

Abstract
Aberrant DNA methylation is widely observed in many cancers. Concurrent DNA methylation of multiple genes occurs in endometrial cancer and is referred to as the CpG island methylator phenotype (CIMP). However, the features and causes of CIMP-positive endometrial cancer are not well understood. To investigate DNA methylation features characteristic to CIMP-positive endometrial cancer, we first classified samples from 25 patients with endometrial cancer based on the methylation status of three genes, i.e. MLH1, CDH1 (E-cadherin) and APC: CIMP-high (CIMP-H, 2/25, 8.0%), CIMP-low (CIMP-L, 7/25, 28.0%) and CIMP-negative (CIMP(-), 16/25, 64.0%). We then selected two samples each from CIMP-H and CIMP(-) classes, and analyzed DNA methylation status of both normal (peripheral blood cells: PBCs) and cancer tissues by genome-wide, targeted bisulfite sequencing. Genomes of the CIMP-H cancer tissues were significantly hypermethylated compared to those of the CIMP(-). Surprisingly, in normal tissues of the CIMP-H patients, promoter region of the miR-663a locus is hypermethylated relative to CIMP(-) samples. Consistent with this finding, miR-663a expression was lower in the CIMP-H PBCs than in the CIMP(-) PBCs. The same region of the miR663a locus is found to be highly methylated in cancer tissues of both CIMP-H and CIMP(-) cases. This is the first report showing that aberrant DNA methylation of the miR-663a promoter can occur in normal tissue of the cancer patients, suggesting a possible link between this epigenetic abnormality and endometrial cancer. This raises the possibility that the hypermethylation of the miR-663a promoter represents an epimutation associated with the CIMP-H endometrial cancers. Based on these findings, relationship of the aberrant DNA methylation and CIMP-H phenotype is discussed.
Keywords: endometrial cancer, CIMP, methylation, epimutation, miR-663a, DNMTs, MiSeq
Introduction
Epigenetic changes are widely observed in various types of cancer. DNA methylation of gene promoter region can suppress expression of cancer-related genes, e.g. tumor suppressor, which may lead to carcinogenesis of many types of cancer including endometrial cancer (1–3). On the other hand, aberrant DNA hypermethylation in normally unmethylated sequences/promoters can be regarded as epimutations. Epimutations are suspected as a cause of some inherited cancer syndrome. Lynch syndrome is one such inherited cancer syndrome related to endometrial cancer, and germline mutations in DNA mismatch repair genes such as MLH1 are major cause of the disease. However, epimutations in MLH1 gene may also cause Lynch syndrome (4–6).
It has been known that DNA methylation likely contributes to both endometrial carcinogenesis and endometrial cancer phenotype (7,8). We have identified aberrant DNA methylations in promoters of various cancer-related genes in endometrial cancer. Such concurrent DNA methylation of multiple genes is observed in colorectal, breast, gastric and endometrial cancers, and this is defined as the CpG island methylator phenotype (CIMP) (9–11), but, methylation status, features and causes of CIMP-positive endometrial cancer have not been well understood. One can assume that cells of those CIMP-positive cancer patients may have aberrant DNA methylation metabolism even in their normal tissues before manifestation of the cancer, which may trigger a series of epigenetic changes leading to carcinogenesis of the CIMP-positive endometrial cancer. Based on this assumption, we investigated DNA methylation status and tried to identify features in normal tissue of CIMP-positive endometrial cancer compared to CIMP-negative endometrial cancer.
CIMP was first identified in colorectal cancer by the study of Toyota et al (12), and in the past decade CIMP-positive colorectal cancer has been related to BRAF mutation, MLH1 methylation and microsatellite instability (MSI) (13,14). However, only a few reports have described CIMP-positive endometrial cancer (10,15), but good CIMP markers for endometrial cancer have not been identified. A recent study also showed that there was no BRAF mutation in endometrial cancer (16,17) and suggested that CIMP markers for colorectal cancer were not suitable for endometrial cancer. In previous studies (18–21), we have shown that promoters of certain tumor suppressor genes were methylated in endometrial cancer tissue, but not in normal endometrium: MLH1, APC and CDH1 were most frequently methylated in endometrial cancer and MLH1 and APC were already methylated in atypical endometrial hyperplasia (AEH). These results suggest that methylation of these gene promoters plays an important role as early event in carcinogenesis of endometrial cancer formation. Thus, we decided to examine methylation status of these three genes as CIMP markers for endometrial cancer in the present study.
The aim of the present study was to investigate genomewide DNA methylation status of both CIMP-positive and negative cancers. In addition, by comparing normal and cancer tissues from these two CIMP classes, we attempted to identify epimutation candidates in CIMP-positive endometrial cancer. The epimutation candidate will be useful as a predictive marker for CIMP-positive endometrial cancer. Further analysis of the epimutation candidate may contribute to understanding the molecular mechanisms underlying CIMP-positive endometrial cancer formation. Furthermore, as DNA methylation is a reversible modification, the results of the present study may contribute to development of 'epigenetic medicine' for cancer prevention.
Materials and methods
DNA and RNA extraction from patient samples
The subjects were 25 Japanese patients diagnosed with endometrial cancer at the Department of Obstetrics and Gynecology, Keio University Hospital from December 2013 to March 2015. Patients aged under 20 years of age were excluded. The 25 patients had an age range of 33–76 years. Clinicopathological data are shown in Table I. Fifty paired peripheral blood and cancer tissue samples were collected from the 25 patients and stored at 4 and −80°C, respectively, until DNA or RNA extraction using an AllPrep DNA/RNA/miRNA Universal kit (Qiagen, Hilden, Germany). The study protocol (no. 2013258) was approved by the Institutional Review Board of Keio University School of Medicine, and the study was performed in compliance with the Declaration of Helsinki. All participants gave written informed consent.
Table I.
Clinicopathological characteristics in endometrial cancer patients.
| Clinicopathological characteristics | Data |
|---|---|
| Age (years) | |
| Average | 55.08 |
| Range | (33–76) |
| Histological type | |
| Endometrioid adenocarcinoma | 25 |
| Differentiation | |
| G1 | 14 |
| G2 | 7 |
| G3 | 4 |
| Stage | |
| I | 17 |
| II | 6 |
| III | 2 |
G1, well-differentiated; G2, moderately differentiated; G3, poorly differentiated.
Bisulfite treatment and methylation specific polymerase chain reaction (MSP)
DNA (1 μg) in a volume of 50 μl was denatured by adding 5.5 μl of 2N NaOH. After incubation of the sample at 37°C for 15 min, 30 μl of 10 mM hydroquinone (Sigma-Aldrich, St. Louis, MO, USA) and 520 μl of 2M sodium bisulfate pH 5.5 (Sigma-Aldrich) were added. The sample was gently mixed and centrifuged briefly, after which the solution was overlaid with 200 μl of mineral oil and incubated at 50°C for 20 h. After incubation, 1 ml of Wizard DNA Clean-up resin (Promega, Madison, WI, USA) was added to the lower layer and mixed for DNA purification. This procedure gave 50 μl of DNA solution, to which 5.5 μl of 3N NaOH was added, and the solution was incubated at 37°C for 20 min. Next, 66 μl of 5N ammonium acetate (Sigma-Aldrich) and 243 μl of 95% ethanol were added and the solution was incubated at −80°C for 1 h. After centrifugation at 20,000 × g for 45 min at 4°C, the DNA pellet was rinsed with 1 ml of 70% ethanol and centrifuged at 20,000 × g for 30 min at 4°C. Precipitated DNA was air dried and resuspended in 20 μl of Milli-Q water. Aliquots of this solution (2 μl) were used as the MSP template. AmpliTaq Gold with 10X PCR Gold Buffer and MgCl2 (Applied Biosystems, Foster City, CA, USA) were used for MSP and the methylation status of each gene was analyzed using a ProFlex PCR System (Applied Biosystems). CpGenome Universal Methylated DNA and Unmethylated DNA (Millipore, Temecula, CA, USA) were used as positive controls for methylated and unmethylated PCR, respectively.
