

Abstract
Spreading depolarization (SD) generates significant alterations in cerebral haemodynamics, which can have detrimental consequences on brain function and integrity. Ketamine has shown an important capacity to modulate SD; however, its impact on SD haemodynamic response is incompletely understood. We investigated the effect of two therapeutic ketamine dosages, a low-dose of 2 mg/kg/h and a high-dose of 4 mg/kg/h, on the haemodynamic response to SD in the gyrencephalic swine brain. Cerebral blood volume, pial arterial diameter and cerebral blood flow were assessed through intrinsic optical signal imaging and laser-Doppler flowmetry. Our findings indicate that frequent SDs caused a persistent increase in the baseline pial arterial diameter, which can lead to a diminished capacity to further dilate. Ketamine infused at a low-dose reduced the hyperemic/vasodilative response to SD; however, it did not alter the subsequent oligemic/vasoconstrictive response. This low-dose did not prevent the baseline diameter increase and the diminished dilative capacity. Only infusion of ketamine at a high-dose suppressed SD and the coupled haemodynamic response. Therefore, the haemodynamic response to SD can be modulated by continuous infusion of ketamine. However, its use in pathological models needs to be explored to corroborate its possible clinical benefit.
Keywords: Arterial reactivity, cerebral blood flow, cerebral blood volume, cerebral haemodynamics, gyrencephalic brain, haemodynamic response, intrinsic optical signal imaging, ketamine, laser-Doppler flowmetry, spreading depolarization
Introduction
Spreading depolarization (SD) corresponds to a massive depolarization wave of neuronal and glial cells that slowly propagates across the cerebral cortex. It is characterized by a near-complete breakdown of ion gradients, neurotransmitter release, increased energy metabolism, water shifts and depression of electrical activity.1–3 Findings in recent years indicate that SDs occurring in the human brain play a role in the pathophysiological basis of cerebrovascular diseases.1,4 In these scenarios, the occurrence of spontaneous SDs developing from the border zone of brain lesions has been associated with neuronal degeneration, infarct size growth and poor clinical outcome. Large alterations in cerebral blood flow (CBF), arterial reactivity and cerebral blood volume (CBV) accompanying the electrocortical activity of SD have been documented;5–11 under pathological conditions, these alterations can have detrimental consequences. In this regard, SDs in tissue at risk can generate an inverse haemodynamic response (spreading ischemia) capable of a severe vasoconstriction, leading to hypoperfusion, oxidative stress, hypoxia and neuronal death.1–4 Still, the mechanisms involved in these cerebrovascular changes during SD remain unclear, and it seems that the vascular responses to SD depend on multiple dilator and constrictor factors by a variety of cells within the cerebrovascular unit.3,12
Theoretically, therapies targeting the noxious haemodynamic changes of SD in brain injury states could contribute to diminishing its pathological effects. In consequence, the therapeutic modulation of harmful vascular responses associated with SDs may have important clinical implications for outcome improvement. In animal models, inhibition of SD initiation and/or propagation13–15 and SD modulation in patients16,17 has been obtained with ketamine, a non-competitive N-methyl-D-aspartate (NMDA) receptor blocker. The NMDA receptor plays a critical role in excitatory synaptic transmission,18,19 which turns it into a pharmacological target for SD modulation. However, despite the effect of ketamine reducing SD occurrence in the acute injured brain of patients, its impact on improving the clinical outcome is unknown. Hence, a better understanding of ketamine’s impact on the haemodynamic response to SD, is of particular interest. In this study, we evaluated the effect of two human equivalent doses of ketamine on the haemodynamic response to SDs assessed through CBV, pial arterial diameter and CBF changes, using intrinsic optical signal (IOS) imaging and Laser-Doppler flowmetry (LDF) in the gyrencephalic swine brain.
Materials and methods
The protocol for the experiments was approved by the Institutional Animal Care and Use Committee in Karlsruhe, Baden-Württemberg, Germany. Experiments were conducted in the Interfaculty Biomedical Research Facility (IBF 347) in accordance with the University of Heidelberg Animal Ethics Policy. The ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines were considered for the study report.
Surgical preparation
Ten male German Landrace swine (28–32 kg), 3 to 4 months of age, were pre-anaesthetized with intramuscular midazolam (8 mg/kg) and azaperone (60 mg/kg). Additionally, 20–30 mg i.v. propofol was administered through a venous line placed in an ear vein. Animals were orally intubated and mechanically ventilated (fraction of inspired oxygen, FiO2 = 0.3). Anaesthesia was maintained with isoflurane (0.8–1%), midazolam perfusion (60 mg in 50 ml ringer solution at a rate of 12–18 ml/h) and whenever an awaking reaction was observed in the animal, a 20–30 mg i.v. bolus of propofol was administered. All animals were deeply sedated throughout the procedure to minimize animal movement, discomfort and pain. Capillary oxygen saturation (SO2) was monitored from the ear and after surgical exposure of the right femoral artery, a 4-French probe was set in place, for permanent monitoring of mean arterial blood pressure (Raumedic AG, Helmbrechts, Germany). Blood-gas analysis (RAPIDpoint 500 Systems, Siemens, Erlangen, Germany) was performed every 3–4 h. Heart rate was kept between 50 and 120 bpm. Mean arterial pressure was maintained at 60–110 mmHg, the core temperature at between 36.5 and 37℃, SO2 > 90%, pCO2 35–45 mmHg, pO2 > 80 mmHg and glucose > 80 mg/dl. All relevant physiological parameters were continuously monitored throughout the experiment. At the conclusion of the experiment, the animals were euthanized with an overdose of KCl under anaesthesia.
Preconditioning and SD induction
Animal heads were firmly held in a custom-made stereotactic frame and an extensive craniotomy and dura mater excision was performed to expose the hemispheres. Following surgical preparation, swine brains were preconditioned for 40 min with an elevated K+ (11 mmol/l) concentration solution, which consisted of adding KCl (7 mmol/l) in a standard Ringer solution (concentration in mmol/l: K 4.0, Na 147, Ca 2.2, Cl 2.2) (B. Braun Medical AG, Melsungen, Germany). This preconditioning solution was warmed to 37℃ before being applied into a well over the exposed cortex. After preconditioning, the exposed brain was protected by a 1–1.5 cm deep paraffin pool, in order to cover the entire visible brain cortex and improve the quality of image acquisition. Paraffin was warmed to 37℃ before being applied and was monitored every hour to ensure that it maintained a temperature of 36.5–37℃. When necessary, paraffin was withdrawn and new paraffin was added. SDs were induced with a small drop (∼2 μl) of KCl (1 mol/l), which was placed directly onto the cortical surface using a modified needle from an insulin syringe. Hemispheres were stimulated every 30 min, with 4 stimulations before medication, 4 stimulations during ketamine infusion and 4 stimulations after, resulting in 12 stimulations in total. In each experiment, stimulations were performed in the same cortex areas, taking into consideration anatomical structures as landmarks.
