of gliomas and 10% of AMLs (3, 7). Nearly all known IDH1/2 alterations are heterozygous missense mutations that confer a neomorphic activity on the encoded enzymes, such that they convert α-KG to (R)-2HG (8). Emerging research indicates that (R)-2HG is an oncometabolite, with pleiotropic effects on cell biology including chromatin methylation and cellular differentiation, although many questions remain about its impact on tumorigenesis and therapy response (9). In addition, the (S)-enantiomer of 2HG was recently found to be produced at high concentrations in renal cell cancer (10) and in response to hypoxia (11, 12). Both (R)- and (S)-2HG appear to exert their regulatory effects via the inhibition of αKG-dependent dioxygenases (13). Emerging data also indicate subsets of breast cancers produce 2HG at high concentrations in the absence of IDH1/2-mutations, thus expanding the clinical relevance of these molecules to other solid tumors (14, 15).
IDH1 and IDH2 small molecule inhibitors, which block the production of (R)-2HG by the mutant enzyme, are being developed and tested in clinical trials for both glioma and AML, with the underlying assumption that blocking IDH neomorphic activity alone will abrogate tumor growth (16). Yet several recent clinical studies suggest that patients with IDH1/2-mutant gliomas and cholangiocarcinomas have longer median survival times than their WT counterparts, which in many cases correlates with a favorable response to conventional radiotherapy and chemotherapy (1, 3, 17–21). These findings have prompted us to hypothesize that exploiting, rather than reverting, the IDH1/2-mutant phenotype might be a more effective therapeutic strategy. We thus sought to further characterize the impact of IDH1/2 mutations to identify alternative therapeutic strategies that could exploit the profound molecular changes associated with 2HG production.
Results
IDH1/2-mutant cells are deficient in DNA double-strand break repair by homologous recombination
Clinical studies suggest a link between IDH1/2 mutations and enhanced chemo- and radio-sensitivity, although the underlying mechanistic basis for this observation is poorly understood (20, 21). We sought to determine whether these sensitivities could arise from intrinsic DSB repair defects, which enhance cells’ susceptibility to DNA-damaging agents (22). We tested two different cell lines engineered to contain a heterozygous arginine (R) to histidine (H) mutation at codon 132 (R132H) in our study: (1) an IDH1-mutant HCT116 cell line generated using recombinant adeno-associated virus (rAAV) targeting, and (2) a HeLa cell line in which we introduced the same mutation by CRISPR/Cas9-based gene targeting. Our IDH1 gene editing strategy is presented in fig. S1A–E. We confirmed that the single cell IDH1-mutant HeLa clone was identical in origin to the parental HeLa cell line that we modified using CRISPR/Cas-based targeting by STR analysis, rather than a contaminant from another IDH1-mutant cell line in culture (tables S1 and 2). In parallel, we created human erythroleukemia (HEL) cell lines with stably integrated, doxycycline (dox)-inducible IDH1/2-WT and -mutant open reading frames (ORFs). We reasoned that the use of multiple matched cell line pairs, each isogenic and differing only by mutant IDH1/2 protein expression, would allow us to precisely test whether these proteins induced a DSB repair defect.
We first confirmed mutant IDH1 protein expression in HCT116 and HeLa cells by western blot analysis (Fig. 1A). We detected an approximately 100-fold increase in (R)-2HG production by LC/MS comparing IDH1 mutant HeLa and HCT116 cells to WT controls (Fig. 1B). We detected 2HG in these cell lines by two other methods for additional validation, including a previously published enzyme-based detection assay and 1H NMR (figs. S1F and S1G, respectively; (23)). We also observed lower concentrations of intracellular NAD+ in IDH1-mutant cells, in keeping with previous reports (24), and we confirmed that the induced (R)-2HG could be suppressed by a known IDH1 inhibitor (figs. S1H and S1I, respectively; (16)). We then confirmed dox-inducible expression of the IDH1 and IDH2 proteins in HEL cells by western blot analysis (Figs. 1C and 1D, respectively), which correlated with increased (R)-2HG production specifically after mutant IDH1/2 protein expression (fig. S1J). Contrary to previous reports suggesting a growth advantage in IDH1/2-mutant cells (16), we observed slightly delayed growth kinetics in IDH1/2-mutant versus WT cells for all four pairs in vitro (figs. S1K–N). No major changes were observed in cell cycle phase distribution, and no differences were observed in the necrotic or apoptotic fractions between IDH1-WT and -mutant cells (figs. S1O and S1P, respectively).
Figure 1. Mutant IDH cells are deficient in DNA DSB break repair.
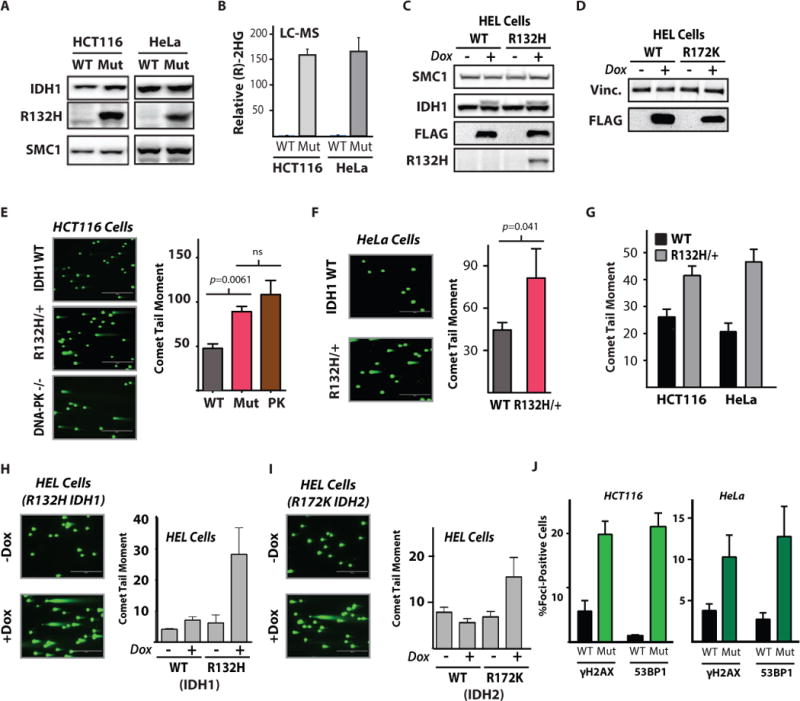
(A) Western blot analysis of IDH1-R132H expression in IDH1-mutant HCT116 and HeLa cells. SMC1 is used as loading control. (B) Quantification of (R)-2HG by LC-MS in WT and R132/+ HeLa and HCT116 cells (n=3, mean +/− SD). (C–D) Western blot analysis of doxycycline-inducible, FLAG-tagged (C) WT or mutant IDH1 and (D) WT or mutant IDH2 expression in HEL cells. (E) Representative images and quantification of neutral comet assays performed 24 h after 5 Gy IR in wild type, IDH1 R123H/+ (Mut), and DNA-PK (PRKDC) knockout HCT116 cells (PK). Scale bar = 400 μm. Statistical analysis by t-test (n=3). (F) Representative images and quantification of neutral comet assay performed 24 h after 5 Gy IR in wild type and IDH1 R123H/+ HeLa cells. Scale bar = 400 μm. Statistical analysis by t-test (n=3). (G) Quantification of neutral comet assay performed in log-phase, wild-type or IDH1 R132H/+ HCT116 and HeLa cells. (H and I) Representative images and quantification of neutral comet assay in HEL cells infected with (H) pSLIK IDH1-WT or pSLIK IDH1 R132H and (I) pSLIK IDH2 WT or pSLIK IDH2-R172K with or without doxycycline induction of the target protein, 7 days after addition of doxycycline or vehicle control. Scale bar = 400 μm (n=3). (J) Quantification of γH2AX and phospho-53BP1foci per nucleus in log-phase, untreated IDH1-WT and IDH1 R132H matched-pair HeLa and HCT116 cells (n=3). For E, F, G, H, I, and J, bars represent mean +/− SEM.
Next, we sought to assess the intrinsic DSB repair capacities of our functionally validated collection of engineered IDH1-WT and -mutant cell lines. We used the neutral comet assay to measure persistence of DSBs after IR, which is a classic approach to assess functional DSB repair activity (25). We detected a markedly reduced capacity to repair DSBs after IR exposure in IDH1-mutant HCT116 cells, which was in the same range as that observed in a genetically matched HCT116 cell line with a homozygous knockout of DNA-PKcs, a key DSB repair gene involved in non-homologous end joining (NHEJ) (Fig. 1E; (26)). Similar results were observed in the HeLa cell line pair (Fig. 1F), and in both cases these differences were correlated with enhanced radiosensitivity in the IDH1-mutant cells, as detected by clonogenic survival assays (figs. S2A and S2B). Mutant IDH1-dependent radiosensitivity was also observed in the HEL cell line in a short-term viability assay; these are suspension cell lines, and thus clonogenic survival assays are not readily feasible (fig. S2C).
We also observed increased persistence of unrepaired DSBs at baseline (in the absence of irradiation) specifically in the IDH1-mutant HCT116 and HeLa cells (Fig. 1G). This phenotype of constitutively increased DSBs could also be induced by transient expression of mutant, but not WT, IDH1 and IDH2 proteins in HEL cells (Figs. 1H and 1I, respectively). Given the marked increase in comet tail moments (~4-fold) after induction of mutant IDH1 protein over-expression in log phase cells, we sought to test whether we could observe the same phenotype in another genetic background. Indeed, we confirmed markedly increased comet tail moments (~4-fold) in the human monocytic cell line, THP1, specifically after expression of mutant IDH1 protein in log phase cells (fig. S2D).
We then performed DNA damage foci studies in log-phase IDH1-WT and -mutant cells to assess whether the increased comet tail moments in the latter cells correlated with larger amounts of DNA damage that could be directly visualized. As shown in Fig. 1J, we detected substantially increased γH2AX and phospho-53BP1 foci, indicative of DNA DSBs, in both HCT116 and HeLa IDH1-mutant cells (representative microscopy images are shown in fig. S2E and S2F). Cells harboring DSB repair defects, including HR gene aberrations, are known to develop increased DSBs in the absence of DNA damaging agent exposure (27). For comparison, we examined DNA damage foci patterns in HR-proficient and -deficient cell lines using the exact protocols and conditions that were used to assay the IDH mutant cells, as above. We found basally elevated DNA damage foci (fig. S2G) and concomitant persistent DNA DSBs measured by neutral comet assay (fig. S2H) in the HR-deficient cell lines (lacking functional BRCA2), which were similar in magnitude to those observed in the IDH1-mutant cell lines.