Each 25 μl PCR reaction mixture contained 1X PCR buffer, 0.8 μM primers, 200 μM dNTP, 3 mM MgCl2 and 1U Taq polymerase. The primers and PCR conditions for MSP analysis were as follows: for MLH1, M-forward, 5′-ACG TAG ACG TTT TAT TAG GGT CGC-3′ and M-reverse, 5′-CCT CAT CGT AAC TAC CCG CG-3′, U-forward, 5′-TTT TGA TGT AGA TGT TTT ATT AGG GTT GT-3′ and U-reverse, 5′-ACC ACC TCA TCA TAA CTA CCC ACA-3′, 95°C for 10 min, 5 cycles at 94°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec, 30 cycles at 94°C for 30 sec, 55°C for 30 sec, 72°C for 30 sec, and 72°C for 10 min; for APC, M-forward, 5′-TAT TGC GGA GTG CGG GTC-3′ and M-reverse, 5′-TCG ACG AAC TCC CGA CGA-3′, U-forward, 5′-GTG TTT TAT TGT GGA GTG TGG GTT-3′ and U-reverse, 5′-CCA ATC AAC AAA CTC CCA ACA A-3′, 95°C for 10 min, 35 cycles at 95°C for 30 sec, 68°C (M) or 66°C (U) for 30 sec, 72°C for 30 sec and 72°C for 10 min. A CpG WIZ E-cadherin amplification kit (Millipore) was used for CDH1 MSP analysis, using methylated and unmethylated primer sets supplied with the kit and PCR conditions of 95°C for 10 min, 35 cycles at 95°C for 45 sec, 60°C for 45 sec, 72°C for 45 sec and 72°C for 10 min. PCR products were separated by electrophoresis on a 3% agarose gel and stained with ethidium bromide.
Based on the MSP analysis, patients with methylation of two of the three genes were defined as CIMP-High (CIMP-H), those with methylation of one of the genes as CIMP-Low (CIMP-L) and those with no methylation as CIMP-negative (CIMP(-)).
DNA methylation analysis using next-generation sequencing (NGS)
A SureSelect Human Methyl-Seq Capture Library and a SureSelect Target Enrichment kit (Agilent Technologies, Santa Clara, CA, USA) were used to investigate the genomewide DNA methylation status. The SureSelect Human Methyl-Seq kit captures 84 Mb of the human genome with 3.7 million individual CpG dinucleotides covering ~91% CpG islands and ~141,000 promoters. We used the kit for DNA library preparation with the SureSelect Methyl-Seq and Postbisulfite Adaptor Tagging Protocol (http://www.chem-agilent.com/pdf/PBAT_SureSelect_Methyl_DraftB_19AUG15.pdf#search='SureSelectPBAT'). In brief, 100 ng of dsDNA measured with a Qubit dsDNA BR Assay kit (Life Technologies, Carlsbad, CA, USA) was diluted in 130 μl of TE buffer and fragmented into 500–600 bp with Covaris S2 (Covaris, Woburn, MA, USA). After sheared DNA purification with AMPure XP beads (Beckman Coulter, Brea, CA, USA), the DNA was hybridized with a biotinylated RNA probe and enriched with Dynabeads MyOne Streptavidin T1 (Life Technologies). Bisulfite conversion was performed using an EZ DNA Methylation-Gold kit (Zymo Research, Irvine, CA, USA) according to the SureSelect-PBAT protocol. After first and second strand synthesis with Klenow fragment (3′→5′ exo-) (NEB, Ipswich, MA, USA), the concentration of library DNA was measured with a Library Quantification kit for Illumina (Kapa Biosystems, Boston, MA, USA). PCR amplification of 7–15 cycles was performed using a GeneAmp PCR System 9700 (Applied Biosystems). After measuring the final concentration of template DNA using qPCR, 10 pM DNA was used for sequencing. PhiX Control v3 (Illumina, San Diego, CA, USA) was spiked at a final concentration of 1.6 pM and 2×75 paired-end sequencing was performed using a MiSeq reagent kit v3 (Illumina).
Data analysis
To avoid low quality reads and contamination by adapter sequences, quality control and trimming were performed using FASTQ Toolkit ver. 2.0.0 (BaseSpace on the Illumina web site). After trimming of adapter sequences, the read pairs for which the 3′ end quality score was <30 were excluded. After adapter sequence trimming, 15 bps were trimmed from both the 5′ and 3′ ends. A quality control check of trimmed read pairs was performed using FASTQC ver. 1.0.0 (BaseSpace on Illumina web site).
Reads were aligned to the reference human genome (hg19) using Bismark ver. 0.14.5, Bowtie2 ver. 2.2.6 and Samtools ver. 1.2 with settings of --pbat, --score_min L, −0.6, −0.6 -D 150 -X 1000. PCR duplicates were removed using default Bismark settings. Methylation calling was also processed using Methylation Extractor in the Bismark module for visualization on IGV. The SureSelect Human Methyl-Seq kit captures 84 Mb of the human genome with 3.7 million individual CpG dinucleotides. Thus, the on-target rate against a designed target sequence was measured using Bedtool ver. 2.19.1. After deduplication by Bismark, the output SAM file was used as input to methylKit ver. 0.9.2. (22) with default settings. Correlation plots and detection and annotation of differentially methylated CpGs (DMCs) and differentially methylated regions (DMRs) were performed in MethylKit. The minimum read coverage was set to 10 to measure DMCs and DMRs. A 25% methylation difference has been shown to induce a 2-fold repression in gene expression (23). Thus, we used a methylation difference >25% and a cut-off of q<0.01. DMRs were identified using a 50-bp window and a 10-bp step size.
Validation of genome-wide bisulfite sequencing
Bisulfite treated DNA was amplified by PCR with the following primers and condition; miR-663aBS forward, 5′-GTTTGTAGAGGA ATTTTTTTTAGTT-3′ and reverse, 5′-ACCACAACCACA AACTCAAC-3′, 95°C for 10 min, 35 cycles at 95°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec and 72°C for 10 min. Other PCR settings were same as MSP. PCR products were separated by electrophoresis on a 3% agarose gel and stained with ethidium bromide, and purified with a NucleoSpin Gel and PCR Clean-up kit (Takara Bio, Tokyo, Japan) according to the manufacturer's instructions. The PCR products were TA cloned by using the pGEM-T Easy Vector System (Promega) according to the manufacturer's instructions. PCR with universal T7 and SP6 primers was performed on transformed colonies and correctly inserted clonal amplicons were sent to The Core Instrumentation Facility in Keio University for sequencing.