Ketamine infusion
Animals were divided into two groups, with five animals each according to the ketamine (Ketanest S® 25 mg/ml) dose. A low-dose of ketamine (2 mg/kg/h) was infused for 2 h from the fifth to ninth stimulation. A higher ketamine dose (4 mg/kg/h) was infused for 2 h from the fifth to ninth stimulation in the other five.
Intrinsic optical signal imaging
The swine cortex was illuminated with a white broad-spectrum LED (400–700 nm). The IOS imaging acquisition system consisted of a CCD camera (Smartec GC1621M, 8 bit greyscale, 1628 × 1236 pixels, MaxxVision GmbH, Stuttgart, Germany) mounted about 25 cm above the exposed cortex with a 25 mm lens (Fujinon HR25HA-1B, 2/3″, 25 mm, 1:1.4, MaxxVision GmbH, Stuttgart, Germany). Calibration of the distances and dimensions in the gyrencephalic brain was performed by placing a metric scale as reference at the beginning of each experiment. This allowed the number of pixels of a known distance to be measured in the cortex and the conversion of pixels into metric units. Only a two-dimensional measurement was performed; therefore, slight variations are expected due to the non-parallel cortex in relation to the image sensor and the distance between the camera and the gyrencephalic cortex. An optical bandpass filter (564 nm, 14 nm FWHM, Schott, Germany) near to the green spectrum was mounted in front of the lens (Figure 1(a)). The wavelength was chosen to be close to that of the isosbestic point of oxygenated and deoxygenated haemoglobin ∼569 nm. Thus, the proportion of back-scattered light corresponds mainly to the total tissue haemoglobin concentration. Image acquisition at one 2-Megapixel 8-bit greyscale frame was acquired every 1–5 s with a computer that provided a live view of the exposed cortex and also showed the amplified image intensity differences from a manually chosen reference image. The images within a predefined time-range could continuously be played back and forth to review the intensity changes caused by the SDs. Thus, we were able to directly observe SDs during the experiments. Images were also saved to disk for later offline post-processing and analysis. Self-developed software based on the open source scientific image processing framework ImageJ (see http://rsbweb.nih.gov/ij/index.html) was used for offline analysis. In the post-processing, images were elastically registered to a representative reference image, which was manually chosen to reduce movement artefacts of the cortex related to breathing, heartbeat and slow brain shifts caused by the craniotomy. This ensured that a specific location in the images always corresponded as precisely as possible to the same location on the cortex during the entire experiment, as previously described.20
Figure 1.

(a) Illustration of the experimental setup. (b) Anatomical surface image of a representative gyrus of the swine brain. The section shows the placing of 2 square-ROIs (ROI-1 and ROI-2) and a line-ROI perpendicular to the arterial axis. (c) Schematic shapes of a variety of haemodynamic responses observed in IOS after KCl-stimulation. (d) Representative example of parenchymal CBV and arterial diameter responses to SD. An example of intensity IOS changes from square-ROIs (ROI-1 and ROI-2) and x-t plots of line-ROIs of arterial response before, during and after 4 mg/kg/h ketamine infusion are shown. A biphasic response of hyperemia/dilation and oligemia/vasoconstriction can be observed. Note that during ketamine infusion, the haemodynamic changes are absent.
Laser-Doppler flowmetry
CBF was continuously recorded using LDF (Periflux System 5000, Perimed, Jarfälla, Sweden) to evaluate the impact of high-dose ketamine (4 mg/kg/h) on CBF responses to SDs. The probe (0.5 mm in diameter) was placed in a peripheral region devoid of large vessels and implanted at a depth of ∼1–3 mm in the opposite hemisphere to that used for the pial arterial analysis. This was performed to avoid any risk of brain damage or bleeding that would prevent an adequate IOS recording for arterial diameter analysis. The signal obtained from the LDF was exported from the Perimed Software to LabChart v.7 (AD Instruments, New South Wales, Australia) for later offline analysis.
Data analysis
Pial arterial diameter analysis
Pial arteries were identified in vivo by their characteristics: smaller diameter, fewer branching points at less acute angles and brighter red colour than veins. They also presented a marked dilation and constriction during SDs. After imaging, post-processing, pial arteries were chosen from the IOS imaging field for the offline analysis according to the following criteria: the diameter should have a width of about 8–20 pixels (0.2–0.5 mm) to allow for sufficiently high resolution when examining the pial arterial diameter changes, should be within a distance of about 10–20 mm from the stimulation point (to avoid the area directly affected by the KCl-stimulation), should be in an area with sufficient brightness and without specular reflections, and should be located perpendicular (90° ± 15°) to the direction of SD propagation. Two pial arteries were selected from each experiment and ImageJ was used for all image analyses. A line-ROI of about 40 pixels (1 mm) was placed perpendicular to the long axis of the pial artery, able to reveal the diameter of the vessel, prior, during and post-SD occurrence, when plotted over the time course of the experiment; it also enabled the timing of the dilation relative to the SD wave to be determined with our software. The peak changes of the pial arterial diameter prior, during and post-SD occurrence were analyzed and compared with the various experimental phases: before, during and after ketamine infusion.
Regions of interest (ROIs) analysis
Two square-ROIs with a size of about 10 × 10 pixels (0.06 mm2) were placed offline adjacent to the previously selected pial artery (line-ROI), touching the boundary sides. Thus, the SD propagation expanded in a perpendicular direction, reaching first ROI-1, then line-ROI, followed by ROI-2 (Figure 1(b)). In the opposite hemisphere to that in which the arterial analysis was performed, another square-ROI of same size was placed next to the LDF probe to correlate it with the LDF-CBF changes and evaluate both responses.
To better visualize the wave front of the propagating SDs during inspection, changes in IOS intensities were amplified by multiplication of the differentiated (along the time axis) IOS signal. A self-developed software based on ImageJ was used to view the large amounts of images and to identify the ROIs. These data were collected and analyzed offline using the LabChart v.7 software.
Intensity profiles extracted from ROIs during SD were analyzed in relation to the relative minimum of the intensity change (peak hyperemic response) and the relative maximum of the intensity change (peak oligemic response) with respect to the baseline intensity. In the result section as well as in tables and figures, the time courses of IOS intensity values are represented inversely for a more intuitive interpretation of the CBV responses to SD; this implies a positive hyperemia and a negative oligemia. Measurement of the time of the minimum of the intensity change (hyperemia) was performed by quantifying its total duration and the duration from the increase of intensity to at least 80% of the recovery was considered for the maximum of the intensity change (oligemia). Differences in CBV responses and their duration during SD occurrence were analyzed and compared with the various experimental phases.
Laser-Doppler flowmetry analysis
The LDF collected data from the Perimed Software were analyzed offline using the LabChart v.7 software. The hyperemia and oligemia analyses were performed in the same way as for the IOS profiles.
Low frequency-vascular fluctuation analysis
Square-ROIs with a size of about 10 × 10 pixels (0.06 mm2) were distributed offline along the hemispheres, to explore and describe the presence of vascular fluctuations associated with haemoglobin concentration changes observed in the IOS field. The collected data and the signals were filtered for the range of frequency 0.01 to 0.1 Hz. Analysis was performed using the LabChart v.7 software.