The results above suggest that IDH1/2-mutant cells harbor an intrinsic DSB repair defect, which prompted us to directly interrogate the integrity of HR and NHEJ in these cells. To this end, we used plasmid reporter assays recently developed by our group, which compare relative DSB repair activity between cell lines or conditions (schematic shown in Fig. 2A;(28)). We observed a marked deficiency in HR in HCT116 and HeLa IDH1-mutant cells compared to their WT counterparts, whereas no differences were observed in the other major DSB repair pathway, NHEJ (Figs. 2B and 2C, respectively). The ability of these assays to report on pathway-specific DSB repair defects have been demonstrated previously(28), and representative data validating the ability to measure NHEJ repair are shown in fig. S2I (validation data for HR are presented below). Once again, we confirmed that this HR defect could be recapitulated by the expression of mutant, but not WT, IDH1 or IDH2 proteins in the HEL cells (Fig. 2D), with no observed effects on NHEJ repair (fig. S2J). As above, we observed normal cell cycle phase distributions, which ruled out the possibility of a confounding effect arising from altered cell cycle phase distribution (fig. S1O) Collectively, these data establish a mutant IDH1/2-dependent HR defect.
Figure 2. The IDH1 R132H-dependent homologous recombination deficiency confers synthetic lethality with PARP inhibition.
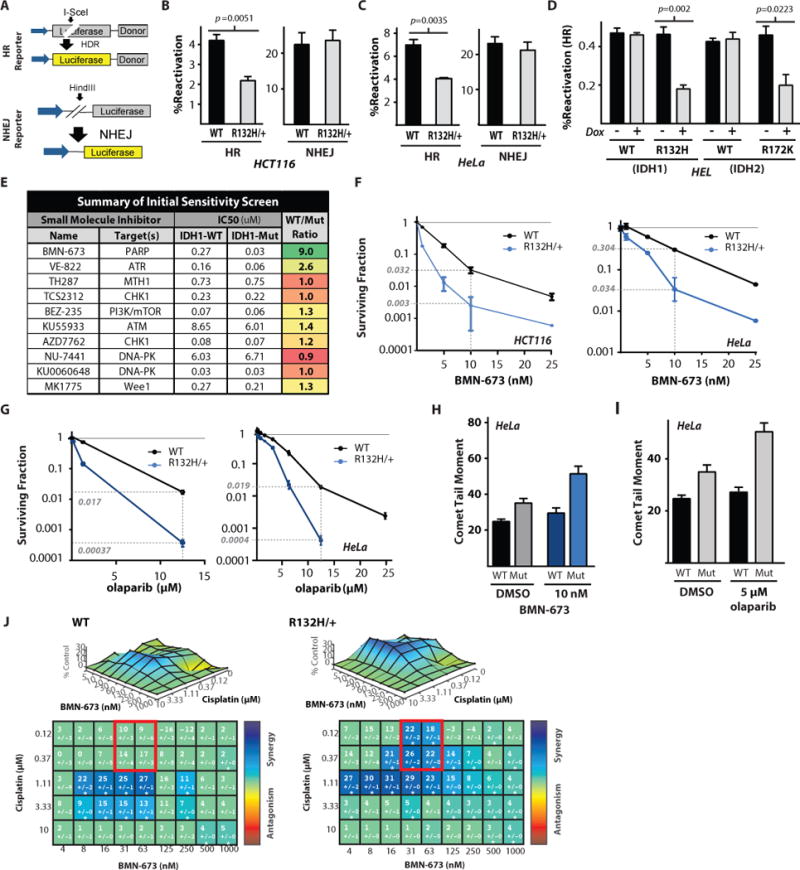
(A) Schematic representation of the luciferase-based reporter assays for HR and NHEJ. Note the inactivating I-SceI gene in the luciferase open reading frame of the HR reporter, allowing restoration of functional luciferase only by homologous recombination at the I-SceI induced double strand break. Pathway-specific, luciferase-based HR and NHEJ reporter assays in the (B) WT and R132H heterozygous HCT116 cells, (C) WT or IDH1 R132H HeLa cells, and (D) doxycycline inducible HEL + WT IDH1, IDH1 R132H, WT IDH2, or IDH2 R172K. Statistical analysis by t-test (n=3). (E) Targeted small molecule screen in the WT and R132H heterozygous HeLa cells identifies a synthetic lethal interaction between IDH1 R132H and the PARP inhibitor BMN-673. (F) Clonogenic survival assays in the IDH1-WT and R132H HCT116 cells or HeLa cells treated with indicated doses of BMN-673 (n=6). (G) Clonogenic survival assays in the IDH1-WT or R132H HCT116 cells or HeLa cells treated with indicated doses of olaparib. Dashed lines indicate surviving fraction at 10 nM BMN-673 in F and 12.5 μM olaparib in G (n=6). (H and I) Quantification of neutral comet assay performed in WT or R132H/+ HeLa cells treated with indicated doses of (H) BMN-673 or (I) olaparib. (J) Synergy surface plots in WT or IDH1 R132H/+ HeLa cells show a mutant specific increase in synergy between BMN-673 and cisplatin (n=3). For B, C, D, H and I, bars represent mean +/− SEM.
IDH1 R132H mutant cells are selectively killed by PARP inhibitors
It is well established that HR defects confer sensitivity to small molecule inhibitors of specific DNA repair pathways via synthetic lethal interactions (29). We thus performed a focused screen for DNA repair inhibitors that might selectively target IDH1-mutant cells, using a high-throughput, short-term cell growth inhibition assay (schematic shown in fig. S3A, and representative data from the primary screen shown in fig. S3B). These studies revealed a marked synthetic lethal interaction between the poly (ADP-ribose) polymerase (PARP) inhibitor, BMN-673, and the IDH1 R132H mutation (Fig. 2E). We also detected a substantial synthetic lethal interaction with the ATR inhibitor, VE-822 (30). Further, we observed synthetic lethal interactions with several other PARP inhibitors, including olaparib, MK-4827, and rucaparib (fig. S3C) in IDH1 R132H-mutant HeLa cells. Synthetic lethality with BMN-673 was also observed in the HCT116 IDH1-mutant cells using the same short-term viability assay (fig. S3D). In both HCT116 and HeLa cell lines, we observed a 10-fold increase in cell killing with BMN-673 in IDH-mutant vs. -WT cells in the viability assays. The magnitude of synthetic lethality was similar to that observed in BRCA2-deficient ovarian cancer cells treated with these PARP inhibitors, as measured under similar short-term assay conditions (fig. S3E). Mutant IDH1-dependent PARP inhibitor synthetic lethality was further confirmed in longer-term clonogenic survival assays based on colony formation in both the HCT116 and HeLa cell line pairs (Fig. 2F). We observed substantial sensitivity of the respective IDH1-mutant cells to BMN-673 in both cases, with an approximately 10-fold decrease in cell survival compared to WT cells at a dose of 10 nM. We observed a 3-fold decrease in survival when IDH1 R132H/+ Hela cells were treated with MK-4827 and rucaparib (figs. S4A and S4B). In the case of another PARP inhibitor, olaparib, both HCT116 and HeLa IDH1-mutant cell lines demonstrated exquisite sensitivity, with relative killing compared WT cells exceeding 45-fold (Fig. 2G). Similarly, olaparib demonstrated a substantial increase (nearly 8-fold) in relative HEL cell kill after expression of the mutant, but not WT, IDH1 protein (fig. S4C). To further extend these findings, we also found that mutant IDH1 protein overexpression was sufficient to confer PARP inhibitor sensitivity in the THP1 cell line, for both olaparib and BMN-673 (figs. S4D and S4E respectively). In parallel, we found that the BMN-673 and olaparib concentrations associated with enhanced killing in IDH1-mutant versus -WT cells also induced an IDH1 R132H mutant-specific increase in DSBs at these same doses (Figs. 2H and 2I, respectively). This particular finding has been observed in BRCA2-deficient tumor cells previously, which further highlights the similarities in the extent of the DSB repair defect observed in IDH1/2-mutant and BRCA-deficient cells (31).
PARP inhibitors are synergistic with platinum-based chemotherapeutics (32), particularly in HR-deficient cells (33). This interaction has formed the basis for a number of clinical trials testing these agents together in BRCA1 and BRCA2-deficient tumors (34, 35). We thus sought to test whether we could detect a similar interaction in IDH1-mutant cells. To this end, we tested a range of doses of cisplatin and BMN-673, either alone or in combination, in HeLa IDH1-WT and –mutant cells, and we measured viabilities using the short-term growth assays presented above. We analyzed the data using a validated, open-access software tool to assess and quantify possible drug combination effects in terms of synergy versus antagonism (36). Using a classical Lowe synergy model, we detected a substantial synergistic interaction between cisplatin and BMN-673 in HeLa cells, which was notably higher in IDH1-mutant vs. -WT cells (36). The synergy scores are presented in both a matrix format and as surface plots in Fig. 2J, and relative cell kill data are presented in figs. S4F and S4G. An example is highlighted by the red squares in the matrix plots in Fig. 2J, specifically at doses of cisplatin which do not affect viability in either cell line when given alone (see fig. S4G). Synergy scores were increased on average by approximately 2-fold under these conditions in IDH1-mutant versus -WT cells (which correlated with an approximately 50% increase in cell kill). Combined together with the functional DSB repair assay presented earlier, these data support the presence of an HR defect in IDH1/2-mutant cells, which renders them sensitive to PARP inhibitors, alone or in combination with cisplatin.
2HG is sufficient to induce mutant IDH1-dependent HR deficiency and PARP inhibitor sensitivity
Next, we investigated the mechanism by which IDH1 mutations confer an HR defect. As discussed earlier, nearly all mutant IDH1/2 proteins acquire neomorphic activity resulting in the production of 2HG, and this oncometabolite is thought to be a major driver of tumorigenesis (9). We thus tested whether 2HG itself could induce the HR defect that is seen in IDH1-mutant cells. We found that treatment of IDH1-WT HeLa cells with (2R)-octyl-α-hydroxyglutarate ((2R)-octyl-2-HG), a modified form of 2HG with high cellular uptake, resulted in a dose-dependent increase in DSBs in log-phase cells, and similar effects were observed with the (S) enantiomer of 2HG (Fig. 3A). We found that 900 μM of exogenously added 2HG, a dose that yields an intracellular concentration of 2HG similar to the actual concentration found in HeLa cells with a heterozygous IDH1-mutation as measured in the same assay (fig. S5A), induced the highest amount of DSBs in these experiments (Fig 3A). The increased comet tail moments after 2HG addition were similar to those observed in IDH1-mutant cells and to those observed in WT cells after 5 Gy of IR (fig. S5B). Of note, treatment of IDH1-mutant HeLa cells with 2HG did not further increase the already greater persistence of DSBs, although IR treatment did increase the amounts of DSBs in these cells. Treatment of plasmid DNA with 2HG in vitro for 8 hours did not induce any DNA cleavage, which ruled out any possible direct effect of this molecule on DNA (fig. S5C). The appearance of unrepaired DSBs after 2HG addition was rapid in HCT116 and HeLa IDH1-WT cells, occurring within 2 hours of exposure of cells to the molecule (fig. S5D and S5E), which is suggestive of a post-translational mechanism of action rather than an effect mediated through changes in transcription. Further, we found that 2HG exposure induced increased rates of DSBs in a range of cell lines with diverse genetic backgrounds, including immortalized astrocytes, primary melanoma cultures, breast cancer cell lines, and U2OS osteosarcoma cells (Figs. 3B, 3C, fig. S5F, and fig. S5G, respectively), suggesting that this is a fundamental effect of this metabolite.