Semi-quantitative RT-PCR and RT-qPCR
cDNA for miR-663a expression analysis was synthesized with 0.5 μg of total RNA using a Mir-X miRNA First-Strand Synthesis kit (Clontech Laboratories, Mountain View, CA, USA). AmpliTaq Gold with 10X PCR Gold Buffer and MgCl2 (Applied Biosystems) were used in the semi-quantitative RT-PCR analysis. PCR amplification was performed using a ProFlex PCR System (Applied Biosystems). Each 25 μl PCR reaction mixture contained 1X PCR buffer, 1.0 μM primers, 200 μM dNTP, 3 mM MgCl2 and 1U Taq polymerase, with 5% dimethyl sulfoxide (DMSO) spiked in the reaction mix for miR-663a analysis. The primers and the PCR conditions for semi-quantitative RT-PCR were forward primer for miR-663a 5′-AGG CGG GGC GCC GCG GGA CCG C-3′, reverse primer for miR-663a and U6 control primers supplied with the kit, 95°C for 10 min, 28 cycles of 95°C for 20 sec, 68°C for 20 sec, 72°C for 20 sec and 72°C for 10 min. PCR products were separated by electrophoresis on a 3% agarose gel and stained with ethidium bromide. Signals were quantified using an E-BOX VX2 system and E-Capt software (Bilber Lourmat, Marne-la-Vallée, France). miR-663a relative expression was calculated using U6 as an internal control.
cDNA for GAPDH, TGF-β, DNMT1, DNMT3a and DNMT3b expression analysis was synthesized with 1 μg of total RNA using a Superscript First-Strand Synthesis system for RT-PCR kit (Invitrogen, Carlsbad, CA, USA), with 1 μl of synthesized First-Strand cDNA as template. Thunderbird SYBR qPCR Mix (Toyobo, Co., Ltd., Tokyo, Japan) was used for RT-qPCR, with 10 μl of PCR reaction mixture containing 1X qPCR mix and 0.3 μM primers. The primers and PCR conditions used for RT-qPCR were as follows: for GAPDH forward, 5′-GAA GGT GAA GGT CGG AGT C-3′ and reverse, 5′-GAA GAT GGT GAT GGG ATT TC-3′; for TGF-β forward, 5′-AGT GGA CAT CAA CGG GTT CAG-3′ and reverse, 5′-CAT GAG AAG CAG GAA AGG CC-3′; for DNMT1 forward, 5′-AAG GGA AGG GCA AGG GAA AAG G-3′ and reverse, 5′-AGA AAA CAC ATC CAG GGT CCG CAG-3′; for DNMT3a forward, 5′-GAT TGA TGC CAA AGA AGT GTC AG-3′ and reverse, 5′-CAT TCA CAG TGG ATG CCA AC-3′; for DNMT3b forward, 5′-AAT GTG AAT CCA GCC AGG AAA GGC-3′ and reverse, 5′-ACT GGA TTA CAC TCC AGG AAC CGT-3′; 95°C for 30 sec, 40 or 50 cycles of 95°C for 5 sec, 60°C for 30 sec. Quantification was performed using a LightCycler® 480 system (Roche Diagnostics, Basel, Switzerland). Expression of TGF-β and DNMTs was calculated by the ΔΔCq method using GAPDH as an internal control.
Results
Defining CIMP classes based on the MSP analysis of MLH1, APC and CDH1
All endometrial cancer tissues were analyzable by MSP (Fig. 1 and Table II). The methylation frequencies in the MLH1, APC and CDH1 promoter regions in these tissues were 32.0% (8/25), 12.0% (3/25) and 0.0% (0/25), respectively. Based on the MSP analysis, we defined CIMP classes as follows: cases with hypermethylation in more than two of the three promoter regions are defined as CIMP-high, whereas cases with no hypermethylation in the corresponding regions are CIMP(-). If only one of the regions is hypermethylated, we defined these cases as CIMP-low. According to these criteria, we identified 2 CIMP-H (8.0%; 2/25), 7 CIMP-L (28.0%; 7/25) and 16 CIMP(-) (64.0%; 16/25).
Figure 1.

MSP analysis of MLH1, APC and CDH1 genes in endometrial cancer tissues. Endometrial cancer samples showed aberrant DNA methylation in MLH1 and APC gene promoter regions. (M), methylated PCR; (U), unmethylated PCR; m, marker; P positive control; N, negative control.
Table II.
Aberrant DNA méthylation of three genes in endometrial cancer.
| No. | Result of MSP
|
Classification of CIMP | Age (years) | Histological type | Differentiation | Stage | ||
|---|---|---|---|---|---|---|---|---|
| hMLH1 | APC | CDH1 | ||||||
| 1 | U | U | U | CIMP (-) | 69 | Endometrioid adenocarcinoma | G3 | IIIc2 |
| 2 | M | U | U | CIMP-L | 47 | Endometrioid adenocarcinoma | G2 | Ia |
| 3a | U | U | U | CIMP (-) | 50 | Endometrioid adenocarcinoma | G2 | Ia |
| 4 | U | U | U | CIMP (-) | 66 | Endometrioid adenocarcinoma | G2 | Ib |
| 5 | M | U | U | CIMP-L | 50 | Endometrioid adenocarcinoma | G1 | Ia |
| 6 | U | U | U | CIMP (-) | 65 | Endometrioid adenocarcinoma | G1 | Ia |
| 7 | U | U | U | CIMP (-) | 70 | Endometrioid adenocarcinoma | G2 | Ia |
| 8 | M | U | U | CIMP-L | 50 | Endometrioid adenocarcinoma | G2 | II |
| 9 | M | U | U | CIMP-L | 53 | Endometrioid adenocarcinoma | G3 | II |
| 10 | U | U | U | CIMP (-) | 76 | Endometrioid adenocarcinoma | G2 | Ia |
| 11 | M | U | U | CIMP-L | 54 | Endometrioid adenocarcinoma | G3 | Ia |
| 12 | U | U | U | CIMP (-) | 63 | Endometrioid adenocarcinoma | G1 | II |
| 13 | U | U | U | CIMP (-) | 42 | Endometrioid adenocarcinoma | G1 | Ia |
| 14 | M | U | U | CIMP-L | 58 | Endometrioid adenocarcinoma | G1 | Ia |
| 15a | M | M | U | CIMP-H | 56 | Endometrioid adenocarcinoma | G2 | Ia |
| 16 | U | M | U | CIMP-L | 56 | Endometrioid adenocarcinoma | G1 | Ia |
| 17 | U | U | U | CIMP (-) | 64 | Endometrioid adenocarcinoma | G3 | II |
| 18 | U | U | U | CIMP (-) | 54 | Endometrioid adenocarcinoma | G1 | II |
| 19 | U | U | U | CIMP (-) | 55 | Endometrioid adenocarcinoma | G1 | IIIc2 |
| 20 | U | U | U | CIMP (-) | 45 | Endometrioid adenocarcinoma | G1 | Ia |
| 21 | U | U | U | CIMP (-) | 65 | Endometrioid adenocarcinoma | G1 | Ib |
| 22 | U | U | U | CIMP (-) | 44 | Endometrioid adenocarcinoma | G1 | Ia |
| 23a | U | U | U | CIMP (-) | 46 | Endometrioid adenocarcinoma | G1 | Ia |
| 24 | U | U | U | CIMP (-) | 46 | Endometrioid adenocarcinoma | G1 | II |
| 25a | M | M | U | CIMP-H | 33 | Endometrioid adenocarcinoma | G1 | Ia |
M, methylated; U, unmethylated;
The samples used for genome-wide bisulfite sequencing.
Genome-wide bisulfite sequencing in CIMP-H and CIMP(-) cases
The SureSelect Human Methyl-Seq method adopting PBAT protocol was used for genome-wide bisulfite targeted sequencing analysis. The SureSelect method captures 84 Mb of the human genome containing 3.7 million individual CpG dinucleotides in theory. The captured regions cover >90% of the CpG islands and ~141,000 regions corresponding to the gene promoters defined by GENCODE. Cancer-specific or tissues-specific differentially methylated regions (DMRs) can also be analyzed by this method. PBAT protocol was used to construct the libraries, as this protocol requires less amount of materials compared to the Methyl-Seq procedures (24). Using this genome-wide analytical method, we investigated DNA methylation status of the paired samples of PBC and cancer from two CIMP-H and two CIMP(-) patients. The two CIMP(-) patients were selected based on patient age, cancer stage and differentiation status. We sequenced total of eight bisulfite-converted DNA libraries and an average of 13.6 million paired end sequence reads were obtained from each library. The mapping rate against the human reference genome (hg19) ranged from 20.4 to 37.9%, which is consistent with the fact that sequences obtained from PBAT library give lower mapping rate compared to those from other bisulfite sequencing libraries (https://sequencing.qcfail.com/articles/). The duplication rate, which varies depending on the number of amplification PCR cycles used, was 15.91–85.93%. Despite the high duplication rate, the average on-target rate for all 8 samples was 84.24% (Table III).