Statistical analysis
Standard descriptive statistics were calculated for all outcome variables of interest. To test differences in means/medians, the (paired) Student’s t-test, the Wilcoxon signed rank test and the Friedman test were used where appropriate. The relationship between the various outcome variables of interest is described using correlation analyses. Descriptive analyses and tests for group differences were performed using the statistical analysis software SPSS 22.0 for Windows (SPSS Inc., Chicago, Illinois, USA).
Additionally, the relative CBV and pial arterial diameter responses to SD were analyzed using linear mixed effects models. We modelled the mean relative change, X, of either the IOS intensity or the arterial diameter for all experimental phases (before, during and after ketamine infusion) and dose combinations, by accounting for the statistical dependencies in the data through the inclusion of random effects for arterial and CBV identity. We calculated model-based P values for differences in relative changes. The adequacy of the normality assumption for the outcomes was checked graphically by QQ-plots, as well as a histogram, and no major violations were detected. The models were fit by restricted maximum likelihood using the package “lme4” in the R language and environment for statistical computing, Version 3.1.1 (2014-07-10) (http://www.R-project.org/), (http://CRAN.R-project.org/package=lme4). Additionally, the R package “lmerTest” was used (http://CRAN.R-project.org/package=lmerTest) to obtain model-based P values.
All statistical tests were two-sided and values of P < 0.05 were considered to be statistically significant. Since the study was exploratory and no adjustment for multiple testing was performed, the P values are to be interpreted descriptively.
Results
Physiological parameters
Systemic physiological parameters were monitored for each swine during the experiment. All parameters were kept within the normal range. Measurements of systemic parameters did not show any significant difference between the various experimental phases and between groups (supplementary Table 1).
Haemodynamic response to SD in the gyrencephalic swine brain
After KCl stimulation, we observed an initial intensity change around the KCl drop in the IOS field, which turned into a multiphasic intensity change in parenchymal reflectance that evolved and propagated throughout the cerebral cortex for ∼15 min. Immediately after stimulation, a biphasic intensity change of a prominent hyperintensity front (decrease of haemoglobin concentration), followed by a hypointensity (increase of haemoglobin concentration), or a triphasic intensity change with a prominent hyperintensity front, followed by a hypointensity and a longer hyperintensity could be observed. The mark hyperintensity front could disappear during its propagation and turn into a biphasic pattern of hypointensity followed by hyperintensity. This was the most common pattern reaching the triad of ROIs (ROI-1, line-ROI, ROI-2). This biphasic pattern could further evolve in more peripheral regions into triphasic or even tetraphasic intensity changes (Figure 1(c)). These patterns coexisted and spread simultaneously in the cerebral cortex on different fronts. The majority of the waves spread in a radial form following gyral anatomy, and in some instances, anatomical structures such as sulci, gyri and large pial vessels represented barriers for its propagation.
During the passage of the SD through ROI-1 (before pial artery) and ROI-2 (after pial artery), a biphasic CBV haemodynamic response of hyperemia followed by oligemia was observed. As expected, pial arteries between square-ROIs exhibited the corresponding vasodilation followed by a vasoconstriction; however, they returned to a baseline more dilated than before (Figure 1(d)). Plots of the estimated densities of the haemodynamic responses indicated a clear reactivity of pial arteries to SD (Figure 2(a) and (b)) and indicated that arteries did not hinder SD passage between ROI-1 and ROI-2 (Figure 3(a) and (b)). Considering both experimental groups before medication, a significant correlation between the parenchymal CBV and pial arterial responses to SD was found (Figure 4(a)). The duration of pial arterial vasodilation was shorter than the parenchymal CBV hyperemic response (2.03 min ± 8 s vs. 3.51 min ± 23 s) (P < 0.001). The relative CBV and pial arterial responses and their duration to SDs for the various experimental phases are shown in Table 1 (A and B).
Figure 2.

Estimated density of arterial diameter changes relative to baseline values in response to SDs for both experimental groups. The density was obtained by kernel density estimation. The vertical axis represents the density, and the x-axis represents the relative change from baseline (in %). The time scale within each SD is given by colour: vasodilation (red), vasoconstriction (blue) and diameter post-SD (grey). (a) Density of the relative diameter change in response to SD before, during and after ketamine treatment for the low-dose group. A separation between vasodilative and vasoconstrictive responses is equally well visible before ketamine administration (first row). During ketamine treatment, vasoconstriction remains unaffected, while vasodilation is clearly reduced (second row). After ketamine administration (third row) responses differ, there is an attenuation of the vasodilative response compared to before the treatment. However, the vasoconstrictive response is still present. A progressive increase in arterial diameter is also observed; note the separation of the post-SD distribution from the dotted vertical lines. (b) Density of the relative diameter change in response to SD before, during and after ketamine treatment for the high-dose group. Note that after medication the vasodilative response is preserved, a clear separation of both responses is visible and a smaller increase in arterial diameter is seen.
Figure 3.

Estimated density of the parenchymal CBV relative change to baseline intensity of ROI-1 and ROI-2 for both experimental groups. The density was obtained by kernel density estimation. The vertical axis represents the density, and the x-axis represents the relative change from baseline. The time scale within each SD is given by the colour: hyperemia (red) and oligemia (blue). (a) Density of ROI-1 and ROI-2 in the low-dose group. Pial arteries did not hinder SD passage between ROI-1 and ROI-2. (b) Density of ROI-1 and ROI-2 in the high-dose group is shown. Note that during the high-dose of ketamine, SDs were blocked.
Figure 4.

Pial arterial diameter changes in response to SDs. (a) Correlation of relative pial arterial diameter changes versus CBV haemodynamic changes registered in IOS changes for the various experimental phases. Note that there is a good correlation between these for the various experimental phases (before, during and after ketamine) (Pearson correlation R = 0.624, P < 0.01; R = 0.684, P < 0.01; R = 0.661, P < 0.01, respectively). (b) Representative example of pial arterial diameter responses to SD. Examples of x-t plots of line-ROIs for the pial arterial response before, during and after 2 mg/kg/h (upper trace) and 4 mg/kg/h (lower trace) ketamine perfusion are shown. A biphasic response of vasodilation and vasoconstriction can be observed. During ketamine at the high-dose, no haemodynamic response is seen. A progressive increase in pial arterial diameter can be observed. (c) Arterial dilatory changes during the experimental phases for the various groups. The diameter (mm) before SD (blue) and the diameter after SD (green) are shown. Note the gradual increase in diameter before the ketamine period. The dotted vertical lines indicate the period during ketamine administration. After the low ketamine dose, an increase in arterial diameter can be observed. In comparison, after the high-dose, arteries presented an increased diameter with a gradual decrease.
Table 1.