Figure 3. 2-Hydroxyglutarate is sufficient to induce homologous recombination deficiency and PARP inhibitor synthetic lethality.
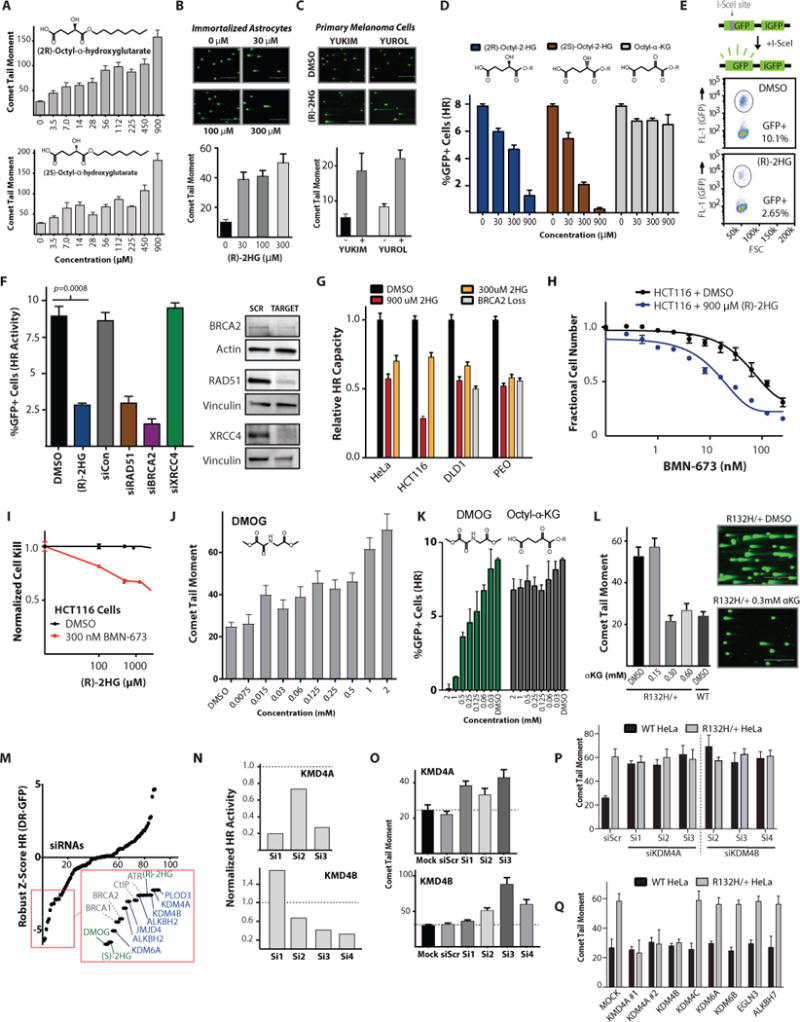
(A) Quantification of neutral comet assay performed in WT HeLa cells after 24 h exposure to indicated amounts of (R)-2-hydroxyglutarate and (S)-2-hydroxyglutarate (n=3). (B) Quantification of neutral comet assay performed in immortalized astrocytes after 24 h exposure to indicated amounts of (R)-2-hydroxyglutarate (n=3). (C) Quantification of neutral comet assay performed in primary melanoma cultures treated with 300 μM (R)-2-hydroxyglutarate for 24 h (n=3). (D) U2OS DR-GFP show a dose response of HR suppression after 24 h exposure to both (2R)-octyl-2-HG and(2S)-octyl-2-HG but not the octyl- α-ketoglutarate control (n=3). (E) Schematic and representative dot plot of the DR-GFP assay results that are quantified in F. (F) Quantification of U2OS DR-GFP assay performed after 7 day culture in 2HG or DMSO control or after siRNA knockdown of the indicated targets, with western blot analysis of siRNA target knockdown. Statistical analysis by t-test (n=3). (G) Relative HR capacity determined by quantification of luciferase-based plasmid HR reporter assay normalized to DMSO-treated WT for each respective cell type in HeLa, HCT116, DLD1, and PEO1 C4-2 cells (WT BRCA2) treated with 900 μM and 300 μM (2R)-octyl-2-HG, assayed in parallel with DMSO-treated WT and DMSO-treated BRCA2 homozygous knockout DLD1 cells and BRCA2 functional-null PEO1 cells (n=3). (H) Short-term growth delay assays in HCT116 WT cells after 4-day culture in 900 μM (2R)-octyl-2-HG or DMSO treated with indicated concentrations of BMN-673 (n=3). (I) Short-term growth delay assay in wild-type HCT116 cells after 4-day culture in 300 nM BMN-673 or DMSO control, treated with increasing concentrations of (2R)-octyl-2-HG, as indicated (n=3). Fractional survival is normalized to survival in either DMSO or 300 nM BMN-673 without (2R)-octyl-2-HG. (J) Quantification of neutral comet assay performed in WT U2OS cells after 24 h exposure to indicated amounts of the α-ketoglutarate-dependent dioxygenase inhibitor, DMOG. (n=3). (K) Quantification of U2OS DR-GFP assay performed after 24 h pretreatment with indicated concentrations of DMOG or α-ketoglutarate (n=3). (L) Quantification and representative images of neutral comet assay performed in IDH1-mutant HeLa cells treated with exogenous octyl-α-ketoglutarate and WT HeLa cells. Scale bar = 400 μm (n=3). (M) Robust Z-Score of normalized HR from the ligand-inducible DR-GFP of 64 smart pool siRNAS targeting α-ketoglutarate dependent dioxygenases compared to the effects of 1 mM (R)-2HG, (S)-2HG, and DMOG, as well as control siRNAs targeting known core DNA repair proteins (n=3). (N-O) Quantitation of (N) ligand-inducible DR-GFP and (O) neutral comet assay (n=3) with deconvoluted siRNAs against KDM4A and KDM4B, each performed 96 h after siRNA transfection in U2OS inducible DR-GFP cells. (P and Q) Quantification of neutral comet assay performed in WT and R132H/+ HeLa cells (n=3) (P) 72 h after transfection with indicated deconvoluted siRNAs and (Q) 24 h after transfection with expression vectors for indicated ORFs. (n=3) Note two independent KDM4A expression constructs KDM4A #1 (pCMV-HA-KDM4A) and KDM4A #2 (pCMV-FLAG-KDM4A). For A, B, C, D, F, G, J, K, O, P, and Q, bars represent mean +/− SEM.
We then tested the effect of 2HG directly on HR using the well-established, chromosomally integrated HR reporter assay, DR-GFP, in IDH1-WT U2OS cells. These cells were chosen because they are widely used as a tool to measure factors that impact HR (37). HR suppression by 2HG was dose-dependent and was observed with both R- and S- enantiomers, whereas no effects were seen with α-KG (Fig. 3D) (schematic and an example of the flow cytometry read-out are shown in Fig. 3E). Suppression of HR by 2HG in the U2OS cells was dramatic, and it approached the extent of suppression seen with siRNAs targeting two key HR genes, RAD51 and BRCA2 (Fig. 3F). In IDH1-WT HCT116 and HeLa cells, exposure to 2HG recapitulated the HR defect that is otherwise seen in the IDH1-mutant versions of these cells (Fig. 3G; compare to Fig. 2B and 2C, respectively).
Returning to the plasmid-based assay, we further determined that treatment with 2HG was able to phenocopy the magnitude of the BRCA-deficient HR phenotype seen in two independent matched cell line pairs with or without inactivating BRCA2 mutations (Fig. 3G; BRCA2 WT and deficient DLD1 pair, and BRCA2 WT and deficient PEO1 C4-2 and PEO1 pair; the expected reduction in HR measured in the BRCA2-deficient cells provides validation for the assay). In keeping with the effect of 2HG itself, on HR capacity, we found that treatment of WT HCT116 cells with 2HG conferred increased PARP inhibitor sensitivity (Figs. 3H and 3I).
We also sought to test the effects of manipulating endogenously produced 2HG on baseline DSBs and HR repair activity. To this end, we treated IDH1-WT and –mutant cells with siRNAs targeting mRNAs for either IDH1 or L-2-hydroxyglutarate dehydrogenase (L2HGDH). Knockdown of L2HGDH, which metabolizes (S)-2HG, was recently shown to increase (S)-2HG in log-phase, normoxic cells (38). As shown in fig. S6A and S6B, IDH1 siRNA knockdown reduced the high amounts of DSBs in IDH1-mutant HeLa cells, with no effects on the IDH-WT cell line, which is consistent with the findings presented above that mutant IDH1-generated (R)-2HG induces baseline high amounts of DSBs. In contrast, siRNAs targeting L2HGDH increased baseline DSBs in both the IDH1-WT and -mutant HeLa cells, in keeping with the effects of (S)-2HG on HR as shown above. Further, treatment of U2OS DR-GFP cells with L2HGDH siRNAs suppressed HR (fig. S6B and S6C). Collectively, these data establish (R)-2HG as the mediator of HR suppression and consequent PARP inhibitor sensitivity in IDH1/2-mutant cells and further demonstrate that the enantiomer, (S)-2HG, which is produced by other pathways (and increases upon knockdown of its respective dehydrogenase), can have similar effects.