Table III.
Summary of mapping of Methyl-Seq Libraries.
| No. of reads (pair) | Mapping
|
Duplication
|
On-target
|
|||||
|---|---|---|---|---|---|---|---|---|
| No. of reads (pair) | Mapping rate (%) | No. of duplicated reads (pair) | Duplication rate (%) | Leftover sequences (single) | No. of reads (single) | On-target rate (%) | ||
| CIMP-H | ||||||||
| no. 15 PBC | 9,247,852 | 1,923,159 | 20.8 | 305,967 | 15.91 | 3,234,384 | 2,795,612 | 86.43 |
| no. 15 cancer | 18,617,848 | 5,638,156 | 30.3 | 4,705,597 | 83.46 | 1,865,098 | 1,466,259 | 78.62 |
| no. 25 PBC | 8,738,237 | 1,785,247 | 20.4 | 300,533 | 16.83 | 2,969,426 | 2,583,578 | 87.01 |
| no. 25 cancer | 18,123,262 | 5,882,527 | 32.5 | 4,536,000 | 77.11 | 2,693,030 | 2,270,036 | 84.29 |
| CIMP (-) | ||||||||
| no. 3 PBC | 12,788,054 | 3,681,561 | 28.8 | 1,185,309 | 32.20 | 4,992,504 | 4,249,381 | 85.12 |
| no. 3 cancer | 13,742,323 | 3,015,433 | 21.9 | 2,591,097 | 85.93 | 848,654 | 705,353 | 83.11 |
| no. 23 PBC | 10,010,082 | 2,477,429 | 24.7 | 541,780 | 21.87 | 3,871,296 | 3,335,224 | 86.15 |
| no. 23 cancer | 17,277,985 | 6,543,732 | 37.9 | 4,182,685 | 63.92 | 4,722,094 | 3,926,204 | 83.15 |
Identification of DMCs and DMRs in CIMP-H and CIMP(-) cases
After alignment of the sequence reads, we calculated methylation % of each read and used the methylKit program to analyze global DNA methylation profiles among the samples. For this analysis, only CpG sites with sequence depth of >10 were used. The average number of the analyzed CpGs here was 35,000 only (range, 4,104–103,761), ~1% of CpG sites covered by the SureSelect method (although the method used would be able to analyze 3.7 million CpGs, due to the small number of sequence reads, we could analyze ~35,000 CpGs on average). The global methylation profiles were similar between the PBC samples and the cancer samples, showing a high positive correlation between CIMP-H and CIMP(-) cancer (Pearson correlation coefficient: 0.89–0.95). As shown in the histograms of Fig. 2, normal tissues, i.e. PBCs, of both CIMP-H and CIMP(-) cases show bimordal distribution with peaks at low (<30%) and high (>90%) DNA methylation. On the contrary, cancer tissues show relatively hypomethylated status, demonstrating a peak at very low (<10%) DNA methylation level. This trend is true for both CIMP-H and CIMP(-), and is consistent with the notion that cancer genome is generally hypomethylated (25,26).
Figure 2.

Scatter plot and correlation of CpG methylation between CIMP-H and CIMP(-) cases. Heat plots show % methylation for pairwise comparisons of four samples. Numbers in the upper right corner denote Pearson correlation coefficients. The histograms on the diagonal are the frequency of % methylation per cytosine for each pool. (A) PBCs; (B) cancer samples.
DMCs and DMRs between the CIMP-H and CIMP(-) samples were also identified based on a q-value <0.01 and a méthylation difference >25%
Table IV shows differentially methylated CpG sites among the samples. For example, in comparison of CIMP-H cancer with CIMP(-) cancer, 9 out of 573 informative CpG sites were found to be significantly hypermethylated in CIMP-H. There is no hypomethylated CpG site in CIMP-H relative to CIMP(-) in this comparison. Six out of these 9 DMCs are located in the gene promoter regions. DMR analysis also showed similar results, demonstrating that the DMRs are always hypermethylated in the CIMP-H cases (Table V). Notably, when PBCs of the CIMP-H cases were compared with those of the CIMP(-), 7 hypermethylated sites were detected in the CIMP-H, of which 4 were in the promoter regions. In contrast to cancer tissues, 8 out of the 14 DMRs detected between CIMP-H and CIMP(-) PBCs are hypomethylated in the CIMP-H cases (Table V). Fig. 3 shows chromosomal distribution of the DMCs and DMRs detected by the methylKit. Seven DMCs detected in the comparison of CIMP-H PBCs with CIMP(-) PBCs are located on chromosomes 20 and 21.
Table IV.
Details of DMCs in each comparison.
| No. of DMC (meth.diff >1%) | (meth.diff >25%, q-value <0.01)
|
||||
|---|---|---|---|---|---|
| Hypermethylated CpGs | Hypomethylated CpGs | Hypermethylated CpGs in promoter region | Hypomethylated CpGs in promoter region | ||
| CIMP-H PBC vs. CIMP(-) PBC | 1,218 | 7 | 0 | 4 | 0 |
| CIMP-H cancer vs. CIMP-H PBC | 745 | 132 | 12 | 67 | 7 |
| CIMP(-) cancer vs. CIMP(-) PBC | 396 | 31 | 8 | 14 | 2 |
| CIMP-H cancer vs. CIMP(-) cancer | 573 | 9 | 0 | 6 | 0 |
meth.diff, percent methylation difference cut-offs.
Table V.
Details of DMRs in each comparison
| No. of DMR (meth.diff >1%) | (meth.diff >25%, q-value <0.01)
|
||||
|---|---|---|---|---|---|
| Hypermethylated regions | Hypomethylated regions | Hypermethylated regions in promoter region | Hypomethylated regions in promoter region | ||
| CIMP-H PBC vs. CIMP(-) PBC | 2,427 | 6 | 8 | 3 | 0 |
| CIMP-H cancer vs. CIMP-H PBC | 269 | 232 | 37 | 114 | 14 |
| CIMP(-) cancer vs. CIMP(-) PBC | 919 | 51 | 17 | 22 | 5 |
| CIMP-H cancer vs. CIMP(-) cancer | 1,311 | 35 | 0 | 13 | 0 |
meth.diff, percent methylation difference cut-offs.
Figure 3.

Chromosomal distribution of the DMCs and DMRs. (A–D) Visualization of the distribution of differentially methylated CpG sites and differentially methylated regions on each chromosome. (A) DMCs in PBCs. (B) DMRs in PBCs. (C) DMCs in cancer tissues. (D) DMRs in cancer tissues. Proportion (%) of DMCs (A and C) or DMRs (B and D) in all the CpG sites per chromosome is depicted as bar graph. In these comparisons, if CIMP-H samples show hypermethylation relative to CIMP(-) cases, the bar is colored pink. If CIMP-H samples show hypomethylation to CIMP(-), the bar is green. (E–H) Annotation of DMCs/DMRs. Classification of the detected DMCs and DMRs into genomic domains such as promoter (blue), exon (orange), intron (green), and intergenic regions (purple). (E) DMCs in PBCs. (F) DMRs in PBCs. (G) DMCs in cancer tissues. (H) DMRs in cancer tissues.