Relative haemodynamic response to SD for the various experimental phases and their duration.
| A | |||||||
|---|---|---|---|---|---|---|---|
| Relative CBV responses to SD |
|||||||
| Group | Dose | Phase | # of measurements | Hyperemia | Oligemia | Time of hyperemia | Time of oligemia |
| First group, n = 5 | 2 mg/kg/h | Before | 80 | 21.3 ± 1.1% | − 10.2 ± 2.8% | 3.02 min ± 23 s | 11.03 min ± 66 s |
| During | 46 | 14.0 ± 1.3% | − 13.9 ± 3.4% | 2.34 min ± 25 s | 8.26 min ± 77 s | ||
| After | 64 | 13.7 ± 1.2% | − 10.6 ± 3.0% | 2.21 min ± 24 s | 9.16 min ± 69 s | ||
| Second group, n = 5 | 4 mg/kg/h | Before | 80 | 18.6 ± 1.1% | − 17.9 ± 2.8% | 4.00 min ± 23 s | 11.06 min ± 66 s |
| During | – | – | – | – | – | ||
| After | 62 | 12.3 ± 1.2% | − 18.5 ± 3.0% | 3.35 min ± 24 s | 8.44 min ± 70 s | ||
| B | |||||||
| Relative arterial diameter responses to SD |
|||||||
| Group |
Dose |
Phase |
# of measurements |
Vasodilation |
Vasoconstriction |
Post-SD |
Time of dilation |
| First group, n = 5 | 2 mg/kg/h | Before | 40 | 39.5 ± 4.9% | − 14.3 ± 2.8% | 4.1 ± 2.2% | 1.90 min ± 8 s |
| During | 23 | 20.4 ± 5.5% | − 13.9 ± 3.3% | 5.5 ± 2.9% | 1.21 min ± 10 s | ||
| After | 32 | 15.3 ± 5.1% | − 11.4 ± 2.9% | 5.7 ± 2.4% | 1.36 min ± 9 s | ||
| Second group, n = 5 | 4 mg/kg/h | Before | 40 | 45.0 ± 4.9% | − 13.3 ± 2.8% | 6.6 ± 2.2% | 2.23 min ± 8 s |
| During | – | – | – | – | – | ||
| After | 31 | 24.1 ± 5.1% | − 13.2 ± 3.0% | 1.7 ± 2.5% | 1.33 min ± 9 s | ||
| C | |||||||
| Relative CBF and CBV responses to SD |
|||||||
| Group |
Dose |
Method |
Phase |
# of SDs |
Hyperemia |
Time of hyperemia |
|
| Second group, n = 4 | 4 mg/kg/h | LDF | Before | 16 | 120.4 ± 59.6% | 5.83 min ± 90 s | |
| During | – | – | – | ||||
| After | 16 | 104.4 ± 50.7% | 3.40 min ± 90 s | ||||
| IOS | Before | 16 | 12.8 ± 4.1% | 3.34 min ± 60 s | |||
| During | – | – | – | ||||
| After | 16 | 9.0 ± 2.3% | 2.55 min ± 60 s | ||||
Values are expressed as mean and standard deviation.
CBF: cerebral blood flow; CBV: cerebral blood volume; IOS: intrinsic optical signal imaging; LDF: laser-Doppler flowmetry; SD: spreading depolarization.
In the opposite hemisphere to that in which the pial arterial analysis was performed, the responses of CBF (represented by LDF) and CBV (represented by IOS) to SDs were evaluated in peripheral regions devoid of large vessels. This was performed only in the high-dose group. One case was discarded due to bleeding and obscuring artefacts. During the passage of the SD through the LDF probe, CBF changes exhibited mostly a monophasic hyperemic response to SDs, while the square-ROI next to the probe exhibited multiphasic responses (Figure 5(a)). We found a significant correlation of the hyperemic response between CBF (LDF) and CBV (IOS) (Figure 5(b)). The duration of the hyperemic response in CBF was found to be longer than that detected in CBV (5.83 min ± 90 s vs. 3.34 min ± 60 s) (P = 0.005). The relative CBF and CBV hyperemic response and their duration to SD for the various experimental phases is shown in Table 1 (C).
Figure 5.

Parenchymal CBF and CBV hyperemic responses to SD registered by LDF and IOS. (a) Examples of blood flow velocity changes in LDF and CBV intensity changes in the opposite hemisphere to that of the pial artery analysis are shown. The LDF probe was placed in a peripheral region devoid of large vessels with a corresponding IOS ROI next to the probe. In IOS, multiphasic intensity changes can be observed. LDF velocity changes show monophasic hyperemic responses. (b) Correlation of hyperemia in IOS CBV changes vs. LDF CBF changes before (Spearman correlation R = 0.647, P < 0.01) and after ketamine (Spearman correlation R = 0.681, P < 0.01). (c) Changes in the percentage of the CBF hyperemic response before and after high-dose ketamine. (d) Duration of hyperemic response in CBF before and after the high-dose of ketamine. Note that after ketamine administration, there is lasting effect on the hyperemia duration. T-bars represent the 95% CI of the values. *P < 0.05.
Additionally to our findings, repetitive SDs evoked by stimulation with KCl every 30 min did not show a progressive attenuation of the hyperemic and oligemic responses in amplitude and in duration, neither in CBV (IOS) nor in CBF (LDF) in response to SD for the subsequent stimulations before ketamine. Nevertheless, repetitive SDs caused a persistent increase in the baseline pial arterial diameter after each passage (4.1 ± 2.2%). Although pial arteries presented an increase in diameter before medication, their capacity to dilate or constrict in response to SD did not diminish (Figure 4(b) and (c)).
Modulation of the haemodynamic response during SDs with ketamine
Ketamine infusion modulated SD induction and propagation, and altered the haemodynamic responses in a dose-dependent manner. The effect of ketamine on SD induction and propagation observed in IOS during the various experimental phases is shown in supplementary Table 2. Ketamine at a higher dose suppressed SD elicitation and the coupled haemodynamic response; therefore only the effect of the low-dose was able to be analyzed. However, evaluation of the baseline pial arterial diameter during ketamine infusion indicated a temporarily SD-independent diameter increase of 14.7%, in comparison with the before ketamine values; this effect disappeared after its administration.
During the 2 h of ketamine infusion at a low-dose, the CBV hyperemic response to SD, in ROI-1 and ROI-2, decreased from 21.3 ± 1.1% to 14.0 ± 1.3%, as did its duration from 3.02 min ± 23 s to 2.34 min ± 25 s (P < 0.001 and P = 0.012, respectively), when compared with the values before medication. Ketamine at a low-dose also decreased the duration of the oligemic response from 11.03 min ± 66 s to 8.26 min ± 77 s (P = 0.001); however, it did not seem to alter the amplitude of the oligemic response, when compared with the responses before medication. In agreement with these results, the corresponding pial arteries exhibited a decrease in vasodilation to SD from 39.5 ± 4.9% to 20.4 ± 5.5% (P < 0.001), and its duration decreased from 1.90 min ± 8 s to 1.21 min ± 10 s (P = 0.008), while vasoconstriction to SD was found to be preserved. However, despite ketamine’s reducing effect on the amplitude of hyperemic response to SDs, it did not hinder the persistent increase in arterial diameter due to the continued presence of SDs. Pial arteries experienced an increase in baseline diameter of 5.5 ± 2.9% during this phase and a diminished capacity to dilate was observed while the constrictive response was preserved during medication (Figure 2(a)).
