

Abstract
Cortical spreading depression is associated with activation of NMDA receptors, which interact with the postsynaptic density protein 95 (PSD-95) that binds to nitric oxide synthase (nNOS). Here, we tested whether inhibition of the nNOS/PSD-95/NMDA receptor complex formation by anti-ischemic compound, UCCB01-144 (Tat-N-dimer) ameliorates the persistent effects of cortical spreading depression on cortical function. Using in vivo two-photon microscopy in somatosensory cortex in mice, we show that fluorescently labelled Tat-N-dimer readily crosses blood-brain barrier and accumulates in nerve cells during the first hour after i.v. injection. The Tat-N-dimer suppressed stimulation-evoked synaptic activity by 2–20%, while cortical blood flow and cerebral oxygen metabolic (CMRO2) responses were preserved. During cortical spreading depression, the Tat-N-dimer reduced the average amplitude of the negative shift in direct current potential by 33% (4.1 mV). Furthermore, the compound diminished the average depression of spontaneous electrocorticographic activity by 11% during first 40 min of post-cortical spreading depression recovery, but did not mitigate the suppressing effect of cortical spreading depression on cortical blood flow and CMRO2. We suggest that uncoupling of PSD-95 from NMDA receptors reduces overall neuronal excitability and the amplitude of the spreading depolarization wave. These findings may be of interest for understanding the neuroprotective effects of the nNOS/PSD-95 uncoupling in stroke.
Keywords: Cortical spreading depression, spreading depolarization, NMDAR, postsynaptic density protein 95, Tat-N-dimer, UCCB01-144, stroke
Introduction
Cortical spreading depression (CSD) is a propagating depolarization wave that causes dramatic failure of brain ion homeostasis and affects neurons and astrocytes, resulting in massive, but spontaneously reversible, changes in energy consumption and cerebral blood flow (CBF).1,2 Non-pathological forms of CSD are associated with development of migraine aura,2 whereas CSDs in the form of peri-infarct depolarizations contribute to infarct expansion in acutely injured human brain cortex.3,4 Therefore, CSD is a potential target for neuroprotective therapeutics.
Uncoupling of the scaffolding protein postsynaptic density protein 95 (PSD-95) from the NMDA-receptor (NMDAR) by the peptide-based dimeric compound UCCB01-144 (Tat-NPEG4(IETDV)2; hereafter referred to as Tat-N-dimer reduces ischemic stroke infarct volume in mice after middle cerebral artery occlusion (MCAO) by up to 40%.5 The Tat-N-dimer is derived from the intracellular C-terminal domain of the NMDAR GluN2B subunit and blocks the PDZ1 and PDZ2 domains of PSD-95. These domains are responsible for multiple protein–protein interactions including the interaction with the NMDAR and neuronal nitric oxide synthase,5,6 which is linked to excitotoxicity in stroke.7
Given that NMDAR activation is crucial for CSD propagation,8,9 we hypothesised that Tat-N-dimer neuroprotection is mediated by modulation of CSD.
Materials and methods
Preparative surgery
All animal experiment protocols complied with the guidelines of the Danish National Ethics Committee for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes and are in compliance with the ARRIVE guidelines. The timeline of surgery and experiments is presented in Figure 1. Total number of 43 (with excluded animals) 10–12 weeks old, 28–30 g male adult wild-type C57Bl/6j mice (Janvier Labs) were anaesthetised by i.p. injection of xylazine (10 mgċkg−1) and ketamine (60 mgċkg−1), and subsequent supplemental doses of ketamine (30 mgċkg−1ċ20 min−1). Tracheotomy was performed for mechanical ventilation (SAR-830; CWE) and monitoring of exhaled CO2 (MicroCapstar End-tidal CO2 monitor; CWE). The left femoral artery was cannulated for arterial blood sampling and measurement of mean arterial blood pressure (MABP). The left femoral vein was cannulated for intravenous (i.v.) administration of protein compounds or physiological saline (control group) and α-chloralose anaesthesia during the measurements. Body temperature was maintained at 37℃ using a rectal thermal probe feedback-regulated heating pad. The skull was exposed and mounted onto a custom-made steel bar. Two craniotomies were performed, a primary craniotomy overlying the right whisker barrel cortex (4 mm diameter, 3 mm lateral, 0.5 mm caudal to bregma) for measurements and a secondary craniotomy (1 mm diameter, 2 mm posterior to the primary craniotomy) for CSD induction. The dura mater was removed and the brain surface covered with 0.75% agarose (type IIIA; Sigma-Aldrich) and aCSF (in mmol/L: NaCl 120, KCl 2.8, NaHCO3 22, CaCl2 1.45, Na2HPO4 1, MgCl2 0.876, glucose 2.55, HEPES 10, pH = 7.4) at 37℃.
