

Abstract
The MinD ATPase is critical to the oscillation of the Min proteins, which limits formation of the Z ring to midcell. In the presence of ATP, MinD binds to the membrane and recruits MinC, forming a complex that can destabilize the cytokinetic Z ring. MinE, which is also recruited to the membrane by MinD, displaces MinC and stimulates the MinD ATPase, resulting in the oscillation of the Min proteins. In this study we have investigated the role of lysine 11, present in the deviant Walker A motif of MinD, and the three residues in helix 7 (E146, S148, and D152) that interact electrostatically with lysine 11. Lysine 11 is required for interaction of MinD with the membrane, MinC, MinE, and itself. In contrast, the three residues in helix 7 that interact with lysine 11 are not required for binding to the membrane or activation of MinC. They are also not required for MinE binding; however, they are required for MinE to stimulate the MinD ATPase. Interestingly, the D152A mutant self-interacts, binds to the membrane, and recruits MinC and MinE in the presence of ADP as well as ATP. This mutant provides evidence that dimerization of MinD is sufficient for MinD to bind the membrane and recruit its partners.
In Escherichia coli the min system spatially regulates cell division by preventing Z ring assembly away from midcell (1, 6). This spatial regulation is accomplished by positioning MinC, an antagonist of FtsZ assembly (13, 16), on the membrane at the poles of the cell (12, 29). This positioning is not static, however, as MinC oscillates between the poles of the cell with a period of 50 s. The oscillation of MinC is driven by the other two Min proteins, MinD and MinE. MinD recruits MinC to the membrane, which potentiates its inhibitory activity by concentrating MinC at the membrane and bestowing on the MinCD complex a higher affinity for a septal component (19). MinD also recruits MinE to the membrane, which induces MinD, and thereby MinC, to oscillate between the cell poles (7, 9, 28). Through this oscillation the time-averaged concentration of MinC on the membrane is highest at the poles and lowest at midcell (18, 20, 24).
The biochemical basis of the Min oscillation is the reversible binding of MinD to the membrane that is regulated by MinE (14). MinD binds cooperatively to the membrane through a C-terminal amphipathic helix in a step that requires ATP and the oligomerization of MinD (11, 15, 21, 25, 35, 36, 38). Consistent with this, MinD polymerizes on vesicles leading to tubulation in vitro (11) and forms spirals on the membrane in vivo (33). The release of MinD from the membrane is induced by MinE, which stimulates the MinD ATPase (14, 21, 34). MinE mutants unable to stimulate the MinD ATPase fail to induce the oscillation, and MinD is bound to the membrane all around the cell. MinE mutants that only partially stimulate the MinD ATPase induce a slower oscillation (14). These results provide a direct correlation between the abilities of MinE to stimulate the MinD ATPase and to induce the oscillation of MinD. Furthermore, the period of the oscillation depends upon the ratio of MinD to MinE and is not influenced by MinC (30).
The recruitment of MinC to the membrane by MinD was first shown in vivo. Green fluorescent protein (GFP)-MinC is present in the cytoplasm unless MinD is present, whereupon it localizes to the membrane (12, 29). Subsequent in vitro studies demonstrated that MinD recruits MinC to phospholipid vesicles in an ATP-dependent manner (17, 21). Analysis of several MinD mutants indicated that ATP binding and the oligomerization of MinD are important for recruiting MinC to the vesicles (39). MinE displaces MinC from the MinC-MinD-vesicle complex in a step preceding ATP hydrolysis. Following ATP hydrolysis, MinD and MinE are also released from the vesicles (17, 21).
MinD is a member of a subgroup of ATPases designated the deviant Walker A motif family, which includes plasmid partition proteins such as ParA and more distantly related proteins such as the Fe protein of the nitrogenase complex (5, 22, 23). The unique feature of this family is an additional lysine located at the amino-terminal end of the Walker A motif. In the Fe protein dimer this lysine reaches across the dimer interface to contact ATP bound to the opposite monomer and is thought to be required for ATPase activity (32).
The crystal structure of a MinD-like protein with ADP bound from an archaeon revealed that the corresponding lysine, lysine 11, interacts electrostatically with three residues present in helix 7 (10). These residues are conserved in the E. coli MinD and are E146, S148, and D152. Analysis of mutations indicated that several of these residues (K11, E146, and D152) are important for the binding and activation of MinC (10). Since the binding of MinC to MinD is complex, involving the binding of ATP, oligomerization, and the membrane, it was not clear which step was affected by these mutations. Also, we thought it possible that altering residues that interact with lysine 11 might mimic the action of MinE, leading to a constitutive MinD ATPase and therefore decreased interaction with MinC. We therefore analyzed these mutations in detail. We find that K11 is required for all MinD activities and that E146, S148, and D152 are required for MinE to stimulate the MinD ATPase but are not required for MinC binding and activation.
MATERIALS AND METHODS
Strains and plasmids.
The E. coli K-12 strain JS964 (MC1061 malP::lacIq Δmin::kan) was used in this study (26). This strain was grown in L broth supplemented with the following antibiotics as needed: ampicillin (100 μg/ml) and spectinomycin (50 μg/ml). Arabinose was used to induce expression from the PBAD promoter, and IPTG (isopropyl-β-d-thiogalactopyranoside; 1 mM) was used to induce expression from the lac promoter. Glucose (0.2%) was used to repress expression of these promoters.
pZH106 contains gfp-minD under the control of the arabinose promoter (12). pSEB104 contains a gfp-ftsZ fusion under the control of the arabinose promoter cloned into pGB2 (3). The araC gene and PBAD promoter were obtained from pBAD18 (8). The gfp-ftsZ fusion was replaced with minCminD or minCminDΔ10 genes present on SacI-HindIII fragments obtained by PCR with primers containing the restriction sites and pJPB210 (26) as a template to give plasmids pCS104CD and pCS104CDΔ10. The inserts were completely sequenced to verify that no mutations had been introduced during amplification. The minE gene was added downstream of minD in pCS104CD by replacing the BstXI-HindIII fragment with a fragment with the same restriction sites from pJPB210 to give pCS104CDE. The plasmid used for overexpression and purification of MinD was pZH115 (11). MinE was overexpressed and purified using pJPB216 (11). MalE-MinC116-231 was purified using the plasmid pZH112 (17). pZH110 (minC) contains the minC gene on a low-copy-number vector under the control of its own promoter (12). JS964 (Δmin) was used as a host for all plasmids.