Haemodynamic response after ketamine infusion
After ending ketamine infusion, a lasting effect on the previously affected responses in the triad of ROIs in both groups was found. The hyperemic amplitude, its duration and duration of oligemia as well as the corresponding pial arterial vasodilation and its duration remained significantly reduced for the following 2 h of stimulation when compared with before medication values. In the low-dose group after medication, the persistent increase of baseline pial arterial diameter due to the continuous SD development was found to be of 5.7 ± 2.4%. This was still associated with a diminished capacity to further dilate and the preservation of the constrictive response (Figure 2(a)). In comparison with the high-dose group after ketamine, the reappearance of depolarizations increased the baseline pial arterial diameter (1.7 ± 2.5%); however, this occurred to a lesser extent than before medication (6.6 ± 2.2%) and than to that observed in the low-dose group in this experimental phase. Plots of the estimated densities of the pial arterial changes to baseline values indicated a preserved capacity to dilate in response to SD (Figure 2(b)).
In the opposite hemisphere to that in which the pial arterial analysis was performed, CBF responses (LDF) were evaluated. After the high-dose infusion, the expected reduction of the hyperemic response was seen. The amplitude of the CBF hyperemic response decreased from 120.4 ± 59.6% to 104.4 ± 50.7% (P = 0.148) and its duration decreased from 5.83 min ± 90 s to 3.40 min ±90 s (P = 0.003). The corresponding amplitude of the CBV hyperemic response (IOS) and its duration, as seen in the opposite hemisphere, were found to be decreased (Figure 5(c) and (d)).
Low frequency-vascular fluctuations
We observed, in the IOS field, CBV fluctuations of enhanced amplitude (5–10% IOS intensity changes) corresponding to changes in haemoglobin concentration. These fluctuations appeared to be also related to diameter changes of superficial arteries (Figure 6(a)). A more detail analysis revealed the frequency of these signals to be between 0.037 and 0.054 Hz. We attribute these to be low frequency-vascular fluctuations (LF-VF, 0.05–0.1 Hz) (Figure 6(b)). During SD, these fluctuations, presented a reduction (3.5–8.5%) or disappearance of the enhanced amplitude. This reduction was clearly observed at the beginning of the SD, which on some occasions continued until the later oligemia. However, in other occasions during the later oligemia or mostly after SD occurrence, a strong enhancement up to 20% of the fluctuations amplitude was observed. We found no significant change in the wave frequency over time. These vascular fluctuations of enhanced amplitude were heterogeneous and differed according to the observed area, time and animal studied; in addition, they were also present despite ketamine administration.
Figure 6.
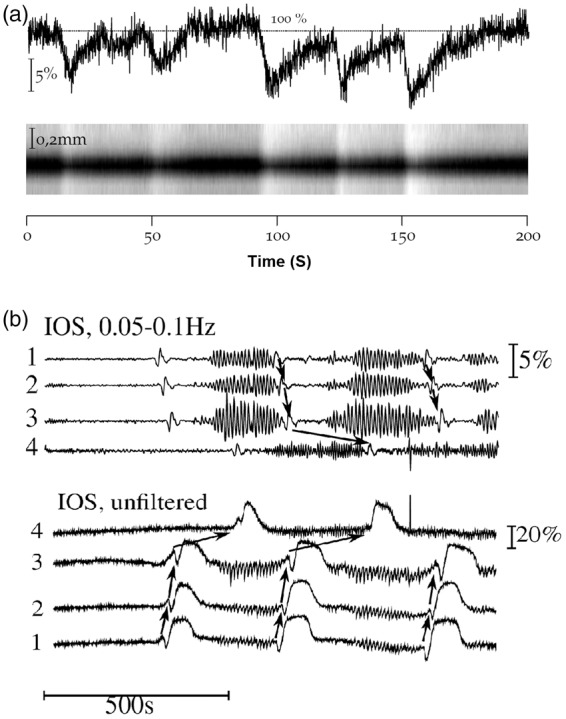
Low frequency-vascular fluctuations. (a) The upper trace shows intensity changes of haemoglobin concentration detected by IOS imaging in a ROI near to a superficial artery. The lower trace exhibits the corresponding changes in arterial diameter (vasomotion) in relation with the vascular fluctuations. (b) Representative example of the first occurrence and transient disappearance of the enhanced amplitude of LF-VF during KCl-evoked SDs. Upper traces show LF-VF at a frequency of 0.05–0.1 Hz. It can be noted that the reduction of the enhanced amplitude of the LF-VFs begins nearly synchronously with SD occurrence. Lower traces exhibit multiphasic intensity changes in IOS in four different ROIs. The arrows show the propagation of the SD.
Discussion
Haemodynamic response to SD in the gyrencephalic brain
In this study, the cerebral haemodynamic profiles of brain tissue affected by SDs induced with KCl in the gyrencephalic swine brain were characterized through CBV, pial arterial diameter and CBF. The haemodynamic response to SD has been extensively studied in various animal models, including mice, rat, rabbit, cat and monkey.3,12 There are few study reports of SDs in the gyrencephalic swine brain;15,20–23 up to now, there is only one study investigating the spatiotemporal propagation, haemodynamic response and vascular changes to SD in the gyrencephalic human brain with IOS and laser-Speckle imaging.24 In our experiments, a variety of haemodynamic responses after KCl stimulation were detected in IOS. Our findings are consistent with the current literature and share similarities with the previous data found in other species,3,6,8–12,25 as well as for the gyrencephalic human cortex, where a heterogeneity of haemodynamic responses has been observed in IOS after malignant hemispheric stroke.24
Different factors may have contributed to the diversity of the haemodynamic responses observed in IOS from which two possible aspects can be taken into consideration: (a) K+ diffusion after KCl stimulation, and (b) a brain region and/or arterial territory haemodynamic dependency. In this model, after brain preconditioning, SDs were experimentally induced with KCl in an attempt to simulate the conditions observed in ischemic tissue where high concentrations of K+ are found. It is well known that increases in K+ are a key factor in the initiation of SD and that moderate K+ elevations cause vasodilation (<20 mmol/l), whereas higher concentrations cause vasoconstriction.26,27 In the areas near to the stimulation point (<10 mm) an initial strong hypoperfusion was observed, which gradually decreased in intensity or disappeared during SD propagation into farther regions.
A more plausible responsible cause for the variety of haemodynamic responses is the evidence indicating that vascular segments and compartments respond differently to SD. In this regard, it has been described that pial arteries dilate while smaller penetrating arteries constrict in response to SD.3,27 Therefore, the existence of a layer-specific haemodynamic response to SD may contribute to explaining in part the presence of the very brief hyperemia before the initial hypoperfusion in the tetraphasic intensity change, suggestive of superimposed responses. The monophasic hyperemic response detected in LDF may be related to the lack of spatial resolution of this technique, being limited to tracking only the superficial blood flow velocity changes or being unable to detect rapid and small flow velocity changes. Similar discrepancies for this technique have been reported by others earlier.3,28 These data strongly suggest the possibility of different mechanisms mediating the response in cortical vessels and the underlying cortex observed by the different techniques.