Figure 1.

Surgical and experimental procedures. All numbers represent time in minutes unless otherwise stated. (a) Surgical and experimental timeline and (b) experimental protocol.
CSD: cortical spreading depression; i.o.: infraorbital; KAc: potassium acetate.
Stimulation electrodes and cerebral probes
For stimulation-evoked neuronal activity, bipolar stimulation electrodes connected to an ISO-flex stimulator (A.M.P.I.) were inserted into the left infraorbital (i.o.) nerve and masticatory muscles.10 For measurement of CBF, a laser-Doppler probe (Perimed) was placed ∼0.3 mm above the cortex and the initial laser-Doppler flow (LDF) response to 5 Hz i.o. stimulation was used to assess neurovascular coupling. Extracellular local field potentials (LFPs) and direct current (DC) were recorded using a single-barrelled borosilicate microelectrode (2.8–3.0 mΩ) containing an Ag/AgCl electrode in aCSF. Tissue partial pressure of oxygen (tpO2) was measured using a calibrated Clark-type oxygen electrode (OX10; Unisense). Both electrodes were inserted in proximity to each other in the somatosensory area ∼150 µm below the brain surface (cortical layer II/III). A reference electrode was inserted into the neck musculature.10 Preserved neurovascular and metabolic coupling under control conditions were taken as indicators of preserved functional integrity of the brain cortex.
Synthesis of compounds
Tat-N-dimer (Tat-NPEG4(IETDV)2), Tat-N-dimer-AA (Tat-NPEG4(IEADA)2), and carboxytetramethylrhodamine (TAMRA)-conjugated Tat-N-dimer (TAMRA-Tat-N-dimer) were synthesised and prepared as HCl salts as previously described.5
Experimental design and post-surgical anaesthesia
The experiments and data analysis for Tat-N-dimer and saline were performed in randomised order (four animal groups, two for saline, and two for Tat-N-dimer to avoid group bias) by investigators blinded to experimental conditions. The Tat-N-dimer-AA negative control was assessed in subsequent experiments. Animals received a single i.v. bolus injection of either physiological saline (Amgros I/S) as control, Tat-N-dimer-AA as a negative control or Tat-N-dimer (both 3 nmolċg animal weight−1, in saline). Following probe insertion, an arterial blood sample was analyzed (ABL700; Radiometer Copenhagen) to ensure blood gases and pH within physiological ranges. Subsequently, anaesthesia was switched to α-chloralose (HBC-complex diluted in saline; Sigma-Aldrich) by i.v. bolus injection (75 mgċkg−1) followed by i.v. continuous infusion (50 mgċkg−1ċh−1). Experimental procedures were performed 30 min after administration of α-chloralose (Figure 1(a)).
Blood-brain barrier (BBB) penetration
To characterise the kinetics of Tat-N-dimer penetration of the BBB and determine the optimal time for commencing experimental procedures after the i.v. administration of the compound, we employed in vivo two-photon microscopy. Imaging was performed with an SP5 upright laser scanning microscope with a 20 × 1.0 N.A. water-immersion objective (Leica Microsystems). Carboxytetramethylrhodamine-conjugated Tat-N-dimer (TAMRA-Tat-N-dimer) fluorescence was elicited with a MaiTai HP Ti:Sapphire two-photon laser (Millennia Pro, Spectra-Physics) at 890 nm and the signal collected by multi-alkali NDD after 560–625 nm band pass filter. Images were exported to ImageJ (v. 1.48u4; NIH) and further analyzed in Prism 6.0 (Graphpad). Fluorescence signals were normalised to 100% (the average of initial fluorescence intensity from region of interest at selected blood vessel).