The plasmid pJK100 (Ptrc minDE-gfp) (J. Kueker and J. Lutkenhaus, unpublished data) was constructed in two steps. The minDE fragment was amplified from plasmid pJPB210 (minCDE) with primers 5′ minD (5′-TAGCATGAATTCGGAGAGAAACTATGGCACGCATTATTGTTGTTAC-3′) and 3′ minE (5′-TCAGTCTAGATTTCAGCTCTTCTGCTTCCGGTAAGGTC-3′) (restriction sites underlined), and cloned into the EcoRI-XbaI sites on the plasmid pWL70 (PBAD gfp) upstream of gfp (37). Then the EcoRI-HindIII fragment containing the minDE-gfp fragment was used to replace the EcoRI-HindIII fragment on pDSW208 (2), resulting in pJK100. pJK110 (Ptrc minE-gfp) was constructed similarly, except that a 5′ minE primer (5′-CCATGATTACGAATTCCCGCTTC-3′) and the 3′ minE primer were used to amplify the minE fragment from plasmid pJPB216 (minE).
Fluorescence microscopy.
The effect of minD mutations on membrane localization of GFP-MinD fusions was examined by fluorescence microscopy as described previously (12). Aliquots of cultures were placed on microscope slides, and cells were photographed with a Nikon fluorescence microscope with a MagnaFire charge-coupled device camera (Optronics). Images were imported into Adobe PhotoShop for assembly.
The localization of MinE-GFP was examined in the absence of MinD and in the presence of wild-type MinD or MinD-D152A. The plasmids pJK100 and pJK110, a derivative carrying D152A, were transformed separately into JS964 (Δmin). Single colonies from plates containing spectinomycin and glucose were inoculated into liquid culture; 20 μM IPTG was added when the optical density at 600 nm reached 0.05. Samples were examined 2 h later as described above.
Isolation of minD mutations.
The minD mutations were made by site-directed mutagenesis. The mutations were introduced into minD contained on the expression plasmid pZH115 by using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, Calif.). The mutations were then moved to pCS104CD or pZH106 by replacing the BstXI-HindIII fragment of these plasmids with the corresponding fragment from pZH115 containing the minD mutations. The D152A mutation was introduced into pJK100 by site-directed mutagenesis.
Phenotypic analysis of minD mutants.
The effect of minD mutations on the ability of MinD to activate MinC to block cell division was examined in two ways. In the first test derivatives of pZH115 (minD) containing various mutations were cotransformed with pZH110 (minC) into JS964 (Δmin) and selected on L broth plates containing ampicillin, spectinomycin, and 0.2% glucose (15). The lack of transformants indicated that the minD mutation could still activate minC. If transformants were obtained, they were analyzed for the extent of filamentation. A minicell phenotype indicated that the minD mutation had inactivated minD, whereas a filamentous phenotype indicated that the mutation had only partially inactivated minD. In a second test derivatives of pCS104CD containing the various mutations were analyzed. Colonies of JS964 (Δmin) containing pCS104CD growing on plates containing spectinomycin were picked into 1 ml of L broth. After vortexing, the cultures were serially diluted and aliquots were spotted onto plates containing spectinomycin with various concentrations of arabinose. The plates were incubated overnight at 37°C and then photographed. Also, cells from the spots were examined by microscopy. Controls included JS964 (Δmin) containing pCS104CDE and pCS104CΔD10.
The effect of minD mutations on the ability of MinD to respond to MinE was assessed with the aide of pJPB216 (minE). JS964 (Δmin) containing derivatives of pCS104CD with various minD mutations was transformed with pJPB216 (minE) or the vector pUC19 by selecting for spectinomycin and ampicillin. Colonies were picked into 1 ml of L broth, and 5-μl aliquots were spotted onto L agar plates containing the appropriate antibiotics and 0.1% arabinose.
Protein purification.
The various MinD mutants were purified according to the protocol used for the wild-type protein (11). One liter of L broth supplemented with ampicillin at 100 μg/ml was used to grow cultures of JS964 (Δmin) with pZH115 containing the mutation of interest. The culture was induced with 1 mM IPTG at an optical density at 600 nm of 0.4 at 37°C. After 3 h the cells were collected by centrifugation, resuspended in 20 ml of 25 mM Tris-HCl (pH 7.5)-20 mM NaCl-1 mM EDTA-2 mM dithiothreitol-10% glycerol, and broken with a French press. After removal of cell debris the lysate was loaded onto a DEAE-cellulose column and eluted with a linear salt gradient (20 to 120 mM NaCl). The peak fractions were pooled and concentrated with a Centricon device (Amicon) with a 10,000-molecular-weight-cutoff membrane. The concentrated sample was loaded onto a Superdex 200HR column equilibrated with 10 mM HEPES-NaOH (pH 7.0)-20 mM NaCl. All of the mutant MinD proteins eluted at the monomer position. The proteins were stored at −80°C after addition of dithiothreitol to 1 mM and glycerol to 10%. MinE purification and the purification of MalE-MinC116-231 were described previously (17).
MinD ATPase and vesicle binding assay.
The MinD ATPase assay was carried out as described previously (14, 39). MinD and MinD mutants (6 μM) and small unilamellar vesicles (400 μg/ml) were mixed with 1 mM [γ-32P]ATP in ATPase buffer (25 mM Tris-HCl [pH 7.5], 50 mM KCl, 5 mM MgCl2). MinE (6 μM, unless specified otherwise) was added to reaction mixtures as indicated. Reaction mixtures were incubated at 30°C, and samples were taken at 10 and 20 min after addition of the labeled ATP. The specific activity of the MinD ATPase was determined from the amount of released 32Pi.
The association of proteins with vesicles was determined by a sedimentation assay (38). MinD mutant proteins (4 or 8 μM as indicated), large multilamellar vesicles (400 μg/ml), and nucleotide (1 mM ADP or ATP) were mixed at room temperature in 50 μl of ATPase buffer. MalE-MinC116-231 (4 μM) and MinE (4 μM) were added as indicated. The reaction mixtures were incubated at room temperature for 10 min, followed by a centrifugation at 10,000 × g at room temperature in a tabletop centrifuge for 2 min. The supernatants were carefully removed, and the pellets were resuspended in 50 μl of sodium dodecyl sulfate (SDS) sample buffer. Ten-microliter aliquots were electrophoresed on SDS-12.5% polyacrylamide gels and stained with Coomassie brilliant blue.
Size-exclusion chromatography.
MinD mutant proteins were analyzed by size-exclusion chromatography on an AKTA-fast protein liquid chromatograph equipped with a Superdex 75HR column. Samples (200 to 250 μl) of MinD mutant protein (0.7 to 1.0 mg/ml) were diluted twofold into Pol buffer (50 mM MES [morpholineethanesulfonic acid; pH 6.5]-50 mM KCl-10 mM MgCl2). ADP or ATP was then added to a final concentration of 1 mM. After incubation at room temperature for 10 min the samples were loaded onto the column and eluted at room temperature with Pol buffer containing either 0.2 mM ADP or ATP.