We found that repetitive SDs evoked by stimulation with KCl in the gyrencephalic brain did not attenuate the amplitude and duration of CBV or CBF responses of subsequent SDs. This finding might be related to the time interval between stimulations of 30 min, which in a well-supplied and oxygenated gyrencephalic brain provided sufficient time to recover. In smaller species, it has been reported that the haemodynamic response of SDs repeated more than 1 h apart resembles that of the naive cortex, in comparison with those occurring within less than 30–45 min3. Therefore, the species-related differences (size, gyrencephaly, angioarchitecture, etc.) might have contributed, by offering more resistance. Conversely, disruption of normal functional CBV responses due to a decrease in vascular reactivity, and spontaneous vasomotion have been described in the rat lissencephalic brain.11
We observe a constant increase in arterial diameter after each consecutive SD, leading to a diminished capacity to further dilate in response to SD. In agreement with these findings, attenuation of vascular responses despite parenchymal SD propagation has been documented after repetitive SDs in various studies.8,10,11,27 The existence of a distinct vascular conduction independent of the parenchymal depolarization changes in lissencephalic brain is also well known.3,8,9 Persistent arteriolar dilation or intense constriction with a decrease or absence of vascular response have been described after repetitive SDs in lissencephalic mice and rat brains. A time interval of 1 h between stimulations appears to be able to preserve the vascular responses to SD in these models.8 The mechanisms implicated in the impairment of arterial smooth muscle function after repetitive SDs are still unclear and further studies addressing this issue are required.
Modulation of the haemodynamic response to SD with ketamine
Little is known about the influence of ketamine on cerebral haemodynamics and of its effect on the haemodynamic response to SDs. In this study, we found that ketamine at a low-dose significantly reduced the hyperemic response, but did not alter the oligemic response to SD; this effect was in total agreement with the observations made in pial arteries. It is possible that lower doses of ketamine are needed to modulate the formation of hyperemic/vasodilative products (NO, H+, lactate, adenosine, K+, proglandine E2 and epoxyeicosatrienoic acids)12,26 due to NMDA receptor activation, than those needed to modulate the oligemic/vasoconstrictive response.29,30 Only ketamine at a high-dose was able to suppress SD induction and consequently the coupled haemodynamic response. In a previous study, we showed how a ketamine bolus of 4 mg/kg was able to block SD propagation within 1–2 min, accompanied by a suppression of the haemodynamic responses.15
It is important to mention that despite ketamine’s capacity, at a low-dose, to reduce the vasodilative response to SD, its administration did not hinder the constant increase in baseline pial arterial diameter after each consecutive SD, which led to a diminished dilative capacity. We hypothesized that this reduction in the arterial diametric response could be a combination of the following factors: (a) the effect of ketamine reducing hyperemia through the reduction of the depolarization per se, (b) a slight relaxing effect of ketamine on cerebral vasculature during the period of administration and (c) the gradual increase in baseline arterial diameter due to the impairment of arterial smooth muscle function after repetitive SDs (probably also responsible for the effect observed after medication). Consequently, arteries reached a maximum dilation and their further capacity to dilate decreased, while the vasoconstrictive function was preserved.
In this study, we observed a dilative effect during high-dose ketamine infusion. Previous studies exploring the effect of ketamine on animal cerebral arteries in in vitro models report a dose-dependent dilation/relaxation with attenuation of the contractile response to high concentrations of K+, thromboxane A2 analogue or endothelin.31–34 However, the mechanism underlying the dilating/relaxing effect of ketamine on cerebral vasculature at high doses might differ from those for SD modulation and reduction of hyperemia/vasodilation, since isolated cerebral arteries have not been found to dilate or constrict in response to the application of glutamate and NMDA in diverse species.12 Nevertheless, it has been shown that superfusion of NMDA in an in vivo mice model induced pial arterial dilation by triggering SD.35
After medication, we observed a lasting dose-dependent effect of ketamine, suggesting the continued presence of ketamine in the brain. In this regard, ketamine has shown a high affinity for brain tissue; this is in part due to its high lipid solubility with a brain-to-plasma ratio of 6.5:1.36 In humans, it is known that the free fraction of ketamine (∼53%) determines the rate of diffusion to a site of action. Ketamine has a slow off-rate or high-trapping block property (∼86%), which implicates a prolonged blockage on its binding site on the NMDA receptor.37 In intensive care patients, the administration of ketamine (2 mg/kg/h for 2 h) has shown an elimination half-life of 4.98 h with a longer half-life of metabolites.38 This may help to explain why for the following 2 h after ketamine infusion the same haemodynamic responses diminished. This finding also suggests that the attenuated dilative capacity after the low-dose, not observed after the high-dose, can be the result of the cumulative effect of SD on pial arterial smooth muscle. Thus, lower doses may not be able to shield the artery smooth muscle effectively from the cumulative depolarization burden, while the high-dose leads to a relatively preserved arterial function by preventing SDs.
Low frequency-vascular fluctuations
Spontaneous vascular fluctuations of low-frequencies reflect neuronal activity and synchrony between brain regions. SDs cause suppression of these fluctuations independent from the haemodynamic response. They have been previously reported in patients with SDs after malignant hemispheric stroke and aneurismal subarachnoid haemorrhage.24,39 We were able to observe such fluctuations superimposed on tissue perfusion associated with haemoglobin and pial arterial diameter changes at low-frequencies of 0.05–0.1 Hz with IOS. The aetiology and physiological significance of LF-VF remains unknown. It has been proposed that they originate from the resting brain activity or due to vasomotion.39 The fact that these fluctuations in our experiments were only suppressed during SD occurrence despite the gradual increases in pial arterial diameter, reduction of the dilative capacity and ketamine infusion, speaks in favour of an oscillatory neuronal activity origin that might be detectable through pial vasculature.
Study limitations
This study has some limitations and some results should be interpreted with caution. First, it is important to note that the study did not involve an independent untreated control group and it is therefore not possible to make any quantitative statements about the haemodynamic differences and amount of fatigue due to repeated SD induction in the swine cortex without ketamine administration. We partly compensated this limitation by letting, every animal serve as its own control by using the phase before medication to control for differences with the following experimental phases between animals. Moreover, the presence of the blocking period during the high ketamine dose indicates a different vascular behaviour between groups after medication. Second, the possible pharmacological interactions between the anaesthetics used (isoflurane, midazolam and propofol) with SD development are not known, but the anaesthetics are known to have an impact on SD susceptibility.17 However, the intention of this study, which used large gyrencephalic brains, was to replicate clinical conditions found in intensive care units as closely as possible.