Pre- and post-CSD neurovascular and neurometabolic coupling: Excitatory postsynaptic potential (EPSP), CBF, and cerebral metabolic rate of oxygen (CMRO2)
Physiological (before CSD) and CSD-suppressed (45 min after CSD onset) neurovascular and neurometabolic responses were evaluated by simultaneous measurement of stimulation-evoked responses of LFP (eLFP), LDF, and tpO2. I.o. stimulation was conducted in three stimulation trains (15 s, 1.5 mA, at 0.5, 1, 2, 3, and 5 Hz) with 2-min intervals between consecutive stimulations (Figure 1(b)).
eLFPs were averaged for individual stimulation frequencies and analyzed using MatLab (MathWorks) to extract the maximum negative amplitude (difference between baseline and first negative peak) reflecting the EPSP.
Cerebral blood flow (CBF) was calculated from LDF measurements normalised to resting baseline (120 s average of the LDF signal 20 s before experimental onset), which were multiplied by 53 mLċ100 g−1ċmin−1.11
Brain tpO2 was converted to mmHg and used with CBF to calculate the CMRO2 as described previously.12 Briefly, CMRO2 is expressed as:
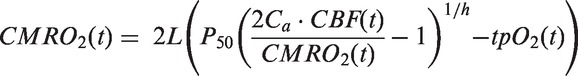
where L is the calculated oxygen diffusibility equalling 4.67 µmolċ100 g−1ċmmHg−1ċmin−1; P50 is the oxygen tension at 50% haemoglobin oxygen saturation equalling 41 mmHg; Ca is the arterial oxygen concentration equalling 8 µmolċml−1; CBF(t) is the CBF at time (t); h is the Hill coefficient of the oxygen-haemoglobin dissociation curve equalling 2.7; tpO2(t) is the tpO2 at time (t).12
CSD: DC shift, CBF, CMRO2, and electrocorticographic (ECoG) recovery
CSD was induced by microinjection of 1 mol/L potassium acetate (KAc) (0.1 s; 10–30 psi; PV830; World Precision Instruments) 150 µm below the brain surface into the posterior parietal cortex of the secondary craniotomy using heat-pulled borosilicate micropipette tips (1 mΩ). Upon induction, the tip was retracted and post-CSD neurovascular and neurometabolic coupling was assessed 45 min after CSD onset (Figure 1(b)).
DC, CBF, and CMRO2 were exported in a time period of 5 min before and 40 min after DC shift. CBF and CMRO2 were further normalised to baseline (average of 1 min before DC shift). AUC and maximum amplitude were calculated using Prism v.6.0 (Graphpad).
Spontaneous brain activity (ECoG) was calculated as the average root mean square (RMS) of the LFP signal from a 5 min period before CSD and eight successive periods of 5 min after DC shift using Spike2 software (CED).
Experimental termination
Following experiments, a second arterial blood sample was collected to analyze the physiological end-state of the animal. Subsequently, the animal was euthanised by i.v. injection of pentobarbital (100 mgċkg−1; Glostrup Hospital).
Data acquisition and signal filtering
All measurements were digitalised (Power 1401; CED) and recorded using Spike2 software (CED). For exhaled %CO2, MABP, LDF, and tpO2, the raw signal was recorded. The pre-amplified (×10) raw total brain electrical signal was filtered into LFP (0.1–1000 Hz) and DC (0–0.1 Hz) bands (CyberAmp 380; Axon Instruments).
Systemic parameters
MABP was calculated as the 2-min average of the signal immediately before experimental onset and euthanasia. Blood pO2, pCO2, and pH were obtained from blood sampling before and after experiments (Figure 1). Brain tpO2 was collected at the resting baseline period 2 min before experimental onset.