RESULTS
Lys11 is a critical residue for MinD.
It was previously reported that mutation of Lys11 resulted in failure of MinD to cooperate with MinC to inhibit division and to bind to MinC in the yeast two-hybrid system (10). Since the binding of MinD to MinC requires that MinD first interact with ATP, which also leads to oligomerization and membrane binding (17), it is possible that the K11A mutation affects a step before the binding of MinC and therefore may not be directly involved in the interaction.
We confirmed that the K11A mutation led to a loss in the ability of MinD to activate MinC to inhibit division. An expression plasmid carrying K11A was introduced into JS964 (Δmin) carrying pZH110 (minC). The resultant transformants displayed a typical minicell phenotype, indicating that K11A could not activate MinC to inhibit division (Table 1). The same results were obtained with a plasmid carrying K16Q previously shown to lack all MinD activities (5, 11). In contrast, no transformants were obtained with an expression plasmid carrying wild-type minD (Table 1). We observed that a GFP-K11A fusion was present in the cytoplasm as previously observed for a GFP-K16Q fusion (11) (Fig. 1A). Thus, K11A would be unable to recruit MinC to the membrane, a necessary step in the activation of MinC.
TABLE 1.
Summary of the behavior of the MinD mutants
| Allele | Cotransformation, MinC + MinDa | Cellular locationb | 0.1% Arabinosec
|
|
|---|---|---|---|---|
| pUC19 | pJPB216 (minE) | |||
| Vector | + | NAd | + | + |
| WTe | − | Membrane | − | + |
| K16Q | + | Cytoplasm | + | + |
| K11A | + | Cytoplasm | + | + |
| D152A | − | Membrane | − | − |
| E146A | − | Membrane | − | − |
| E146A/D152A | − | Membrane | − | − |
| S148A/D152A | − | Membrane | − | − |
| AAA | − | Membrane | − | − |
| D154A | + | Membrane | + | + |
| N45A | − | Membrane | − | + |
| K163A/S164A | − | Membrane | − | + |
Derivatives of pZH115 containing the indicated minD alleles were cotransformed into JS964 (Δmin) with pZH110 (minC). −, no transformants were obtained; +, transformants were obtained.
Derivatives of pZH106 (gfp-minD) containing the indicated minD alleles in JS964 (min) were examined by fluorescence microscopy after induction of the fusion with 0.01% arabinose for 2 h.
JS964 (Δmin) containing pCS104CD with the indicated minD alleles and either the vector pUC19 or pJBP216 (minE) was spotted on plates, and the ability to form colonies was assessed. +, colonies were formed; −, colonies were not formed.
NA, not applicable.
WT, wild type.
FIG. 1.

Analysis of K11A. (A) The K11A mutation was introduced into pZH106, resulting in the GFP-K11A fusion under the control of the PBAD promoter and introduced into JS964 (Δmin). After induction with 0.01% arabinose for 2 h cells were examined by fluorescence microscopy and compared to JS964 carrying pZH106 expressing GFP-MinD. (B) The ability of K11A to bind to phospholipid vesicles was examined. K11A or MinD (6 μM) was incubated with phospholipid vesicles (400 μg/ml) in ATPase buffer (25 mM Tris-HCl [pH 7.5], 50 mM KCl, 5 mM MgCl2) in the presence of 1 mM ATP or ADP. The vesicles were pelleted, and the amount of protein bound was determined by analyzing the pellets by SDS-PAGE. (C) Determination of the ATPase activity of K11A. K11A or MinD (4 μM) was incubated in ATPase buffer with phospholipid vesicles (400 μg/ml) and 1 mM [γ-32P]ATP with or without MinE. The amount of 32Pi released was determined.
To further explore the deficiency of K11A, we studied the mutant protein. K11A did not bind appreciably to phospholipid vesicles in vitro (Fig. 1B) and had a basal ATPase that could not be stimulated by MinE (Fig. 1C). The presence of a basal ATPase suggested that K11A was properly folded. Support for this was obtained by examining the sensitivity of K11A to trypsin. Sensitivity to trypsin was blocked by ADP and ATP, indicating that K11A is properly folded in the presence of either nucleotide (data not shown). Thus, K11A behaves like K16Q and several other mutants (D120G, D38A, and S121T [39]) that fail to activate MinC. D120G, D38A, S121T, and K16Q fail to self-interact in the yeast two-hybrid system, suggesting that they cannot dimerize (39). If this interpretation is correct, it suggests that a failure to dimerize is common among mutants with residues altered that interact with ATP.
Previously we observed by size-exclusion chromatography that MinD dimerized in the presence of ATP but not ADP (17). To determine if K11A was able to dimerize, we analyzed it by size-exclusion chromatography (Fig. 2). It eluted as a monomer with ADP. With ATP it also eluted as a monomer, although there was a slight shift in the peak, suggesting that it may undergo a change in conformation. K16Q was also examined, and it also failed to dimerize with ATP (data not shown). This result is consistent with its failure to self-interact in the yeast two-hybrid system. Together the results indicate that a common deficiency in mutants that are affected in binding or responding to ATP is their inability to self-interact, which is required for membrane association and activation of MinC.
FIG. 2.

Size-exclusion chromatography of MinD mutants. Purified proteins were analyzed by size-exclusion chromatography on a Superdex 75HR column. The proteins were loaded onto the column at 10 μM. The proteins were incubated at room temperature for 10 min in elution buffer (50 mM MES [pH 6.5], 50 mM KCl, 10 mM MgCl2) supplemented with 1 mM ADP or ATP before loading. The nucleotide concentration in the elution buffer was reduced to 0.2 mM during chromatography.
Residues interacting with K11 are not required to activate MinC.
The structure of the MinD-like protein from Pyrococcus furiosus revealed that Lys11 interacts with three residues in helix 7 (10) (Fig. 3). These residues are conserved in the E. coli MinD and are E146, S148, and D152. It was observed that mutation of either E146 or D152 led to a decreased ability of MinD to bind MinC and inhibit division (10). To confirm this result, we made the E146A and D152A mutations. We also constructed mutants in which two or three of these interacting residues were changed to alanine. As a control, we mutated D154, which lies within helix 7 but points away from K11. This residue is not as highly conserved and is a methionine in the P. furiosus MinD-like protein.
FIG. 3.

