

Abstract
Nucleic acid oxidation plays a vital role in the etiology and treatment of diseases, as well as aging. Reagents that oxidize nucleic acids are also useful probes of the biopolymers’ structure and folding. Radiation scientists have contributed greatly to our understanding of nucleic acid oxidation using a variety of techniques. During the past two decades organic chemists have applied the tools of synthetic and mechanistic chemistry to independently generate and study the reactive intermediates produced by ionizing radiation and other nucleic acid damaging agents. This approach has facilitated resolving mechanistic controversies and lead to the discovery of new reactive processes.
1. INTRODUCTION
Within physical organic chemistry, independent generation of reactive intermediates is a powerful method for proving their intermediacy in chemical processes and unambiguous characterization of their reactivity.1 Photochemistry is often the method of choice and under appropriate conditions the use of lasers and spectroscopic methods (eg ultraviolet (UV)–visible absorption, infrared) together enables their direct observation and kinetic characterization.2,3 In the absence of laser flash photolysis, product analysis, sometimes in conjunction with isotopic labelling, and competitive kinetics experiments of reactive intermediates generated under steady-state conditions have shed valuable light on their reactivity. For instance, investigations of independently generated carbon-centred reactive species, including radicals, radical ions and carbenes, have enhanced our understanding of the effects of substituents on reactivity, the effects of structure on ground state spin states and the effects of the latter on reactivity.4–6 A greater understanding of the connection between reactive intermediate structure and reactivity facilitates their use in organic synthesis and novel materials.7–11 During the past two decades, our understanding of oxidative damage of nucleic acids (DNA, RNA) has been greatly improved by independently generating reactive intermediates. Experiments carried out on nucleosides and oligonucleotides have resolved mechanistic controversies and uncovered novel chemical pathways.12–16
The importance of nucleic acid damage in aging, disease development, as well as the treatment of cancer, provides a part of the motivation for such investigations.17–21 However, nucleic acid oxidation is also useful for determining RNA structure and its folding dynamics, and may have applications in material science.22–26 Reactive oxygen species, especially hydroxyl radical (HO•), play a role in the therapeutic aspects of nucleic acid damage and disease etiology.17–19,27 Hydroxyl radical is also a powerful probe for determining biomacromolecular interactions and RNA folding dynamics.28–31 Much has been learned about the reactivity of HO• with DNA and RNA using various forms of ionizing radiation in conjunction with product analysis and various spectroscopic methods, including electron paramagnetic resonance (EPR) to detect radical intermediates.17,32 These investigations are limited by the lack of control over HO• reactivity, resulting in heterogeneous mixtures of reactive intermediates. Consequently, the formation of putative reactive intermediates produced by HO• from synthetic precursors both simplifies elucidating the chemistry of this important reactive oxygen species, and facilitates revealing complexities hidden by the formation of multiple reactive species within the biopolymers.
2. RADICAL FORMATION IN NUCLEIC ACIDS
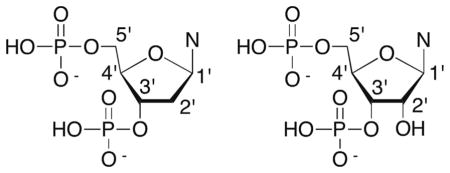
Radicals are directly produced in nucleic acids predominantly via hydrogen atom abstraction from the carbohydrate moiety or radical addition to the nucleobase ρ-bonds.17,32 Hydroxyl radical is produced by metal complexes, most notably Fe•EDTA, and is a major source of DNA damage by γ-radiolysis.32,33 Hydrogen atom abstraction by diffusible species such as HO• is believed to be governed by solvent accessibility and not bond dissociation energies due to the radical’s high reactivity. Solvent accessibility favors reaction at the C4′- and C5′-positions (Fig. 1A).34 Computational experiments also favor C5′-hydrogen atom abstraction in DNA, followed by reaction at C4′ and C1′ (Table 1).35 However, the C1′-hydrogen atom is buried in the minor groove and inaccessible to diffusible species such as HO•. Small molecules that bind in the minor groove of DNA, many of which have antitumor activity can access the C1′-position, as well as the hydrogen atoms bonded to the C4′- and C5′-carbons.36 Hydrogen atoms bonded to the C2′-carbon in DNA (Fig. 1) have considerably stronger bond dissociation energies (Table 1), and HO• is the rare exception of an oxidant that reacts at this position. Another example is the σ-radical derived from 5-halo-2′-deoxyuridines.37–43 The C3′-hydrogen atom (Fig. 1B) is abstracted less frequently due to the smaller number of oxidants that bind in the major groove of DNA, and possibly a surprisingly high calculated bond dissociation energy (BDE) (Table 1). The weaker bond strengths of the C2′- and C3′-carbon–hydrogen bonds in RNA (Table 1) compared to DNA are the greatest differences in potential hydrogen atom abstraction sites between the two biopolymers.
Figure 1.

Structure of duplex DNA from the perspective of the (A) minor groove and (B) major groove. Selected hydrogen atoms are highlighted in black. Reproduced from pdb: 2dau.
Table 1.
Calculated average bond dissociation free energies (kcal/mol) 35
| ||
|---|---|---|
| Bond | DNA | RNA |
| C1′-H | 93.4 | 92.6 |
| C2′-H | 97.0 | 86.5 |
| C3′-H | 97.0 | 92.6 |
| C4′-H | 93.8 | 93.3 |
| C5′-H | 91.3 | 91.3 |
Direct hydrogen atom abstraction occurs less frequently from the nucleobases, despite the expected modest carbon–hydrogen bond dissociation energy of the carbon–hydrogen bonds in the methyl groups of thymidine and 5-methyl-2′-deoxycytidine due to resonance stabilization of the incipient radicals. The respective radicals are also formed by deprotonation of the nucleobase radical cations, intermediates involved in electron transfer that are produced via one-electron oxidation. Amine radicals are also postulated as intermediates produced from the spontaneous decomposition of chloramines that arise from reactions of nucleosides with hypochlorous acid.44 However, the majority of nucleobase radical intermediates arise from the addition to nucleobase ρ-bonds. In fact, this is the kinetically preferred pathway for HO•. Although estimates vary, nucleobase addition may account for more than 90% of the reactions between nucleic acids and HO•.17 Significantly more data are available concerning the reactivity of pyrimidine nucleobases than purines. In fact, as discussed later, many questions regarding the reactivity of purine nucleobases remain.
3. THE NORRISH TYPE I PHOTOREACTION
A considerable number of examples described below in which nucleic acid radicals are independently generated take advantage of the α-photo-cleavage (Norrish type I) of ketones (Scheme 1 ).45 Most of the examples that will be cited involve photolysis of t-butyl or benzyl ketones (R′). This is consistent with the general quantum yield efficiency and rate constant for α-cleavage of the triplet excited state ketone. Consideration of the rate constant for decarbonylation of the ground state acyl radical is also relevant, with the efficiency of decarbonylation correlating with radical stability.46
Scheme 1.

Norrish type 1 photocleavage of ketones.
4. C1′-RADICAL GENERATION, REACTIVITY AND RELATED MECHANISTIC IMPLICATIONS
4.1 C1′-Radical Formation
Abstraction of the C1′-hydrogen atom in duplex DNA by diffusible species (eg HO•) is limited by its position in the minor groove (Fig. 1A), despite the relatively modest C1′-H BDE (Table 1). The solvent inaccessibility of the C1′-hydrogen is overcome by DNA oxidizing agents that bind in the minor groove. For instance, the antitumor agent neocarzinostatin (NCS), which forms a biradical upon reductive activation in the minor groove abstracts hydrogen atoms from the C1′-position.47 Activated forms of some coordination compounds, such as manganese porphyrins (MnPy) and copper bis-phenanthroline (Cu•OP2) also abstract the C1′-hydrogen atom.48–50
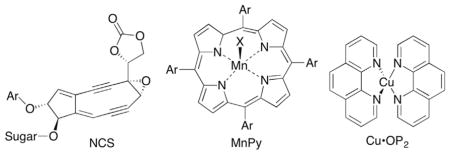
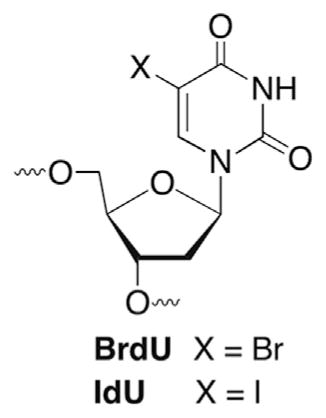
The C1′-radical is believed to form indirectly via reactions of initially formed intermediates. For instance, photolysis of menadione (2-methyl-1,4-napthoquinone, MD) in the presence of 2′-deoxycytidine (dC) produces the pyrimidine radical cation (1, Scheme 2 ).51,52 The radical cation of dC is proposed to yield the C1′-radical (2) upon deprotonation, which ultimately leads to 2-deoxyribonolactone (L), presumably via a mechanism discussed in more detail below. This process has not been detected in DNA, possibly because 1 is too short lived for deprotonation to compete with hole migration (electron transfer). Other pathways that do not involve radical cations produce 2-deoxyribonolactone via C1′-hydrogen atom abstraction by nucleobase radical adducts and are discussed in more detail below. Irradiation of DNA containing 5-bromo-(BrdU) or 5-iodo-2′-deoxyuridine (IdU) yields the highly reactive σ-radical (uracil-5-yl radical), which abstracts the C1′-hydrogen (and C2′-hydrogen) atom from the 5′-adjacent nucleotide.37,38,53,54 (This topic is discussed in more detail below in the section concerning C2′-radical reactivity.)
Scheme 2.

Pyrimidine radical cation formation and reactivity.
4.2 C1′-Radical Reactivity
The mechanism for transformation of the C1′-radical to 2-deoxyribonolactone (L) under aerobic conditions was examined using a photochemical precursor to generate 3 directly (Scheme 3 ).55–58 2′-Deoxyuridin-1′-yl radical (3) was generated via Norrish type I photocleavage of 4. Steady-state and laser flash photolysis experiments supported transformation of the C1′-radical into 2-deoxyribonolactone (L) under aerobic conditions via the carbocation (6). Superoxide formation was detected spectrophotometrically during steady-state generation of 3 from 4. However, the use of competitive kinetics using thiol and isotopic labelling (H218O) in conjunction with one another under steady-state photolysis conditions resulted in a gross underestimation of the rate constant for superoxide elimination from 5.57 Using laser flash photolysis, Newcomb detected the release of superoxide via its reduction of tetranitromethane, which is observed directly (350 nm).58 Deconvolution of these data yielded a rate constant for superoxide elimination from 5 of ~1 × 104 s−1, which is comparable to the rate constants reported for similarly substituted peroxyl radicals.59 Although comparable experiments were not reported in DNA, thiol trapping of 5 would not be expected to compete with superoxide elimination, suggesting that release of the reactive oxygen species will accompany 2-deoxyribonolactone (L) formation from 2′-deoxyuridin-1′-yl radical (3) under aerobic conditions.
Scheme 3.

2-Deoxyribonolactone (L) formation from a C1′-radical.
Laser flash photolysis of 4 under anaerobic conditions provided rate constants for thiol trapping of 2′-deoxyuridin-1′-yl radical (3, Scheme 4 ) by following the decay of the 3 at 320 nm β-Mercaptoethanol (BME), cysteine and glutathione (GSH) reacted with 3 between 2 and 4 × 106 M−1s−1.58 The rate constants for thiol trapping of 3 are slightly lower than those typically reported for reactions with other alkyl radicals, and may be a consequence of the stabilization of 2′-deoxyuridin-1′-yl radical (3) by two α-heteroatoms.60 These absolute rate constants were corroborated in single- and double-stranded DNA by competitive kinetic studies under aerobic conditions in which BME (and separately dithiothreitol) concentration was varied and the ratio of α,β-2′-deoxyuridine versus 2-deoxyribono-lactone (L) used to estimate the rate constant thiol trapping of 3.61 (Please note that reactive intermediates are referred to by the same descriptor whether they are monomeric or within biopolymers throughout this chapter.) The stereoselectivity of thiol reduction of 3 was also determined in single- and double-stranded DNA by generating the radical under anaerobic conditions. Product ratios were determined by HPLC analysis of nucleosides released upon enzymatic digestion of the DNA. β-2′-Deoxyuridine was favoured over the anomer by BME and dithiothreitol in single- and double-stranded DNA. The ratio of β-2′-deoxyuridine (β-dU) to α-2′-deoxyuridine (α-dU) varied between 4.1 and 4.5 in single-stranded DNA, but increased to between 6.2 and 8.3 in double-stranded substrates. Preferential formation of β-dU from 3 is relevant to the role of thiols as radioprotecting agents.62,63 Thiols not only need to compete with O2 for radicals but they need to restore the nucleic acids to guard against formation of potential pro-mutagenic nucleotides, such as α-dU.
Scheme 4.

Formation of α,β-2′-deoxyuridine from 2′-deoxyuridin-1′-yl radical (3).
4.3 Utility of C1′-Radical Generation as a Source of 2-Deoxyribonolactone in Mechanistic Studies
Independent generation of 2′-deoxyuridin-1′-yl radical (3) from 4 and its transformation into 2-deoxyribonolactone (L) under aerobic conditions provided a valuable tool for mechanistic studies on nucleic acid damage, although more efficient methods for generating L were subsequently developed.64–67 As noted above, a variety of DNA damaging agents abstract the C1′-hydrogen atom, ultimately producing L. The lactone is an example of an alkali-labile lesion, indicating that it yields strand breaks upon treatment with mild base. In fact, L is so labile that it yields strand breaks upon incubation in aqueous buffer, albeit with a half-life on the order of days.68,69 The copper bis-phenanthroline complex (Cu•OP2) was a notable exception. Although C1′-oxidation was the predominant pathway proposed by its pioneering discoverer, David Sigman, the copper complex yielded direct strand breaks (Scheme 5 ).70 Despite the absence of L in DNA damaged by Cu•OP2, the formation of 5-methylene-2-furanone (8), which could be construed to arise from 2-deoxyribonolactone, as well as a labile intermediate detected by gel electrophoresis that could be butenolide 7 were consistent with C1′-oxidation.50,71
Scheme 5.

Direct strand scission by Cu(OP)2.
The absence of L and formation of direct strand breaks when DNA is treated with Cu•OP2 was investigated by several laboratories. Several reports by Sigman implied that 2-deoxyribonolactone (L) was an intermediate en route to strand scission.49,50 However, subsequent mechanistic studies using C1′-deuterated DNA and 18O-labelling led to a mechanism that avoids formation of L (Scheme 6 ).72,73 Although this mechanism still began with C1′-hydrogen atom abstraction, the initially formed radical was oxidized to the carbocation, which then yielded a ketene acetal (9) that undergoes hydrolysis of the allylic 3′-phosphate to yield a strand break. Formation of 9 to explain 18O incorporation from H218O was unnecessary because of the reactivity of 3 described above (Scheme 3 ).
Scheme 6.

Sigman’s proposal for DNA strand scission by Cu(OP)2.
Experimental doubt was cast on the kinetic viability of 9 using a model system in which 11 was independently synthesized by oxidizing 10 (Scheme 7 ).74 The nucleoside model (11) was stable for days under physiologically relevant pH and temperature. This model study provided an alternative explanation for the formation of direct strand breaks by Cu•OP2 and the absence of L. β-Elimination from 12 was first-order in Cu•OP2 and the rate constant was such that an effective molarity of 10 M in an intramolecular reaction would yield a half-life of <1 min for L in DNA.74 Hence, the model study suggested that while a variety of oxidants damage DNA by abstracting the C1′-hydrogen, the differing products (direct strand breaks versus the alkali-labile lesion 2-deoxyribonolactone, L) were a consequence of the instability of L in the presence of Cu•OP2.
Scheme 7.