Inclusion criteria
The following criteria were used to ensure proper measurements of brain activity: (a) MABP stable and above 70 mmHg; (b) blood gas values within physiological range (pO2 ∼ 120–140 mmHg, pCO2 ∼ 40 mmHg); (c) a functional cortical hyperemic response (≥10% LDF increase upon 5 Hz i.o. stimulation prior to α-chloralose injection).
Statistical analysis
Statistical analyses were performed in Prism 6.0 (Graphpad). Pearson's normality test was used and group responses (paired: before/after CSD, or unpaired: control/Tat-N-dimer/Tat-N-dimer-AA) were compared by either t-test or two-way ANOVA with Sidak correction for multiple comparisons. For all tests, α = 0.05 and p-values are indicated as not significant (ns) for p ≥ α, or asterisks (*, t-test) and hash symbols (#, two-way ANOVA), where * denote p < α; ** and ##p < 0.01; *** and ###p < 0.001; ####p < 0.0001.
Results
Tat-N-dimer penetrates the BBB
BBB penetration is essential for the action of Tat-N-dimer within the brain and has previously been demonstrated in situ to occur within 1 h.5 I.v.-injected fluorophore (TAMRA) -conjugated Tat-N-dimer readily crossed the BBB and approached saturation within 1 h after i.v. injection, thus validating the presence of the compound in the brain tissue at the time of experiments. Following injection, we observed a decrease of compound fluorescence in blood vessels over time accompanied by a gradual increase of fluorescence in the brain tissue with clear indication of drug accumulation in cell bodies (Figure 2, Supplementary Movie I).
Figure 2.

In vivo blood-brain barrier penetration of TAMRA-Tat-N-dimer. (a) Two-photon imaging of whisker barrel cortex in living mice showing accumulation of i.v.-delivered carboxytetramethylrhodamine (TAMRA)-labelled Tat-N-dimer in the brain tissue, as determined by the increase of compound-derived fluorescence over time (left panel = 1; right panel = 54 min post-i.v. delivery). Images are represented in the rainbow 8-bit colour scale (arbitrary fluorescence units); scale bar = 50 µm. Square ROIs represent areas sampled for fluorescence intensity measurement in (b). (b) Average fluorescence in ROI#1 (blood vessel) and ROI#2 (brain tissue) over time.
Tat-N-dimer does not affect systemic parameters
We tested whether Tat-N-dimer induces systemic effects by comparing systemic parameters between control and Tat-N-dimer-treated groups. There was no significant difference between control (saline) and Tat-N-dimer groups in MABP, pO2, pCO2, tpO2, nor blood pH (nControl = 12, nTat-N-dimer = 12) (Supplementary Figure I).
Tat-N-dimer ameliorates CSD and preserves spontaneous brain activity
We examined the effect of the Tat-N-dimer on the DC shift during CSD (Figure 3(a)), which occurred in both groups with the same frequency in response to KAc injection (12/18 saline control, 12/17 Tat-N-dimer), suggesting that Tat-N-dimer did not cause major changes in the threshold for CSD induction. However, the average amplitude of the DC shift in the Tat-N-dimer group (−8.384 ± 0.70 mV, n = 12) was significantly lower compared with the saline-injected animals (−12.44 ±0.65 mV, n = 12, p = 0.0004, Figure 3(b)). Furthermore, the AUC of the DC potential shift was significantly reduced in Tat-N-dimer-treated animals (638.7 ±84.04 mVċs, n = 12) compared with saline control (969.6 ± 66.72 mVċs, n = 12, p = 0.0054) (Figure 3(c)).
Figure 3.

Effect of Tat-N-dimer on CSD DC shift and ECoG recovery. For all graphs, values are indicated for control (saline, blue), Tat-N-dimer-AA (negative control compound, orange), and Tat-N-dimer (red) groups. (a) Average of DC shift traces. (b,c) Bar graphs of means ± SEM of (b) DC shift amplitudes and (c) DC shift AUCs. (d) Change in spontaneous brain activity (ECoG) during CSD plotted as group means ± SEM. * denotes p < 0.05; **p < 0.01; ***p < 0.001; ###p < 0.001; ns = not significant.