The 2HG-induced BRCAness occurs via inhibition of specific αKG-dependent dioxygenases
Both the (R) and (S) forms of 2HG are thought exert their effects primarily via direct inhibition of αKG-dependent dioxygenases, which prompted us to test whether we could induce a similar HR defect via treatment with a known inhibitor of these proteins, dimethyloxalylglycine (DMOG; (39)). DMOG is a structural analog of αKG in which the -CH2-moiety has been replaced by an -NH-, and it acts as a competitive inhibitor of αKG-dependent dioxygenases (13, 39). As shown in Fig. 3J, treatment of IDH1-WT U2OS DR-GFP cells with DMOG resulted in a dose-dependent increase in baseline DSBs, which correlated with dose-dependent HR suppression (Fig 3K). No major changes in cell cycle phase distribution were observed after treatment with DMOG (or with the octyl ester forms of 2HG or αKG), and thus these effects cannot be explained by a confounding G1/G0-arrest phenotype (Figure S6D). Next, we tested if exogenous αKG could rescue the IDH1-R132H dependent DNA repair defect. Exogenous αKG was able to suppress the persistence of DNA DSBs in IDH1 R132H/+ HeLa cells down to amounts observed in WT cells (Figure 3L). Additionally, exogenous αKG was able to rescue the HR suppression of 2HG in the DR-GFP assay (fig. S7A).
Next, we performed a focused siRNA screen targeting all major αKG-dependent dioxygenases, to identify the protein(s) involved in the 2HG-induced HR suppression phenotype. We used a U2OS DR-GFP cell line optimized for use in 96- and 384-well microplate screening campaigns that was recently published by our group (40). The genes that we targeted with pooled siRNAs (4 siRNAs per gene) are shown in table S3. This screen pointed to a handful of αKG-dependent dioxygenase genes, the knockdown of which yielded Z-scores (for the HR suppression phenotype) that clustered with the effects of exposure to (R)-2HG, (S)-2HG, and DMOG (Fig. 3M). Deconvolution of the corresponding siRNA pools narrowed the candidate list to two key αKG-dependent dioxygenase genes, KDM4A and KDM4B, based on the criteria that three or more siRNAs targeting these two genes substantially reduced HR activity compared to scrambled control siRNAs (Figs. 3N). Each of the active siRNAs (fig. S7B) also substantially amplified the baseline increased DSBs in IDH1-WT U2OS cells and HeLa cells, which served as an orthogonal validation of specificity (Fig. 3O and 3P). Notably, each of the 3 siRNAs targeting KDM4A and KDM4B were unable to further increase the DNA DSB persistence in IDH1 R132H cells, suggesting an epistatic relationship (Fig 3P). Expression of the KDM4A ORFs from two independent constructs, as well as expression of the ORF encoding KDM4B, were able to rescue the DNA DSB repair defect in IDH1 R132H cells, but had no effect on IDH1-WT cells (Fig. 3Q and fig. S7C and S7D), as measured by the neutral comet assay. Conversely, ORFs for the αKG-dependent dioxygenases KDM4C, KDM6A, KDM6B, EGLN3, and ALKBH7 were unable to rescue the DNA DSB repair defect in IDH mutant cells (Fig. 3Q and fig. S7D). We then tested whether we could induce a similar phenotype in the same IDH1-WT cells with a small molecule inhibitor of KDM4A/KDM4B, NSC-636819. As shown in fig. S7E, treatment of HCT116 IDH-WT cells with this compound induced a dose-dependent increase in DSBs, which approached the amount seen in untreated HCT116 IDH-mutant cells. We observed this phenotype at doses ranging from 12.5–50 μM, which is within the reported range for the activity of this agent in cell culture studies (41, 42). NSC-636819 treatment in HCT116 IDH-mutant cells did not further increase comet tail moments, suggesting an epistatic interaction with the mutant IDH1 gene. Additionally, we observed a decrease in HR measured by the U2OS DR-GFP assay after treatment with NSC-636819 (fig. S7F). Both KDM4A and KDM4B are histone lysine demethylases that play roles in the orchestration of DSB repair and recruitment of repair factors to sites of DNA damage (43, 44), providing the mechanistic basis to link 2HG inhibition of these dioxygenases to attenuation of DNA repair.
The DSB repair defect and the PARP inhibitor vulnerability in IDH1-mutant cells are reversed by treatment with inhibitors of mutant IDH
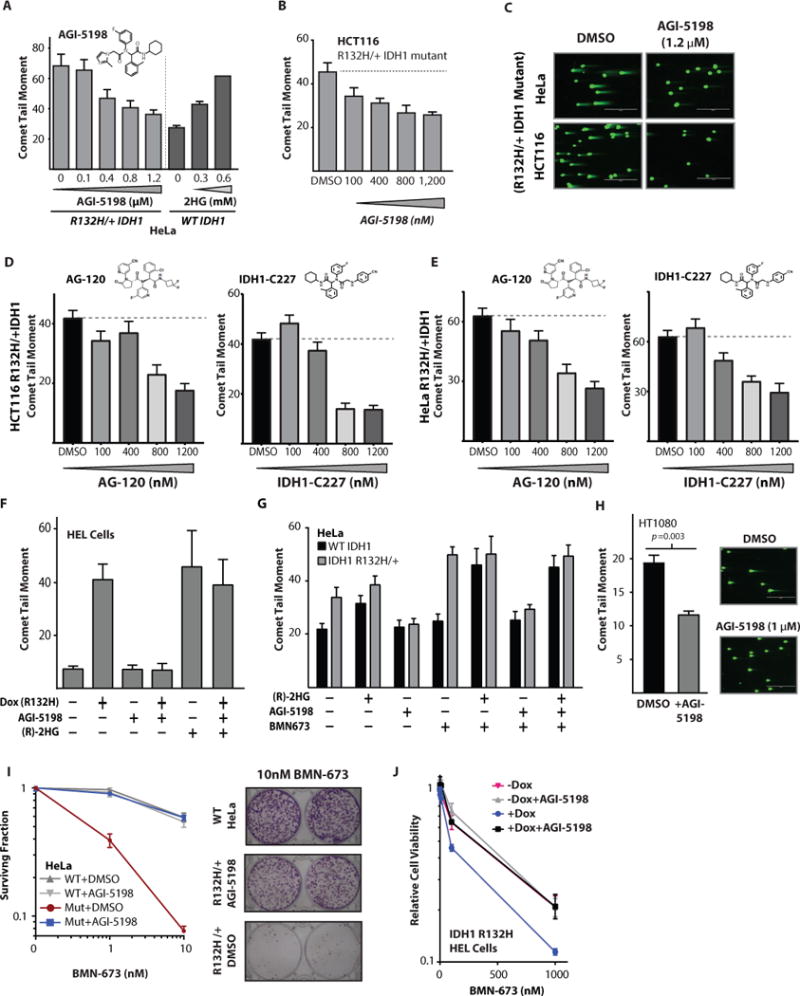
Next, we sought to test whether small molecule inhibitors of the mutant IDH1 enzyme, which potently block the production of 2HG, could block the DSB repair deficiency phenotype. Treatment with a selective IDH1 R132H inhibitor, AGI-5198, reduced the amounts of baseline DSBs in IDH1-mutant HeLa cells in a dose-dependent manner and did so nearly to the extent seen in IDH-WT cells (Fig. 4A). AGI-5198 also reduced baseline DSBs in HCT116 IDH1-mutant cells (Fig. 4B). Representative images from the corresponding comet assays for both IDH1-mutant lines with and without AGI-5198 treatment are shown in Fig. 4C. We next sought to rule out the possibility that off-target effects of AGI-5198 might be mediating the reversal of the baseline increased DSBs in IDH1-mutant cells. To this end, we confirmed a similar phenotype with two other structurally distinct inhibitors of mutant IDH1, AG-120 and IDH1-C227, in both IDH1-mutant HCT116 and HeLa cells (Figs. 4D and 4E, respectively). We also demonstrated that the increased DSBs produced by induction of mutant IDH1 protein expression in HEL cells could be fully reversed by AGI-5198. This reversal could be overcome with simultaneous treatment of cells with (R)-2HG (Fig. 4F), to thereby provide increased 2HG independently of the inhibited mutant enzyme. Similarly, the greater persistence of PARP inhibitor-induced DSBs in IDH1-mutant HeLa cells was reversed by AGI-5198 (Fig. 4G). In contrast, AGI-5198 was unable to prevent increases in DSBs that were induced by exogenously added 2HG (in both IDH1-WT and -mutant HeLa cells), demonstrating that the rescue effect of AGI-5198 in this experiment is specific for inhibition of the IDH1-mutant enzyme (Fig. 4G).
Figure 4. Inhibition of mutant IDH1 reverses the DSB repair defect and PARP inhibitor vulnerability.
(A) Quantification of neutral comet assay performed in R132H/+ HeLa cells treated with indicated doses of AG1-5198 or WT HeLa cells treated with indicated doses of (R)-2HG (n=3). (B) Quantification of neutral comet assay performed in R132H/+ HCT116 cells treated with indicated doses of AG1-5198 (n=3). (C) Representative images from quantification shown in (A–B). Scale bar = 400 μm. (D–E) Quantification of neutral comet assays performed in (D) HCT116 IDH1 R132H/+ cells (n=3) and (E) HeLa IDH1 R132H/+ cells (n=3) treated with indicated amounts of the mutant IDH1 inhibitors AG120 and IDH1-C227. (F) Quantification of neutral comet assay in HEL cells, performed after the indicated conditions of induction of IDH1 R132H and treatment with 1.2 μM AGI-5198 and 1 mM (R)-2HG (n=3). (G) Quantification of neutral comet assay. WT or R132H/+ Hela cells were cultured in 1.2 μM AGI-5198 or DMSO control for 96 h, then supplemented with 300 μM (2R)-octyl-2-HG or DMSO control for 24 h before being split and treated with 10 nM BMN-673 for 48 h with continued exposure to 1.2 μM AGI-5198, 300 μM (2R)-octyl-2-HG, or DMSO control, then collected for analysis (n=3). (H) Quantification of neutral comet assay in HT1080 cells harboring an endogenous IDH1 R132C mutation treated with 1 μM AGI-5198 or DMSO control. Statistical analysis by t-test (n=3). (I) Quantification and representative images of clonogenic survival assays of WT and R132H/+ (Mut) HeLa Cells treated with the indicated concentrations of BMN-673 in the presence or absence of AGI-5198. HeLa cells were cultured in 1.2 μM AGI-5198 or DMSO control for 96 h before seeding for clonogenic survival, and exposure to AGI-5198 was maintained throughout the clonogenic survival assay. Representative images are HeLa seeded at 2400 cells per well and treated with 10 nM BMN and 1.2 μM AGI-5198 or DMSO control (n=6). (J) Short term viability assay in HEL cells with dox inducible IDH1 R132H treated with either 1 μM AGI-5198 or DMSO control and indicated doses of BMN-673 (n=4). For A, B, D, E, F, G, and H, bars represent mean +/− SEM.