Model studies on Cu(OP)2 cleavage of DNA.
Sugiyama put forth a completely different mechanism to explain the lack of 2-deoxyribonolactone formation from Cu•OP2.75 Using hexanucleotide duplexes and electron spray ionization-mass spectrometry (ESI-MS) to analyze products, Sugiyama confirmed that Cu•OP2 forms L in relatively high yield but the lactone does not yield strand breaks via β-elimination in these short DNA substrates. Furthermore, no evidence for a 1′,2′-dehydronucleotide (eg 9) was observed. The authors detected strand scission products consistent with C4′- and C5′-hydrogen atom abstraction, which led them to conclude that these were the positions that Cu•OP2 reacted with to yield direct strand breaks. The significance of the stability of L in these substrates was questioned because duplexes containing the lactone lesion melted below the temperature at which the aforementioned experiments were carried out.12 The Cu•OP2 would not be expected to bind to the single-stranded oligonucleotides and catalyze β-elimination.76
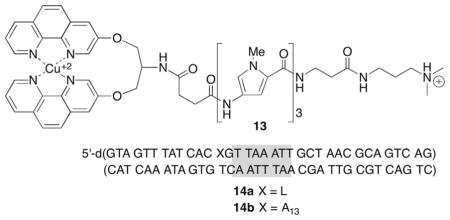
The latest mechanistic proposal was derived from experiments that combine the ability to independently synthesize duplex DNA containing 2-deoxyribonolactone (L) at a specific position from 2′-deoxyuridin-1′-yl (3) with sequence-selective delivery of Cu•OP2.12,77 The metal complex was delivered to the position where L was produced by tethering the metal complex to a minor groove DNA-binding molecule (13).78,79 The half-life for L in a duplex (14a) bound by minor groove-binding distamycin tethered to Cu•OP2 was 20.6 min (kElim = 5.6 ± 0.7 × 10−4 s−1), and was ~100 times shorter than when the metal complex was not delivered to the lesion. Cu•OP2-induced strand scission at 2-deoxyribonolactone was supported by experiments containing dA at the position where L was independently generated in an otherwise identical duplex. Oxidation at this position (A13 in 14b) by the distamycin-Cu•OP2 conjugate (13) was recorded by measuring the combined yields of alkali-labile lesions (presumably mostly L) and direct strand breaks as a function of time. The overall rate constant for oxidation (kOx = 1.9 ± 0.6 × 10−5 s−1) was slower than the rate constant measured for cleavage at L incorporated at the comparable position. In addition, fitting the growth and decay of alkali-labile lesions as a function of time to a sequential mechanism yielded a rate constant for decay of these lesions (kDecay = 4.7 ± 0.9 × 10−4 s−1, Eq. [1]) that is within experimental error of the independently measured rate constant for cleavage at L (kElim). Unless, more than one alkali-labile lesion reacts with similar rate constants, the similarity in kDecay and kElim indicates that 2-deoxyribonolactone (L) is the pre-dominant alkali-labile lesion that is cleaved in the presence of Cu•OP2. Similarly, the rate constant for the formation of the alkali-labile lesions (kGrow = 1.8 ± 0.4 × 10−5 s−1, Eq. [1]) was within error of the overall rate constant for oxidation (kOx). This latter point supports the suggestion that alkali-labile lesion formation accounts for the majority of Cu•OP2-mediated DNA oxidation. In summary, the ability to independently generate 2′-deoxyuridin-1′-yl radical (3) at a defined position within DNA enabled carrying out kinetic experiments on 2-deoxyribonolactone (L) reactivity that support the original mechanism put forth by Sigman explaining why Cu•OP2 produces direct strand breaks following C1′-hydrogen atom abstraction, whereas other damaging agents yield L as a final product following the same initial oxidation event.
| [1] |
4.4 Probing DNA Repair Enzyme Activity Using Independently Generated 2′-Deoxyuridin-1′-yl Radical (3)
Independent formation of 2-deoxyribonolactone (L) in DNA using ketone 4 was also useful for examining the interactions of the DNA lesion with repair enzymes. Base excision repair (BER) is a general pathway used by cells to replace damaged nucleotides with the correct native one (Scheme 8 ).80–82 (Tomas Lindahl received a share of the 2015 Nobel Chemistry Prize for his work on BER.) BER typically proceeds through abasic site (AP) intermediates that are produced by glycosylases, which recognize damaged nucleobases (NDam). Some glycosylases are bifunctional and are equipped with the ability (lyase activity) to induce β-elimination, or even β,δ-elimination of the AP sites. The primary BER pathway in mammalian cells involves a similar lyase reaction by DNA polymerase β (Pol β) following hydrolysis of the 5′-phosphate of the AP site by apurinic endonuclease 1 (Ape1) to produce dRP. Pol β then translocates the DNA to its polymerase active site and fills in the now missing nucleotide before passing the substrate off to DNA ligase, which stitches the DNA together.
Scheme 8.

Base excision repair.
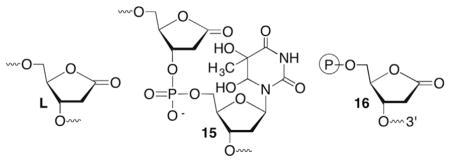
Iminium ions (Schiff bases) are common intermediates in the lyase reactions carried out by various repair enzymes (Scheme 9 ). Consideration of elementary organic chemistry raised the question concerning how BER enzymes that possess lyase activity would cope with 2-deoxyribonolactone (L). Biochemically, this is potentially important because failure to correctly repair damaged DNA can lead to mutagenesis and/or cell death. Indeed, incubation of duplex DNA containing 2-deoxyribonolactone (L) produced from photolysis of 4 revealed that none of a series of eight BER glycosylases incise the lesion.83,84 In addition, the presence of L when part of a tandem lesion 15 (defined as two or more contiguously damaged nucleotides) inhibited repair of the accompanying damaged nucleotide as well.85 Moreover, 2-deoxyribonolactone (L) irreversibly inhibits one of the enzymes, endonuclease III (Nth) by forming cross-links with the nucleophilic lysine residue that is responsible for Schiff base formation.83,84 This was the first demonstration that a DNA lesion inactivates a repair enzyme, a process that has since been characterized for a small number of other damaged nucleotides and enzymes.86–90
Scheme 9.

DNA damage upon irradiation of 5-halopyrimidines.
2-Deoxyribonolactone (L) inactivation of Nth may be of minor importance because excision of an incised AP site by Pol β is the primary pathway for removing this lesion after incision by the phosphodiesterase, apurinic endonuclease I (Ape 1). Similar to the interaction with Nth, incised 2-deoxyribonolactone (16) cross-links to Pol β in a manner that is dependent upon the presence of the lysine residue believed to be responsible for Schiff base formation.91 Recently, evidence was provided for cross-linking between 16 and Pol β in mammalian cells.92 This latest report illustrates the possibility that the BER enzyme inactivation by DNA lesions provides a chemical basis for the cytotoxic effects of the therapeutic agents and other modalities that produce them.93
5. C2′-RADICAL GENERATION AND REACTIVITY IN DNA, RIBONUCLEOSIDES AND RNA
5.1 C2′-Radical Formation Following Irradiation of 5-Halopyrimidine Nucleotides in DNA
As mentioned above, C2′-hydrogen atom abstraction in DNA is a rare occurrence due to the strong carbon–hydrogen bond (Table 1) and relative inaccessibility of the hydrogen atoms to diffusible species, such as HO•.34 C2′-Radical formation is indirectly detected when DNA containing 5-bromo-(BrdU) or 5-iodo-2′-deoxyuridine (IdU) is irradiated. UV irradiation generates the uracil-5-yl radical (17) via photoinduced electron transfer from a 2′-deoxyguanosine within the duplex (Scheme 10 ).53 The efficiency of σ-radical (17) generation is highly sequence dependent due to competition between halide ion loss and back electron transfer.38,54 The uracil-5-yl radical (17) abstracts hydrogen atoms from the C1′- and C2′-positions of the 5′-adjacent nucleotides (Scheme 10 ). Hydrogen atom abstraction from these positions by 17 was determined using deuterium isotope effects, tritium transfer and product studies.40–43,53 2-Deoxyribonolactone (L) formation is indicative of C1′-oxidation. Whether the initially formed C1′-radical is oxidized via the distal oxidized purine or by O2 as described above (Section 4.2) is uncertain, as H2O is the ultimate source of the carbonyl oxygen in either scenario.38,57,58 Formation of the C2′-oxidized abasic site (18) is attributed to O2 trapping of the radical produced upon C2′-hydrogen atom abstraction.41 The details for obtaining 18 from the peroxyl radical are uncertain, including the identity of the reducing agent. A Criegée rearrangement is drawn here but the original report suggested a different mechanism.41 Independent generation of the C2′-radical (20) from 2′-iodo-2′-deoxyuridine (19, Scheme 11 ) in DNA or a nucleoside provided 18 but 2-deoxyribonolactone (L) was also observed. Photolysis of C1′-deuterated 19 indicated that the lactone was formed via oxidation of the C2′-radical (21) and subsequent 1,2-hydride rearrangement (Scheme 11 ).94 Although not proposed at the time, one could invoke a photoinduced single electron transfer mechanism for the generation of 20 analogous to that substantiated for BrdU. In that situation the oxidized purine nucleotide formed upon irradiation could carry out the oxidation of the C2′-radical in DNA. Moreover, this chemistry suggests that C2′-hydrogen atom abstraction by a uracil-5-yl radical (17) could yield the formal C1′-oxidation product (L) in DNA.
Scheme 10.

DNA damage upon irradiation of 5-halopyrimidines.
Scheme 11.

C2′-DNA radical reactivity.
The reactivity of the uracil-5-yl radical (17) is strongly dependent upon the DNA structure and is also affected by whether the complementary strand is RNA.95,96 The reactivity of the C2′-radical is also affected by the nucleic acid structure and the distance between it and the electron-deficient nucleobase.97,98 The C2′-oxidized abasic site (18) is observed in B-DNA, whereas the respective ribonucleotides are produced in Z-DNA. The product distribution obtained upon irradiating BrdU and/or IdU in DNA has been used by Sugiyama to probe nucleic acid structure.99 As discussed below, the chemistry is different still in RNA100
5.2 Generation and Reactivity of the 2′-Radical in RNA
The significant radical stabilizing energy of a hydroxyl group weakens the C2′-carbon–hydrogen bond in RNA relative to that in DNA and may be enhanced due to hydrogen bonding to the 3′-phosphate (Table 1).35,101 Stabilized radicals containing β-leaving groups undergo heterolytic fragmentation to produce radical cations, a process that is facilitated in polar solvents such as water.102–106
Norrish type I photocleavage has been used to generate the adenosin-2′-yl radical (22) and uridin-2′-yl radical (23) from 24 and 25, respectively (Scheme 12 ).107–109 The reactivity of each radical was examined with thiol. Trapping of 22 in aqueous buffer by GSH yielded adenine and a 3.5:1 ratio of arabinoadenosine and adenosine, although the yield was not reported.107 A competition study in which the ratio of reduction products (arabinoadenosine + adenosine) versus adenine were measured as a function of [GSH] suggested that the rate constant for loss of the nucleobase was ~2 × 105 M−1 s−1. This ratio of deglycosylation versus thiol trapping products is significantly different than what was observed from 23. When 25 was photolyzed in phosphate buffered (pH 7.2) saline (100 mM), uracil (56–59% based upon unrecovered 25) was the only product detected in the presence of BME (0.25 M).108 The inability of thiol to trap 23 in aqueous buffer suggests that loss of uracil is significantly faster than that of adenine. Thiol trapping (BME, 0.1 M) of 23 was observed in H2O and greater yields of arabinouridine were obtained as increasing amounts of acetonitrile were added. Another difference between 23 and 22 was that arabinouridine was the only reduction product formed, which is typical of reductions of nucleoside radicals.110–112
Scheme 12.

Photochemical generation and reactivity of ribonucleoside 2′-radicals.
Uridin-2′-yl radical (23) was also independently generated from 25 in single- and double-stranded RNA.109 The radical rapidly yields direct strand breaks in an O2-independent manner (Scheme 13 ). Thiol (BME, 1 M) also has no effect on strand scission. Assuming rate constants for the reactions of O2 (kO2 = 2 × 109 M−1s−1) or BME (kBME = 1–10 × 106 M−1s−1)113 with 23 and the respective concentrations of the traps ([O2] = 0.2 mM, [BME] = 1 M), the rate constant for strand scission was estimated to be >106 M−1s−1. Based upon precedents in DNA (more below when the chemistry of C4′-hydrogen atom abstraction is discussed), and the general reactivity of alkyl radicals containing β-leaving groups noted above, strand scission was postulated to occur via heterolytic cleavage of the C3′-carbon–oxygen bond in single- and double-stranded substrate to form an olefin cation radical (26, Scheme 13 ).106 Product analyses using gel electrophoresis in conjunction with enzymatic and chemical reactivity are consistent with this. The 3′-fragment is composed solely of a 5′-phosphate terminus (27). The major products detected at the 3′-terminus of the 5′-fragment are the phosphate (28) and 2′-keto-3′-deoxyuridine (29). The ratios of these products depend upon O2 and thiol concentration. The ketone product (29) dominates (>3:1) under anaerobic conditions, even at low thiol concentration (5 mM). It is not known whether the initially formed radical cation (26) undergoes deprotonation (30), followed by hydrogen atom transfer to the α-keto radical or is reduced directly to the enol (31), which tautomerizes to 29 (Scheme 14 ). Radical cation 26 may also be reduced by guanosine within the RNA. This should be thermodynamically favourable. However, no evidence has been presented in support of this. 3′-Phosphate (28) formation is favoured over 29 under aerobic conditions but its mechanism of formation is less obvious. One speculative mechanism that requires further investigation involves O2 trapping of the α-keto radical (30, Scheme 14 ). Other questions, including uracil loss in the oligonucleotides, which was not reported on also need to be addressed.
Scheme 13.

Direct RNA strand scission from a C2′-radical.
Scheme 14.

Product formation following RNA strand scission.
5.3 C2′-Radical Formation and Reactivity Following Irradiation of 5-Bromouridine in RNA
Recently, the photochemistry of 5-bromouridine (BrU) was examined in a series of sequences in which BrU was flanked on its 5′-side by either adenosine or guanosine (Scheme 15 ).100 In contrast to studies involving BrdU, no evidence for C1′-hydrogen atom abstraction by the uracil-5-yl radical (32) was obtained. Conformational differences between duplex RNA and DNA could contribute to this difference, but certainly the more favorable C2′-carbon–hydrogen BDE in RNA than DNA (Table 1) plays a role. The final product resulting from C2′-oxidation (characterized by MS) is also different in RNA than DNA. The C2′-oxidized abasic site (18, Scheme 11 ) is not observed, nor do the authors report any strand scission resulting from heterolytic cleavage of the 3′-phosphate (Schemes 13 and 14). The sole C2′-oxidation product observed in RNA when the adjacent nucleotide is adenosine or guanosine is the 2′-keto purine (33). The authors attribute 33 to oxidation of the C2′-radical by the one-electron oxidized guanosine that provided the initial electron used to reduce BrU. Based upon the reactivity of uridin-2′-yl radical (23) described above, electron transfer to the oxidized purine must occur faster than 106 s−1 in order to prevent strand scission.
Scheme 15.