To ascertain that the observed effect of Tat-N-dimer could be attributed specifically to the PSD-95-binding elements of the compound, we synthesised and tested negative control compound, the Tat-N-dimer-AA. The Tat-N-dimer-AA is structurally identical to the Tat-N-dimer except that the two C-terminal amino acids, Val and Thr have both been replaced with Ala. The resulting compound (Tat-NPEG4(IEADA)2; referred to as Tat-N-dimer-AA) does not bind PSD-95,13–15 but possesses the dimeric structure similar to Tat-N-dimer including the cell-penetrating Tat moiety. CSD was induced in Tat-N-dimer-AA-treated animals with similar frequency (7/9) to Tat-N-dimer and saline controls. There was no difference in the average DC shift between saline and Tat-N-dimer-AA-treated animals (−11.79 ± 1.41 mV, n = 7, p = 0.6381) and Tat-N-dimer displayed lower average DC shift amplitude compared with Tat-N-dimer-AA negative control (p = 0.0273, Figure 3(b)). Furthermore, there was no difference in the average DC shift AUC between saline and Tat-N-dimer-AA group (992.9 ±98.23 mVċs, n = 7, p = 0.8415) and the average DC shift AUC in Tat-N-dimer-treated animals was lower compared with Tat-N-dimer-AA negative control (p = 0.0166, Figure 3(c)).
Next, we examined the effect of Tat-N-dimer on the ECoG activity during the first 40 min of recovery following CSD (Figure 3(d)). The animals that exhibited spontaneous CSD during recovery period (saline n = 1, Tat-N-dimer n = 1) or distorted LFP signal due to measurement artefacts (Tat-N-dimer n = 2) were excluded. There was no significant difference in basal ECoG activity (pre-CSD) between saline control (n = 11) or Tat-N-dimer-AA (n = 7) and Tat-N-dimer (n = 9, p = 0.0638, p = 0.5215, respectively). In all experimental groups, CSD caused a rapid decrease in average ECoG amplitude compared with pre-CSD. Notably, following DC shift, Tat-N-dimer-treated animals exhibited more preserved ECoG activity (0.6722 ±0.04251) compared with saline (0.5241 ± 0.04962, p = 0.0361) with no difference between Tat-N-dimer-AA (0.5255 ± 0.07019, p = 0.9878) and saline control. Importantly, the overall ECoG activity measured over time in the post-CSD recovery period was less suppressed in the Tat-N-dimer group compared with saline control and Tat-N-dimer-AA (p = 0.0004, p < 0.0001, respectively, two-way ANOVA), with no difference in post-CSD ECoG activity measured over time between Tat-N-dimer and Tat-N-dimer-AA control (p = 0.4069, two-way ANOVA).
The observed effect of the Tat-N-dimer on ECoG was not reflected in the increase in CBF or CMRO2 that occurs during the DC shift, nor the prolonged reduction of CBF, which follows the onset of CSD (Table 1). Two animals from saline control and one from Tat-N-dimer group were excluded due to unstable CBF or tpO2 signal during CSD.
Table 1.
Tat-N-dimer does not affect CBF and CMRO2 changes during CSD.
| n | Hyperemia |
Oligemia |
CMRO2 |
||||
|---|---|---|---|---|---|---|---|
| Amplitude (% increase above baseline) | AUC (% change) | Amplitude (% decrease below baseline) | AUC (% change) | Amplitude (% increase above baseline) | AUC (% change) | ||
| Control saline | 9 | 26.14 ± 3.75 | 2688.83 ± 728.68 | −35.69 ± 4.08 | 43261.75 ± 7619.06 | 98.97 ± 14.79 | 18643.9 ± 4907.55 |
| Tat-N-dimer | 8 | 39.43 ± 11.12 | 3556.40 ± 1122.05 | −33.77 ± 6.24 | 44287.40 ± 11318.58 | 68.80 ± 5.59 | 14800.88 ± 2176.64 |
| Unpaired t-test | p = 0.3097, ns | p = 0.5371, ns | p = 0.8036, ns | p = 0.9420, ns | p = 0.0846, ns | p = 0.4891, ns | |
Values are average ± SEM.