Next, the DMCs and DMRs were classified into annotated genomic domains such as promoters, introns, exons and intergenic regions (Fig. 3E–H). Here the promoter regions are defined as +1000 to −1000 bp from the transcription start sites (TSS). Fifty-seven percent of the DMCs (4/7) and 21% of DMRs (3/14) detected in the PBC group comparisons were found to be located in promoter regions (Fig. 3E and F and Tables IV and V). Since methylation of promoter region DNA suppresses gene expression, expression of genes with methylated promoters may be already suppressed in the CIMP-H PBCs. Moreover, all 4 hypermethylated DMCs and 1 of the 3 hypermethylated DMRs in the CIMP-H PBCs were located on the MIR663A host gene of human chromosome 20 (Fig. 4 and Tables VI and VII). MIR663A host gene harbors the miR-663a microRNA sequence. There are two other hypermethylated DMRs with gene annotation of microRNA 3648-1 and MTR-1-P. A previous genome-wide human epigenome study showed that the miR-663a region is normally hypomethylated in all the tissues tested (27). These results suggested that the identified differential DNA methylation is an aberrant modification occurring in the CIMP-H patients and may represent a candidate of epimutation.
Figure 4.

Visualization of % methylation in the miR-663a region in all eight libraries. The blue peak indicates the % methylation at each CpG site (0–100%). Arrows indicate hypermethylated DMCs/DMRs in CIMP-H PBCs compared to CIMP(-) PBCs.
Table VI.
Annotation of hypermethylated DMCs (CIMP-H PBCs vs. CIMP(-) PBCs).
| Feature.name | Chromosome | Feature. strand | Start | End | q-value | Meth.diff | Gene name |
|---|---|---|---|---|---|---|---|
| NR_040095 | 20p11.1 | – | 26190118 | 26190118 | 0.007784562 | 33.33333 | MIR663A host gene |
| NR_040095 | 20p11.1 | – | 26190161 | 26190161 | 0.000695757 | 26.59274 | MIR663A host gene |
| NR_040095 | 20p11.1 | – | 26190239 | 26190239 | 0.000907851 | 26.28239 | MIR663A host gene |
| NR_040095 | 20p11.1 | – | 26190246 | 26190246 | 0.007784562 | 26.42045 | MIR663A host gene |
meth.diff, percent methylation difference cut-offs.
Table VII.
Annotation of hypermethylated DMRs (CIMP-H PBCs vs. CIMP(-) PBCs).
| Feature.name | Chromosome | Feature. strand | Start | End | q-value | Meth.diff | Gene name |
|---|---|---|---|---|---|---|---|
| NR 037421 | 21 | + | 98225451 | 9825500 | 0.000009127 | 26.02800 | microRNA 3648-1 |
| NM_032285 | 19p13.2 | + | 13875071 | 13875120 | 0.005663958 | 25.67164 | MTR-1-P |
| NR_040095 | 20p11.1 | – | 26189381 | 26189430 | 0.000312766 | 25.48611 | MIR663A host gene |
meth.diff, percent methylation difference cut-offs.
There were 67 hypermethylated DMCs and 114 hypermethylated DMRs in the CIMP-H cancer group compared to the CIMP-H PBC group, whereas only 14 hypermethylated DMCs and 22 hypermethylated DMRs in the CIMP(-) cancer group compared to the CIMP(-) PBC group. The number of hypermethylated DMCs/DMRs in the CIMP-H group was ~5-fold (4.8-fold for DMCs, 5.3-fold for DMRs) higher than that in the CIMP(-) group (Tables IV and V). These results suggest that the CIMP-H cases appear to gain hypermethylation in gene promoters during endometrial cancer development, and that in general the CIMP-H cases have elevated DNA methylation in promoter regions compared to the CIMP(-) cases.
Validation of methylation status in miR-663a promoter region
As described above, MIR663A host gene that includes miR-663a promoter region is hypermethylated in the PBCs of the CIMP-H cases, though this region is known to be hypomethylated in PBCs of normal individuals. Human miR-663a is a microRNA possibly involved in tumorigenesis (28,29). Therefore, aberrant DNA methylation in the miR-663a promoter may be involved in CIMP-H endometrial cancer formation. We next validated methylation status of miR-663a promoter region in the CIMP-H and the CIMP(-) samples by conventional bisulfite sequencing. Consistent with the genomewide bisulfite sequencing, the CIMP-H PBCs showed higher level of methylation than the CIMP(-) PBCs. Furthermore, the correspondig region in the cancer tissues of the CIMP-H and the CIMP(-) cases demonstrated that this region is almost completely methylated (Fig. 5).
Figure 5.

Bisulfite sequencing of PBC and cancer from CIMP-H and CIMP(-) samples. No. 3 patient samples were used as representative data of CIMP(-). No. 25 patient samples were used as representative data of CIMP-H. Methylated CpGs are shown as black and unmethylated CpGs as white circles.
Expression analysis of miR-663a and its target genes
The average miR-663a expression level in the CIMP-H PBCs (0.54) was lower than that in the other PBCs (CIMP-L, 0.81, CIMP(-), 0.70; Fig. 6A). This suggests that miR-663a expression in the CIMP-H PBCs is reduced by the methylation of the promoter region. We also analyzed expression of TGF-β, a possible target gene of miR-663a, using RT-qPCR. However, there was no significant difference in TGF-β mRNA expression between the CIMP-H and the CIMP(-) PBCs (Fig. 6B), with an average expression level of 1.4 in each group. However, the average TGF-β expression in CIMP-H cancer samples (5.4) was higher than that in the CIMP(-) cancer samples (4.9) (Fig. 6C).
Figure 6.
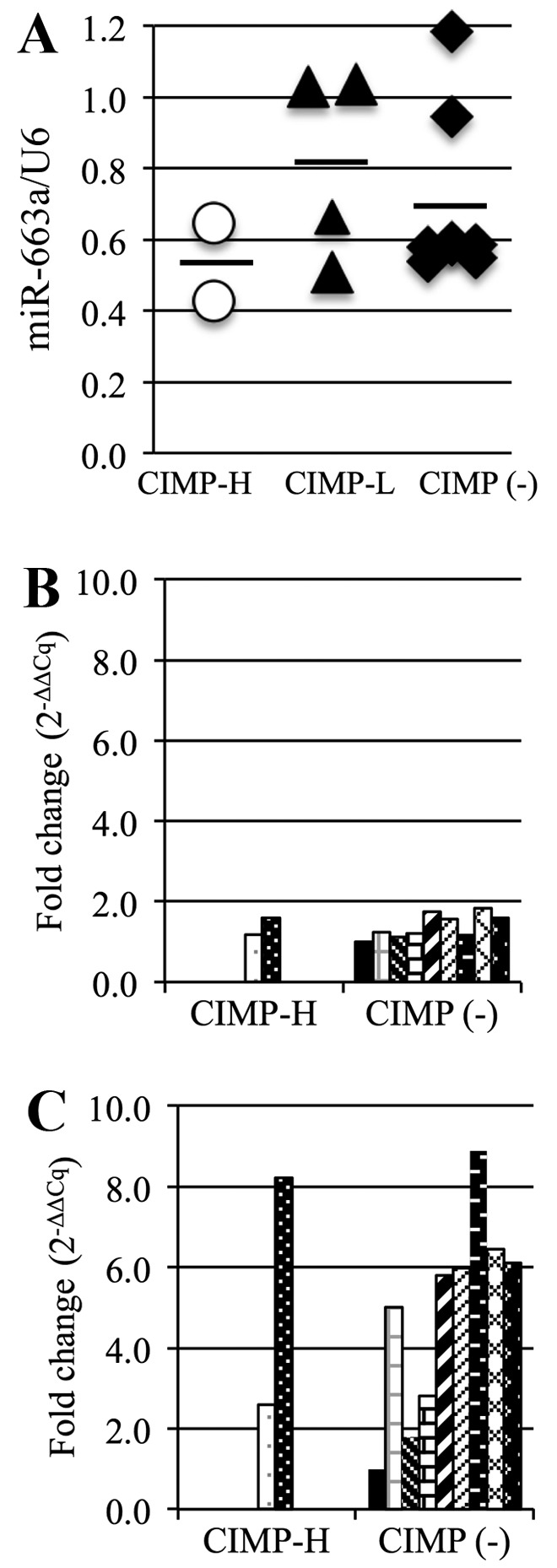
miR-663a and TGF-β expression analysis. (A) miR-663a expression in all 3 PBC groups analyzed by semi-quantitative RT-PCR. Each dot represents 1 patient, bars indicate average. (B and C) TGF-β expression in CIMP-H and CIMP(-) PBCs (B) and CIMP-H and CIMP(-) cancer samples (C).