RNA damage upon irradiation of 5-bromouridine.
6. C3′-RADICAL GENERATION AND REACTIVITY IN DNA
6.1 C3′-Radical Formation Following Irradiation of Transition Metal Coordination Complexes
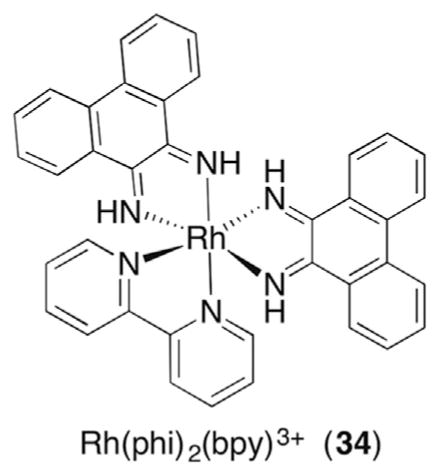
The C3′-hydrogen exposure to diffusible species is considerably less than hydrogen atoms at the C4′- or C5′-positions (Fig. 1B) and the calculated carbon–hydrogen BDE is surprisingly high given its substitution by a phosphate group (Table 1). In addition, the C3′-hydrogen lies in the major groove of DNA, while most small molecules bind in the minor groove. Consequently, molecules that abstract the C3′-hydrogen atom from DNA are largely confined to coordination compounds. The Barton group has described a number of Rh complexes (eg Rh(phi)2(bpy)3+, 34) that bind in the major groove and oxidatively damage DNA upon photoexcitation.114,115 C3′-Hydrogen atom abstraction (35) is supported by the binding preference and product analysis. Strand scission was observed under aerobic and anaerobic conditions (Scheme 16 ). 3′-Fragments containing 5′-phosphate termini were formed exclusively regardless of whether O2 was present or not, as were free nucleobases. The 5′-fragments were composed of a mixture of 3′-phosphate and 3′-phosphoglycoaldehyde (37) termini. The remaining three carbons of the 2′-deoxyribose ring were released in the form of base propenoic acids (38) and were only detected under anaerobic conditions. Peroxyl radical formation (36) from the C3′-radical and subsequent Criegée rearrangement of hydroperoxide reduction product are consistent with the O2 dependence of 37 and base propenoic acid (38) generation. Under anaerobic conditions, the C3′-radical (35) ultimately must be oxidized and trapped by H2O (39). However, as is often the case with studies in nucleic acids, it is unclear what oxidizes the C3′-radical. Similarly, it is unknown what reduces the peroxyl radical to the hydroperoxide (36).
Scheme 16.

Photoinduced C3′-oxidation by rhodium coordination complexes.
6.2 Independent Generation and Reactivity of Thymidin-3′-yl Radical (35)
The observations and mechanistic proposals put forth by Barton have largely been corroborated by experiments in which thymdin-3′-yl (35) was independently generated via Norrish type I photocleavage from 40a,b.116–119 A preliminary report established that 35 produced in single-stranded oligonucleotides was efficiently trapped by GSH under anaerobic conditions. The reduction products were accompanied by small amounts of strand scission but the mechanistic source of the fragmentation was not ascertained.116 Strand scission resulting in the formation of fragments containing phosphate groups at their termini (Scheme 17 ) was the major pathway when 35 was generated under anaerobic conditions in the absence of thiol. This was consistent with the chemistry discovered by Barton (Scheme 16 ), and the metastable ketone (39) was proposed as an intermediate, although the authors note that the source of the oxidant of 35 is unknown. In addition, an unspecified yield of cleavage product resulting from oxidation of the 3′-adjacent nucleotide was observed and it was suggested that radical transfer occurs via hydrogen atom abstraction by 35.
Scheme 17.

Independent generation of a C3′-radical in DNA.
Greater detail was provided using mass spectrometry to characterize products when 35 was generated in single-stranded oligonucleotides via Norrish type I photocleavage under aerobic conditions and dilute GSH (6 mM).117 The major products were consistent with those detected by Barton. For instance, the majority of 3′-fragments contained 5′-phosphate termini (82%) and the majority of the 5′-fragments (78%) were attributable to the 3′-keto thymidine (35, Scheme 16 ). Ketone 39 and elimination product 41 were observed in a combined 21% yield. The 3′-phosphate product, which also presumably arises from 35 was the major product (57%). In contrast to Barton’s experiments (Scheme 16 ), the 3′-phosphoglycoaldehyde (37) was only formed in 9% yield under aerobic conditions, and the authors did not report the base propenoic acid (38) that would be expected to accompany formation of this product. This difference may indicate that the coordination complexes influence the Criegée rearrangement, possibly by acting as an acid catalyst. Alternatively, the GSH present in the experiments where 35 is independently generated from 40 may reduce the hydroperoxide derived from 36 (Scheme 16 ) before it can rearranges, which would result in greater amounts of 39.
The C3′-peroxyl radical (36) derived from 35 was also suggested to abstract hydrogen atoms in an intranucleotidyl and internucleotidyl manner (Scheme 18 ). (The detailed reactivity of the subsequently formed radicals is discussed in Sections 7.2–7.5 and 8.2 and 8.3.) Intranucleotidyl abstraction of the C4′-hydrogen atom is proposed to explain the formation of small amounts (4%) of phosphoglycolate (42), leaving <10% of the 5′-fragment unaccounted for. A greater yield of 3′-fragments (18%) is attributed to 5′-hydrogen atom abstraction from the 3′-adjacent nucleotide. The diagnostic C5′-aldehyde (6%, 43) and 5′-phosphorylated 3′-fragment lacking the 3′-adjacent nucleotide (12%) are observed by high pressure liquid chromatography (HPLC) and matrix assisted laser desorption ionization-fime of flight (MALDI-TOF) MS. The latter is presumably formed via elimination from 43.120 The proposed sites of hydrogen atom abstraction are consistent with O2 trapping from the α-face of 35. These product observations were corroborated in a later study in which the GSH concentration was varied from 0 to 30 mM where there was a complex product dependence on thiol.118 More limited studies have been carried out in which 35 was generated in double-stranded substrates, and it is unknown how this more conformationally restricted environment affects the reactivity of the C3′-radical (35) and subsequently formed intermediates.119
Scheme 18.

Intra- and internucleotidyl hydrogen atom abstraction by a C3′-peroxyl radical (36).
7. C4′-RADICAL GENERATION AND REACTIVITY IN DNA AND RNA
7.1 C4′-Radical Formation
Due to its accessibility on the outer edge of the minor groove (Fig. 1A) and its relatively favourable C–H bond dissociation energy (Table 1), the C4′-hydrogen is abstracted by a number of DNA damaging agents, including HO• and the antitumor agent bleomycin.34,121 Several members of the enediyne family of antitumor agents also form the C4′-radical.122–124
7.2 C4′-Radical Reactivity in DNA
There are three general reaction pathways associated with the C4′-radical in DNA (44, Scheme 19 ). Much of the mechanistic details for two of these pathways were worked out using bleomycin as a means for generating the radical.125 Bleomycin is a peptide that coordinates iron. The resulting complex carries out redox chemistry in the presence of O2, and selectively abstracts the C4′-hydrogen atom from nucleic acids. The coordination complex, and in particular the metal is believed to be involved in subsequent steps.126 Inferential support for the latter is based upon metal-independent methods for generating the C4′-radical, such as γ-radiolysis.103 However, C4′-oxidation in DNA by other reagents has not been examined as thoroughly as that by bleomycin. Studies carried out using bleomycin made extensive use of isotopic labelling. The C4′-radical has also been independently generated in DNA and RNA from stable photochemical precursors. It has been produced most frequently from a t-butyl ketone (45), although a phenyl selenide (46) was also employed in some investigations.
Scheme 19.

Independent generation and reactivity of a C4′-radical (45).
C4′-hydrogen atom abstraction yields the alkaline-labile, oxidized abasic site (C4-AP). Isotopic labelling ruled out bleomycin formation of C4-AP via an oxygen rebound-type mechanism, and instead oxidation of 44 and trapping of the carbocation (47) by H2O was invoked (Scheme 20 ).127,128 However, the species responsible for oxidizing 44 is uncertain. Other damaging agents, such as the enediynes produce C4-AP as well via hydrogen atom abstraction. However, in these instances the mechanism proceeding from 44 to C4-AP is also unknown. (As discussed in Section 7.3, it is unlikely that superoxide elimination from 48 yields 47.) Strand breaks are produced at C4-AP upon mild treatment with hydrazine or 0.1 M NaOH at 37 C.129,130 This molecule has rich chemistry in its own right. It is an irreversible inhibitor of the vital DNA repair enzyme DNA polymerase β.90 In addition, strand scission at C4-AP is significantly accelerated in nucleosomes.131,132 The histone proteins, which are modified during the process, catalyze strand scission.
Scheme 20.

C4-AP generation and reactivity.
The formation of direct strand breaks containing 3′-phosphoglycolate termini (42) in the 5′-fragment is a signature product indicative of C4-oxidation (Scheme 21 ). (Please note that as described in Section 6.2 C4′-oxidation can be a secondary process when the C3′-hydrogen atom is abstracted.) The 3′-DNA fragment contains a phosphate group at its 5′-terminus and the remaining three carbons of the 2′-deoxyribose that underwent oxidation are accounted for by the formation of base propenals. Kinetic isotope effects of C4′-isotopically labelled substrates (3H, 2H) definitively documented rate limiting hydrogen atom abstraction from this position by bleomycin.121,133,134 A key step in the overall transformation is a putative Criegée rearrangement of an intermediate hydroperoxide, which is believed to be catalyzed by the metallopeptide. As mentioned previously, this is another instance in which the reducing agent responsible for hydroperoxide formation from the peroxyl radical is unidentified. One possibility is that a Fe+3 species could be the reductant, which would reactivate bleomycin. Bleomycin reactivation while bound to DNA has been proposed as a means for producing some double-strand breaks.135 Nonetheless, the steps leading up to and including the formation of 48 are widely accepted.
Scheme 21.

Phosphoglycolate formation from a C4′-peroxyl radical (48).
A greater number of proposals were proffered explaining formation of the final observed products following the Criegée rearrangement. Extensive experimentation that included determination of the sequence in which strand scission, 3′-phosphoglycolate (42), 5′-phosphate and base propenal (50) release occur gave rise to the currently accepted mechanism (Scheme 21 ).126 This mechanism is consistent with incorporation of a single 18O atom from 18O2 into the glycolate. Importantly, the 18O is not from the activated bleomycin but comes from 18O2 trapping of the C4′-radical.136 In contrast, the 18O incorporated into the base propenal carbonyl is derived from H218O.126 In addition, elegant experiments in which the C2′-position was stereoselectively labelled with 3H revealed that the 2′-pro-R proton is removed en route to base propenal and that deprotonation occurs prior to DNA strand scission.137,138
Direct strand breaks attributable to the C4′-radical are produced that yield 3′- and 5′-phosphate termini instead of 3′-phosphoglycolate termini and base propenal (Scheme 22 ). This phenomenon was originally observed in DNA subjected to γ-radiolysis under deoxygenated conditions.103 Interestingly, the mechanism was originally proposed by von Sonntag based upon the products derived from the 2′-deoxyribose component of the DNA. The phosphate termini, while undoubtedly produced by one or more of the myriad pathways populated upon γ-radiolysis of DNA were proposed to rationalize the formation of the sugar degradation products. As seen below, this pathway proved prescient and the intermediate formation of olefin radical cations (eg 51) was immensely useful for studying electron transfer in DNA (Section 7.5).
Scheme 22.

DNA strand scission via olefin radical cation formation.
7.3 Independent Generation and Reactivity of C4′-Radicals in DNA
Strong evidence for the von Sonntag mechanism was obtained by independently generating C4′-radicals (Scheme 19 ) via Norrish type I photocleavage of 45 and to a lesser extent phenyl selenide 46.139,140 Initial studies under anaerobic conditions on monomeric models and single-stranded oligonucleotides containing 45 or 46 supported initial 3′-phosphate cleavage, followed by regioselective addition of H2O to regenerate a C4′-radical that then eliminated the 5′-phosphate (Scheme 22 ). Subsequent studies confirmed preferential H2O addition to the C3′-position of 51 to yield a diastereomeric mixture of 52.141 The products were characterized by MALDI-TOF MS, which was useful for subsequent 18O studies under aerobic conditions.
Under aerobic conditions, the authors observed an ion whose m/z corresponded to the hydroperoxide (eg 49) derived from O2 of the initially formed C4′-radical (44). 18O2 labelling, which showed that two 18O atoms were incorporated, corroborated this assignment. Interestingly, the respective hydroperoxide was detected from the oligonucleotide containing 45 but not 46. The latter precursor yielded a strand scission product that based upon MS analysis in conjunction with 18O2 and H218O labelling was proposed to be the hydroperoxide containing a 3′-hydroxyl group (53, Scheme 23 ). The difference in products from the two precursors was suggested to be due to formation of a radical pair from 46, which prevented O2 trapping of the initially formed C4′-radical.140 Both hydroperoxides produce the 3′-phosphoglycolate (42) signature product of C4′-oxidation (Schemes 21 and 23). The cleaved oligonucleotide containing a 3′-hydroxyl group was proposed to undergo a Grob-type fragmentation (Scheme 23 ), whereas the intact DNA containing hydroperoxide was believed to undergo a Criegée rearrangement as discussed above for bleomycin-induced strand scission (Scheme 21 ). Unfortunately, kinetic studies on 3′-phosphoglycolate formation were not reported, as this would have been useful for evaluating the role of the metallated bleomycin in the respective rearrangement.
Scheme 23.

Phosphoglycolate formation via Grob fragmentation.
C4′-Radical generation from 45 and 46 under anaerobic conditions in the presence of GSH yielded the expected diastereomeric mixture of reduction products consisting of the native nucleotide and the C4′-epimer (54, Scheme 24 ).106 Hydrogen atom delivery from the α-face to restore the naturally occurring stereochemistry of the DNA was slightly favoured in single-stranded substrates (≤2:1), but much more so (<8:1) when the C4′-radical was generated in duplex DNA. Competition studies between strand scission and GSH (kGSH) trapping provided estimates of the rate constant for strand scission from DNA containing C4′-radical via phosphate elimination and concomitant radical cation (51, Scheme 24 ) formation. The ratio of hydrogen atom trapping products (native nucleoside + 54) relative to strand scission was measured as a function of GSH concentration. In single-stranded DNA the rate constant (kCleave) was estimated to be from ~0.8 to 1.9 × 103 s−1. Moreover, the C4′-radical was observed to yield strand breaks ~10-fold more slowly in double-stranded DNA (kCleave ~0.2–2.1 × 103 s−1). Although fundamentally the same type of process described above for strand scission from the C2′-radical in RNA, strand scission from the C4′-radical in DNA is at least 1000-times slower.109
Scheme 24.

Competitive kinetic determination of rate constant for C4′-radical fragmentation.
The rate constants for strand scission via phosphate elimination from the C4′-radical in DNA are also considerably slower than the expected rate of radical trapping by O2 (kO2 ~2 × 109 M−1s−1, [O2] ~0.2 mM). However, strand scission products ascribable to phosphate elimination from the C4′-radical were reported. This apparent discrepancy was resolved by competitive kinetic studies with GSH under aerobic conditions from which O2 trapping was determined to be reversible (Scheme 25 ).142 In contrast to the discussion above regarding elimination of superoxide from a C1′-peroxyl radical (See Section 4.2), the C4′-peroxyl radical fragments much more slowly (kO2 ~ 1 s−1) and does so homolytically. However, the estimated rate constant is consistent with that for related radicals containing a single oxygen substituent.59
Scheme 25.