AUC: area under curve; ns: not significant.
Tat-N-dimer reduces EPSP amplitude
Next, we examined the effects on stimulation-evoked synaptic, vascular and oxygen responses in the whisker barrel cortex before CSD and 45 min after CSD. EPSPs arise as a consequence of synaptic activity and reflect neuronal excitation. Tat-N-dimer group (n = 9) exhibited mild decrease of EPSP amplitude both before and after CSD (pbefore = 0.0019, pafter = 0.0002, two-way ANOVA) compared with saline control (n = 11) (Figure 4) indicating an overall suppression of neuronal activity by the compound. A similar effect was found for Tat-N-dimer-AA mutant peptide (n = 6), which reduced EPSP amplitude before CSD, however, had no significant effect on EPSP following CSD (pbefore < 0.0001, pafter = 0.2361, two-way ANOVA). One animal from Tat-N-dimer-AA group was excluded due to unstable LFP signal.
Figure 4.
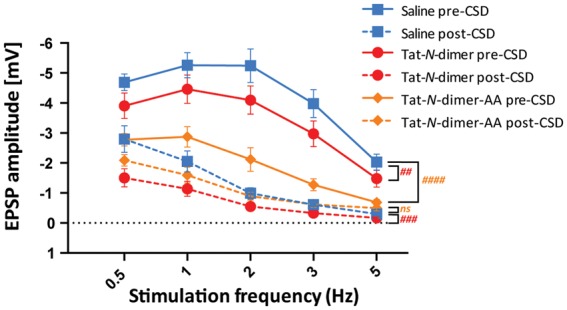
Effect of Tat-N-dimer on EPSP amplitude during stimulation-evoked responses. Group response mean amplitudes and AUC ± SEM are shown for control (saline, blue), Tat-N-dimer-AA (negative control compound, orange), and Tat-N-dimer (red) groups before (solid) and after (dashed) CSD. Significance levels were marked for Tat-N-dimer vs. saline in red and Tat-N-dimer-AA vs. saline in orange, ##p < 0.01, ###p < 0.001, ####p < 0.0001, ns = not significant.
Tat-N-dimer does not affect cerebral blood flow and CMRO2
The intimate functional coupling between neuronal activity-dependent oxygen consumption and oxygen supply by increased CBF is essential for normal brain function and thus of significant clinical interest.16 Therefore, we evaluated the effects of Tat-N-dimer on haemodynamic and metabolic responses to stimulation. However, the presence of the compound had no significant effect on CBF or CMRO2 responses to somatosensory stimulation neither before nor after CSD (Supplementary Figure II).
Discussion
To assess the potential effects of the PSD-95–NMDAR interaction inhibitor, Tat-N-dimer, on synaptic function as well as vascular and metabolic responses, we performed in vivo measurements in the mouse brain whisker barrel cortex during CSD and somatosensory stimulation. Using two-photon microscopy, we showed that fluorescently labelled Tat-N-dimer penetrates the BBB and is present in the brain without affecting systemic parameters. Tat-N-dimer significantly decreased the DC potential amplitude and preserved residual ECoG activity during CSD, while CSD-associated changes in CBF and CMRO2 remained unaffected. In line with these results, we further demonstrated that the compound significantly suppressed both pre- and post-CSD stimulation-evoked EPSPs yet preserved the activity-dependent CBF and CMRO2 responses.