Analysis of DNMT expression in CIMP-H and CIMP(-) patient samples
TGF-β induces global DNA methylation through upregulation of DNMTs (30). Thus, we suspected that DNMT expression may contribute to the development of CIMP-H methylation phenotype. The average mRNA expression levels for all three DNMTs was lower in the CIMP-H PBCs than in the CIMP(-) PBCs, but the average levels of DNMT1 and DNMT3b in the CIMP-H cancer samples was higher than that in the CIMP(-) cancer samples (Fig. 7). The average mRNA levels in the CIMP-H and the CIMP(-) cases were 2.7 and 3.4 for DNMT1, 2.1 and 2.7 for DNMT3a, and 1.1 and 2.8 for DNMT3b in PBCs; and 3.1 and 2.4 for DNMT1, 2.6 and 2.7 for DNMT3a, and 2.7 and 1.3 for DNMT3b in cancer samples. These results suggest that expression of DNMTs, especially DNMT3b, are linked to the CIMP-H endometrial cancer phenotype.
Figure 7.

Analysis of DNMT expression in PBCs and cancer samples. (A) DNMT1, 3a, 3b in CIMP-H and CIMP(-) PBCs measured by RT-qPCR. Each dot represents one patient, bars are averages. (B) DNMT1, 3a, 3b in CIMP-H and CIMP(-) cancer samples measured by RT-qPCR. Each dot represents one patient, bars are averages.
Discussion
Endometrial cancer is known to have a CIMP phenotype (10,15,31), similarly to other cancers. There have been several reports on the relationship of CIMP-positive cancer with genetic mutations and clinicopathological features (9,11,32), but the mechanism of carcinogenesis in CIMP-positive cancer is still unknown. The present study is the first to focus on the cause of CIMP-positive endometrial cancer and demonstrated for the first time that aberrant DNA methylation occurs in the miR-663a promoter region in normal tissue, i.e. PBCs, of patients with CIMP-H endometrial cancer. The miR-663a promoter was fully methylated in the cancer tissues of both CIMP-H and CIMP(-) cases examined in this study. The miR-663a promoter region is known to be unmethylated in all the tissues of normal individuals (33), and miR-663a is thought to be involved in the formation of certain cancers. Therefore, it is possible that aberrant DNA methylation in the miR-663a promoter is involved in endometrial cancinogenesis.
Methylation levels of miR-663a promoter DNA is higher in the CIMP-H than in the CIMP(-) cases, suggesting that aberrant DNA methylation may be associated with the CIMP-H phenotype, and that it could serve as an epigenetic marker for endometrial cancer diagnosis or prediction. We do not know at present that this epimutation in the miR-663a promoter occurs de novo during development (primary epimutation), or is involved in a genetic mutation (secondary epimutation). Primary epimutations are defined as constitutional epimutations that are independent of genetic mutation. On the other hand, secondary epimutations are caused by genetic mutation in other loci controlling its epigenetic modification (34). To address these issues, it will be important to analyze genetic alterations of regions adjacent to the miR-663a locus and to perform systematic survey of the miR-663a epimutation in larger number of patients in future.
The proportion of CIMP-H, CIMP-L and CIMP(-) endometrial cancer in our samples were 8.0, 28.0 and 64.0%, respectively. The frequencies of MLH1, APC and CDH1 methylation were consistent with previous studies (15,35), but the frequency of CIMP-positive endometrial cancer was found to be lower in this study than in the previous studies, in which 49 or 75% of the cases were found to be CIMP-H (6,7). This is probably due to the use of different CIMP markers in each study. Thus, further investigation is required to identify the best CIMP markers with higher detection power for CIMP-H phenotype. In this study, MLH1, APC and CDH1 were used for definition of the CIMP phenotype because we have shown that these genes are frequently methylated in endometrial cancer (19), of which MLH1 promoter is most frequently methylated in endometrial carcinogenesis. MLH1 and APC methylation is also observed in AEH. Here we demonstrated that the CIMP-H cases defined by MLH1 and APC had hypermethylated DMCs and DMRs compared to CIMP(-) endometrial cancer, suggesting usefulness of MLH1 and APC for detection of CIMP-H endometrial cancers.
Genome-wide bisulfite sequencing was performed to identify DMCs and DMRs in CIMP-H endometrial cancer. Since total number of sequence reads obtained in this study were low, we could analyze only ~35,000 CpGs on average. Despite such low number of analyzed CpGs, we identified aberrant DNA methylation in the miR-663a promoter of CIMP-H PBCs, which is normally unmethylated. Therefore, deeper sequencing analysis of the CIMP-H normal tissues is likely to reveal more aberrant DNA methylation or epimutation.
DNA methylation status of miR-663a promoter region in both CIMP-H and CIMP(-) cases was validated by conventional bisulfite sequencing. Expression analysis of miR-663a and its target genes revealed that miR-663a expression in the CIMP-H normal tissue was lower than that in the CIMP(-) normal tissue. Since peripheral blood cells and endometrium are of the same mesoderm origin, the DNA methylation status of PBCs is likely to share those of normal endometrium and other mesoderm-derived tissues.
miRNAs are small non-coding RNAs that regulate expression of gene products through translational inhibition or cleavage of target mRNAs. miRNAs have been associated with oncogenesis and tumor progression in many cancer types (36–40), and miR-663a has roles in cell proliferation, immunity and cancer, and acts as a tumor suppressor miRNA in gastric cancer (41). miR-663a is downregulated by promoter methylation in breast, hepatocellular and other cancers (42–44). The CpG island located 1 kb upstream of the pre-miR-663 sequence shows promoter activity (45), and a DMR identified in the comparison of CIMP-H and CIMP(-) PBC was included in this region.
TGF-β is a direct target of miR-663a (46) and has oncogenic activities and is upregulated in endometrial cancer (47). We found no inverse correlation between miR-663a and TGF-β expression levels detected by qRT-PCR in the PBCs. However, miR-663a has been shown to inhibit TGF-β expression at the post-transcriptional level (29). We could not analyze the TGF-β protein level because of the limitation in the amounts of cancer samples in this study. We also analyzed expression of DNMTs using RT-qPCR. TGF-β induces DNMT expression in cancers (48,49) and DNMTs are upregulated in CIMP-positive cancers (50–52). In particular, the DNMT3b expression level was higher in CIMP-H than in CIMP(-) endometrial cancer. This suggests that DNMT upregulation might contribute to the development of CIMP-positive endometrial cancer.
In summary, this is the first report of aberrant DNA methylation in the miR-663a promoter region in normal tissue of patients with CIMP-H endometrial cancer. Both peripheral blood and endometrium originated from the same germ layer, i.e. mesoderm. Therefore, miR-663a methylation may be an epimutation candidate in the development of endometrial cancer. To evaluate whether miR-663a methylation is a primary or a secondary epimutation, a larger-scale analysis is required, and there is also a need to show a role of miR-663a in endometrial cancer using a functional assay. However, the present study is significant in showing a potential basis for the development of CIMP-H endometrial cancer, and this finding may contribute to the prevention or the therapy for CIMP-H endometrial cancer using miR-663a demethylation.