Reversible peroxyl radical (48) formation.
7.4 Double-Strand Cleavage via a Single C4′-Radical
Molecules that produce double-strand breaks are rare and highly valued due to the biological significance of this form of DNA damage.135,143–147 Hence, the possibility that a single radical can result in a double-strand break via interstrand hydrogen atom transfer by one or more intermediates is very interesting. Radiation scientists debated this possibility in the 1990s due the observation that double-strand break yield varied linearly with dose (which HO• yield is proportional to) at low doses of radiation.148,149 Ultimately, the formation of locally high HO• concentrations (‘spurs’), which led to clusters of DNA lesions and in turn double-strand breaks, became widely accepted.150–152 Recently, Taverna Porro discovered a chemical pathway by which a single C4′-radical yields double-strand breaks in an O2-dependent manner.13,153
Reversible peroxyl radical formation from the C4′-radical (Scheme 25 ) was crucial for providing an explanation for the O2-dependent production of double-strand breaks from this species.13,153 Generation of 44 from 45 in duplex DNA yielded double-strand breaks under aerobic but not anaerobic conditions. ESI-MS analysis and deuterium kinetic isotope effects indicated that complementary strand scission resulted from C4′-hydrogen atom abstraction at nucleotides opposite the three most proximal 5′-adjacent nucleotides (Scheme 26 ). This produces double-strand breaks biased in the 3′-direction, which is consistent with reaction in the minor groove of right-handed helical DNA.154 Double-strand scission is made possible via cleavage of the initial strand containing a C4′-radical via the von Sonntag mechanism. As described by Giese, the radical cation (51) yields a second peroxyl radical via sequential addition of H2O and O2.140,141 Four different peroxyl radicals can be produced from 51 with those resulting from H2O addition at C3′-favoured.141 Mechanistic studies using oligonucleotide substrates that contain the C4′-radical precursor (45, Scheme 19 ) at their 3′-termini provided an independent method for producing the C3′-hydration product of 55.153 This radical did not yield opposite strand cleavage, suggesting that the (minor) regioisomeric C4′-hydration product that yields the C3′-peroxyl radical(s) (56) is responsible for interstrand hydrogen atom abstraction.
Scheme 26.

Double-strand break formation from a C4′-radical (44).
7.5 The Role of Independent C4′-Radical Generation in Understanding Electron Transfer in DNA
Although this subject has been reviewed, one would be remiss not to briefly mention how independent C4′-radical generation in DNA contributed to this important problem.155,156 The efficiency, distance and sequence dependence of DNA hole migration was vigorously debated in the 1990s.20,157–160 Elucidation of the rules that govern this process are useful to this day as DNA electron transfer is a continued source of chemical innovation.26,161–165 Briefly, Barton reported the rapid, long-range (40 Å) transfer of electrons between two metal complexes bound to DNA.166–168 In addition, initial reports indicated that in contrast to electron transfer in proteins there was very small dependence of hole migration on distance. These observations raised the possibility that a DNA duplex could behave like a molecular wire in its ability to conduct electrons. Although one can transfer electrons over large distances through DNA, it is now accepted that the efficiency is strongly dependent on sequence.169
Giese made significant contributions to this contested subject by taking advantage of 44 generation from 45 (Scheme 19 ). Irreversible formation of olefin cation radical 51 from 44 (Scheme 22 ) was critical for the success of this approach. The reduction potential of the radical cation (51) is sufficient to oxidize 2′-deoxyguanosine. Hence, C4′-radical generation provides a site-specific means for irreversibly introducing a hole in DNA. Using chemically synthesized oligonucleotides containing 45, Giese was able to demonstrate the importance of sequence on hole transfer efficiency. The interested reader is referred to the references noted above to learn more about this topic.
7.6 C4′-Radical Reactivity in RNA
Bleomycin also cleaves RNA, and it has been postulated that this contributes to the biological activity of the antitumour agent.170–173 However, the details of the chemistry, including C4′-hydrogen atom abstraction are not as well understood as in DNA. Giese independently generated a C4′-radical in single-stranded RNA (57, Scheme 27 ) under aerobic and anaerobic conditions via Norrish type I photocleavage of 58.174 The rate constant for strand scission (kCleave, Scheme 27 ) from 58 was estimated by measuring the ratio of thiol trapping products (59, 60) versus phosphate cleavage product as a function of GSH concentration. By assuming that the thiol traps 57 with a bimolecular rate constant kTrap = 1 × 107 M−1s−1, kCleave was estimated to be 5 × 102 s−1. This is slightly more than threefold slower than the comparable cleavage step in DNA, assuming that the rate constants for GSH trapping of the DNA and RNA C4′-radicals are equal.106 The decrease in reactivity of 57 compared to the comparable DNA radical is modest compared to a related model study containing phosphate triesters (instead of phosphate diesters that are present in DNA and RNA) that was carried out in aqueous alcohol solvent (as opposed to H2O).175 The modest difference in the rate constants for phosphate elimination from 57 compared to its DNA counterpart (44, Scheme 24 ) was attributed to solvation of the radical cation by the aqueous solvent, such that the proximal 2′-hydroxyl group in RNA had a small effect on the intermediate’s stability.174
Scheme 27.

Competitive kinetic determination of rate constant for C4′-radical fragmentation in RNA.
Aerobic photolyses yielded the expected fragmentation products following peroxyl (62) radical formation, a 5′-fragment containing a 3′-phosphoglycolate (42) and a 3′-fragment containing a 5′-phosphate terminus (Scheme 28 ).174 The ratio of these products was ~3:1 in favor of phosphoglycolate 42 over phosphate. This is distinct from what happens in DNA where 42 forms concomitantly with base propenal (50) and phosphate product (Scheme 21 ). Although the authors present a mechanism rationalizing the formation of these products, evidence for the formation of the crucial product (61) is not presented. Presumably, elimination from the Criegée product (63) is slower than in DNA due to the presence of the hydroxyl group. This enables H2O to trap the carbocation and altering the final decomposition pathway to phosphoglycolate (42).
Scheme 28.

Alternative phosphoglycolate (42) formation pathway in RNA.
8. C5′-RADICAL GENERATION AND REACTIVITY IN DNA
8.1 C5′-Radical Formation
The C5′-hydrogen atoms are even more accessible to diffusible species than the C4′-position (Fig. 1A) and this site has been suggested to be the preferred position for hydrogen atom abstraction by HO•.34 A variety of minor groove-binding DNA oxidizing agents abstract the C5′-hydrogen atom, including manganese porphyrins and the enediyne antitumor antibiotics, resulting in strand scission.120,176–178 C5′-Deuterium-labelled DNA substrates were commonly used for detecting hydrogen atom abstraction from this position by the antitumour antibiotics.123 Frequently, the deuterium was used to reduce strand scission and/or shift reaction to another nucleotide position (eg C4′) resulting in different products, both of which were detected by gel electrophoresis.177–179 However, in some instances the deuterium was even traced to its incorporation into the final product obtained from the antitumor agent.180
8.2 C5′-Radical Reactivity in DNA
The major product formed following C5′-oxidation is the C5′-aldehyde (43), which releases furfural from DNA upon heating (Scheme 29 ). Under most experimental conditions 43 is believed to arise via the peroxyl radical (65) (see below). 18O-Labelling was employed to obtain additional information on the mechanism for formation of 43 resulting from manganese porphyrin oxidation. However, the results were inconclusive and it was proposed that this was due to rapid exchange of the aldehyde in H2O.181 In contrast, 18O-labelling experiments indicated that O2 was the source of oxygen in 43 when neocarzinostatin reacts with DNA.182 These experiments also ruled out oxidation of the initially formed C5′-radical (64) to a carbocation. The aldehyde was reduced in the neocarzinostatin experiments to prevent solvent exchange of the carbonyl oxygen. However, reduction was unsuccessful in preventing equilibration in experiments using the manganese porphyrin.
Scheme 29.

C5′-Oxidation of DNA.
Various proposals have been put forth to explain how peroxyl radical 65 is transformed into the aldehyde (43, Scheme 29 ), including the dimerization of two peroxyl radicals (not shown).183 The tetroxide pathway is improbable in DNA because of the unlikeliness that two peroxyl radicals will collide. In addition, the 18O-labelling experiments carried out using neocarzinostatin as a source of 65 suggest that superoxide elimination is also unlikely.182 Ferric ion was reported to rapidly oxidize (k ~4 × 109 M−1s−1) the nucleoside 2′-deoxyadenosin-5′-yl radical (64).184 Although this could be a viable pathway under anoxic conditions, 18O-labelling indicates that this process also does not compete with formation of 65 without adding exogenous metal ion. Hence, the most general pathway to 43 from 64 under aerobic conditions would appear to require reduction of the peroxyl radical (65), most likely by thiol.
The putative hydroperoxide (66) from 65 generated by reaction with neocarzinostatin has also been proposed to undergo a Criegée rearrangement to yield a strand break concomitantly with free base release and a 5′-fragment containing a labile formate bonded to the 3′-terminal phosphate (67, Scheme 30 ).185 The 3′-fragment contains chemically unstable 5′-(2-phosphoryl-1,4-dioxobutane) (DOB) at its 5′-terminus.186,187 Like C4-AP (Section 7.2), DOB irreversibly inhibits DNA polymerase β and yields histone proteins containing modified lysines in nucleosomes.87–89,188 The yield of free base obtained from neocarzinostatin reactions varies from 16% to 20% depending upon the thiol that activates the antibiotic.186 The DOB yield determined by measuring the stabilized reduction product (68) is about half this and may be due to reaction of the lesion with nucleophilic thiol.187,189
Scheme 30.

Direct strand breaks from C5′-oxidation of DNA.
Under anaerobic conditions, the distinctive 5′,8-cyclonucleos(t)ide DNA lesions (69, 70) are produced from 64 of purine nucleos(t)ides(Scheme 31 ).107,184,190–192 The 5′,8-cyclonucleotides have significant effects on replication and repair.193–196 They are strong replication blocks and are repaired via the nucleotide excision repair pathway instead of BER. The repair pathway is unusual for modified nucleotides formed via oxidative stress. The mechanism and kinetics of their formation are described below (Scheme 32 ).
Scheme 31.

Cyclonucleoside formation from C5′-radicals.
Scheme 32.

Cyclization and oxidation of a C5′-radical.
8.3 Independent Generation and Reactivity of C5′-Radicals
The C5′-radical has not yet been independently generated in oligonucleotides but the respective 2′-deoxy- and ribonucleoside radicals have been produced via Norrish type I photocleavage and a less conventional manner using pulse radiolysis.107,184,190,192,197 Pulse radiolysis of various 8-bromopurine nucleosides provided access to the C5′-radicals via initial reaction with the solvated electron, which yields the C8-σ-radicals on the sub-microsecond timescale (Scheme 32 ). The C8-purine σ-radicals are believed to rapidly abstract the C5′-hydrogen atom providing 64. The investigators typically monitor the growth of the subsequent C8-cycloadduct (71) on the microsecond timescale by absorption spectroscopy. C5′-radical cyclization rate constants range from ~1 × 104 s−1 to ~1 × 106 s−1.107,190,192 However, it should be noted that the upper range of these rate constants was estimated using competitive kinetics by generating a protected form of 2′-deoxyguanosin-5′-yl radical via Norrish type I generation in tetrahydrofuran (THF) solution.190 These rate constants suggest that cyclization should be competitive with O2 trapping of the C5′-radicals, and this was demonstrated for 2′-deoxyadenosin-5′-yl radical (64) that was generated via pulse radiolysis of the corresponding 8-bromopurine nucleoside (Scheme 32 ).184 5′-Aldehyde yields correlated with increased O2 concentration at the expense of cyclonucleoside product formation (Scheme 32 ). C5′-Radical cyclization of pyrimidines was not detected. In addition, no mention of free base release or other evidence for DOB formation was reported. There is still quite a bit to learn about the reactivity of the C5′-nucleos(t)ide radicals, particularly within the biopolymer.
9. NUCLEOBASE RADICAL GENERATION AND REACTIVITY IN DNA AND RNA
9.1 Nucleobase Radical Formation
Nucleobase radicals are largely the domain of HO• and to a lesser extent hydrogen atoms, that are produced by ionizing radiation.17,32 Hydroxyl radical is also produced by metal complexes such as Fe•EDTA.33 Unlike the various molecules mentioned above that bind in the minor groove and abstract hydrogen atom(s) from the 2′-deoxyribose backbone, HO• addition to the nucleobases is the major pathway for this species. Estimates for the partitioning of HO• reactivity with nucleic acids range from ~80% to more than 90% addition to nucleobase π-bonds. The electrophilic HO• preferentially adds to the C5-position of pyrimidines, with the ratio of the resulting C6- and C5-radicals ranging from 2:1 to 4–5: 1.17,198–200 Hydrogen atom abstraction from the methyl group of thymidine and 5′-methyl-2′-deoxycytidine is also a minor pathway for HO• and other diffusible species.201,202 Direct hydrogen atom abstraction form nitrogen–hydrogen bonds in purines have also recently been proposed but this has been questioned.203–205 In general, the reaction of HO• with purines is less well understood than with pyrimidines.
Hydroxyl radical formation via ionizing radiation is typically referred to as the indirect effect because HO• is the product of initial H2O ionization. The energy released upon γ-radiolysis can also interact directly with nucleic acids, and the corresponding direct effect of ionizing radiation accounts for approximately one-half of the induced damage. The direct effect of ionizing radiation initially produces radical cations, which as briefly mentioned when describing the reactivity of C4′-radicals, are responsible for electron transfer (‘hole migration’) in DNA (Section 7.5). The radical cations also give rise to other forms of DNA damage. The purines are more readily oxidized than the pyrimidines, and dG has a more favorable redox potential than does dA.206 Hydration of the nucleobase radical cations (eg 72, Scheme 33 ), followed by deprotonation, yields the same radical species resulting from HO• addition, which yield products such as thymidine glycol (Tg).207 In contrast, deprotonation produces the formal hydrogen atom abstraction products (eg 73), which yield O2 trapping products (eg 74, 75).208 This pathway has been observed in thymidine (Scheme 33 ) and 5-methyl-2′-deoxycytidine using photosensitization and illustrates that the direct and indirect (HO•) effects of ionizing radiation produce many of the same products.209–211 The purine radical cations tautomerize212 in competition with deprotonation to yield the neutral radicals whose direct formation, as mentioned above, is a topic of current interest. The neutral purine radicals, formal products of hydrogen atom abstraction, are believed to be the thermodynamic sinks during electron transfer and are unreactive with O2.213 Neutral nucleobase radicals are also believed to result from decomposition of the respective chloramines, which are generated during oxidative stress.44
Scheme 33.