We also confirmed these findings in a cell line that harbors an endogenous IDH1 mutation by showing that baseline increased DSBs could be reversed by treatment with a small molecule inhibitor of the mutant protein. For this, we used HT1080 cells, a human sarcoma cell line which harbors a naturally occurring heterozygous R132C IDH1 mutation. We confirmed that this cell line produces high concentrations of (R)-2HG that are suppressed by AGI-5198 (fig. S7G), in keeping with prior work (45). As shown in Fig. 4H, we found that treatment of H1080 cells with AGI-5198 resulted in a reduction in mean comet tail moments.
In addition, we found that AGI-5198 treatment reverts the PARP inhibitor sensitivity seen in IDH1-mutant HeLa cells (Fig. 4I). Similarly, we found that the PARP inhibitor sensitivity conferred by induced mutant IDH1 protein expression in HEL cells could be reversed by AGI-5198 (Fig. 4J). We also tested the HT1080 cells (with an endogenous R132C IDH1 mutation), and we found that AGI-5198 also substantially reduced the sensitivity of this cell line to both olaparib and BMN-673 (figs. S7H and S7I, respectively). Collectively, these data indicate that the DSB repair defect and the PARP inhibitor sensitivity caused by IDH1 mutations are effectively reversed by treatment with a selective inhibitor of the mutant IDH1 protein.
NAD+ levels do not play a role in mutant IDH1/2-induced PARP inhibitor sensitivity
Because it has been reported that IDH1-mutant cells can harbor low NAD+ concentrations (24), and because PARP enzymes use NAD+ as a substrate (46), we examined NAD status under the conditions of our experiments. As shown in fig. S7J, BMN-673 did not suppress NAD+ in either WT or mutant HeLa cells. Also, treatment of wild-type HeLa with 2HG (at concentrations that mediate suppression of HR and confer PARP inhibitor sensitivity, as shown earlier) did not alter NAD concentrations. In contrast, treatment with FK866, an inhibitor of nicotinamide phosphoribosyltransferase (NAMPT) that depletes NAD+ (47), substantially reduced NAD+ concentrations in both cell lines, and served as a positive control for the ability to detect reduced NAD+ content in this assay. Collectively, these data rule out a role for altered NAD in the observed effects of 2HG on DSB repair function.
Patient-derived gliomas show IDH1 mutation-associated and 2HG-mediated HR suppression and PARP inhibitor sensitivity
Next, we sought to further establish the clinical relevance of our findings using a collection of early-passage, patient-derived IDH1-WT and -mutant glioma cell lines available at our institution (clinical characteristics are shown Fig. 5A). IDH1 mutations were confirmed by sequencing as reported previously (48), and we also confirmed that we could detect 2HG production in samples harboring an IDH1 R132H or R132C mutation (Fig. 5B). We detected increased baseline persistence of DSBs by comet assay in the primary glioma cell lines harboring IDH1 mutations, and we demonstrated that we could induce this phenotype in the WT glioma cell lines by the addition of 2HG. Representative images from the corresponding comet assays are shown in Fig. 5C, and quantitative data for each cell line are presented in Fig. 5D. The higher amounts of DSBs in the IDH1-mutant primary glioma cell lines (and also IDH1-WT cells treated with 2HG) correlated with increased γH2AX and 53BP1 foci (Figs. 5E and S8A, respectively). We transfected two IDH1-WT and two IDH1-mutant cell lines with our HR reporters, and in both cases we detected reduced HR capacities in the latter samples (Fig. 5F). Finally, although these are early-passage primary cells obtained directly from fresh glioma resection tissue, we nonetheless were able to perform clonogenic survival assays on a subset of them. We were able to detect PARP inhibitor sensitivity in two IDH1-mutant primary glioma cultures compared to two IDH1-WT cultures by clonogenic survival, and once again we found that 2HG exposure could recapitulate PARP inhibitor sensitivity in WT cultures (Fig. 5G).
Figure 5. IDH1 mutations confer repair deficiency and PARP inhibitor sensitivity in human glioma cells and mouse tumor models.
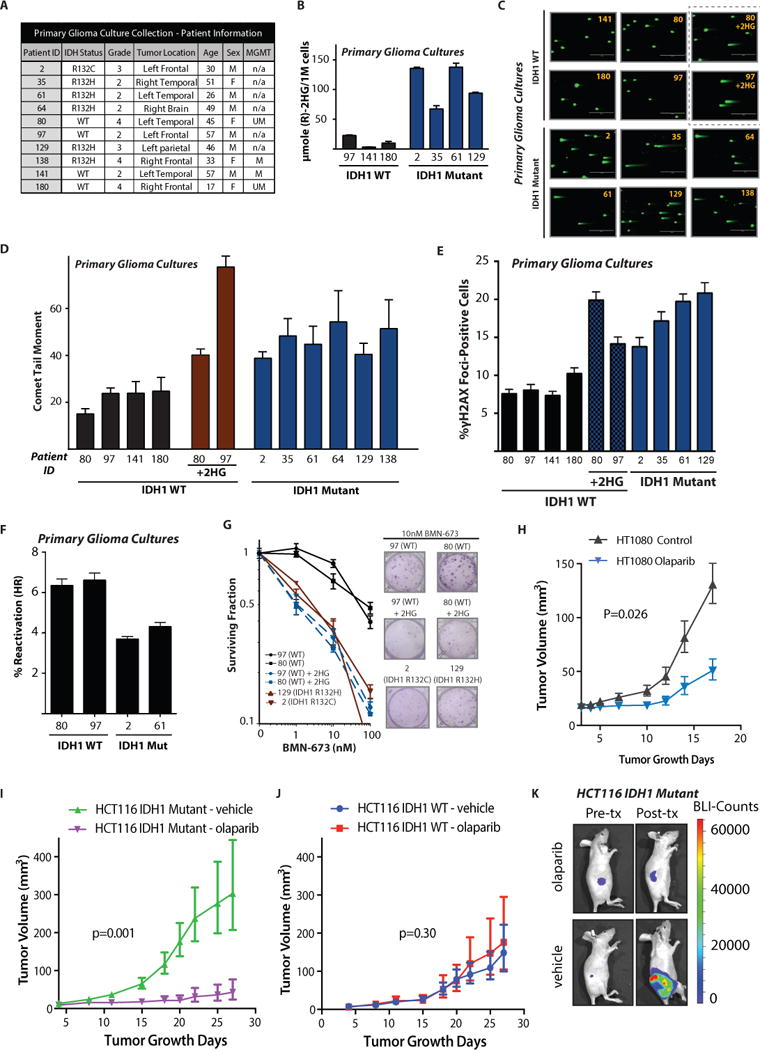
(A) Patient information and clinical characteristics for the primary, patient-derived glioma culture collection. (B) Quantification of (R)-2HG production in IDH mutant primary glioma cultures (n=3). (C) Representative images and (D) quantification of neutral comet assays performed on primary, patient-derived glioma cultures (n=3). Scale bar = 400 μm. For comparison, primary cultures 80 and 97 were grown in media supplemented or not with 300 μM (2R)-octyl-2-HG for 7 days as indicated. (E) Quantification of γH2AX foci in primary glioma cultures treated the same way as in (C–D) (n=3). (F) Host cell reactivation assay for HR in primary glioma cultures. (G) Quantification and representative images of clonogenic survival assays in response to BMN-673 performed in primary glioma cultures. Gliomas 80 and 97 in black are IDH1 WT. Gliomas 129 and 2 in brown are IDH1 mutant. WT gliomas 80 and 97 were also cultured in 300 μM (2R)-octyl-2-HG for 7 days before seeding for the clonogenic survival assay and were maintained in 2HG throughout the assay; these are in blue (n=4). (H) Tumor volume growth curves for IDH1 R132C/+ HT1080 xenografts treated daily with either 50 mg/kg olaparib or vehicle control (ANOVA p=0.026) starting 4 days after implantation (n=9 per group). (I and J) Murine tumor volume growth curves for (I) IDH1 R132H heterozygous (ANOVA P = 0.001) (n=9 per group) and (J) IDH1 WT HCT116 xenografts (ANOVA P = 0.30) (n=9 per group) after treatment with 50 mg/kg olaparib once daily five days per week versus vehicle control. Treatment was initiated on day 4 after tumor implantation. (K) Representative images of in vivo luciferase activity in HCT116 IDH1 R132H heterozygous xenografts before and after olaparib treatment. For B, D, E, and F, bars represent mean +/− SEM.
The PARP inhibitor, olaparib, selectively inhibits the growth of IDH1-R132H mutant tumor xenografts
We next tested the extent to which IDH1 mutant-dependent PARP inhibitor sensitivity could be recapitulated in vivo using 3 independent subcutaneous xenograft tumor models. We chose to conduct in vivo efficacy studies with olaparib, because it showed close to a 50-fold difference in IDH1-mutant sensitivity in vitro. Moreover, olaparib is currently in clinical trials and is FDA-approved for numerous cancers, and thus the pharmacokinetics and pharmacodynamics of this agent are well established. The purpose of our study was to determine whether we could detect a statistically significant, mutant IDH1-dependent difference in PARP inhibitor response in vivo.
To this end, we first implanted HT1080 cells harboring an endogenous IDH1 R132C mutation into the flanks of nude mice, and we observed a significant tumor growth delay in mice receiving olaparib treatment (beginning four days after tumor cell implantation) compared to vehicle control (ANOVA p=0.026; Figure 5H). Next, we sought to further confirm that this is an IDH mutant specific effect. To this end, we implanted HCT116 IDH1-WT and R132H-mutant tumor cells into the flanks of nude mice, and, after confirmation of tumor formation by bioluminescence imaging (BLI), treated the mice with olaparib at 50 mg/kg daily (versus vehicle control in both cases). We observed a striking (8-fold) and significant (ANOVA p=0.001; Fig. 5I) difference in tumor growth delay specific to the olaparib-treated IDH1-mutant xenograft as compared to the vehicle control. In contrast, exposure of HCT116 IDH1-WT xenografts to olaparib did not result in a statistically significant growth delay compared to vehicle (ANOVA p=0.30; Fig. 5J). Bioluminescence imaging demonstrated a marked reduction in the BLI signal in the IDH1-mutant xenografts after treatment of mice with olaparib as compared to vehicle control (Fig. 5K), whereas no differences in the BLI signal were observed in the IDH1-WT xenografts whether the mice were treated with olaparib or not (fig. S8B). We next sought to test a second genetically matched pair of xenograft models, the CRISPR-engineered IDH1-mutant HeLa cells and parental wild-type line. Treatment with olaparib induced a mutant-specific effect in this model as well, significantly inhibiting the growth of the IDH1-mutant HeLa tumor by 3-fold (ANOVA p=0.007; fig. S8C), without affecting the WT isogenic control tumor (ANOVA p=0.90; fig. S8D). In addition, we harvested untreated HCT116 IDH1-mutant tumor xenografts and untreated IDH WT xenografts and assayed for 2HG by 1H NMR, confirming that the IDH1-mutant tumors produced higher amounts of 2HG in vivo, as expected (fig. S8E).