CSD is assumed to contribute to infarct expansion in ischemic stroke, traumatic brain injury, and subarachnoid haemorrhage and therefore offers an attractive therapeutic target.3,4,17,18 While uncoupling of PSD-95 by Tat-N-dimer has previously been demonstrated to reduce ischemic infarct volume by up to 40% after MCAO in mice,5 its neuroprotective mechanism is largely unexplored. Here, we report that the Tat-N-dimer ameliorates CSD, and hypothesise that the observed preservation of ECoG activity following CSD is linked with the previously reported Tat-N-dimer neuroprotection.4
PSD-95 interacts with multiple proteins at the postsynaptic membrane.6 The observed effects of uncoupling the interactions of the PSD-95 PDZ1 and PDZ2 domains by the Tat-N-dimer may therefore arise either directly (i.e. loss of protein interaction) or indirectly (i.e. loss of downstream modulation). A decrease in the DC shift may arise as a consequence of increased membrane resistance due to either direct or indirect effects of PSD-95 uncoupling, for example by destabilisation of transmembrane ion channels or by modulation of ion channel activity. As the massive tissue depolarization during the DC shift affects transmembrane ion channels globally,19 further studies are needed to clarify the molecular basis of our observations.
The mechanism by which PSD-95 uncoupling causes suppression of neuronal excitation after CSD compared with saline and Tat-N-dimer-AA remains unknown. AMPA receptor (AMPAR) conductivity is the primary determinant of EPSP amplitude20,21 and AMPAR recruitment and stabilisation at postsynaptic membranes depends upon interaction with the PDZ1 and PDZ2 domains of PSD-95 via the adaptor protein Stargazin.22–24 Furthermore, postsynaptic AMPAR activity is efficiently inhibited by a biomimetic ligand derived from Stargazin, which binds to and blocks PSD-95 PDZ1 and PDZ2,25 reminiscent of the mechanism originally demonstrated for the Tat-N-dimer.5 A decrease in the number of AMPARs at the postsynaptic membrane caused by Tat-N-dimer uncoupling of AMPAR-Stargazin complexes and PSD-95 could therefore account for the post-CSD observed effects.
The Tat-N-dimer reduction of DC shift and EPSP amplitudes were associated with preserved CBF and CMRO2 responses to CSD and somatosensory stimulation. This finding may suggest that the mechanisms affected by the Tat-N-dimer do not influence brain vascular and metabolic control. It is possible that the relatively modest decrease in EPSP amplitude observed here (2–20%) was insufficient to cause detectable differences in CBF and CMRO2 responses between treated and non-treated animals. Alternatively, the compound may uncouple CBF and CMRO2 from neuronal activity by an as yet unknown mechanism. Our findings are, however, encouraging in a clinical context, as adequate delivery of glucose and oxygen to depolarised tissue is essential for normal brain function and for counteracting metabolic dishomeostasis during CSD.
In summary, this study assessed the influence of PSD-95–NMDAR interaction inhibition on CSD. We demonstrated that the Tat-N-dimer ameliorated the DC shift evoked by CSD and mitigated the reduction in ECoG activity following CSD, without affecting the associated CBF and CMRO2 responses. Importantly, the Tat-N-dimer did not affect any systemic parameters and CBF, neither before nor after CSD.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Novo Nordisk & Novozymes, Lundbeck Foundation, Brødrene Hartmanns Fond, NORDEA Foundation Grant to the Center for Healthy Aging at the University of Copenhagen, Danish Medical Research Council, and Foundation Leducq.
Supplementary Material
Acknowledgements
We would like to acknowledge Claus Mathiesen and Micael Lønstrup for their excellent assistance with electrophysiology and animal surgery, respectively.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: AB and KS are founders and shareholders of Avilex Pharma. ISR, KK, and ML declare no conflict of interest.