Pyrimidine radical cation formation and reactivity.
9.2 Nucleobase Radical Reactivity
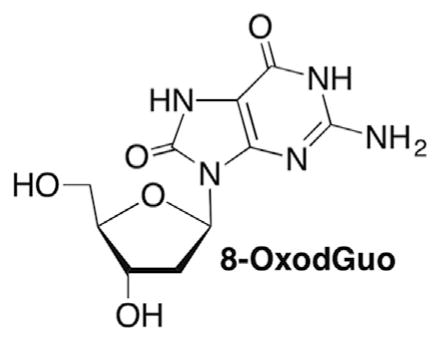
A great deal of research has been carried out on the formation of various modified nucleotides following direct ionization or reaction with HO• under aerobic and anaerobic conditions.214,215 Pyrimidine nucleosides yield the hydrates (78, 79) and glycols (Tg) via regioisomeric HO• adducts (76, 77) under anaerobic and aerobic conditions, respectively (Scheme 34 ). The cytidine molecules are unstable upon saturation of the π-bond and hydrolyze to the corresponding modified uridines.216 The reaction of dG is especially rich because the initial two-electron oxidized product, 8-oxodGuo is more reactive than the native nucleoside and produces a variety of DNA lesions.217,218 Many of the DNA lesions produced by oxidation of native nucleotides are mutagenic.219–221
Scheme 34.

HO· addition to thymidine.
To produce a strand break, a nucleobase radical or respective peroxyl radical must abstract a hydrogen atom from the carbohydrate backbone. Approximately 40% of the reactions between HO• and RNA result in strand scission.222 Since a minimum of 80% of HO• reactions occur with the nucleobases, at least 20% of the nucleobase radicals must ultimately transfer spin to the ribose ring.223 In contrast, strand scission in DNA following HO• to a nucleobase is significantly less efficient (≤5%) and nucleobase (peroxyl) radicals do not have to react with a 2′-deoxyribose ring to account for direct strand scission.224 A variety of mechanisms have been proposed for the requisite spin transfer from nucleobases to the (2′-deoxy)ribose that is required for strand scission, some of which involve initial one-electron oxidation of the nucleobase followed by hydration to form the formal HO• adduct.199,200,225–229
5-(2′-Deoxyuridinyl)methyl radical (74) and the respective radical from 5-methyl-2′-deoxycytidine have not been proposed to yield strand breaks. However, 74 and other nucleobase radicals have been implicated in reactions with adjacent nucleotides, resulting in two contiguously damaged nucleotides (eg 15). Such lesions are typically referred to as tandem lesions.230–235 Tandem lesions are a subset of clustered lesions, which are defined as two or more damaged nucleotides within one to two helical turns. Clustered lesions are of particular interest to radiation scientists because of their formation via HO• spurs and the difficulties that they present for DNA repair.85,236–239 In addition, 74 has been invoked as an intermediate in DNA electron transfer where it arises from deprotonation of the radical cation (Scheme 33 ).240,241 Stable products resulting from O2 trapping of 74 (76, 77) are observed under these conditions.
9.3 Independent Generation and Reactivity of DNA Nucleobase Radical Adducts
Several DNA and RNA pyrimidine nucleobase radicals have been generated from ketone precursors by the Norrish type I cleavage, aryl sulfides and phenyl selenides (Scheme 35 ). Formal hydrogen atom addition adducts of thymidine (80) and 2′-deoxyuridine (83) have been produced using these types of precursors, as has the C5-thymidine HO• adduct (76, Scheme 35 ).46,233,242–251 In each instance, the chemical integrity of the photochemical precursor with respect to generating the respective radical is characterized at the nucleoside level using standard product analysis conditions.
Scheme 35.

Independent generation of pyrimidine radical adducts.
Direct strand breaks or alkali-labile lesions are not observed from 76, 80 or 83 under anaerobic conditions. (Reminder: Radicals are referred to using the same descriptor whether they are nucleosides or in biopolymers.) Small amounts of direct strand breaks are detected at the 5′-adjacent nucleotide when the formal C6-hydrogen atom adduct of thymidine (80) is produced under aerobic conditions.243 Strand scission is reduced approximately fourfold when the 5′-adjacent nucleotide is deuterated at the C1′-position.242 Deuteration of the C2′-, or C4′-positions has no effect suggesting that peroxyl radical 86 selectively abstracted the C1′-hydrogen atom from the 5′-adjacent nucleotide (Scheme 36 ). Additional support for internucleotidyl C1′-hydrogen atom abstraction was obtained when the tandem lesion containing 2-deoxyribonolactone (15) was observed in low yield when 80 was generated from 82 in a dinucleotide under aerobic conditions (Scheme 36 ).233 It is possible that the observed direct strand scission was an artifact of sample handling, as C1′-oxidation does not typically result in this product. Direct strand scission could also have resulted from hydrogen atom abstraction from another position that was not detected using deuterium labelling.
Scheme 36.

Tandem lesion formation from 5,6-dihydrothymidin-5-yl radical (80).
Independent generation of C6-radicals 76 and 83 (Scheme 35 ) failed to yield any direct strand scission under aerobic conditions.244,247,249 These radicals do produce a variety of alkali-labile tandem lesions that were detected by denaturing polyacrylamide gel electrophoresis and mass spectrometry. The peroxyl radical of the formal C5-hydrogen atom adduct of 2′-deoxyuridine (87) produces tandem lesions by reacting with the 5′- and 3′-adjacent nucleobases, and abstracting hydrogen atom(s) from the 5′-adjacent nucleotide but not the 3′-adjacent nucleotide (Scheme 37 ). Selective C1′-hydrogen atom abstraction from the 5′-adjacent nucleotide was based upon several observations, including a significant observed 2H kinetic isotope effect (KIE) (4.4 ± 0.1). Additional information was gleaned from chemical fingerprinting using a series of reactions that are diagnostic for 2-deoxyribonolactone (L), reaction with a biotinylated sensor that selectively tags L, as well as observation of 2-deoxyribonolactone upon MS analysis of photolyzed single-stranded dodecamer and trinucleotides containing 84.244,247,252,253 MS experiments also indicated that L is formed at the original site of the radical, presumably via intranucleotidyl C1′-hydrogen atom abstraction.244 There is no evidence for hydrogen atom abstraction from the 2′-deoxyribose ring of the 3′-adjacent nucleotide. The absence of reactivity at this carbohydrate component is consistent with the greater distance from the peroxyl radical center (87).247 Reaction at the adjacent nucleotides is also dependent upon the peroxyl radical configuration at C6 and the conformation about the glycosidic bond (Scheme 37 ). Kinetic evidence suggested that the syn- and anti-conformations of 6R-87 and 6S-87 are involved in tandem lesion formation (Scheme 37 ), and that proximity dictated by the right handed α-helical duplex structure strongly influences reactivity (Fig. 2).244,245,247 Molecular modelling reveals that the C1′-hydrogen atom of the 5′-adjacent nucleotide is accessible to anti-5R-87 (Fig. 2A). In contrast to 5R-87, the C1′-hydrogen atom of the 3′-adjacent nucleotide is not readily observable when viewed from the major groove when anti-5S-87 is present (Fig. 2B).
Scheme 37.

Tandem lesion formation from 5,6-dihydro-2′-deoxyuridin-6-yl peroxyl radical (87).
Figure 2.
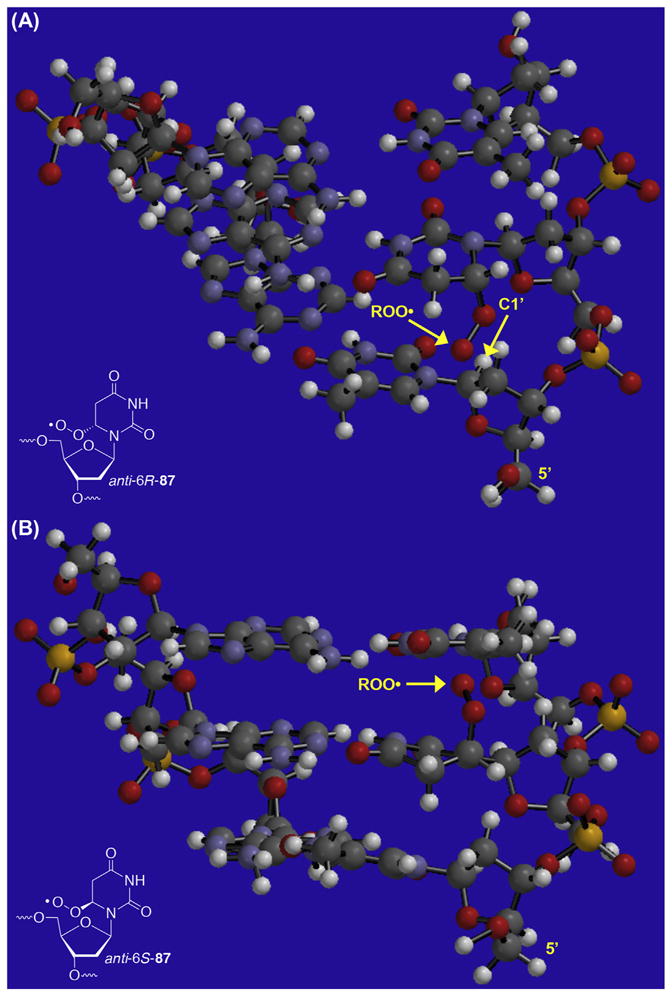
Molecular modelling of duplex DNA containing epimers of 5,6-dihydro-2′-deoxyuridin-6-yl peroxyl radical (87) in the sequence 5′-T·87·T/A·A·A. (A) anti-5R-87; (B) anti-5S-87.
Incorporation of 5,6-dihydrothymidine (dHT) at the 5′-adjacent nucleotide to where 87 is generated significantly reduced the contribution of piperidine-labile lesions at that position, which are indicative of nucleobase lesions, indicating that the majority of 5′-tandem lesions involved addition to the π-bond.247 Some of the tandem lesions detected by MS are attributable to peroxyl radical addition to adjacent nucleotides.244,247 Competitive kinetics using GSH as a competitor provided an estimate of the rate constants for reaction of 87 with the 5′-adjacent nucleotide. The rate constant for reaction with the adjacent nucleotide was almost 30 times greater for a 5′-dG (1.2 ± 0.2 × 10−1 s−1) than with a 5′-dT (4.4 ± 0.6 × 10−3 s−1).244,249 This is consistent with the more electron-rich nature of dG and its relatively favorable oxidation potential compared to thymidine.206
Although tandem lesions were detected from the peroxyl radical (88, Scheme 38 ) of the C5-hydroxyl radical adduct of thymidine (76) and some were characterized by MS (eg 89, 90, Scheme 38 ), the reactivity of this peroxyl radical was considerably different than that of 87.249 For instance, there was no evidence for intramolecular hydrogen atom abstraction or from the 5′-adjacent nucleotide. In addition, competitive kinetics using GSH indicated that 88 reacted with a 5′-adjacent dG approximately one-half as fast (7.3 ± 0.9 × 10−2 s−1) as did 87. The slower reactivity of 88 than 87 is consistent with the rate constants for the respective nucleobase radicals (Scheme 35 ). The thymidine C5-hydroxyl radical adduct (76, Scheme 35 ) reacts with β-mercaptoethanol (BME) unusually slowly (k = 4.7–5.7 ± 0.1 × 105 M−1s−1) and considerably more slowly than does 83 (k = 8.8 ± 0.5 × 106 M−1s−1).246,248 Although it is not known why 88 reacts more slowly and selectively within DNA than does 87, the possibility that disubstitution at C5 of the pyrimidine increases steric hindrance and/or disrupts base stacking making it more difficult for the former to access the adjacent nucleotide was considered.249 The disruption of base stacking by 5,6-dihydrothymidines that are disubstituted at C5 is an outcome of computational studies.254,255
Scheme 38.

Tandem lesion formation from 5,6-dihydro-thymidin-6-yl peroxyl radical (88).
Independent generation of 88 was also used to test a proposal that pyrimidine peroxyl radicals contribute to DNA electron transfer by oxidizing purine nucleotides (Fig. 3).256 Independent generation of 88 at a defined site of a carefully chosen duplex sequence provides a facile means for detecting electron transfer by monitoring DNA damage at distal sites. The 5′-dGGG sequence is often used as a tool for monitoring DNA oxidation (Fig. 3). Outer sphere oxidation of dG by a DNA peroxyl radical was expected to be uphill by ~0.23 eV.206,257 Although a variety of oxidation products were detected, independent generation in a variety of duplex sequences that are frequently used to probe for electron transfer provided no evidence for this pathway.249,258–260
Figure 3.

Probing for DNA electron transfer via independent generation of a nucleobase peroxyl radical (88). X = 85b (Scheme 35 ), ROO· = 88 (Scheme 38 ), G+· = oxidized guanine.
Overall, independent generation of HO• (and formal H•) addition products indicate that these radicals and their respective peroxyl radicals do not yield direct strand breaks. However, the respective peroxyl radicals produce a variety of tandem lesions. Although it is not possible to generate the analogous 2′-deoxycytidine radicals due to anticipated hydrolysis of the 5,6-dihydropyrimidine precursors, consideration of sterics suggests that their reactivity will be similar to the 2′-deoxyuridine adducts described above, which are more reactive than that of thymidine.
9.4 Independent Generation and Reactivity of RNA Nucleobase Radical Adducts
The formal C5- (91) and C6-hydrogen atom adducts (92) of uridine have been generated from the respective t-butyl ketones (93, 94, Scheme 39 ).261–264 Direct strand breaks were observed at the 5′-adjacent ribonucleotides when 91 or 92 were generated (Scheme 40 ).261–263 The C6-radical (91) also yielded intranucleotidyl cleavage, whereas a low level of cleavage at the nucleotide where 92 was generated was attributed to an artifact. Strand scission was as much as four-fold more efficient in duplex RNA than in single-stranded substrates. In addition, direct strand scission was at least seven-fold more efficient under anaerobic conditions than when O2 was present.
Scheme 39.

Independent generation of uridine radical adducts.
Scheme 40.

Direct strand scission from nucleobase radicals in RNA.
Strand scission products were characterized by gel electrophoresis with the aid of chemical and enzymatic reactions, and MALDI-TOF MS. Radicals 91 and 92 gave rise to common products (Scheme 40 ). In both instances a significant deuterium isotope effect was observed when the C2′-position of the 5′-adjacent nucleotide was labelled. Radical 92 yielded a KIE = 3.6 ± 0.7 but no effect when the C3′-position was deuterated.261 5′-Internucleotide strand scission via 91 was even more strongly affected by C2′-deuteration.263 The mechanism for strand scission was proposed to involve C2′-hydrogen atom abstraction (23), followed by heterolytic cleavage of the 3′-phosphate (Scheme 13 ). As discussed above (See: Generation and reactivity of the 2′-radical in RNA, Section 5.2), how the radical cation is transformed into the respective ketone (29, Scheme 14 ) and 3′-phosphate termini products is uncertain. The possibility that 26 generated from 91 is reduced by a guanosine within the RNA duplex was examined using a variety of sequences but no evidence for this process was obtained.262
Independent generation of 91 and 92 was carried out prior to reports on C2′-radical generation.109 At that time it was uncertain whether more efficient strand scission under anaerobic conditions was due to O2 trapping of 23 in competition with strand scission or less efficient hydrogen atom abstraction by the corresponding nucleobase peroxyl radicals (95, 96). It was also uncertain whether C2′-hydrogen atom abstraction or fragmentation from 23 was the rate determining step in strand scission. The recent independent generation of 23 from 24 (Scheme 12 ) revealed that O2 trapping does not compete with strand scission from this reactive intermediate and that hydrogen atom abstraction by the nucleobase radicals (91, 92) must be the rate determining step.109 Competitive kinetics revealed that 92 abstracts the C2′-hydrogen atom from the 5′-adjacent nucleotide almost 25-times faster than does 91.261,262 Computational studies corroborated these findings.261 The C5-carbon–hydrogen bond in N1-methyl-5,6-dihydrouracil (97) was determined to be at least 2.8 kcal/mol stronger (95.3 kcal/mol) than the C6-carbon–hydrogen bond.
The results are consistent with γ-radiolysis experiments that showed that RNA is significantly more susceptible to strand scission than is DNA.223,224 Furthermore, the mechanism of strand scission is in line with expectations set by computational studies that indicate that the C2′-hydrogen atom, whose respective carbon–hydrogen bond is on average 4.8 kcal/mol weaker than any other in a ribonucleotide, should be the most readily abstracted.35 However, mechanistic questions remain. How the 3′-phosphate product is formed is still unclear. In addition, the transformation of 26 into the 5′-fragment containing a 3′-terminal 3′-deoxy-2′-ketouridine (29) is also unknown.
9.5 Independent Generation and Reactivity of DNA 5-(2′-Deoxyuridinyl)methyl and 5-(2′-Deoxycytidinyl) methyl Radicals
5-(2′-Deoxycytidinyl)methyl radical (98) was produced from the respective phenyl sulfide (99) via 254 nm photolysis.265,266 Initial studies in dinucleotides revealed that 98 adds to the C8-position of dG to form intrastrand cross-linked products (Scheme 41 ). These products were also observed when 98 was generated from 99 in chemically synthesized oligonucleotides, and when DNA was subjected to γ-radiolysis.266 In DNA, 100 is favored over 101 and is even detected, albeit in almost 20-fold lower yield when 98 is generated under aerobic conditions.
Scheme 41.