Location of Lys11 and the interacting triad. The E. coli MinD was modeled using the crystal structure of the MinD-like protein from P. furosius containing ADP. In the structure Lys11 interacts with three residues in helix 7, including D152, S148, and E146.
We were also interested in additional residues that might be involved in the interaction between MinD and MinC or MinE. We therefore mutated the highly conserved N45, which lies within the switch I region which we previously found to be important for binding MinC (39). The two residues, G42 and R44, that we found previously to be important for MinC binding lie near N45, which is highly conserved among all MinD proteins. We also mutated K163 and S164, as these residues are present in gram-negative bacterial MinD proteins but not in gram-positive bacterial MinD proteins. Only gram-negative organisms have MinE (31).
The mutations were made by site-directed mutagenesis in pZH115 (minD) and recovered in JS964 (Δmin). This is an important step since the basal expression of MinD, and especially that of some of the mutants, is quite toxic in a wild-type strain (data not shown). To test the effect of these mutations on the ability to activate MinC, plasmids containing the various alleles were transformed into JS964 (Δmin) containing pZH110 (minC). Although plasmids containing K16Q or K11A produced transformants (see above), most of the others did not (Table 1). Surprisingly, plasmids carrying D152A, E146A, or combinations of these mutations with S148A did not yield transformants, indicating that they were capable of activating MinC. In fact a plasmid in which all three of these interacting residues were changed to alanines (designated AAA) did not yield transformants. Also, N45A did not yield transformants, indicating that this highly conserved residue in the switch I region is not required for MinC activation, although several neighboring residues are required (39). The double mutant K163A S164A did not yield transformants, indicating that these residues are also not required for MinC activation. However, D154A, a mutation lying within helix 7 and constructed as a control for the other helix 7 mutations, yielded transformants with a minicell phenotype, indicating that D154 is required for MinC activation. Although these results suggest that D152, S148, E146, N45, E163, and S164 are not required for MinC activation, the levels of MinC and MinD are probably higher than the physiological level in this test system, and this may mask the possibility that the corresponding mutants are attenuated to some extent.
We therefore chose a test system in which minC and minD are expressed in cis at physiological levels to better evaluate the effects of these mutations. This system utilizes the plasmid pCS104CD, which contains the minC and minD genes cloned downstream of the arabinose promoter in a low-copy-number vector. This plasmid can be maintained in a min deletion strain (JS964) since the basal level of expression is extremely low. The addition of arabinose leads to expression of minCD, filamentation, and cell death. Colony formation was reduced over 103 in the presence of 0.2% arabinose (Fig. 4), and immunoblot analysis revealed that the expression of minC and minD was similar to the physiological levels at this concentration (data not shown). In contrast, JS964 (Δmin) containing pCS104CDE, which has minE cloned downstream of minCD, forms colonies on 0.2% arabinose. Also, JS964 with pCS104CDΔ10, which expresses MinDΔ10 that lacks the carboxy-terminal 10 amino acids required for membrane binding (15, 27), formed colonies on 0.2% arabinose (Fig. 4). This latter result illustrates the importance of membrane binding by MinD in the activation of MinC.
FIG. 4.

Effect of minD mutations on the ability of MinD to activate MinC to inhibit colony formation. All plasmids were derivatives of pCS104CD, which carries minC and minD under the control of the PBAD promoter on a low-copy-number plasmid. Colonies of JS964 (Δmin) carrying the various plasmids were picked and resuspended in L broth. The cultures were serially diluted (10-fold each step), and 5 μl was spotted on plates carrying various arabinose concentrations. The minD mutation carried on the plasmid is indicated to the right of the figure. From the top, the first is wild-type (WT) minD, the second contains the minE gene downstream of minD, the third contains minDΔ10 (missing the carboxy-terminal 10 amino acids), and the others carry the mutations isolated in this study.
The various minD mutations were transferred to pCS104CD, the resultant plasmids were introduced into JS964, and the strains were tested for sensitivity to arabinose (Fig. 4; only arabinose concentrations of 0.003 and 0.2% are shown). JS964 (Δmin) with plasmids carrying D152A, E146A, AAA, D152AE146A, D152AS148A, K163AS164A, or N45A was unable to form colonies at 0.2% arabinose (Fig. 4). In contrast, JS964 (Δmin) carrying a plasmid derivative with D154A grew, indicating that MinC was not activated by this mutant to inhibit cell division. Although these results indicated that the helix 7 mutants, except for D154A, were able to activate MinC, we tested them at lower arabinose concentrations to determine if they were attenuated with respect to wild-type MinD. The lowest concentration of arabinose that visibly reduced the growth of JS964 (Δmin) containing wild-type minD was 0.003%. The growth of JS964 (Δmin) containing D152A or E146A was also diminished at 0.003% arabinose, as was the growth of JS964 (Δmin) containing minD alleles with multiple mutations including the triple mutant AAA (Fig. 4). Examination of the morphology of the cells revealed that the reduced growth correlated with an extreme filamentation. These results demonstrate that these minD alleles are not attenuated and the three residues that interact with Lys11 are not required for activation of MinC. In contrast D154 is required for activation of MinC.
Since most of the above mutants were able to activate MinC, we expected them to be able to bind to the membrane. To confirm this, the mutations were introduced into a GFP-MinD fusion contained on plasmid pZH106 and examined by fluorescence microscopy. As expected, the various mutant proteins were present on the membrane similar to the wild-type GFP-MinD fusion. Thus, E146A, D152A, and the double and triple mutants (Fig. 5; Table 1), as well as the switch I mutant (N45A; data not shown), were all able to bind to the membrane. Interestingly, D154A, which does not activate MinC, was also present on the membrane.
FIG. 5.