Acknowledgments
The present study was supported by a grant to M.Y. and K.B. from JSPS KAKENHI Grants-in-Aid for Scientific Research (C) (grant nos. 15K10727 and 16K11154), and in part by a grant to K.A. from JSPS KAKENHI Grant-in-Aid for Scientific Research on Innovative Areas (26112515 and 16H01225).
Abbreviations
- PBC
peripheral blood cell
- CIMP
CpG island methylator phenotype
- CIMP-H, -L, (-)
CIMP-high, -low, negative
- MSI
microsatellite instability
- PCR
polymerase chain reaction
- RT-PCR
reverse transcription polymerase chain reaction
- RT-qPCR
reverse transcription-quantitative polymerase chain reaction
- MSP
methylation specific polymerase chain reaction
- DMC
differentially methylated CpG
- DMR
differentially methylated region
References
- 1.Fiolka R, Zubor P, Janusicova V, Visnovsky J, Mendelova A, Kajo K, Lasabova Z, Plank L, Danko J. Promoter hypermethylation of the tumor-suppressor genes RASSF1A, GSTP1 and CDH1 in endometrial cancer. Oncol Rep. 2013;30:2878–2886. doi: 10.3892/or.2013.2752. [DOI] [PubMed] [Google Scholar]
- 2.Guida M, Sanguedolce F, Bufo P, Di Spiezio Sardo A, Bifulco G, Nappi C, Pannone G. Aberrant DNA hypermethylation of hMLH-1 and CDKN2A/p16 genes in benign, premalignant and malignant endometrial lesions. Eur J Gynaecol Oncol. 2009;30:267–270. [PubMed] [Google Scholar]
- 3.Kanaya T, Kyo S, Maida Y, Yatabe N, Tanaka M, Nakamura M, Inoue M. Frequent hypermethylation of MLH1 promoter in normal endometrium of patients with endometrial cancers. Oncogene. 2003;22:2352–2360. doi: 10.1038/sj.onc.1206365. [DOI] [PubMed] [Google Scholar]
- 4.Crépin M, Dieu MC, Lejeune S, Escande F, Boidin D, Porchet N, Morin G, Manouvrier S, Mathieu M, Buisine MP. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat. 2012;33:180–188. doi: 10.1002/humu.21617. [DOI] [PubMed] [Google Scholar]
- 5.Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013;15:25–35. doi: 10.1038/gim.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cini G, Carnevali I, Quaia M, Chiaravalli AM, Sala P, Giacomini E, Maestro R, Tibiletti MG, Viel A. Concomitant mutation and epimutation of the MLH1 gene in a Lynch syndrome family. Carcinogenesis. 2015;36:452–458. doi: 10.1093/carcin/bgv015. [DOI] [PubMed] [Google Scholar]
- 7.Bischoff J, Ignatov A, Semczuk A, Schwarzenau C, Ignatov T, Krebs T, Küster D, Przadka-Rabaniuk D, Roessner A, Costa SD, et al. hMLH1 promoter hypermethylation and MSI status in human endometrial carcinomas with and without metastases. Clin Exp Metastasis. 2012;29:889–900. doi: 10.1007/s10585-012-9478-0. [DOI] [PubMed] [Google Scholar]
- 8.Romero-Pérez L, López-García MÁ, Díaz-Martín J, Biscuola M, Castilla MÁ, Tafe LJ, Garg K, Oliva E, Matias-Guiu X, Soslow RA, et al. ZEB1 overexpression associated with E-cadherin and microRNA-200 downregulation is characteristic of undifferentiated endometrial carcinoma. Mod Pathol. 2013;26:1514–1524. doi: 10.1038/modpathol.2013.93. [DOI] [PubMed] [Google Scholar]
- 9.Park SY, Kook MC, Kim YW, Cho NY, Jung N, Kwon HJ, Kim TY, Kang GH. CpG island hypermethylator phenotype in gastric carcinoma and its clinicopathological features. Virchows Arch. 2010;457:415–422. doi: 10.1007/s00428-010-0962-0. [DOI] [PubMed] [Google Scholar]
- 10.Zhang QY, Yi DQ, Zhou L, Zhang DH, Zhou TM. Status and significance of CpG island methylator phenotype in endometrial cancer. Gynecol Obstet Invest. 2011;72:183–191. doi: 10.1159/000324496. [DOI] [PubMed] [Google Scholar]
- 11.Weisenberger DJ, Levine AJ, Long TI, Buchanan DD, Walters R, Clendenning M, Rosty C, Joshi AD, Stern MC, Le Marchand L, et al. for the Colon Cancer Family Registry Association of the colorectal CpG island methylator phenotype with molecular features, risk factors, and family history. Cancer Epidemiol Biomarkers Prev. 2015;24:512–519. doi: 10.1158/1055-9965.EPI-14-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–845. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 14.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 15.Whitcomb BP, Mutch DG, Herzog TJ, Rader JS, Gibb RK, Goodfellow PJ. Frequent HOXA11 and THBS2 promoter methylation, and a methylator phenotype in endometrial adenocarcinoma. Clin Cancer Res. 2003;9:2277–2287. [PubMed] [Google Scholar]
- 16.Kawaguchi M, Yanokura M, Banno K, Kobayashi Y, Kuwabara Y, Kobayashi M, Nomura H, Hirasawa A, Susumu N, Aoki D. Analysis of a correlation between the BRAF V600E mutation and abnormal DNA mismatch repair in patients with sporadic endometrial cancer. Int J Oncol. 2009;34:1541–1547. doi: 10.3892/ijo_00000283. [DOI] [PubMed] [Google Scholar]
- 17.Peterson LM, Kipp BR, Halling KC, Kerr SE, Smith DI, Distad TJ, Clayton AC, Medeiros F. Molecular characterization of endometrial cancer: a correlative study assessing microsatellite instability, MLH1 hypermethylation, DNA mismatch repair protein expression, and PTEN, PIK3CA, KRAS, and braf mutation analysis. Int J Gynecol Pathol. 2012;31:195–205. doi: 10.1097/PGP.0b013e318231fc51. [DOI] [PubMed] [Google Scholar]
- 18.Banno K, Kisu I, Yanokura M, Masuda K, Kobayashi Y, Ueki A, Tsuji K, Yamagami W, Nomura H, Susumu N, et al. Endometrial cancer and hypermethylation: Regulation of DNA and MicroRNA by epigenetics. Biochem Res Int. 2012;2012:738274. doi: 10.1155/2012/738274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banno K, Yanokura M, Susumu N, Kawaguchi M, Hirao N, Hirasawa A, Tsukazaki K, Aoki D. Relationship of the aberrant DNA hypermethylation of cancer-related genes with carcinogenesis of endometrial cancer. Oncol Rep. 2006;16:1189–1196. [PubMed] [Google Scholar]
- 20.Yanokura M, Banno K, Kawaguchi M, Hirao N, Hirasawa A, Susumu N, Tsukazaki K, Aoki D. Relationship of aberrant DNA hypermethylation of CHFR with sensitivity to taxanes in endometrial cancer. Oncol Rep. 2007;17:41–48. [PubMed] [Google Scholar]
- 21.Yanokura M, Banno K, Susumu N, Kawaguchi M, Kuwabara Y, Tsukazaki K, Aoki D. Hypermethylation in the p16 promoter region in the carcinogenesis of endometrial cancer in Japanese patients. Anticancer Res. 2006;26(2A):851–856. [Google Scholar]
- 22.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avraham A, Cho SS, Uhlmann R, Polak ML, Sandbank J, Karni T, Pappo I, Halperin R, Vaknin Z, Sella A, et al. Tissue specific DNA methylation in normal human breast epithelium and in breast cancer. PLoS One. 2014;9:e91805. doi: 10.1371/journal.pone.0091805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136. doi: 10.1093/nar/gks454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez H, Opalinska J, Zhou L, Sohal D, Fazzari MJ, Yu Y, Montagna C, Montgomery EA, Canto M, Dunbar KB, et al. Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis. PLoS Genet. 2011;7:e1001356. doi: 10.1371/journal.pgen.1001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. Roadmap Epigenomics Consortium: Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi C, Wang Q, Wang L, Huang Y, Li L, Liu L, Zhou X, Xie G, Kang T, Wang H, et al. MiR-663, a microRNA targeting p21WAF1/CIP1, promotes the proliferation and tumorigenesis of nasopharyngeal carcinoma. Oncogene. 2012;31:4421–4433. doi: 10.1038/onc.2011.629. [DOI] [PubMed] [Google Scholar]