Tandem lesion formation from 5-(2′-deoxycytidinyl)methyl radical (98).
5-(2′-Deoxyuridinyl)methyl radical (102) was independently generated from a methoxy substituted arylsulfides (103a–c) and phenyl selenide (104) via 350 nm irradiation (Scheme 42 ).267–269 The Norrish type I photo-cleavage of 105 also produced 102.270 Thiol and O2 radical trapping products were observed under the appropriate anaerobic and aerobic conditions when monomeric 102 was generated.267 Although intrastrand cross-links analogous to those from 98 (Scheme 40 ) were not detected, interstrand cross-links with the opposing dA (Scheme 44 ) were formed upon photolysis of duplex DNA containing 103c or 104 (Scheme 41 ).268,271,272 The formation of interstrand cross-links from 102 was surprising given the absence of a similar product from 98, as well as in DNA electron transfer experiments where products attributable to 5-(2′-deoxyuridinyl)methyl radical (102) were observed.241,273,274
Scheme 42.

Independent generation of 5-(2′-deoxycytidinyl)methyl radical (102).
Scheme 44.

Interstrand cross-link formation from 104.
Another surprise was that interstrand cross-links were also formed independently of O2. However, this was initially rationalized by demonstrating that O2 reacted reversibly with 102 (Scheme 43 ).268 Nonlinear regression analysis of the ratio of thymidine to oxygenated products (eg 107) as a function of GSH concentration provided an estimated rate constant for GSH trapping of 102 (kGSH = 6.9 × 106 M−1s−1) consistent with expectations for reaction of an alkyl radical with the thiol.60 The accuracy of the thiol trapping rate constant validated the extracted rate constant for O2 loss from 106 to reform 102 (kO2 = 3.4 s−1), which is consistent with rate constants reported for O2 loss from other peroxyl radicals.59,142
Scheme 43.

Reversible O2 trapping of 5-(2′-deoxycytidinyl)methyl radical (102).
The isolated cross-link resulted from formal addition of the radical to the N6-amine of dA, an improbable process. It was proposed that 102 adds to the N1-position, and following a second one-electron oxidation, that the N1-alkylated product (108) rearranges to the final product (109, Scheme 44 ).275,276 Rearrangement of the initially formed N1-alkylation product to 109 was supported by nuclear magnetic resonance (NMR) experiments using momomeric substrates, as well as doubly-13C and 15N labelled DNA substrate.272 The same product was formed from 104 under mild oxidative conditions (eg NaIO4, H2O2, 1O2) and the mechanism involving oxidation to 110 and its rapid rearrangement to 111 (Scheme 44 ) was supported by NMR experiments using 15N and 13C labelled DNA substrates.272,277,278 Oxidatively induced cross-linking from 104 has proved useful in a variety of applications, including single nucleotide polymorphism detection, triple helical (triplex) DNA detection and as a tool for examining DNA–protein interactions.279–281
Despite the interesting and useful chemistry emanating from 104 (and 103c), the attribution of DNA interstrand cross-links from 5-(2′-deoxyuridinyl)methyl radical (102) was inconsistent with DNA electron transfer experiments in that formation of the same radical intermediate failed to yield cross-links.21,240,273,274 This discrepancy was resolved by Weng who determined that in addition to generating 102 upon photolysis, 104 and 103c also produce the carbocation (112, Scheme 45 ).282 Carbocation 112 may arise via heterolytic cleavage of the excited state precursor or electron transfer with the initially formed radical pair.283 The carbocation is trapped by nucleophiles, including methoxyamine (113) and t-butyl thiol. The latter is important because it helps explain why initial studies supported interstrand cross-link formation from 5-(2′-deoxyuridinyl)methyl radical (102).268 Importantly, while methoxyamine had no effect on the yield of radical trapping products (eg 107, Scheme 43 ), it quenched DNA interstrand cross-link formation upon irradiation of DNA containing 103c in a concentration-dependent manner. In contrast, Tempo derivative 115 had no effect on cross-linking but did trap 102 (114) when it was produced in oligonucleotides. These experiments clearly indicated that, although 104 and 103c produce 5-(2′-deoxyuridinyl)methyl radical (102), the precursors also produce the carbocation (112), and it is this latter species that is responsible for DNA interstrand cross-link formation. Computational experiments on free nucleobases corroborated these findings.282 Addition of (5-uracil)methyl radical to adenine was determined to face a 50 kJ mol−1 barrier, whereas reaction of the corresponding carbocation analogous to 112 was barrierless.
Scheme 45.

Interstrand cross-link formation via a carbocation (112).
9.6 Independent Generation and Reactivity of Neutral Purine Radicals
Limited data are available on the species that result from formal hydrogen atom abstraction from either dA or dG. As mentioned above, these radicals play a pivotal role in DNA electron transfer and may also be important species that are produced directly from the respective chloramines via oxidative stress (See Sections 2 and 7.5). Photolyses of 117 and 118 were proposed to produce the formal N1-hydrogen atom abstraction product from dG (116, Scheme 46 ).284,285 Data from 118 were limited to quantification of dG and the proposed generation of 117 is complicated by a required rearrangement to 119 and subsequent photolysis.284 Independent synthesis of 118 was reported to produce 116 upon Pyrex filtered photolysis, and is supported by transient spectroscopy.285 Studies on the formation of 116 from 118 in oligonucleotides have not yet been reported.
Scheme 46.

Independent generation of the N1-hydrogen atom abstraction product of dG.
The N6-aminyl nucleoside radical of dA (120) has also been produced from a photochemical precursor (121) but not within DNA (Scheme 47 ).286 Photolysis (315 ± 2 nm) of phenylhydrazone 121 in the presence of GSH (50 mM) provides a 73% yield of dA. In the absence of excess thiol, the yield of dA is reduced (32%) as expected and is accompanied by the radical recombination product resulting from the dimerization of 120.
Scheme 47.