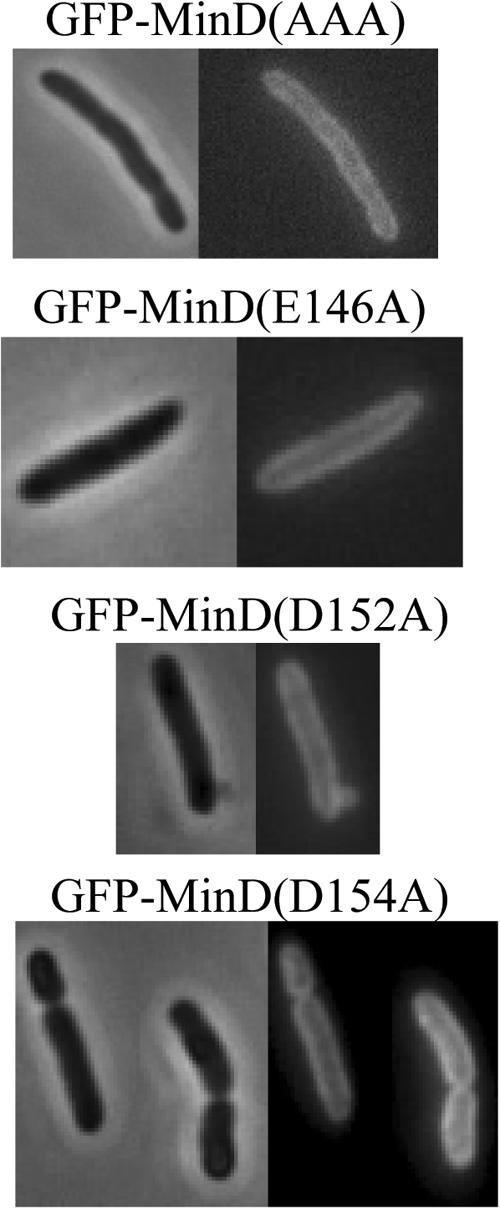
Effect of minD mutations on the ability of MinD to bind to membranes. JS964 (Δmin) containing derivatives of pZH106 (gfp-minD) carrying various minD mutations was examined by fluorescence microscopy. Samples were taken 2 h after addition of 0.01% arabinose.
Residues interacting with Lys11 are necessary for responding to MinE.
To determine if any of the mutations constructed above affected the interaction with MinE, pJPB216, which contains minE under lac promoter control, was introduced into JS964 (Δmin) containing the various pCS104CD derivatives. The basal expression of MinE was sufficient to suppress division inhibition caused by MinCD induced with 0.1% arabinose (Table 1). Interestingly, the inhibition of division caused by plasmids carrying mutations in helix 7 could not be suppressed by MinE, although inhibition caused by N45A and the double mutant K163AS164A was completely suppressed. These results demonstrate that the three residues in helix 7 that interact with Lys11 are essential for responding to MinE. Since D154A was unable to activate MinC, its sensitivity to MinE could not be tested.
Effect of MinE on the ATPase activity of MinD mutants.
MinD expresses a basal ATPase activity that is stimulated about 10-fold in the presence of phospholipid vesicles and MinE (14, 21, 34). This stimulation requires the C-terminal tail of MinD, indicating that MinE stimulation requires that MinD be bound to the membrane (15). The in vivo results revealed that altering the residues that interacted with Lys11 resulted in mutants that were still able to activate MinC but were no longer responsive to MinE. This latter result suggests that MinE would not stimulate the ATPase activity of these MinD mutants. To better understand the effects of the mutations altering residues in helix 7, we purified the mutant proteins and tested them for MinE stimulation. The wild-type MinD ATPase is stimulated by MinE as previously reported (14, 34) with maximal stimulation requiring at least 1 μM MinE (Fig. 6). The basal ATPase activity (in the absence of MinE) of the triple AAA mutant was similar to that of wild-type MinD. This result indicates that simply uncoupling Lys11 from the interacting residues in helix 7 does not lead to a constitutive ATPase. In addition, the AAA mutant was very poorly stimulated by MinE (Fig. 6). This poor stimulation is consistent with MinE being unable to suppress the inhibition of cell division caused by AAA in the presence of MinC.
FIG. 6.

Effect of mutations affecting residues in helix 7 on MinE's ability to stimulate MinD ATPase. MinD, AAA, D152A, or D154A (6 μM) was incubated in ATPase buffer. MinE was added at various concentrations, and the ATPase activity was measured by the release of 32Pi from [γ-32P]ATP. WT, wild type.
D152A was also examined and found to have a basal ATPase that was similar to that of the wild type but also poorly stimulated by MinE (Fig. 6). Finally we examined the ability of MinE to stimulate D154A. This mutant did not activate MinC, so we were unable to examine its response to MinE in vivo. D154A expressed a basal ATPase similar to that of the wild type but was also poorly stimulated by MinE. Thus, this mutant cannot activate MinC or be stimulated by MinE. The other helix 7 mutants could not be stimulated by MinE but activated MinC normally.
Examining the interaction of MinD mutants with phospholipid vesicles, MinC, and MinE.
The behavior of D152A, the double mutant E146A D152A and the triple mutant (AAA), which lacks all three residues that interact with Lys11, was examined for interaction with phospholipid vesicles. In this assay the MinD mutant proteins were incubated with phospholipid vesicles and the amount of bound protein was assessed by SDS-polyacrylamide gel electrophoresis (PAGE) after recovery of the vesicles by sedimentation. The triple mutant bound to phospholipid vesicles to a greater extent in the presence of ATP than in the presence of ADP, behavior similar to that of wild-type MinD (Fig. 7A, lanes 1 and 2). When MinC or MinE was added to the reaction, they were recruited to the vesicles in the presence of ATP but not ADP, indicating that these three residues of helix 7 are not required for MinC or MinE binding (Fig. 7A, lanes 3 to 6). In addition, MinE did not cause significant reduction in the level of AAA bound to the vesicles, consistent with its inability to stimulate the ATPase of this mutant (Fig. 7A, lanes 5 and 6). The amount of MinD152, MinC, and MinE in the pellet with ATP is comparable to the amount observed with MinD in the presence of ATPγS (data not shown).
FIG. 7.

Interaction of MinD mutants with phospholipid vesicles, MinC, and MinE. (A) AAA (4 μM) was incubated with phospholipid vesicles (400 μg/ml) in ATPase buffer in the presence of 1 mM ADP or ATP. Either no additions were made or MalE-MinC116-231 (4 μM), MinE (4 μM), or both (4 μM) were added, and the samples were incubated at room temperature for 10 min. The samples were centrifuged to sediment the vesicles, and the pellets were analyzed by SDS-PAGE. (B) D152A (4 μM) was incubated with phospholipid vesicles (400 μg/ml) in ATPase buffer in the presence of 1 mM ADP or ATP or no nucleotide. Either no additions were made or MalE-MinC116-231 (4 μM), MinE (4 μM) or both (4 μM each) were added, and the amount of proteins bound to vesicles was determined after centrifugation and analysis of the pellets by SDS-PAGE.
Interestingly, when both MinC and MinE were added to the reaction MinC was bound to the AAA-vesicle complex but MinE was not. This result is in sharp contrast to results obtained with wild-type MinD in the presence of ATPγS (17, 21). In the presence of this nonhydrolyzable analog MinE cannot remove MinD from the MinD-vesicle complex. When both MinE and MinC are added, MinE is the preferred binding partner. As shown here the AAA mutant can bind either MinC or MinE but has a clear preference for MinC when both are added.