Patient-derived AML cells show IDH1 mutation-associated and 2HG-mediated HR suppression and PARP inhibitor sensitivity
As an additional test of clinical relevance, we sought to determine whether we could detect a mutant IDH1/2-dependent DSB repair defect in primary bone marrow cultures derived from AML patients with tumors harboring IDH1 and IDH2 gene mutations. The clinical characteristics of four specimens from our institution are shown in Fig. 6A. We were able to successfully maintain and expand these cultures for several passages, and we confirmed log-phase proliferation (representative cell cycle plots are shown in Fig. 6B for two samples). Similar to the results obtained with the primary glioma cell lines, we detected IDH-mutation associated increases in DSBs in a matched IDH1-WT and –mutant pair of primary samples, and similar results were obtained with a matched IDH2-WT and –mutant pair (Fig. 6C; representative comet images shown in Fig. 6D). We also detected increased radiosensitivity in both the IDH1- and IDH2-mutant AML cells compared to their WT counterparts (Fig. 6E), which correlated with prolonged DSBs 24 h after IR (Fig. 6F).
Figure 6. Patient-derived primary acute myeloid leukemia samples show a 2HG-dependent DSB repair defect.
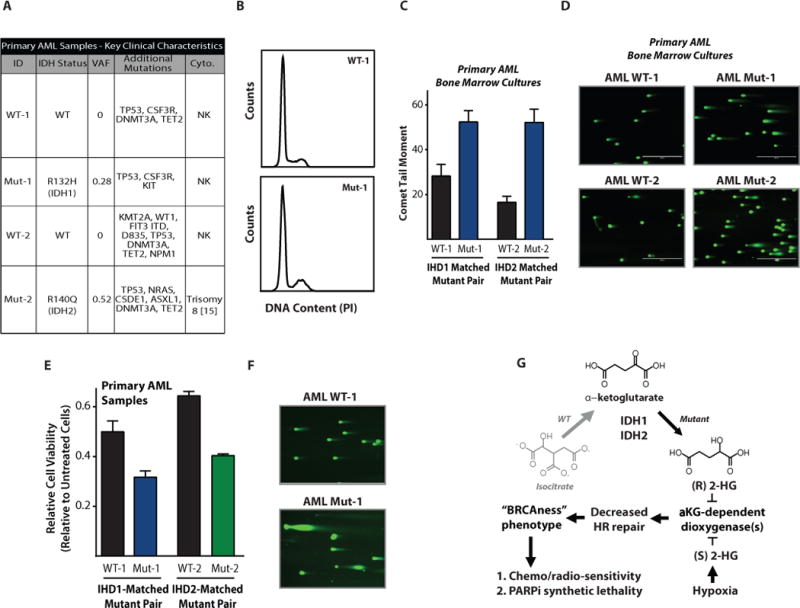
(A) Patient information and clinical characteristics for the matched pairs of primary AML samples. (B) Cell cycle analysis of the IDH1 WT and R132H primary AML samples. (C) Quantification and (D) representative images of neutral comet assays performed on primary AML patient cells (n=3). Scale bar = 400 μm. (E) Radiation survival of primary AML cultures assayed by a short term viability assay 48 h after 5 Gy IR (n=3). (F) Representative images of neutral comet assay performed 24 h after 5 Gy IR on the primary AML cells WT-1 and Mut-1 (IDH1 R132H), indicating differential persistence of DNA DSBs after IR. Scale bar = 400 μm. (G) Proposed mechanism by which the (R)-2-hydroxyglutarate produced by IDH1 R132H (or (S)-2HG produced by hypoxia) induces an HR-deficient “BRCAness” phenotype and subsequent vulnerability to PARP inhibition. For C and E, bars represent mean +/− SEM.
Discussion
In summary, we report here that IDH1/2 mutations induce an HR defect that renders tumor cells sensitive to PARP inhibition. We validated this phenotype across five genetically diverse cell line pairs that were engineered to express either the WT or the mutant IDH1/2 proteins, and we confirmed the observed DSB repair defect using multiple orthogonal functional assays. The IDH1-dependent PARP inhibitor sensitivities were profound, and approached a 50-fold difference compared to IDH1-WT cells, with the FDA-approved PARP inhibitor, olaparib. In addition, we demonstrated multiple aspects of the IDH1/2-induced BRCAness phenotype in a range of clinically relevant models, including patient-derived glioma cell lines, primary AML bone marrow cultures, a human sarcoma cell line carrying a naturally occurring IDH1 R132C mutation in cell culture and as a xenograft in mice, and two genetically matched pairs of tumor xenografts in mice. Mechanistically, this phenotype can be entirely recapitulated by exposure to either 2HG enantiomer, and it cannot be explained by the alterations in NAD+ that have been seen in IDH1/2-mutant cancers (24). Our data suggest that 2HG-induced HR suppression is mediated by direct inhibition of αKG-dependent dioxygenases, in particular KDM4A and KDM4B. We found that treatment with a mutant IDH1-specific small molecule inhibitor that potently suppresses 2HG production (similar to drugs in clinical trials) reversed the observed HR defect and eliminated the associated PARP inhibitor sensitivity. We demonstrated this reversal in both our mutant IDH1/2 cell lines and in a cell line harboring an endogenous IDH1 mutation. Reversal of the mutant IDH1-associated DSB repair defect was confirmed using three different small molecule inhibitors of the mutant protein, and also with siRNAs targeting the IDH1 gene, thus ruling out potential off-target effects of the inhibitors. A proposed mechanism of action is summarized in Fig. 6G.
Both IDH1 mutant-induced (R)-2HG and hypoxia-induced (S)-2HG suppress α-KG-dependent dioxygenases, resulting in profound epigenetic reprogramming in cells (9, 11). We have previously shown that hypoxia suppresses HR, driving genetic instability and conferring a BRCAness phenotype in hypoxic tumor cells (49–51). It is tempting to speculate that the findings reported here may provide a shared mechanism by which hypoxia and IDH1/2 mutations promote genetic instability and tumor progression: through induction of 2HG and consequent suppression of HR, thereby also bestowing a vulnerability to PARP inhibition that can be therapeutically exploited. Clinical trials such as NCT01116648 are now testing synthetic lethal targeting of hypoxia-induced HR suppression by combining cediranib, an angiogenesis inhibitor that induces transient hypoxia, with PARP inhibitors as a therapeutic strategy, further highlighting the clinical relevance of the results presented here.
IDH1-mutant gliomas are chemo- and radiosensitive, although the mechanism underlying this enhanced sensitivity has been elusive (20). The findings presented here that IDH1/2 mutations induce “BRCAness” provide a basis for this sensitivity. Because multiple PARP inhibitors are currently in clinical trials and one is FDA-approved for HR-deficient cancers (olaparib), our data suggest an urgent need to test these agents in IDH1-mutant gliomas and in other cancers with IDH1/2 mutations. Emerging evidence reveals that IDH1/2 mutations that generate (R)-2HG are also found in numerous other tumor types, including cholangiocarcinoma, chondrosarcoma, AML, melanoma, gastric cancer, colorectal cancer, liver cancer, and others (52). Furthermore, substantial 2HG production has been reported in a subset of breast cancers in the absence of IDH1/2 mutations (14, 15), and the (S) enantiomer is produced in some renal cell cancers (10). The (S) enantiomer is also generated in hypoxic cancer cells as noted above (11, 12). Because our data establish 2HG as necessary and sufficient for the induced BRCAness phenotype, it is likely that these other classes of tumors will be also susceptible to PARP inhibition. 2HG can be detected non-invasively by a number of approaches, including LC/MS analyses of blood and urine specimens (53), as well as MR spectroscopy (54), and thus we suggest that 2HG could serve as a biomarker to identify HR-deficient tumors (independent of BRCA1/2 mutations) that will respond to PARP inhibitors.
Although the findings reported here have therapeutic implications, there are nonetheless some limitations to our work that warrant further study. For example, we did not study PARP inhibitor activity against IDH1/2-mutant glioma xenografts in vivo. We did demonstrate mutant IDH1-dependent PARP inhibitor sensitivity in primary, patient-derived glioma cells ex vivo, but these lines do not form tumors in mice, and so we were unable to test their response in vivo. A demonstration of the in vivo sensitivity of IDH1 mutant gliomas to PARP inhibition would be valuable to further support the direct translation of our findings into the clinic specifically for IDH1/2-mutant gliomas. Nonetheless, we have presented consistent data using a panel of IDH1-WT and -mutant tumor cell lines in a series of in vitro and in vivo assays, which taken together point to a tissue-agnostic suppression of HR repair and induction of PARP inhibitor sensitivity by 2HG and/or IDH mutation. Furthermore, we confirmed that 2HG exposure in immortalized human astrocytes induces a marked DSB repair defect. In addition, several PARP inhibitors penetrate the blood-brain barrier, including olaparib (55) and veliparib (56, 57), which further supports the eventual testing of PARP inhibitors against IDH1/2-mutant gliomas.
Small molecule inhibition of oncogenic kinases is a pillar of precision medicine in modern oncology (58), and this approach has been extrapolated to treat IDH1/2-mutant cancers with small molecule inhibitors of the neomorphic activity of the mutant protein (59). In the work reported here, we demonstrate that IDH1/2-mutations induce an unexpected HR defect and consequent vulnerability to PARP inhibition, and we propose that this vulnerability should be exploited rather than inhibited.
Materials and methods
Study design
The objective of the present study was to characterize IDH1/2 mutant cells and identify a mechanistic basis for synthetic lethal drug interactions. The sample sizes of the experiments were selected on the basis of previous experience. Data collection was stopped at a priori defined time points for all experiments. Animal experiments were performed in a confirmatory fashion with an a priori hypothesis and a priori endpoints for tumor growth delay. All in vitro experiments were carried out in biological triplicate and data are presented as +/− SEM. In vivo experiments were carried out in a blinded fashion with animals randomly assigned to treatment and control groups, and in vitro experiments were not blinded.