Independent generation of the N6-hydrogen atom abstraction product of dA.
10. SUMMARY AND FUTURE CONSIDERATIONS
Although the ability to independently generate reactive intermediates has increased our understanding of nucleic acid damage processes, the brief description of neutral purine radical generation (Section 9) illustrates that unanswered questions remain regarding the roles of these reactive intermediates in DNA. Nucleobase radical cations, the family of species produced via the direct effect of ionizing radiation have also never been independently generated in nucleic acids. Furthermore, little is known about the effects of protein binding on the reactivity of nucleic acid reactive intermediates.287,288 Other gaps in our knowledge were pointed out throughout the above review. Clearly, there is still much to be done.
Acknowledgments
I am grateful for continuous support from the National Institute of General Medical Science (GM-054996) for our research on the mechanisms of nucleic acid oxidation.
References
- 1.Berson JA. A new class of non-kekule molecules with tunable singlet-triplet energy spacings. Acc Chem Res. 1997;30:238–244. [Google Scholar]
- 2.Scaiano JC. Nanosecond Laser Flash Photolysis: a Tool for Physical Organic Chemistry, Reactive Intermediate Chemistry. John Wiley & Sons, Inc; 2004. pp. 847–871. [Google Scholar]
- 3.Wang Y, Hadad CM, Toscano JP. Solvent dependence of the 2-naphthyl(carbomethoxy)carbene singlet-triplet energy gap. J Am Chem Soc. 2002;124:1761–1767. doi: 10.1021/ja012139v. [DOI] [PubMed] [Google Scholar]
- 4.Burdzinski G, Platz MS. Ultrafast kinetics of carbene reactions. Wiley Ser React Intermed Chem Biol. 2014;7:166–192. [Google Scholar]
- 5.Newcomb M. Radical Kinetics and Clocks, Encyclopedia of Radicals in Chemistry, Biology and Materials. John Wiley & Sons Ltd; 2012. pp. 107–124. [Google Scholar]
- 6.Johnston LJ, Schepp NP. Kinetics and mechanisms for the reactions of alkene radical cations. Adv Electron Transfer Chem. 1996;5:41–102. [Google Scholar]
- 7.Maiti A, Gerken JB, Masjedizadeh MR, Mimieux YS, Little RD. From dimerization, to cycloaddition, to atom transfer cyclization: the further chemistry of TMM diradicals. J Org Chem. 2004;69:8574–8582. doi: 10.1021/jo048717i. [DOI] [PubMed] [Google Scholar]
- 8.Little RD, Ott MM. A diradical route to bioactive natural products and their analogs. Stud Nat Prod Chem. 2000;22:195–243. [Google Scholar]
- 9.Allan AK, Law Carroll G, Little RD. The versatile trimethylenemethane diyl. Diyl trapping reactions. Retrospective and new modes of reactivity. Eur J Org Chem. 1998:1–12. [Google Scholar]
- 10.Vaz MGF, Cassaro RAA, Akpinar H, Schlueter JA, Lahti PM, Novak MA. A cobalt pyrenylnitronylnitroxide single-chain magnet with high coercivity and record blocking temperature. Chem Eur J. 2014;20:5460–5467. doi: 10.1002/chem.201304852. [DOI] [PubMed] [Google Scholar]
- 11.Seber G, Brook DJR, Taylor PS, Cassaro RAA, Lahti PM. Magnetostructural effects of changing spin unit structure and molecular connectivity on 1H-benzimidazole functionalized radicals. Polyhedron. 2014;76:36–44. [Google Scholar]
- 12.Bales BC, Pitié M, Meunier B, Greenberg MM. A minor groove binding copper-phenanthroline conjugate produces direct strand breaks via β-elimination of 2-deoxyribonolactone. J Am Chem Soc. 2002;124:9062–9063. doi: 10.1021/ja026970z. [DOI] [PubMed] [Google Scholar]
- 13.Taverna Porro ML, Greenberg MM. DNA double strand cleavage via interstrand hydrogen atom abstraction. J Am Chem Soc. 2013;135:16368–16371. doi: 10.1021/ja409513q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giese B, Wessely S, Spormann M, Lindemann U, Meggers E, Michel-Beyerle ME. On the mechanism of long-range electron transfer through DNA. Angew Chem Int Ed. 1999;38:996–998. doi: 10.1002/(SICI)1521-3773(19990401)38:7<996::AID-ANIE996>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Meggers E, Michel-Beyerle ME, Giese B. Sequence dependent long range hole transport in DNA. J Am Chem Soc. 1998;120:12950–12955. [Google Scholar]
- 16.Meggers E, Kusch D, Spichty M, Wille U, Giese B. Electron transfer through DNA in the course of radical-induced strand cleavage. Angew Chem Int Ed. 1998;37:460–462. doi: 10.1002/(SICI)1521-3773(19980302)37:4<460::AID-ANIE460>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 17.von Sonntag C. Free-Radical-Induced DNA Damage and Its Repair. Berlin: Springer-Verlag; 2006. [Google Scholar]
- 18.Cadet J, Loft S, Olinski R, et al. Biologically relevant oxidants and terminology, classification and nomenclature of oxidatively generated damage to nucleobases and 2-deoxyribose in nucleic acids. Free Rad Res. 2012;46:367–381. doi: 10.3109/10715762.2012.659248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutagen. 2015;763:212–245. doi: 10.1016/j.mrrev.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Genereux JC, Barton JK. Mechanisms for DNA charge transport. Chem Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanvah S, Joseph J, Schuster GB, Barnett RN, Cleveland CL, Landman U. Oxidation of DNA: damage to nucleobases. Acc Chem Res. 2010;43:280–287. doi: 10.1021/ar900175a. [DOI] [PubMed] [Google Scholar]
- 22.Clatterbuck Soper SF, Dator RP, Limbach PA, Woodson SA. In vivo X-ray footprinting of pre-30s ribosomes reveals chaperone-dependent remodeling of late assembly intermediates. Mol Cell. 2013;52:506–516. doi: 10.1016/j.molcel.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behrouzi R, Roh JH, Kilburn D, Briber RM, Woodson SA. Cooperative tertiary interaction network guides RNA folding. Cell. 2012;149:348–357. doi: 10.1016/j.cell.2012.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlatterer JC, Brenowitz M. Complementing global measures of RNA folding with local reports of backbone solvent accessibility by time resolved hydroxyl radical footprinting. Methods. 2009;49:142–147. doi: 10.1016/j.ymeth.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang L, Palma JL, Bruot C, Mujica V, Ratner MA, Tao N. Intermediate tunnelling–hopping regime in DNA charge transport. Nat Chem. 2015;7:221–226. doi: 10.1038/nchem.2183. [DOI] [PubMed] [Google Scholar]
- 26.Xuefeng G, Gorodetsky AA, Hone J, Barton JK, Nuckolls C. Conductivity of a single DNA duplex bridging a carbon nanotube gap. Nat Nanotech. 2008;3:163–167. doi: 10.1038/nnano.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Neill P, Wardman P. Radiation chemistry comes before radiation biology. Int J Radiat Biol. 2009;85:9–25. doi: 10.1080/09553000802640401. [DOI] [PubMed] [Google Scholar]
- 28.Trauger JW, Dervan PB. Footprinting methods for analysis of pyrrole-imidazole polyamide/DNA complexes. Methods Enzymol. 2001;340:450–466. doi: 10.1016/s0076-6879(01)40436-8. [DOI] [PubMed] [Google Scholar]
- 29.Jain SS, Tullius TD. Footprinting protein-DNA complexes using the hydroxyl radical. Nat Protoc. 2008;3:1092–1100. doi: 10.1038/nprot.2008.72. [DOI] [PubMed] [Google Scholar]
- 30.Tullius TD, Greenbaum JA. Mapping nucleic acid structure by hydroxyl radical cleavage. Curr Opin Chem Biol. 2005;9:127–134. doi: 10.1016/j.cbpa.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Ralston CY, Sclavi B, Sullivan M, et al. Time-resolved synchrotron X-ray footprinting and its application to RNA folding. Methods Enzymol. 2000;317:353–368. doi: 10.1016/s0076-6879(00)17024-7. [DOI] [PubMed] [Google Scholar]
- 32.von Sonntag C. The Chemical Basis of Radiation Biology. London: Taylor & Francis; 1987. [Google Scholar]
- 33.Pogozelski WK, McNeese TJ, Tullius TD. What species is responsible for strand scission in the reaction of Fe•EDTA2− and H2O2 with DNA? J Am Chem Soc. 1995;117:6428–6433. [Google Scholar]
- 34.Balasubramanian B, Pogozelski WK, Tullius TD. DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. Proc Nat Acad Sci USA. 1998;95:9738–9743. doi: 10.1073/pnas.95.17.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M-J, Liu L, Wei K, Fu Y, Guo Q-X. Significant effects of phosphorylation on relative stabilities of DNA and RNA sugar radicals: remarkably high susceptibility of H-2′ abstraction in RNA. J Phys Chem B. 2006;110:13582–13589. doi: 10.1021/jp060331j. [DOI] [PubMed] [Google Scholar]
- 36.Xi Z, Goldberg IH. DNA-damaging enediyne compounds. In: Kool ET, editor. Comprehensive Natural Products Chemistry. Amsterdam: Elsevier; 1999. pp. 553–592. [Google Scholar]
- 37.Tashiro R, Sugiyama H. Reactivity of 5-halopyrimidines in nucleic acids. Wiley Ser React Intermed Chem Biol. 2009;2:163–189. [Google Scholar]
- 38.Watanabe T, Tashiro R, Sugiyama H. Photoreaction at 5′-(G/C)AAbr UT-3′ sequence in duplex DNA: efficient generation of uracil-5-yl radical by charge transfer. J Am Chem Soc. 2007;129:8163–8168. doi: 10.1021/ja0692736. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe T, Bando T, Xu Y, Tashiro R, Sugiyama H. Efficient generation of 2′-deoxyuridin-5-yl at 5′-(G/C)AAXUXU-3′ (X = Br, U) sequences in duplex DNA under UV irradiation. J Am Chem Soc. 2005;127:44–45. doi: 10.1021/ja0454743. [DOI] [PubMed] [Google Scholar]
- 40.Sugiyama H, Fujimoto K, Saito I. Evidence for intrastrand C2′ hydrogen abstraction in photoirradiation of 5-halouracil-containing oligonucleotides by using stereospecifically C2′-deuterated deoxyadenosine. Tetrahedron Lett. 1996;37:1805–1808. [Google Scholar]
- 41.Sugiyama H, Tsutsumi Y, Fujimoto K, Saito I. Photoinduced deoxyribose C2′ oxidation in DNA. Alkali-dependent cleavage of erythrose-containing sites via a retroaldol reaction. J Am Chem Soc. 1993;115:4443–4448. [Google Scholar]
- 42.Cook GP, Chen T, Koppisch AT, Greenberg MM. The effects of secondary structure and O2 on the formation of direct strand breaks upon UV irradiation of 5-bromo-deoxyuridine-containing oligonucleotides. Chem Biol. 1999;6:451–459. doi: 10.1016/s1074-5521(99)80063-5. [DOI] [PubMed] [Google Scholar]
- 43.Cook GP, Greenberg MM. A novel mechanism for the formation of direct strand breaks upon anaerobic photolysis of duplex DNA containing 5-bromodeoxyuridine. J Am Chem Soc. 1996;118:10025–10030. [Google Scholar]
- 44.Hawkins CL, Davies MJ. Hypochlorite-induced damage to DNA, RNA, and polynucleotides: formation of chloramines and nitrogen-centered radicals. Chem Res Toxicol. 2002;15:83–92. doi: 10.1021/tx015548d. [DOI] [PubMed] [Google Scholar]
- 45.Turro NJ. Modern Molecular Photochemistry. Menlo Park, CA: Benjamin Cummings; 1978. [Google Scholar]
- 46.Barvian MR, Greenberg MM. Independent generation of 5,6-dihydrothymid-5-yl and investigation of its ability to effect nucleic acid strand scission via hydrogen atom abstraction. J Org Chem. 1995;60:1916–1917. [Google Scholar]
- 47.Meschwitz SM, Schultz RG, Ashley GW, Goldberg IH. Selective abstraction of deuterium from C-1′ of the C residue in AGC•ICT by the radical center at C-2 of activated neocarzinostatin chromophore: structure of the drug/DNA complex responsible for bistranded lesion formation. Biochemistry. 1992;31:9117–9121. doi: 10.1021/bi00153a001. [DOI] [PubMed] [Google Scholar]
- 48.Pitié M, Bernadou J, Meunier B. Oxidation of carbon-1′ of DNA deoxyriboses by the Mn-TMPYP/KHSO5 system results from a cytochrome p-450-type hydroxylation reaction. J Am Chem Soc. 1995;117:2935–2936. [Google Scholar]
- 49.Thederahn TB, Kuwabara MD, Larsen TA, Sigman DS. Nuclease activity of 1,10-phenanthroline-copper: kinetic mechanism. J Am Chem Soc. 1989;111:4941–4946. [Google Scholar]
- 50.Goyne TE, Sigman DS. Nuclease activity of 1,10-phenanthroline-copper ion. Chemistry of deoxyribose oxidation. J Am Chem Soc. 1987;109:2846–2848. [Google Scholar]
- 51.Decarroz C, Wagner JR, Cadet J. Specific deprotonation reactions of the pyrimidine radical cation resulting from the menadione mediated photosensitization of 2′-deoxycytidine. Free Radic Res Commun. 1987;2:295–301. doi: 10.3109/10715768709065295. [DOI] [PubMed] [Google Scholar]
- 52.Shaw AA, Cadet J. Direct effects of γ-radiation on 2′-deoxycytidine in frozen aqueous solution. Int J Radiat Biol. 1996;70:1–6. doi: 10.1080/095530096145265. [DOI] [PubMed] [Google Scholar]
- 53.Sugiyama H, Saito I. Highly sequence selective photoreaction of 5-bromouracil-containing deoxyhexanucleotides. J Am Chem Soc. 1990;112:6720–6721. [Google Scholar]
- 54.Chen T, Cook GP, Koppisch AT, Greenberg MM. Investigation of the origin of the sequence selectivity for the 5-halo-2′-deoxyuridine sensitization of DNA to damage by UV-irradiation. J Am Chem Soc. 2000;122:3861–3866. [Google Scholar]
- 55.Goodman BK, Greenberg MM. Independent generation and reactivity of 2′-deoxy-urid-1′-yl. J Org Chem. 1996;61:2–3. [Google Scholar]
- 56.Tronche C, Goodman BK, Greenberg MM. DNA damage induced via independent generation of the radical resulting from formal hydrogen atom abstraction from the C1′-position of a nucleotide. Chem Biol. 1998;5:263–271. [PubMed] [Google Scholar]
- 57.Tallman KA, Tronche C, Yoo DJ, Greenberg MM. Release of superoxide from nucleoside peroxyl radicals, a double-edged sword? J Am Chem Soc. 1998;120:4903–4909. [Google Scholar]
- 58.Emanuel CJ, Newcomb M, Ferreri C, Chatgilialoglu C. Kinetics of 2′-deoxyuridin-1′-yl radical reactions. J Am Chem Soc. 1999;121:2927–2928. [Google Scholar]
- 59.von Sonntag C, Schuchmann H-P. The elucidation of peroxyl radical reactions in aqueous solution with the help of radiation-chemical methods. Angew Chem Int Ed. 1991;30:1229–1253. [Google Scholar]
- 60.Newcomb M. Competition methods and scales for alkyl radical reaction kinetics. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 61.Hwang J-T, Greenberg MM. Kinetics and stereoselectivity of thiol trapping of deoxyuridin-1′-yl in biopolymers and their relationship to the formation of premutagenic α-deoxynucleotides. J Am Chem Soc. 1999;121:4311–4315. [Google Scholar]
- 62.Savoye C, Swenberg C, Hugot S, et al. Thiol WR-1065 and disulphide WR-33278, two metabolites of the drug ethyol (WR-2721), protect DNA against fast neutron-induced strand breakage. Int J Radiat Biol. 1997;71:193–202. doi: 10.1080/095530097144319. [DOI] [PubMed] [Google Scholar]
- 63.Prise KM, Gilles NE, Whelan A, Newton GL, Fahey RC, Michael BD. Role of charge in the radioprotection of E. coli by thiols. Int J Radiat Biol. 1995;67:393–401. doi: 10.1080/09553009514550451. [DOI] [PubMed] [Google Scholar]
- 64.Kotera M, Bourdat A-G, Defrancq E, Lhomme J. A highly efficient synthesis of oligodeoxyribonucleotides containing the 2′-deoxyribonolactone lesion. J Am Chem Soc. 1998;120:11810–11811. [Google Scholar]
- 65.Kotera M, Roupioz Y, Defrancq E, et al. The 7-nitroindole nucleoside as a photochemical precursor of 2′-deoxyribonolactone: access to DNA fragments containing this oxidative abasic lesion. Chem Eur J. 2000;6:4163–4169. doi: 10.1002/1521-3765(20001117)6:22<4163::aid-chem4163>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 66.Crey-Desbiolles C, Lhomme J, Dumy P, Kotera M. 3-Nitro-3-deaza-2′-deoxyadenosine as a versatile photocleavable 2′-deoxyadenosine mimic. J Am Chem Soc. 2004;126:9532–9533. doi: 10.1021/ja047976m. [DOI] [PubMed] [Google Scholar]
- 67.Lenox HJ, McCoy CP, Sheppard TL. Site-specific generation of deoxyribonolactone lesions in DNA oligonucleotides. Org Lett. 2001;3:2415–2418. doi: 10.1021/ol016255e. [DOI] [PubMed] [Google Scholar]
- 68.Zheng Y, Sheppard TL. Half-life and DNA strand scission products of 2-deoxyribonolactone oxidative DNA damage lesions. Chem Res Toxicol. 2004;17:197–207. doi: 10.1021/tx034197v. [DOI] [PubMed] [Google Scholar]
- 69.Roupioz Y, Lhomme J, Kotera M. Chemistry of the 2-deoxyribonolactone lesion in oligonucleotides: cleavage kinetics and products analysis. J Am Chem Soc. 2002;124:9129–9135. doi: 10.1021/ja025688p. [DOI] [PubMed] [Google Scholar]
- 70.Sigman DS. Nuclease activity of 1,10-phenanthroline-copper ion. Acc Chem Res. 1986;19:180–186. [Google Scholar]
- 71.Kuwabara M, Yoon C, Goyne TE, Thederahn T, Sigman DS. Nuclease activity of 1,10-phenanthroline-copper ion: reaction with CGCGAATTCGCG and its complexes with netropsin and EcoR1. Biochemistry. 1986;25:7401–7408. doi: 10.1021/bi00371a023. [DOI] [PubMed] [Google Scholar]
- 72.Meijler MM, Zelenko O, Sigman DS. Chemical mechanism of DNA scission by (1,10-phenanthioline)copper carboxyl oxygen of 5-methylenefuranose is derived from water. J Am Chem Soc. 1997;119:1135–1136. [Google Scholar]
- 73.Zelenko O, Gallagher J, Sigman DS. Scission of DNA with bis(1,10-phenanthroline) copper without intramolecular hydrogen migration. Angew Chem Int Ed. 1997;36:2776–2778. [Google Scholar]
- 74.Chen T, Greenberg MM. Model studies indicate that copper phenanthroline induces direct strand breaks via β-elimination of the 2′-deoxyribonolactone intermediate observed in enediyne mediated DNA damage. J Am Chem Soc. 1998;120:3815–3816. [Google Scholar]