Third, the monitoring techniques applied here might also present some limitations. The use of IOS and LDF together has been shown to be useful for the evaluation of the haemodynamic response to SD in various experimental models. Its correlation with electrophysiology, laser-Speckle flowmetry and intravascular dye imaging has been proven.20,21,24,40 However, IOS intensities can be influenced not only by haemoglobin concentration, so interpretation as a measure of CBV should be considered and cannot be excluded without measuring additional wavelengths or using other monitoring techniques.41,42 In our setting, intraparenchymal LDF would have had the disadvantage of causing focal bleeding preventing an adequate IOS recording. Additionally, the movement-prone environment could have led to changes in the depth of the probe throughout the experiment generating artefacts. Also, the absence of LDF for the low-dose group limits our understanding of the modulation of the CBF responses under this dose. Nevertheless, based on the IOS measurements in the low-dose group, it would also be expected to exhibit a partial reduction of CBV, similar to that observed after the high-dose. Some advantages for continuous perfusion monitoring in an experimental setting have been described with the use of Laser-Speckle flowmetry, due to its widespread availability. In a previous study, however, we detected a decrease in resolution and signal-to-noise ratio when studying the gyrencephalic swine brain due to brain movement.20 We reduced these movement artefacts by using elastic registration in the image post-processing, which enabled us to better visualize the haemodynamic parenchymal and pial arterial changes to SD.
Fourth, we are not able to rule out the impact of the large craniotomy on regional CBF. Studies addressing its impact indicate mixed effects on CBF in normal brain and in brains with oedema and injury. In normal brain, a craniotomy can produce a decrease in CBF, oedema formation, impairment of cerebrovascular pressure reactivity and non-restoration of brain aerobic metabolism. However, under advanced oedema and increased intracranial pressure, a large craniotomy, can have a favourable effect on brain perfusion.43–45 In this study, we would expect a decrease in intracranial pressure and if at all only a minimal reduction of cerebral perfusion pressure with brain oedema formation that might have an impact on the haemodynamic responses of the SDs observed. It has been reported that craniotomy influences parenchymal IOS characteristics, as well as surface arterial diameter changes in response to SD.9
The limitations of this study, however, do not weigh severely on the characterization of the haemodynamic response in the gyrencephalic swine brain. Ketamine’s effect on this response provides a valuable insight into the haemodynamic consequences when using an NMDA receptor antagonist as a potential target for SD suppression. Further studies are required to understand the various facets of the haemodynamic response in the gyrencephalic brain and the potential benefits and limitations of its pharmacological modulation under physiological and pathological conditions.
Clinical implications
The concept that SDs in the gyrencephalic brain are associated with variable haemodynamic responses and pial arterial diameter changes has already been demonstrated in the clinical setting of decompressive craniectomy for ischemic stroke by the group of Woitzik and colleagues.24 We used a similar methodology to understand the pharmacological modulation with ketamine of the haemodynamic response in a gyrencephalic swine brain model. The importance of a gyrencephalic animal model such as swine relies on its resemblance in many aspects to the human brain, which may facilitate the extrapolation of the results in particular when used for therapeutic approaches. Recently, the use of ketamine in SD research has been of interest due to the increasing amount of clinical evidence showing SD modulation in brain injured patients.16,17 However, the effect of ketamine targeted to block SD on the clinical outcome in unknown. An important step forward for the identification and/or development of therapeutic options is the characterization of the haemodynamic effect of pharmacological interventions, because therapies targeting the harmful vascular/haemodynamic responses associated with SD could counteract their pathological effects. Here, we have shown how a lower dose of ketamine, capable of modulating SD in animals and in humans, mainly attenuates the hyperemic response. This is essential for the metabolic recovery from SD, and decreasing it might be detrimental under pathological conditions. These results suggest that only higher doses of ketamine (i.e. ∼4 mg/kg/h) may be appropriate to completely hinder SDs and their coupled haemodynamic response. Whether or not this might offer any clinical benefit is not known; hence, more work is needed before a potential therapeutic scheme can be suggested.
Conclusions
To summarize, the haemodynamic response during SD can be modulated by continuous infusion of ketamine. Lower ketamine doses suppressed the hyperemic/vasodilative response to SD. Only high ketamine doses were able to block SD induction and the coupled haemodynamic response. Frequent SDs caused a persistent increase in the baseline pial arterial diameter, which can lead to a diminished capacity to further dilate. This was prevented with the high-dose. The use of ketamine in pathological gyrencephalic swine brain models needs to be explored. It may be that SD modulation under pathological circumstances differs from that observed under normal conditions. More studies are required to understand the various facets of the haemodynamic response in the gyrencephalic brain under pathological conditions and their interaction with pharmacological therapies.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: RSP was supported by the National Council of Science and Technology (CONACyT), Mexico. ES was supported by the Postdoc Fellowship Program of the Faculty of Medicine Heidelberg. HS was supported by the CONACyT and by the Council of Science and Technology of San Luis Potosi (COPOCyT), Mexico.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
RSP: Designed the project, conducted experiments, analyzed the data, performed statistical analyses and wrote the manuscript. ES: Designed the project, conducted experiments, analyzed the data and critically reviewed the manuscript. MS: Developed the software, performed the technical design of IOS, conducted experiments and critically reviewed the manuscript. KK and CS: Performed statistical analyses, developed linear mixed effect models and critically reviewed the manuscript. HS: Analyzed the data. AWU: Critically reviewed the manuscript. OWS: Provided scientific support and critically reviewed the manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data.