Clonogenic survival assay
Cells in culture were irradiated at varying doses of ionizing radiation. Four to six hours after irradiation, they were trypsinized, washed, counted, and seeded in 6-well plates in triplicate at 3-fold dilutions ranging from 9000 to 37 cells per well. Depending on colony size, these plates were kept in the incubator for 10 to 14 days. After incubation, colonies were washed in PBS, stained with crystal violet, and counted and quantified. For drug treatments, cells were counted and diluted in media containing various concentrations of drug. They were then immediately seeded in 6-well plates in triplicate at 3-fold dilutions, ranging from 9000 to 37 cells per well. These plates were kept in the incubator for 10 to 14 days, after which they were washed in PBS, stained with crystal violet, and quantified. Data are presented as the mean +/− SD. A list of SF50 values for all clonogenic survival assays testing PARP inhibitors is shown in table S4.
Neutral comet assays
Neutral comet assays were performed per manufacturer’s protocol (Trevigen). Briefly, cells were trypsinized, washed with PBS, and replicates were suspended in LM Agarose (Trevigen). Neutral electrophoresis was conducted at 21 V for 1 h in the CometAssay Electrophoresis System (Trevigen). Data were collected with an EVOS FL microscope (Advanced Microscopy Group) and analyzed using Open Comet software(60). Data are presented as the mean +/− SEM for 3 biological replicates with more than 100 cells analyzed per replicate. Statistical analysis was by t-test.
Luciferase-based host-cell reactivation reporter assays for HDR and NHEJ
The HR luciferase reporter has been previously reported(28, 61) and was generated by cloning an inactivating I-SceI recognition site into the BstBI site 56 amino acids into the firefly luciferase gene in the gWIZ.Luciferase vector (Gelantis), and cloning a promoterless copy of the firefly luciferase open reading frame 700 base pairs downstream in reverse orientation as a donor template for HR. A DSB in the firefly luciferase gene was induced by I-SceI digestion and confirmed by electrophoresis. Linearized plasmid was transfected into cells to measure HR as a function of luciferase activity (firefly luciferase activity can only be restored by HR, which removes the inactivating I-SceI site). The NHEJ assay has been previously reported(50, 62) To assay NHEJ, a HindIII-mediated DSB was generated between the promoter and the coding region of the firefly luciferase gene in the pGL3-Control Vector (Promega) and confirmed by electrophoresis. After transfection of linearized plasmid, repair of the DSB by NHEJ restores firefly luciferase activity. All reporter assays were performed in 12-well format by seeding 7 × 104 cells per well 24 h before transfection and transfecting 1 μg of reporter or positive control vector and 50 ng Renilla luciferase vector per well. For HR, cells were analyzed 48 h after reporter transfection, and for NHEJ, cells were analyzed 24 h after reporter transfection. Luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega) for all samples and normalized to Renilla luciferase signal to control for transfection efficiency, to a positive control luciferase expression vector gWIZ.luciferase for HR, and to pGl3-Control for NHEJ. Data are presented as the mean of 3 biological replicates +/− SEM. Statistical analysis was by t-test.
U2OS DR-GFP reporter assays
These reporter assays were carried out as previously described (37, 63). To test the effect of 2HG, cells in culture were treated for the indicated times with (2R)-octyl-2-HG, and then 106 cells were transfected in triplicate with 4 μg pI-SceI using the Amaxa Nucleofector II and Nucleofection Kit V (Lonza) per manufacturer’s protocol. 72 h after transfection, cells were analyzed for GFP expression by flow cytometry, and the data were analyzed using FlowJo software to calculate %GFP-positive cells. Data are presented as the mean of 3 biological replicates +/− SEM. Statistical analysis was by t-test.
For the siRNA screen of the alpha-ketoglutarate-dependent dioxygenases, ligand inducible DR-GFP with siRNA reverse transfection was performed as previously described (64). ON-Target plus smart pool siRNAs (GE Dharmacon) were selected, targeting 64 of the known alpha-ketoglutarate dependent dioxygenases (along with, for comparison, siRNAs targeting selected DNA repair factors and siRNAs targeting IDH1 and L2HGDH). The siRNAs were reverse transfected into cells to a final concentration of 20 nM. Cell cycle analysis for all samples was performed in parallel with the DR-GFP assay to rule out false positive DR-GFP results because of cell cycle arrest. Potential hits were deconvoluted by performing the inducible DR-GFP assay with the four individual RNA oligonucleotides comprising the smart pool, and cell cycle analysis was performed again in parallel for each siRNA oligonucleotide. Robust Z-Scores were calculated for each sample.
Short-term, high-throughput growth delay assays
Cells were plated in 96-well black-walled plates (Costar) at a concentration of 2500 cells per well and allowed to adhere at room temperature for 60 min before return to the incubator. For growth delay assays containing (2R)-octyl-2HG, cells were cultured with the indicated concentration for 10 days before plating. After 24 h, the media were changed, and indicated drugs dissolved in either DMSO or DMF (cisplatin only) were added in quadruplicate at varying concentrations. For synergy experiments, cells were replica-plated and drugs added in single wells at the indicated concentrations. At 96 h after the addition of drugs, cells were washed in PBS, fixed in 70% ethanol, and stained with Hoechst at 1 μg/mL. The plates were then imaged on a Cytation 3 automated imager (BioTek), and cells were counted using CellProfiler (http://cellprofiler.org/). For synergy experiments, experiments were analyzed for synergistic interactions by the Loewe synergy and antagonism method using Combenefit (http://www.cruk.cam.ac.uk/research-groups/jodrell-group/combenefit).
Cell viability assays
Adherent cells were seeded at a density of 2500 cells per well, and suspension cells at a density of 5000 cells per well in solid white 96 well plates (Costar) and incubated under indicated conditions in sextuplicate. Cell viability was assayed using the Cell Titer Glo Kit (Promega) per manufacturer’s protocol, and data are presented as mean +/− SEM.
In vivo olaparib efficacy studies
Female athymic nu/nu mice (Envigo/Harlan) were used for all in vivo xenograft studies. All studies were approved by the Yale University IACUC. Mice were quarantined for at least 1 week before experimental manipulation. Human luciferase-expressing HCT116 cells (with and without IDH1 mutation), HeLa cells (with and without IDH1 mutation), and HT1080 cells (with native IDH1 R132C mutation) were implanted subcutaneously (2 × 106 cells in 0.1 cc PBS) in the right flank. Mice were visually observed daily, and tumors were measured three times per week by calipers to determine tumor volume using the formula: . Olaparib (Selleckchem) was solubilized in DMSO and diluted with 10% (w/v) 2-hydroxy-propyl-beta-cyclodextrin (Sigma) to obtain the desired concentration. Olaparib was delivered via intraperitoneal injection (50 mg/kg) once daily, five days per week, starting four days after tumor implantation. Growth curves were compared statistically using ANOVA for repeated measures.
Bioluminescence imaging (BLI)
BLI was carried out using the IVIS Spectrum In Vivo imaging system (Perkin Elmer) according to the manufacturer’s protocol. Imaging was performed once weekly during the tumor growth delay experiments, starting before the first treatment. Mice were anesthetized, and D-luciferin was injected intraperitoneally. Imaging was carried out 15 minutes after luciferin injection. After image acquisition, a region of interest (ROI) was circumscribed for each tumor, and a corresponding tumor-free ROI was circumscribed to generate a background-corrected bioluminescence flux value. Bioluminescence values from representative animals in each treatment group were compared, and luminescence images were prepared using a standard scale for all mice.
Statistical analysis
Data are presented as means ± SEM and compared using Student’s t test, or ANOVA with repeated measures when appropriate. All tests were two-sided. Statistical analyses were carried out using GraphPad Prism and STATA software. A p value of less than 0.05 was considered statistically significant.
Supplementary Material
Fig. S1. Establishment and characterization of IDH mutant and wild-type cell models.
Fig. S2. Characterization of the DNA DSB repair defect in IDH mutant cells.
Fig. S3. Cell growth and viability assays in response to DNA repair and DNA damage response inhibitors.
Fig. S4. Increased sensitivity of IDH mutant cells to PARP inhibitors alone and in combination with cisplatin.
Fig. S5. Additional characterization of the impact of 2HG on DNA DSB repair.
Fig. S6. Manipulation of IDH and L2HGDH by siRNAs and impact on DNA DSB repair.
Fig. S7. αKG rescues 2HG induced HR defect, and 2HG alters DNA repair through KDM4A/B and not NAD+.
Fig. S8. Additional glioma cell and xenograft data.
Table S1. STR profile of IDH1 WT parental HeLa cells.
Table S2.STR profile of IDH1 R132H/+ HeLa cell sub-clone.
Table S3. List of siRNAs targeting alpha-ketoglutarate dependent dioxygenases and selected DNA repair proteins.
Table S4. Surviving fraction 50% (SF50) values for clonogenic survival assays.
One-sentence summary.
The oncometabolite, 2-hydroxyglutarate, renders IDH1/2 mutant cancer cells deficient in homologous recombination and confers vulnerability to synthetic lethal targeting with PARP inhibitors.
Acknowledgments
We thank Andreas von Deimling for generously providing us with the HGDH enzyme, T. Taniguchi for providing us with the PEO1, PEO4, and dPEO1 C4-2 cells, R. Majeti for the THP1 cells, and T. Chan for providing us with immortalized astrocytes. We apologize to those whose work we cannot list due to the reference limitations for a publication in this journal.
Funding.
This work was supported by the NIH (R01ES005775, R01CA168733, and R01 CA177719 to PMG), by the American Cancer Society (Research Scholar Grant to RSB), and by the Connecticut Department of Public Health (RFP 2014-0135 to SH). Nathaniel Robinson is a Howard Hughes Medical Institute Medical Research Fellow. Susan Scanlon was supported by NIH Medical Scientist Program Training Grant T32GM007205 and NIH NIGMS training grant T32GM007223. The work on this paper used Metabolomics Core Services supported by grant U24 DK097153 of NIH Common Funds Project to the University of Michigan.
Footnotes
Author contributions.
P.L.S., and C.D.C., contributed experiments, scientific hypotheses, data analysis, and compiling of the manuscript. N.D.R., S.E.S., K.R.P., Y.F., R.K.S, D.C.H., N.R.F., Y.S., K.M., H.dF., R.dG., Y.V.S., and M.K. contributed to experiments. M.G., H.B. and G.A.B. contributed data analysis. S.H., P.M.G., and R.S.B. designed experiments. P.L.S., C.D.C., P.M.G., and R.S.B. wrote the manuscript.
Competing interests.
Ranjit Bindra and Peter Glazer are inventors on US patent application No. 62/344,678 submitted by Yale University, which covers compositions and methods for targeting and treating homologous recombination-deficient tumors.
Materials and data availability.
The in-house software written in MATLAB (Mathworks) is available for download by directly contacting Dr. Robin de Graaf (http://mrrc.yale.edu/faculty/robin_degraaf.profile). For all other reagents and data requests, please contact Dr. Ranjit Bindra (ranjit.bindra@yale.edu).