- 29.Li Q, Cheng Q, Chen Z, Peng R, Chen R, Ma Z, Wan X, Liu J, Meng M, Peng Z, et al. MicroRNA-663 inhibits the proliferation, migration and invasion of glioblastoma cells via targeting TGF-β1. Oncol Rep. 2016;35:1125–1134. doi: 10.3892/or.2015.4432. [DOI] [PubMed] [Google Scholar]
- 30.Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP, Matei D. TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics. 2014;9:1461–1472. doi: 10.4161/15592294.2014.971608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suga Y, Sugai T, Uesugi N, Kawasaki T, Fukagawa T, Yamamoto E, Ishida K, Suzuki H, Sugiyama T. Molecular analysis of isolated tumor glands from endometrial endometrioid adenocarcinomas. Pathol Int. 2015;65:240–249. doi: 10.1111/pin.12274. [DOI] [PubMed] [Google Scholar]
- 32.Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, Sinicrope FA, Rosty C, Buchanan DD, Potter JD, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87.e2. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, TontiFilippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hitchins MP. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat Rev Cancer. 2015;15:625–634. doi: 10.1038/nrc4001. [DOI] [PubMed] [Google Scholar]
- 35.Muraki Y, Banno K, Yanokura M, Kobayashi Y, Kawaguchi M, Nomura H, Hirasawa A, Susumu N, Aoki D. Epigenetic DNA hypermethylation: Clinical applications in endometrial cancer (Review) Oncol Rep. 2009;22:967–972. doi: 10.3892/or_00000523. [DOI] [PubMed] [Google Scholar]
- 36.Chen QY, Jiao DM, Wang J, Hu H, Tang X, Chen J, Mou H, Lu W. miR-206 regulates cisplatin resistance and EMT in human lung adenocarcinoma cells partly by targeting MET. Oncotarget. 2016;7:24510–24526. doi: 10.18632/oncotarget.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng S, Zhu X, Fan B, Xie D, Li T, Zhang X. miR-p19a-3p targets PMEPA1 and induces prostate cancer cell proliferation, migration and invasion. Mol Med Rep. 2016;13:4030–4038. doi: 10.3892/mmr.2016.5033. [DOI] [PubMed] [Google Scholar]
- 38.Ge X, Liu X, Lin F, Li P, Liu K, Geng R, Dai C, Lin Y, Tang W, Wu Z, et al. MicroRNA-421 regulated by HIF-1α promotes metastasis, inhibits apoptosis, and induces cisplatin resistance by targeting E-cadherin and caspase-3 in gastric cancer. Oncotarget. 2016;7:24466–24482. doi: 10.18632/oncotarget.8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma M, He M, Jiang Q, Yan Y, Guan S, Zhang J, Yu Z, Chen Q, Sun M, Yao W, et al. MiR-487a promotes TGF-β1-induced EMT, the migration and invasion of breast bancer bells by directly targeting MAGI2. Int J Biol Sci. 2016;12:397–408. doi: 10.7150/ijbs.13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L, Wang W, Li X, He S, Yao J, Wang X, Zhang D, Sun X. MicroRNA-155 promotes tumor growth of human hepatocellular carcinoma by targeting ARID2. Int J Oncol. 2016;48:2425–2434. doi: 10.3892/ijo.2016.3465. [DOI] [PubMed] [Google Scholar]
- 41.Pan J, Hu H, Zhou Z, Sun L, Peng L, Yu L, Sun L, Liu J, Yang Z, Ran Y. Tumor-suppressive miR-663 gene induces mitotic catastrophe growth arrest in human gastric cancer cells. Oncol Rep. 2010;24:105–112. doi: 10.3892/or_00000834. [DOI] [PubMed] [Google Scholar]
- 42.Lehmann U, Hasemeier B, Christgen M, Müller M, Römermann D, Länger F, Kreipe H. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol. 2008;214:17–24. doi: 10.1002/path.2251. [DOI] [PubMed] [Google Scholar]
- 43.Potapova A, Albat C, Hasemeier B, Haeussler K, Lamprecht S, Suerbaum S, Kreipe H, Lehmann U. Systematic crossvalidation of 454 sequencing and pyrosequencing for the exact quantification of DNA methylation patterns with single CpG resolution. BMC Biotechnol. 2011;11:6. doi: 10.1186/1472-6750-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan-Fang T, Jian N, Jun L, Na W, Pei-Fang X, Wen-Li Z, Dong W, Li P, Jian W, Xing F, et al. The promoter of miR-663 is hypermethylated in Chinese pediatric acute myeloid leukemia (AML) BMC Med Genet. 2013;14:74. doi: 10.1186/1471-2350-14-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, Wang LL, Li YH, Gao XN, Liu Y, Yu L. Effect of CpG island methylation on microRNA expression in the k-562 cell line. Biochem Genet. 2012;50:122–134. doi: 10.1007/s10528-011-9478-9. [DOI] [PubMed] [Google Scholar]
- 46.Hong Q, Yu S, Geng X, Duan L, Zheng W, Fan M, Chen X, Wu D. High concentrations of uric acid inhibit endothelial cell migration via miR-663 which regulates phosphatase and tensin homolog by targeting transforming growth Factor-beta1. Microcirculation. 2015;22:306–314. doi: 10.1111/micc.12200. [DOI] [PubMed] [Google Scholar]
- 47.Narkiewicz J, Lapinska-Szumczyk S, Zurawa-Janicka D, Skorko-Glonek J, Emerich J, Lipinska B. Expression of human HtrA1, HtrA2, HtrA3 and TGF-beta1 genes in primary endometrial cancer. Oncol Rep. 2009;21:1529–1537. doi: 10.3892/or_00000385. [DOI] [PubMed] [Google Scholar]
- 48.Kogure T, Kondo Y, Kakazu E, Ninomiya M, Kimura O, Shimosegawa T. Involvement of miRNA-29a in epigenetic regulation of transforming growth factor-beta-induced epithelialmesenchymal transition in hepatocellular carcinoma. Hepatol Res. 2014;44:907–919. doi: 10.1111/hepr.12188. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Q, Chen L, Helfand BT, Jang TL, Sharma V, Kozlowski J, Kuzel TM, Zhu LJ, Yang XJ, Javonovic B, et al. TGF-β regulates DNA methyltransferase expression in prostate cancer, correlates with aggressive capabilities, and predicts disease recurrence. PLoS One. 2011;6:e25168. doi: 10.1371/journal.pone.0025168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanai Y, Ushijima S, Kondo Y, Nakanishi Y, Hirohashi S. DNA methyltransferase expression and DNA méthylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int J Cancer. 2001;91:205–212. doi: 10.1002/1097-0215(200002)9999:9999<::AID-IJC1040>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 51.Roll JD, Rivenbark AG, Jones WD, Coleman WB. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol Cancer. 2008;7:15. doi: 10.1186/1476-4598-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nosho K, Shima K, Irahara N, Kure S, Baba Y, Kirkner GJ, Chen L, Gokhale S, Hazra A, Spiegelman D, et al. DNMT3B expression might contribute to CpG island methylator phenotype in colorectal cancer. Clin Cancer Res. 2009;15:3663–3671. doi: 10.1158/1078-0432.CCR-08-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]