Authors' contributions
KK, ML, and ISR designed the study; ISR and KK performed the experiments and data analysis; ISR, KK, ML, AB, and KS conceived the manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Lauritzen M, Dreier JP, Fabricius M, et al. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 2011; 31: 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron 2015; 86: 902–922. [DOI] [PubMed] [Google Scholar]
- 3.Busch E, Gyngell ML, Eis M, et al. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab 1996; 16: 1090–1099. [DOI] [PubMed] [Google Scholar]
- 4.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011; 17: 439–447. [DOI] [PubMed] [Google Scholar]
- 5.Bach A, Clausen BH, Moller M, et al. A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1-2 and protects against ischemic brain damage. Proc Natl Acad Sci USA 2012; 109: 3317–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Husi H, Ward MA, Choudhary JS, et al. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci 2000; 3: 661–669. [DOI] [PubMed] [Google Scholar]
- 7.Sattler R, Xiong Z, Lu WY, et al. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999; 284: 1845–1848. [DOI] [PubMed] [Google Scholar]
- 8.Marrannes R, Willems R, De Prins E, et al. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res 1988; 457: 226–240. [DOI] [PubMed] [Google Scholar]
- 9.Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab 1992; 12: 223–229. [DOI] [PubMed] [Google Scholar]
- 10.Jessen SB, Brazhe A, Lind BL, et al. GABAA Receptor-Mediated Bidirectional Control of Synaptic Activity, Intracellular Ca2+, Cerebral Blood Flow, and Oxygen Consumption in Mouse Somatosensory Cortex In Vivo. Cerebral cortex 2014; 25: 2594–2609. [DOI] [PubMed] [Google Scholar]
- 11.Zhu XH, Zhang Y, Tian RX, et al. Development of (17)O NMR approach for fast imaging of cerebral metabolic rate of oxygen in rat brain at high field. Proc Natl Acad Sci USA 2002; 99: 13194–13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gjedde A, Johannsen P, Cold GE, et al. Cerebral metabolic response to low blood flow: possible role of cytochrome oxidase inhibition. J Cereb Blood Flow Metab 2005; 25: 1183–1196. [DOI] [PubMed] [Google Scholar]
- 13.Kornau HC, Schenker LT, Kennedy MB, et al. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 1995; 269: 1737–1740. [DOI] [PubMed] [Google Scholar]
- 14.Aarts M, Liu Y, Liu L, et al. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 2002; 298: 846–850. [DOI] [PubMed] [Google Scholar]
- 15.Bach A, Chi CN, Olsen TB, et al. Modified peptides as potent inhibitors of the postsynaptic density-95/N-methyl-D-aspartate receptor interaction. J Med Chem 2008; 51: 6450–6459. [DOI] [PubMed] [Google Scholar]
- 16.Attwell D, Buchan AM, Charpak S, et al. Glial and neuronal control of brain blood flow. Nature 2010; 468: 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Back T, Ginsberg MD, Dietrich WD, et al. Induction of spreading depression in the ischemic hemisphere following experimental middle cerebral artery occlusion: effect on infarct morphology. J Cereb Blood Flow Metab 1996; 16: 202–213. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura H, Strong AJ, Dohmen C, et al. Spreading depolarizations cycle around and enlarge focal ischaemic brain lesions. Brain 2010; 133(Pt 7): 1994–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 2001; 81: 1065–1096. [DOI] [PubMed] [Google Scholar]
- 20.Ouardouz M, Durand J. Involvement of AMPA receptors in trigeminal postsynaptic potentials recorded in rat abducens motoneurons in vivo. Eur J Neurosci 1994; 6: 1662–1668. [DOI] [PubMed] [Google Scholar]
- 21.Ballerini L, Bracci E, Nistri A. Desensitization of AMPA receptors limits the amplitude of EPSPs and the excitability of motoneurons of the rat isolated spinal cord. Eur J Neurosci 1995; 7: 1229–1234. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Chetkovich DM, Petralia RS, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000; 408: 936–943. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi Y, Shi SH, Esteban JA, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 2000; 287: 2262–2267. [DOI] [PubMed] [Google Scholar]
- 24.Schnell E, Sizemore M, Karimzadegan S, et al. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA 2002; 99: 13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sainlos M, Tigaret C, Poujol C, et al. Biomimetic divalent ligands for the acute disruption of synaptic AMPAR stabilization. Nat Chem Biol 2011; 7: 81–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.