References and notes
- 1.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiao Y, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nature genetics. 2013;45:1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krauthammer M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nature genetics. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amary MF, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. The Journal of pathology. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 7.Clark O, Yen K, Mellinghoff IK. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clinical Cancer Research. 2016;22:1837–1842. doi: 10.1158/1078-0432.CCR-13-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shim EH, et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014;4:1290–1298. doi: 10.1158/2159-8290.CD-13-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Intlekofer AM, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015;22:304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell metabolism. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terunuma A, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smolkova K, Dvorak A, Zelenka J, Vitek L, Jezek P. Reductive carboxylation and 2-hydroxyglutarate formation by wild-type IDH2 in breast carcinoma cells. Int J Biochem Cell Biol. 2015;65:125–133. doi: 10.1016/j.biocel.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 16.Rohle D, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bleeker FE, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119:487–494. doi: 10.1007/s00401-010-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molenaar RJ, et al. The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014;16:1263–1273. doi: 10.1093/neuonc/nou005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang P, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran AN, et al. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol. 2014;16:414–420. doi: 10.1093/neuonc/not198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cairncross JG, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32:783–790. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson SP, Helleday T. DNA REPAIR. Drugging DNA repair. Science. 2016;352:1178–1179. doi: 10.1126/science.aab0958. [DOI] [PubMed] [Google Scholar]
- 23.Balss J, et al. Enzymatic assay for quantitative analysis of (D)-2-hydroxyglutarate. Acta neuropathologica. 2012;124:883–891. doi: 10.1007/s00401-012-1060-y. [DOI] [PubMed] [Google Scholar]
- 24.Tateishi K, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell. 2015;28:773–784. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 26.Ruis BL, Fattah KR, Hendrickson EA. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol Cell Biol. 2008;28:6182–6195. doi: 10.1128/MCB.00355-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murfuni I, et al. Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet. 2013;9:e1003910. doi: 10.1371/journal.pgen.1003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czochor JR, Sulkowski P, Glazer PM. miR-155 Overexpression Promotes Genomic Instability by Reducing High-fidelity Polymerase Delta Expression and Activating Error-Prone DSB Repair. Molecular Cancer Research. 2016;14:363–373. doi: 10.1158/1541-7786.MCR-15-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fece de la Cruz F, Gapp BV, Nijman SM. Synthetic lethal vulnerabilities of cancer. Annu Rev Pharmacol Toxicol. 2015;55:513–531. doi: 10.1146/annurev-pharmtox-010814-124511. [DOI] [PubMed] [Google Scholar]
- 30.Foote KM, Lau A, Nissink JW. Drugging ATR: progress in the development of specific inhibitors for the treatment of cancer. Future Med Chem. 2015;7:873–891. doi: 10.4155/fmc.15.33. [DOI] [PubMed] [Google Scholar]
- 31.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108:3406–3411. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donawho CK, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 33.Kortmann U, et al. Tumor growth inhibition by olaparib in BRCA2 germline-mutated patient-derived ovarian cancer tissue xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:783–791. doi: 10.1158/1078-0432.CCR-10-1382. [DOI] [PubMed] [Google Scholar]
- 34.Lee JM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. Journal of the National Cancer Institute. 2014;106:dju089. doi: 10.1093/jnci/dju089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oza AM, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. The Lancet Oncology. 2015;16:87–97. doi: 10.1016/S1470-2045(14)71135-0. [DOI] [PubMed] [Google Scholar]
- 36.Di Veroli GY, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32:2866–2868. doi: 10.1093/bioinformatics/btw230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-mediated increases in l-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell metabolism. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baader E, Tschank G, Baringhaus KH, Burghard H, Gunzler V. Inhibition of prolyl 4-hydroxylase by oxalyl amino acid derivatives in vitro, in isolated microsomes and in embryonic chicken tissues. The Biochemical journal. 1994;300(Pt 2):525–530. doi: 10.1042/bj3000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goglia AG, et al. Identification of novel radiosensitizers in a high-throughput, cell-based screen for DSB repair inhibitors. Mol Cancer Ther. 2015;14:326–342. doi: 10.1158/1535-7163.MCT-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang LY, et al. KDM4A Coactivates E2F1 to Regulate the PDK-Dependent Metabolic Switch between Mitochondrial Oxidation and Glycolysis. Cell Rep. 2016;16:3016–3027. doi: 10.1016/j.celrep.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu CH, et al. KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor. J Med Chem. 2014;57:5975–5985. doi: 10.1021/jm500249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mallette FA, et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young LC, McDonald DW, Hendzel MJ. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J Biol Chem. 2013;288:21376–21388. doi: 10.1074/jbc.M113.491514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, et al. Treatment with a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PloS one. 2015;10:e0133813. doi: 10.1371/journal.pone.0133813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alano CC, et al. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ju HQ, et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. 2016;379:1–11. doi: 10.1016/j.canlet.2016.05.024. [DOI] [PubMed] [Google Scholar]
- 48.Bai H, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48:59–66. doi: 10.1038/ng.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu Y, Chu A, Turker MS, Glazer PM. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol Cell Biol. 2011;31:3339–3350. doi: 10.1128/MCB.01121-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bindra RS, Glazer PM. Basal repression of BRCA1 by multiple E2Fs and pocket proteins at adjacent E2F sites. Cancer Biol Ther. 2006;5:1400–1407. doi: 10.4161/cbt.5.10.3454. [DOI] [PubMed] [Google Scholar]
- 51.Scanlon SE, Glazer PM. Hypoxic stress facilitates acute activation and chronic downregulation of fanconi anemia proteins. Mol Cancer Res. 2014;12:1016–1028. doi: 10.1158/1541-7786.MCR-13-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mondesir J, Willekens C, Touat M, de Botton S. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. Journal of blood medicine. 2016;7:171–180. doi: 10.2147/JBM.S70716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Babakoohi S, Lapidus RG, Faramand R, Sausville EA, Emadi A. Comparative Analysis of Methods for Detecting Isocitrate Dehydrogenase 1 and 2 Mutations and Their Metabolic Consequence, 2-Hydroxyglutarate, in Different Neoplasms. Applied immunohistochemistry & molecular morphology : AIMM. 2016 doi: 10.1097/PAI.0000000000000342. [DOI] [PubMed] [Google Scholar]
- 54.Emir UE, et al. Noninvasive Quantification of 2-Hydroxyglutarate in Human Gliomas with IDH1 and IDH2 Mutations. Cancer research. 2016;76:43–49. doi: 10.1158/0008-5472.CAN-15-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anthony AJ, Chalmers J, Swaisland Helen, Stewart William, Halford Sarah ER, Molife L Rhoda, Hargrave Darren R, McCormick Alex. Results of stage 1 of the oparatic trial: A phase I study of olaparib in combination with temozolomide in patients with relapsed glioblastoma. JCO. 2014;32:abstr 2025. [Google Scholar]
- 56.Su JM, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro-oncology. 2014;16:1661–1668. doi: 10.1093/neuonc/nou103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muscal JA, et al. Plasma and cerebrospinal fluid pharmacokinetics of ABT-888 after oral administration in non-human primates. Cancer Chemother Pharmacol. 2010;65:419–425. doi: 10.1007/s00280-009-1044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–1789. doi: 10.1172/JCI76094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27:599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- 60.Gyori BM, Venkatachalam G, Thiagarajan P, Hsu D, Clement MV. OpenComet: an automated tool for comet assay image analysis. Redox biology. 2014;2:457–465. doi: 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chatterjee G, Jimenez-Sainz J, Presti T, Nguyen T, Jensen RB. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic acids research, gkw242. 2016 doi: 10.1093/nar/gkw242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhuang J, et al. Checkpoint Kinase 2–Mediated Phosphorylation of BRCA1 Regulates the Fidelity of Nonhomologous End-Joining. Cancer research. 2006;66:1401–1408. doi: 10.1158/0008-5472.CAN-05-3278. [DOI] [PubMed] [Google Scholar]
- 63.Stachelek GC, et al. YU238259 Is a Novel Inhibitor of Homology-Dependent DNA Repair That Exhibits Synthetic Lethality and Radiosensitization in Repair-Deficient Tumors. Mol Cancer Res. 2015;13:1389–1397. doi: 10.1158/1541-7786.MCR-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bindra RS, Goglia AG, Jasin M, Powell SN. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013;41:e115. doi: 10.1093/nar/gkt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakanishi K, et al. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakai W, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer research. 2009;69:6381–6386. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singleton B, et al. Molecular and biochemical characterization of xrs mutants defective in Ku80. Molecular and cellular biology. 1997;17:1264–1273. doi: 10.1128/mcb.17.3.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan SM, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nature medicine. 2015;21:178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turcan S, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis CA, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Molecular cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Balss J, et al. Enzymatic assay for quantitative analysis of (D)-2-hydroxyglutarate. Acta neuropathologica. 2012;124:883–891. doi: 10.1007/s00401-012-1060-y. [DOI] [PubMed] [Google Scholar]
- 72.Surovtseva YV, et al. Characterization of Cardiac Glycoside Natural Products as Potent Inhibitors of DNA Double-Strand Break Repair by a Whole-Cell Double Immunofluorescence Assay. Journal of the American Chemical Society. 2016;138:3844–3855. doi: 10.1021/jacs.6b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chowdhury GM, Jiang L, Rothman DL, Behar KL. The contribution of ketone bodies to basal and activity-dependent neuronal oxidation in vivo. Journal of Cerebral Blood Flow & Metabolism. 2014;34:1233–1242. doi: 10.1038/jcbfm.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Establishment and characterization of IDH mutant and wild-type cell models.
Fig. S2. Characterization of the DNA DSB repair defect in IDH mutant cells.
Fig. S3. Cell growth and viability assays in response to DNA repair and DNA damage response inhibitors.
Fig. S4. Increased sensitivity of IDH mutant cells to PARP inhibitors alone and in combination with cisplatin.
Fig. S5. Additional characterization of the impact of 2HG on DNA DSB repair.
Fig. S6. Manipulation of IDH and L2HGDH by siRNAs and impact on DNA DSB repair.
Fig. S7. αKG rescues 2HG induced HR defect, and 2HG alters DNA repair through KDM4A/B and not NAD+.
Fig. S8. Additional glioma cell and xenograft data.
Table S1. STR profile of IDH1 WT parental HeLa cells.
Table S2.STR profile of IDH1 R132H/+ HeLa cell sub-clone.
Table S3. List of siRNAs targeting alpha-ketoglutarate dependent dioxygenases and selected DNA repair proteins.
Table S4. Surviving fraction 50% (SF50) values for clonogenic survival assays.