References
- 1.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011; 17: 439–447. [DOI] [PubMed] [Google Scholar]
- 2.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 2014; 15: 379–393. [DOI] [PubMed] [Google Scholar]
- 3.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauritzen M, Dreier JP, Fabricius M, et al. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2011; 31: 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lauritzen M. Long-lasting reduction of cortical blood flow of the brain after spreading depression with preserved autoregulation and impaired CO2 response. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 1984; 4: 546–554. [DOI] [PubMed] [Google Scholar]
- 6.Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2009; 29: 1517–1527. [DOI] [PubMed] [Google Scholar]
- 7.Ayata C. Spreading depression and neurovascular coupling. Stroke J Cereb Circ 2013; 44: S87–S89. [DOI] [PubMed] [Google Scholar]
- 8.Brennan KC, Beltrán-Parrazal L, López-Valdés HE, et al. Distinct vascular conduction with cortical spreading depression. J Neurophysiol 2007; 97: 4143–4151. [DOI] [PubMed] [Google Scholar]
- 9.Chang JC, Shook LL, Biag J, et al. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain J Neurol 2010; 133: 996–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci Off J Soc Neurosci 2007; 27: 4036–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guiou M, Sheth S, Nemoto M, et al. Cortical spreading depression produces long-term disruption of activity-related changes in cerebral blood volume and neurovascular coupling. J Biomed Opt 2005; 10: 11004. [DOI] [PubMed] [Google Scholar]
- 12.Busija DW, Bari F, Domoki F, et al. Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol 2008; 86: 379–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernándéz-Cáceres J, Macias-González R, Brozek G, et al. Systemic ketamine blocks cortical spreading depression but does not delay the onset of terminal anoxic depolarization in rats. Brain Res 1987; 437: 360–364. [DOI] [PubMed] [Google Scholar]
- 14.Gorelova NA, Koroleva VI, Amemori T, et al. Ketamine blockade of cortical spreading depression in rats. Electroencephalogr Clin Neurophysiol 1987; 66: 440–447. [DOI] [PubMed] [Google Scholar]
- 15.Sánchez-Porras R, Santos E, Schöll M, et al. The effect of ketamine on optical and electrical characteristics of spreading depolarizations in gyrencephalic swine cortex. Neuropharmacology 2014; 84: 52–61. [DOI] [PubMed] [Google Scholar]
- 16.Sakowitz OW, Kiening KL, Krajewski KL, et al. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke J Cereb Circ 2009; 40: e519–e522. [DOI] [PubMed] [Google Scholar]
- 17.Hertle DN, Dreier JP, Woitzik J, et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain J Neurol 2012; 135: 2390–2398. [DOI] [PubMed] [Google Scholar]
- 18.Aiba I, Shuttleworth CW. Sustained NMDA receptor activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons. J Physiol 2012; 590: 5877–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou N, Rungta RL, Malik A, et al. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2013; 33: 1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schöll M, Gramer M, Santos E, et al. Comparison of laser speckle flowmetry and intrinsic optical signal imaging in gyrencephalic swine brain during cortical spreading depolarisations. Biomed Tech (Berl) 2012; 57 (Sl-1, Track-B): 323–326.25854660 [Google Scholar]
- 21.Santos E, Schöll M, Sánchez-Porras R, et al. Radial, spiral and reverberating waves of spreading depolarization occur in the gyrencephalic brain. NeuroImage 2014; 99: 244–255. [DOI] [PubMed] [Google Scholar]
- 22.Bowyer SM, Tepley N, Papuashvili N, et al. Analysis of MEG signals of spreading cortical depression with propagation constrained to a rectangular cortical strip. II. Gyrencephalic swine model. Brain Res 1999; 843: 79–86. [DOI] [PubMed] [Google Scholar]
- 23.Mun-Bryce S, Wilkerson AC, Papuashvili N, et al. Recurring episodes of spreading depression are spontaneously elicited by an intracerebral hemorrhage in the swine. Brain Res 2001; 888: 248–255. [DOI] [PubMed] [Google Scholar]
- 24.Woitzik J, Hecht N, Pinczolits A, et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology 2013; 80: 1095–1102. [DOI] [PubMed] [Google Scholar]
- 25.Leão AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol 1944; 7: 359–390. [DOI] [PubMed] [Google Scholar]
- 26.Ayata C, Shin HK, Salomone S, et al. Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab 2004; 24: 1172–1182. [DOI] [PubMed] [Google Scholar]
- 27.Unekawa M, Tomita Y, Toriumi H, et al. Hyperperfusion counteracted by transient rapid vasoconstriction followed by long-lasting oligemia induced by cortical spreading depression in anesthetized mice. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2015; 35: 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unekawa M, Tomita M, Tomita Y, et al. Sustained decrease and remarkable increase in red blood cell velocity in intraparenchymal capillaries associated with potassium-induced cortical spreading depression. Microcirculation 2012; 19: 166–174. [DOI] [PubMed] [Google Scholar]
- 29.Shin HK, Dunn AK, Jones PB, et al. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2006; 26: 1018–1030. [DOI] [PubMed] [Google Scholar]
- 30.Fordsmann JC, Ko RWY, Choi HB, et al. Increased 20-HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci Off J Soc Neurosci 2013; 33: 2562–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavazzuti M, Porro CA, Biral GP, et al. Ketamine effects on local cerebral blood flow and metabolism in the rat. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 1987; 7: 806–811. [DOI] [PubMed] [Google Scholar]
- 32.Fukuda S, Murakawa T, Takeshita H, et al. Direct effects of ketamine on isolated canine cerebral and mesenteric arteries. Anesth Analg 1983; 62: 553–558. [PubMed] [Google Scholar]
- 33.Wendling WW, Daniels FB, Chen D, et al. Ketamine directly dilates bovine cerebral arteries by acting as a calcium entry blocker. J Neurosurg Anesthesiol 1994; 6: 186–192. [DOI] [PubMed] [Google Scholar]
- 34.Kamel IR, Wendling WW, Chen D, et al. N-methyl-D-aspartate (NMDA) antagonists–S(+)-ketamine, dextrorphan, and dextromethorphan–act as calcium antagonists on bovine cerebral arteries. J Neurosurg Anesthesiol 2008; 20: 241–248. [DOI] [PubMed] [Google Scholar]
- 35.Ayata C, Moskowitz MA. Cortical spreading depression confounds concentration-dependent pial arteriolar dilation during N-methyl-D-aspartate superfusion. Am J Physiol Heart Circ Physiol 2006; 290: H1837–1841. [DOI] [PubMed] [Google Scholar]
- 36.Orser BA, Pennefather PS, MacDonald JF. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. J Am Soc Anesthesiol 1997; 86: 903–917. [DOI] [PubMed] [Google Scholar]
- 37.Sleigh J, Harvey M, Voss L, et al. Ketamine – More mechanisms of action than just NMDA blockade. Trends Anaesth Crit Care 2014; 4: 76–81. [Google Scholar]
- 38.Hijazi Y, Bodonian C, Bolon M, et al. Pharmacokinetics and haemodynamics of ketamine in intensive care patients with brain or spinal cord injury. Br J Anaesth 2003; 90: 155–160. [DOI] [PubMed] [Google Scholar]
- 39.Dreier JP, Major S, Manning A, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009; 132: 1866–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bere Z, Obrenovitch TP, Kozák G, et al. Imaging reveals the focal area of spreading depolarizations and a variety of hemodynamic responses in a rat microembolic stroke model. J Cereb Blood Flow Metab 2014; 34: 1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Obrenovitch TP, Chen S, Farkas E. Simultaneous, live imaging of cortical spreading depression and associated cerebral blood flow changes, by combining voltage-sensitive dye and laser speckle contrast methods. NeuroImage 2009; 45: 68–74. [DOI] [PubMed] [Google Scholar]
- 42.Yin C, Zhou F, Wang Y, et al. Simultaneous detection of hemodynamics, mitochondrial metabolism and light scattering changes during cortical spreading depression in rats based on multi-spectral optical imaging. NeuroImage 2013; 76: 70–80. [DOI] [PubMed] [Google Scholar]
- 43.Bor-Seng-Shu E, Figueiredo EG, Fonoff ET, et al. Decompressive craniectomy and head injury: brain morphometry, ICP, cerebral hemodynamics, cerebral microvascular reactivity, and neurochemistry. Neurosurg Rev 2013; 36: 361–370. [DOI] [PubMed] [Google Scholar]
- 44.Schaller B, Graf R, Sanada Y, et al. Hemodynamic and metabolic effects of decompressive hemicraniectomy in normal brain, An experimental PET-study in cats. Brain Res 2003; 982: 31–37. [DOI] [PubMed] [Google Scholar]
- 45.Slotty PJ, Kamp MA, Beez T, et al. The influence of decompressive craniectomy for major stroke on early cerebral perfusion. J Neurosurg 2015; 123: 59–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.