The E146AD152A double mutant behaved similarly to the triple mutant in the in vivo assays. It inhibited division as efficiently as the triple mutant and was unresponsive to MinE. The experiments with phospholipid vesicles were repeated using the E146AD152A mutant, and the results were identical to those with the triple mutant (data not shown). The double mutant bound to vesicles in an ATP-dependent fashion and recruited MinC or MinE. MinE did not cause a reduction in the amount of MinD bound. When both MinC and MinE were present, MinC was again the preferred binding partner. Thus, this double mutant behaves similarly to the triple mutant.
D152A also bound to phospholipid vesicles in the presence of ATP. Surprisingly, however, it also bound to vesicles in the presence of ADP (Fig. 7B, compare lanes 2 and 3). Binding of D152A to vesicles was nucleotide dependent, as only a background level of the protein was detected in the absence of nucleotide (Fig. 7B, lane 1). Also, D152A was not in the pellet in the absence of vesicles (data not shown). D152A recruited MinC to vesicles in the presence of ATP or ADP but not without nucleotide (Fig. 7B, lanes 6 to 8). MinE was also recruited to vesicles by this mutant in the presence of nucleotides (Fig. 7B, lanes 4 and 5). When both MinE and MinC were added, MinC was the preferred binding partner, similar to the behavior of AAA and in contrast to that of wild-type MinD (Fig. 7B, lanes 9 and 10).
D154A bound to vesicles in the presence of ATP but was unable to recruit MinC (data not shown). This result was consistent with the in vivo result, since D154A was unable to activate MinC. Similarly to the other mutants D154A also recruited MinE to vesicles.
Localization of MinE-GFP in the presence of D152A.
The above results demonstrate that the ATPase of the helix 7 mutants was not stimulated by MinE; however, the mutants were able to recruit MinE to vesicles. To assess this latter capability in vivo, we utilized a construct that expressed MinE-GFP in the presence or absence of MinD or D152A. In the absence of MinD, the fluorescence due to MinE-GFP was present in the cytoplasm (Fig. 8A) whereas in the presence of MinD the fluorescence was present in polar caps and rings as previously described (Fig. 8B) (7, 9, 33). In contrast, in the presence of D152A the fluorescence was at the membrane all around the cell (Fig. 8C). This result is consistent with D152A recruiting MinE-GFP to the membrane. It is also consistent with MinE being unable to stimulate D152A ATPase and induce oscillation.
FIG. 8.

Localization of MinE-GFP in the presence of D152A. MinE-GFP was localized in the absence of MinD (A), the presence of MinD (B), or the presence of D152A (C). JS964 containing pJK110 (minE-gfp), pJK100 (minDE-gfp), or pJK100A (minDD152AE-gfp) was grown in L broth and induced with 20 μM IPTG.
Size-exclusion chromatography of D152A.
The above analysis of D152A was surprising in that ADP, as well as ATP, promoted binding to the membrane and recruitment of MinC and MinE. Previous work with various MinD mutants has shown that the ability of MinD to self-interact correlates with its ability to bind to the membrane and recruit MinC (39). One possible explanation for the behavior of D152A is that both ADP and ATP are able to promote self-interaction. This behavior would be quite distinct from that observed with wild-type MinD, which dimerizes only with ATP (17). It would also be distinct from K11A, which is a monomer with both ADP and ATP (Fig. 2).
D152A was analyzed under the same conditions used for K11A. Interestingly, D152A showed a propensity to dimerize under these conditions (Fig. 2). D152A shifted toward the dimer position in the presence of both ADP and ATP, although the shift was more pronounced with ATP than with ADP. In the absence of nucleotide D152A was a monomer, demonstrating that dimerization, like membrane binding, is nucleotide dependent (data not shown).
Interestingly, the AAA mutant did not dimerize with ADP although it contains the D152A mutation (data not shown). The additional mutations present in AAA appear to restore the ATP dependency. The E146A mutation appears to be responsible, as the S148AD152A mutant dimerizes with ADP (data not shown). Together, these results further extend the correlation among dimerization, membrane binding, and recruitment of MinC and MinE.
DISCUSSION
The main goal of this study was to analyze the roles of lysine 11 within the deviant Walker A motif and the interacting triad in helix 7 in MinD function. We confirmed that lysine 11 is required for MinD to activate MinC, but in contrast to a published report (10), we found that the interacting triad in helix 7 is not required. This triad is required for MinD to respond to MinE. In vitro analysis of these mutants supports a model in which dimerization of MinD leads to stable membrane binding and recruitment of MinC and MinE.
Lysine 11 and the interacting triad.
Our results confirmed that K11 was essential for MinD to activate MinC. Size-exclusion chromatography revealed that K11A is unable to dimerize, suggesting that this is the underlying deficiency of this mutant, since this step appears to be required for MinD to bind to the membrane and activate MinC. Thus, K11A would be similar to other MinD mutants, such as K16Q, D38A, and D120G, that do not display self-interaction in the yeast two-hybrid system and are therefore thought not to dimerize (39). Consistent with this interpretation, we observed that K16Q also failed to dimerize by size-exclusion chromatography.
In contrast to the results obtained with K11A, we were unable to confirm the previous report (10) that the residues in helix 7 that interact with Lys11 are necessary for MinD to activate MinC. Mutants in which one, two, or all three residues that comprise the triad were changed to alanines were just as effective as the wild-type MinD in activating MinC. The main effect of these alanine substitutions was a dramatic defect in responding to MinE.
MinC and MinE interaction with MinD.
The mutants that we examined in this study with substitutions in helix 7 were unable to respond to MinE. Although these mutants still bound MinE, the inhibition of division that they caused in the presence of MinC was not suppressed by MinE and their ATPase could not be stimulated by MinE. The ability of these mutants to bind MinE in vitro was confirmed in vivo for D152A, which recruited MinE-GFP to the membrane. Although MinE still bound to the helix 7 mutants, our results indicate that some aspect of the binding was affected. With wild-type MinD bound to the vesicles in the presence of ATPγS, MinE is the preferred binding partner, as it displaces MinC. However, with the helix 7 mutants that we examined, MinC is the preferred binding partner, as it can displace MinE. This preferential binding of MinC by these mutants would explain why they inhibit division even in the presence of MinE.
Although the triad of residues in helix 7 is not involved in MinC binding or activation, we discovered that D154 was required. D154A was unable to activate MinC in vivo although it could bind to the membrane. Consistent with this, D154A bound to vesicles in vitro but was unable to recruit MinC. The switch I mutants, G42A and R44G, that we reported earlier (39) bind to the membrane and respond to MinE but are unable to activate or bind MinC. Also, a switch II mutant, I125E, did not bind MinC. Mapping the corresponding residues on the P. furiosus MinD-like protein reveals that they are spatially quite separated (Fig. 9A). One possibility is that these residues affect the conformation of the protein at some distance and therefore affect MinC binding. On the other hand, mapping these residues onto the dimer model proposed by Lutkenhaus and Sundaramoorthy (23) reveals that they are clustered and could comprise a site for interaction with MinC (Fig. 9B). The switch I residues from one monomer are juxtaposed to the switch II residue and the helix 7 residues of the other monomer.
FIG. 9.

Mapping of residues required for interaction with MinC. The MinD residues that have been found to affect MinC binding (isolated in this study and another [39]), but not the ability of MinD to bind to the membrane or dimerize, are indicated on the monomer (A) and the proposed dimer model (B) of the P. furiosus MinD (23). In the dimer model one monomer is colored green and the other is tan.
MinE stimulation of the MinD ATPase.
One possibility that we set out to explore in this study was that disrupting the linkage of lysine 11 to the triad of residues in helix 7 would lead to a constitutive ATPase activity, perhaps mimicking MinE action and leading to the reported decrease in interaction with MinC (10). Unexpectedly, however, as we have shown in this study the triad in helix 7 is not required for MinC activation. Nonetheless, our studies revealed that simply uncoupling Lys11 from its interacting residues is not sufficient to yield a constitutive high level of ATPase activity. Instead, the loss of these residues yields a MinD that cannot be stimulated by MinE, although the corresponding mutants still bind MinE, ruling out a simple explanation for their deficiency. Our results suggest a model in which the binding of ATP leads to MinD dimerization and membrane binding. Subsequent binding of MinE to a dimer of MinD results in conformational changes in MinD that results in stimulation of the ATPase activity. For this stimulation to occur, the residues in helix 7 examined in this study have to be intact.
MinD binding to the membrane.
It is well established that MinD binds to the membrane through a C-terminal amphipathic helix (15, 36, 39). Several models have been proposed to account for the mechanism of binding. In the initial models it was suggested that ATP binding by MinD would result in the release of the tethered C-terminal helix, making it available for membrane binding (15, 36). Later this model was amended by taking into account the observation that the C-terminal helix is disordered in the crystal structure of the MinD from Archaeoglobus fulgidus (4). In the revised model (35), the zipper model, the membrane targeting sequence is disordered and forms a helix only upon contact with the membrane. Although a single helix is insufficient to tether the E. coli MinD to the membrane, the cooperative polymerization of MinD, which requires ATP, would lead to a polyvalent MinD polymer that would be stably tethered to the membrane.
In one of the original models it was suggested that MinD dimerization triggers MinD binding by making the C-terminal helix available and providing bivalency (15). This model is consistent with the results presented here including the ADP-dependent binding of D152A to vesicles and the ability of ADP to promote dimerization of this mutant in vitro. We used size-exclusion chromatography with an elution buffer at pH 6.5 to assess dimerization. Recently, we have observed that dimerization is reduced at a higher pH and favored by the removal of the C-terminal helix. Thus, in vivo the membrane may help to sequester the C-terminal helix away from the dimer interface and therefore promote dimerization at the membrane, similar to the proposal of Szeto et al. (35). In our model (Fig. 9), however, only dimerization, and not polymerization, is required. Although we think that MinD eventually polymerizes on the membrane, which is reflected in the tubulation of vesicles observed in vitro (11) and the spirals observed in vivo (33), we think that this is a distinct step after the initial dimerization. In favor of this two-step model is the observation that ATPγS promotes vesicle binding but not tubulation (11) and that tubulation of vesicles by D152A is observed only with ATP (data not shown), although vesicle binding is promoted by both ADP and ATP. Our interpretation of these observations is that MinD with ATPγS and D152A with ADP, although able to bind to the membrane, do not fully mimic MinD with ATP bound and therefore do not polymerize. Szeto et al. (35) have shown that a bivalent MinD membrane targeting sequence is sufficient for membrane binding consistent with the above model.
Binding of MinD to vesicles or the membrane requires ATP (11, 21); however, D152A was able to bind to vesicles and recruit MinC or MinE in the presence of ADP. This result implies that ADP is able to induce the necessary conformational changes to activate this mutant. Our finding that ADP promotes dimerization of D152A extends the correlation between the abilities of MinD to dimerize and to bind to the membrane and recruit MinC and MinE. The behavior of D152A suggests that the binding of ATP to MinD results in breaking the electrostatic interaction between Lys11 and D152, resulting in dimerization.
Acknowledgments
This work was supported by grant GM2974 from the National Institutes of Health. This publication was also made possible by NIH grant no. 5 P20 RR016443 from the COBRE Program of the National Center for Research Resources.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
REFERENCES
- 1.Bi, E., and J. Lutkenhaus. 1993. Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J. Bacteriol. 175:1118-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen, J. C., D. S. Weiss, J. M. Ghigo, and J. Beckwith. 1999. Septal localization of FtsQ, an essential cell division protein in Escherichia coli. J. Bacteriol. 181:521-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Churchward, G., D. Belin, and Y. Nagamine. 1984. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene 31:165-171. [DOI] [PubMed] [Google Scholar]
- 4.Cordell, S. C., and J. Lowe. 2001. Crystal structure of the bacterial cell division regulator MinD. FEBS Lett. 492:160-165. [DOI] [PubMed] [Google Scholar]
- 5.de Boer, P. A., R. E. Crossley, A. R. Hand, and L. I. Rothfield. 1991. The MinD protein is a membrane ATPase required for the correct placement of the Escherichia coli division site. EMBO J. 10:4371-4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Boer, P. A., R. E. Crossley, and L. I. Rothfield. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56:641-649. [DOI] [PubMed] [Google Scholar]
- 7.Fu, X., Y. L. Shih, Y. Zhang, and L. I. Rothfield. 2001. The MinE ring required for proper placement of the division site is a mobile structure that changes its cellular location during the Escherichia coli division cycle. Proc. Natl. Acad. Sci. USA 98:980-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guzman, L., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hale, C. A., H. Meinhardt, and P. A. de Boer. 2001. Dynamic localization cycle of the cell division regulator MinE in Escherichia coli. EMBO J. 20:1563-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayashi, I., T. Oyama, and K. Morikawa. 2001. Structural and functional studies of MinD ATPase: implications for the molecular recognition of the bacterial cell division apparatus. EMBO J. 20:1819-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu, Z., E. P. Gogol, and J. Lutkenhaus. 2002. Dynamic assembly of MinD on phospholipid vesicles regulated by ATP and MinD. Proc. Natl. Acad. Sci. USA 99:6671-6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu, Z., and J. Lutkenhaus. 1999. Topological regulation of cell division in Escherichia coli involves rapid pole to pole oscillation of the division inhibitor MinC under the control of MinD and MinE. Mol. Microbiol. 34:82-90. [DOI] [PubMed] [Google Scholar]
- 13.Hu, Z., and J. Lutkenhaus. 2000. Analysis of MinC reveals two independent domains involved in interaction with MinD and FtsZ. J. Bacteriol. 182:3965-3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu, Z., and J. Lutkenhaus. 2001. Topological regulation of cell division in E. coli. Spatiotemporal oscillation of MinD requires stimulation of its ATPase by MinE and phospholipid. Mol. Cell 7:1337-1343. [DOI] [PubMed] [Google Scholar]
- 15.Hu, Z., and J. Lutkenhaus. 2003. A conserved sequence at the C-terminus of MinD is required for binding to the membrane and targeting MinC to the septum. Mol. Microbiol. 47:345-355. [DOI] [PubMed] [Google Scholar]
- 16.Hu, Z., A. Mukherjee, S. Pichoff, and J. Lutkenhaus. 1999. The MinC component of the division site selection system in Escherichia coli interacts with FtsZ to prevent polymerization. Proc. Natl. Acad. Sci. USA 96:14819-14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu, Z., C. Saez, and J. Lutkenhaus. 2003. Recruitment of MinC, an inhibitor of Z-ring formation, to the membrane in Escherichia coli: role of MinD and MinE. J. Bacteriol. 185:196-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, K. C., Y. Meir, and N. S. Wingreen. 2003. Dynamic structures in Escherichia coli: spontaneous formation of MinE rings and MinD polar zones. Proc. Natl. Acad. Sci. USA 100:12724-12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson, J. E., L. L. Lackner, and P. A. de Boer. 2002. Targeting of DMinC/MinD and DMinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ rings. J. Bacteriol. 184:2951-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruse, K. 2002. A dynamic model for determining the middle of Escherichia coli. Biophys. J. 82:618-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lackner, L. L., D. M. Raskin, and P. A. de Boer. 2003. ATP-dependent interactions between Escherichia coli Min proteins and the phospholipid membrane in vitro. J. Bacteriol. 185:735-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leipe, D. D., Y. I. Wolf, E. V. Koonin, and L. Aravind. 2002. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317:41-72. [DOI] [PubMed] [Google Scholar]
- 23.Lutkenhaus, J. , and M. Sundaramoorthy. 2003. MinD and role of the deviant Walker A motif, dimerization and membrane binding in oscillation. Mol. Microbiol. 48:295-303. [DOI] [PubMed] [Google Scholar]
- 24.Meinhardt, H., and P. A. de Boer. 2001. Pattern formation in Escherichia coli: a model for the pole-to-pole oscillations of Min proteins and the localization of the division site. Proc. Natl. Acad. Sci. USA 98:14202-14207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mileykovskaya, E., I. Fishov, X. Fu, B. D. Corbin, W. Margolin, and W. Dowhan. 2003. Effects of phospholipid composition on MinD-membrane interactions in vitro and in vivo. J. Biol. Chem. 278:22193-22198. [DOI] [PubMed] [Google Scholar]
- 26.Pichoff, S., B. Vollrath, C. Touriol, and J. P. Bouche. 1995. Deletion analysis of gene minE which encodes the topological specificity factor of cell division in Escherichia coli. Mol. Microbiol. 18:321-329. [DOI] [PubMed] [Google Scholar]
- 27.Ramirez-Arcos, S., J. Szeto, J. A. Dillon, and W. Margolin. 2002. Conservation of dynamic localization among MinD and MinE orthologues: oscillation of Neisseria gonorrhoeae proteins in Escherichia coli. Mol. Microbiol. 46:493-504. [DOI] [PubMed] [Google Scholar]
- 28.Raskin, D. M., and P. A. de Boer. 1997. The MinE ring: an FtsZ-independent cell structure required for selection of the correct division site in E. coli. Cell 91:685-694. [DOI] [PubMed] [Google Scholar]
- 29.Raskin, D. M., and P. A. de Boer. 1999. MinDE-dependent pole-to-pole oscillation of division inhibitor MinC in Escherichia coli. J. Bacteriol. 181:6419-6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raskin, D. M., and P. A. de Boer. 1999. Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc. Natl. Acad. Sci. USA 96:4971-4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothfield, L., S. Justice, and J. Garcia-Lara. 1999. Bacterial cell division. Annu. Rev. Genet. 33:423-438. [DOI] [PubMed] [Google Scholar]
- 32.Schindelin, H., C. Kisker, J. L. Schlessman, J. B. Howard, and D. C. Rees. 1997. Structure of ADP × AIF4(−)-stabilized nitrogenase complex and its implications for signal transduction. Nature 387:370-376. [DOI] [PubMed] [Google Scholar]
- 33.Shih, Y. L., T. Le, and L. Rothfield. 2003. Division site selection in Escherichia coli involves dynamic redistribution of Min proteins within coiled structures that extend between the two cell poles. Proc. Natl. Acad. Sci. USA 100:7865-7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suefuji, K., R. Valluzzi, and D. RayChaudhuri. 2002. Dynamic assembly of MinD into filament bundles modulated by ATP, phospholipids, and MinE. Proc. Natl. Acad. Sci. USA 99:16776-16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szeto, T. H., S. L. Rowland, C. L. Habrukowich, and G. F. King. 2003. The MinD membrane targeting sequence is a transplantable lipid-binding helix. J. Biol. Chem. 278:40050-40056. [DOI] [PubMed] [Google Scholar]
- 36.Szeto, T. H., S. L. Rowland, L. I. Rothfield, and G. F. King. 2002. Membrane localization of MinD is mediated by a C-terminal motif that is conserved across eubacteria, archaea, and chloroplasts. Proc. Natl. Acad. Sci. USA 99:15693-15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, L., and J. Lutkenhaus. 1998. FtsK is an essential cell division protein that is localized to the septum and induced as part of the SOS response. Mol. Microbiol. 29:731-740. [DOI] [PubMed] [Google Scholar]
- 38.Zhou, H., and J. Lutkenhaus. 2003. Membrane binding by MinD involves insertion of hydrophobic residues within the C-terminal amphipathic helix into the bilayer. J. Bacteriol. 185:4326-4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou, H., and J. Lutkenhaus. 2004. The switch I and II regions of MinD are required for binding and activating MinC. J. Bacteriol. 186:1546-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]