- 75.Oyoshi T, Sugiyama H. Mechanism of DNA strand scission induced by (1,10-phenan-throline)copper complex: major direct DNA cleavage is not through 1′,2′-dehydro-nucleotide intermediate nor beta-elimination of forming ribonolactone. J Am Chem Soc. 2000;122:6313–6314. [Google Scholar]
- 76.Marshall LE, Graham DR, Reich KA, Sigman DS. Cleavage of deoxyribonucleic acid by the 1,10-phenanthroline-cuprous complex. Hydrogen peroxide requirement and primary and secondary structure specificity. Biochemistry. 1981;20:244–250. doi: 10.1021/bi00505a003. [DOI] [PubMed] [Google Scholar]
- 77.Bales BC, Kodama T, Weledji YN, Pitie M, Meunier B, Greenberg MM. Mechanistic studies on DNA damage by minor groove binding copper-phenanthroline conjugates. Nucleic Acids Res. 2005;33:5371–5379. doi: 10.1093/nar/gki856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pitié M, Van Horn JD, Brion D, Burrows CJ, Meunier B. Targeting the DNA cleavage activity of copper phenanthroline and Clip-Phen to A•T tracts via linkage to a poly-n-methylpyrrole. Bioconjugate Chem. 2000;11:892–900. doi: 10.1021/bc000050t. [DOI] [PubMed] [Google Scholar]
- 79.Pitié M, Burrows CJ, Meunier B. Mechanisms of DNA cleavage by copper complexes of 3-Clip-Phen and of its conjugate with a distamycin analogue. Nucleic Acids Res. 2000;28:4856–4864. doi: 10.1093/nar/28.24.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilson SH, Beard WA, Shock DD, et al. Base excision repair and design of small molecule inhibitors of human DNA polymerase β. Cell Mol Life Sci. 2010;67:3633–3647. doi: 10.1007/s00018-010-0489-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wilson DM, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair. 2007;6:544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 82.Greenberg MM. Abasic and oxidized abasic site reactivity in DNA: enzyme inhibition, crosslinking, and nucleosome catalyzed reactions. Acc Chem Res. 2014;47:646–655. doi: 10.1021/ar400229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kroeger KM, Hashimoto M, Kow YW, Greenberg MM. Cross-linking of 2-deoxyribonolactone and its β-elimination product by base excision repair enzymes. Biochemistry. 2003;42:2449–2455. doi: 10.1021/bi027168c. [DOI] [PubMed] [Google Scholar]
- 84.Hashimoto M, Greenberg MM, Kow YW, Hwang J-T, Cunningham RP. The 2-deoxyribonolactone lesion produced in DNA by neocarzinostatin and other DNA damaging agents forms cross-links with the base-excision repair enzyme endo-nuclease III. J Am Chem Soc. 2001;123:3161–3162. doi: 10.1021/ja003354z. [DOI] [PubMed] [Google Scholar]
- 85.Imoto S, Bransfield LA, Croteau DL, Van Houten B, Greenberg MM. DNA tandem lesion repair by strand displacement synthesis and nucleotide excision repair. Biochemistry. 2008;47:4306–4316. doi: 10.1021/bi7021427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakano T, Terato H, Asagoshi K, et al. DNA-protein cross-link formation mediated by oxanine. A novel genotoxic mechanism of nitric oxide-induced DNA damage. J Biol Chem. 2003;278:25264–25272. doi: 10.1074/jbc.M212847200. [DOI] [PubMed] [Google Scholar]
- 87.Guan L, Greenberg MM. Irreversible inhibition of DNA polymerase β by an oxidized abasic lesion. J Am Chem Soc. 2010;132:5004–5005. doi: 10.1021/ja101372c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guan L, Bebenek K, Kunkel TA, Greenberg MM. Inhibition of short patch and long patch base excision repair by an oxidized abasic site. Biochemistry. 2010;49:9904–9910. doi: 10.1021/bi101533a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stevens AJ, Guan L, Bebenek K, Kunkel TA, Greenberg MM. DNA polymerase λ inactivation by oxidized abasic sites. Biochemistry. 2013;52:975–983. doi: 10.1021/bi301592x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jacobs AC, Kreller CR, Greenberg MM. Long patch base excision repair compensates for DNA polymerase β inactivation by the C4′-oxidized abasic site. Biochemistry. 2011;50:136–143. doi: 10.1021/bi1017667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.DeMott MS, Beyret E, Wong D, et al. Covalent trapping of human DNA polymerase β by the oxidative DNA lesion 2-deoxyribonolactone. J Biol Chem. 2002;277:7637–7640. doi: 10.1074/jbc.C100577200. [DOI] [PubMed] [Google Scholar]
- 92.Quinones JL, Thapar U, Yu K, Fang Q, Sobol RW, Demple B. Enzyme mechanism- ~ based, oxidative DNA–protein cross-links formed with DNA polymerase β in vivo. Proc Nat Acad Sci USA. 2015;112:8602–8607. doi: 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Greenberg MM. Looking beneath the surface to determine what makes DNA damage deleterious. Curr Opin Chem Biol. 2014;21:48–55. doi: 10.1016/j.cbpa.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sugiyama H, Fujimoto K, Saito I. Stereospecific 1,2-hydride shift in ribonolactone formation in the photoreaction of 2′-iododeoxyuridine. J Am Chem Soc. 1995;117:2945–2946. [Google Scholar]
- 95.Sugiyama H, Fujimoto K, Saito I. Preferential C1′ hydrogen abstraction by a uracilyl radical in a DNA–RNA hybrid. Tetrahedron Lett. 1997;38:8057. [Google Scholar]
- 96.Kawai K, Saito I, Kawashima E, Ishido Y, Sugiyama H. Intrastrand 2′-β hydrogen abstraction of 5′-adjacent deoxyguanosine by deoxyuridin-5-yl in Z-form DNA. Tetrahedron Lett. 1999;40:2589–2592. [Google Scholar]
- 97.Kawai K, Saito I, Sugiyama H. Conformation-dependent photochemistry of 5-halouracil-containing DNA: Stereospecific 2′-hydroxylation of deoxyribose in Z-form DNA. J Am Chem Soc. 1999;121:1391–1392. [Google Scholar]
- 98.Tashiro R, Ohtsuki A, Sugiyama H. The distance between donor and acceptor affects the proportion of C1′ and C2′ oxidation products of DNA in a BrU-containing excess electron transfer system. J Am Chem Soc. 2010;132:14361–14363. doi: 10.1021/ja106184w. [DOI] [PubMed] [Google Scholar]
- 99.Xu Y, Tashiro R, Sugiyama H. Photochemical determination of different DNA structures. Nat Protoc. 2007;2:78–87. doi: 10.1038/nprot.2006.467. [DOI] [PubMed] [Google Scholar]
- 100.Morinaga H, Kizaki S, Takenaka T, et al. Photoreactivities of 5-bromouracil-containing RNAs. Bioorg Med Chem. 2013;21:466–469. doi: 10.1016/j.bmc.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 101.Coote ML, Lin CY, Beckwith ALJ, Zavitsas AA. A comparison of methods for measuring relative radical stabilities of carbon-centered radicals. Phys Chem Chem Phys. 2010;12:9597–9610. doi: 10.1039/c003880f. [DOI] [PubMed] [Google Scholar]
- 102.Behrens G, Koltzenburg G, Schulte-Frohlinde D. Model reactions for the degradation of DNA-4′ radicals in aqueous solution. Fast hydrolysis of α-alkoxyalkyl radicals with a leaving group in β-position followed by radical rearrangement and elimination reactions. Z Naturforsch C J Biosci. 1982;37C:1205–1227. [Google Scholar]
- 103.Dizdaroglu M, Von Sonntag C, Schulte-Frohlinde D. Strand breaks and sugar release by γ-irradiation of DNA in aqueous solution. J Am Chem Soc. 1975;97:2277–2278. doi: 10.1021/ja00841a051. [DOI] [PubMed] [Google Scholar]
- 104.Horner JH, Bagnol L, Newcomb M. Kinetics of radical heterolysis reactions forming alkene radical cations. J Am Chem Soc. 2004;126:14979–14987. doi: 10.1021/ja046089g. [DOI] [PubMed] [Google Scholar]
- 105.Cozens FL, O’Neill M, Bogdanova R, Schepp N. Dynamics of ionization reactions of β-substituted radicals. Substituents and solvent effects. J Am Chem Soc. 1997;119:10652–10659. [Google Scholar]
- 106.Giese B, Dussy A, Meggers E, Petretta M, Schwitter U. Conformation, lifetime, and repair of 4′-DNA radicals. J Am Chem Soc. 1997;119:11130–11131. [Google Scholar]
- 107.Chatgilialoglu C, Duca M, Ferreri C, et al. Selective generation and reactivity of 5′-adenosinyl and 2′-adenosinyl radicals. Chem Eur J. 2004;10:1249–1255. doi: 10.1002/chem.200305488. [DOI] [PubMed] [Google Scholar]
- 108.Paul R, Greenberg MM. Independent generation and reactivity of the uridin-2′-yl radical. J Org Chem. 2014;79:10303–10310. doi: 10.1021/jo501916r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Paul R, Greenberg MM. Rapid RNA strand scission following C2′-hydrogen atom abstraction. J Am Chem Soc. 2015;137:596–599. doi: 10.1021/ja511401g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kawashima E, Aoyama Y, Sekine T, et al. Sonochemical and triethylborane-induced tin deuteride reduction for the highly diastereoselective synthesis of (2′R)-2′-deoxy [2′-2H]ribonucleoside derivatives. J Org Chem. 1995;60:6980–6986. [Google Scholar]
- 111.Robins MJ, Wilson JS, Hansske F. Nucleic acid related compounds. 42. A general procedure for the efficient deoxygenation of secondary alcohols. Regiospecific and stereoselective conversion of ribonucleosides to 2′-deoxynucleosides. J Am Chem Soc. 1983;105:4059–4065. [Google Scholar]
- 112.Iino T, Shuto S, Matsuda A. Stereoselective synthesis of (2′S)-2′-C-alkyl-2′-deoxyuridines. Nucleosides Nucleotides. 1996;15:169–181. [Google Scholar]
- 113.Newcomb M, Horner JH, Shahin H. Rate constants for aminyl radical reactions. Tetrahedron Lett. 1993;34:5523–5526. [Google Scholar]
- 114.Sitlani A, Long EC, Pyle AM, Barton JK. DNA photocleavage by phenanthrenequinone diimine complexes of rhodium(III): shape-selective recognition and reaction. J Am Chem Soc. 1992;114:2303–2312. [Google Scholar]
- 115.Pyle AM, Long EC, Barton JK. Shape-selective targeting of DNA by phenanthrenequinone diiminerhodium(III) photocleaving agents. J Am Chem Soc. 1989;111:4520–4522. [Google Scholar]
- 116.Bryant-Friedrich AC. Generation of a C-3′-thymidinyl radical in single-stranded oligonucleotides under anaerobic conditions. Org Lett. 2004;6:2329–2332. doi: 10.1021/ol0493453. [DOI] [PubMed] [Google Scholar]
- 117.Lahoud GA, Hitt AL, Bryant-Friedrich AC. Aerobic fate of the C-3′-thymidinyl radical in single-stranded DNA. Chem Res Toxicol. 2006;19:1630–1636. doi: 10.1021/tx060174f. [DOI] [PubMed] [Google Scholar]
- 118.Zaidi R, Bryant-Friedrich AC. The effect of reductant levels on the formation of damage lesions derived from a 2-deoxyribose radical in ssDNA. Radiat Res. 2012;177:565–572. doi: 10.1667/rr2733.1. [DOI] [PubMed] [Google Scholar]
- 119.Amato NJ, Bryant-Friedrich AC. The impact of structure on oxidatively generated DNA damage products resulting from the C3′-thymidinyl radical. ChemBioChem. 2013;14:187–190. doi: 10.1002/cbic.201200723. [DOI] [PubMed] [Google Scholar]
- 120.Pratviel G, Pitié M, Bernadou J, Meunier B. Furfural as a marker of DNA cleavage by hydroxylation at the 5′ carbon of deoxyribose. Angew Chem Int Ed. 1991;30:702–704. [Google Scholar]
- 121.Kozarich JW, Worth L, Frank BL, Christner DF, Vanderwall DE, Stubbe J. Sequence-specific isotope effects on the cleavage of DNA by bleomycin. Science. 1989;245:1396–1399. doi: 10.1126/science.2476851. [DOI] [PubMed] [Google Scholar]
- 122.Stubbe J, Kozarich JW, Doyle TU. Unmasking the chemistry of DNA cleavage by the esperamicins: Modulation of 4′-hydrogen abstraction and bistranded damage by the fucose-anthranilate moiety. J Am Chem Soc. 1992;114:8763. [Google Scholar]
- 123.Kappen LS, Goldberg IH, Frank BL, et al. Neocarzinostatin-induced hydrogen atom abstraction from C-4′ and C-5′ of the T residue at a d(GT) step in oligonucleotides: shuttling between deoxyribose attack sites based on isotope selection effects. Biochemistry. 1991;30:2034–2042. doi: 10.1021/bi00222a005. [DOI] [PubMed] [Google Scholar]
- 124.Frank BL, Worth L, Jr, Christner DF, et al. Isotope effects on the sequence-specific cleavage of DNA by neocarzinostatin: kinetic partitioning between 4′- and 5′-hydrogen abstraction at unique thymidine sites. J Am Chem Soc. 1991;113:2271–2275. [Google Scholar]
- 125.Stubbe J, Kozarich JW. Mechanisms of bleomycin-induced DNA degradation. Chem Rev. 1987;87:1107–1136. [Google Scholar]
- 126.McGall GH, Rabow LE, Ashley GW, Wu SH, Kozarich JW, Stubbe J. New insight into the mechanism of base propenal formation during bleomycin-mediated DNA degradation. J Am Chem Soc. 1992;114:4958–4967. [Google Scholar]
- 127.Rabow LE, McGall GH, Stubbe J, Kozarich JW. Identification of the source of oxygen in the alkaline-labile product accompanying cytosine release during bleomycin-mediated oxidative degradation of d(CGCGCG) J Am Chem Soc. 1990;112:3203–3208. [Google Scholar]
- 128.Rabow LE, Stubbe J, Kozarich JW. Identification and quantitation of the lesion accompanying base release in bleomycin-mediated DNA degradation. J Am Chem Soc. 1990;112:3196–3203. [Google Scholar]
- 129.Sugiyama H, Kawabata H, Fujiwara T, Dannoue Y, Saito I. Specific detection of C-4′ hydroxylated abasic sites generated by bleomycin and neocarzinostatin in DNA. J Am Chem Soc. 1990;112:5252–5257. [Google Scholar]
- 130.San Pedro JMN, Beerman TA, Greenberg MM. DNA damage by C1027 involves hydrogen atom abstraction and addition to nucleobases. Bioorg Med Chem. 2012;20:4744–4750. doi: 10.1016/j.bmc.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bennett RAO, Swerdlow PS, Povirk LF. Spontaneous cleavage of bleomycin-induced abasic sites in chromatin and their mutagenicity in mammalian shuttle vectors. Biochemistry. 1993;32:3188–3195. doi: 10.1021/bi00063a034. [DOI] [PubMed] [Google Scholar]
- 132.Zhou C, Sczepanski JT, Greenberg MM. Histone modification via rapid cleavage of C4′-oxidized abasic sites in nucleosome core particles. J Am Chem Soc. 2013;135:5274–5277. doi: 10.1021/ja400915w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wu JC, Kozarich JW, Stubbe J. Mechanism of bleomycin: evidence for a rate-determining 4′-hydrogen abstraction from poly(dA-dU) associated with the formation of both free base and base propenal. Biochemistry. 1985;24:7562–7568. doi: 10.1021/bi00347a009. [DOI] [PubMed] [Google Scholar]
- 134.Wu JC, Stubbe J, Kozarich JW. Mechanism of bleomycin: evidence for 4′-ketone formation in poly(dA-dU) associated exclusively with free base release. Biochemistry. 1985;24:7569–7573. doi: 10.1021/bi00347a010. [DOI] [PubMed] [Google Scholar]
- 135.Absalon MJ, Wu W, Kozarich JW, Stubbe J. Sequence-specific double-strand cleavage of DNA by Febleomycin. 2. Mechanism and dynamics. Biochemistry. 1995;34:2076–2086. doi: 10.1021/bi00006a030. [DOI] [PubMed] [Google Scholar]
- 136.McGall GH, Rabow LE, Stubbe J, Kozarich JW. Incorporation of 18O into glycolic acid obtained from the bleomycin-mediated degradation of DNA. Evidence for 4′-radical trapping by 18O2. J Am Chem Soc. 1987;109:2836–2837. [Google Scholar]
- 137.Burger RM, Projan SJ, Horwitz SB, Peisach J. The DNA cleavage mechanism of iron-bleomycin. Kinetic resolution of strand scission from base propenal release. J Biol Chem. 1986;261:15955–15959. [PubMed] [Google Scholar]
- 138.Ajmera S, Wu JC, Worth L, Rabow LE, Stubbe J, Kozarich JW. DNA degradation by bleomycin: evidence for 2′R-proton abstraction and for carbon-oxygen bond cleavage accompanying base propenal formation. Biochemistry. 1986;25:6586–6592. doi: 10.1021/bi00369a037. [DOI] [PubMed] [Google Scholar]
- 139.Giese B, Beyrich-Graf X, Erdmann P, et al. Cleavage of single-stranded 4′-oligonucleotide radicals in the presence of O2. J Am Chem Soc. 1995;117:6146–6147. [Google Scholar]
- 140.Giese B, Beyrich-Graf X, Erdmann P, Petretta M, Schwitter U. The chemistry of single-stranded 4′-DNA radicals: influence of the radical precursor on anaerobic and aerobic strand cleavage. Chem Biol. 1995;2:367–375. doi: 10.1016/1074-5521(95)90217-1. [DOI] [PubMed] [Google Scholar]
- 141.Meggers E, Dussy A, Schäfer T, Giese B. Electron transfer in DNA from guanine and 8-oxoguanine to a radical cation of the carbohydrate backbone. Chem Eur J. 2000;6:485–492. doi: 10.1002/(sici)1521-3765(20000204)6:3<485::aid-chem485>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 142.Dussy A, Meggers E, Giese B. Spontaneous cleavage of 4′-DNA radicals under aerobic conditions: apparent discrepancy between trapping rates and cleavage products. J Am Chem Soc. 1998;120:7399–7403. [Google Scholar]
- 143.Sugiura Y, Matsumoto T. Some characteristics of DNA strand scission by macromolecular antitumor antibiotic C-1027 containing a novel enediyne chromophore. Biochemistry. 1993;32:5548–5553. doi: 10.1021/bi00072a008. [DOI] [PubMed] [Google Scholar]
- 144.Zein N, Sinha AM, McGahren WJ, Ellestad GA. Calicheamicin γ1: an antitumor antibiotic that cleaves double-stranded DNA site specifically. Science. 1988;240:1198–1201. doi: 10.1126/science.3240341. [DOI] [PubMed] [Google Scholar]
- 145.Povirk LF, Han YH, Steighner RJ. Structure of bleomycin-induced DNA double-strand breaks: predominance of blunt ends and single-base 5′ extensions. Biochemistry. 1989;28:5808–5814. doi: 10.1021/bi00440a016. [DOI] [PubMed] [Google Scholar]
- 146.Steighner RJ, Povirk LF. Bleomycin-induced DNA lesions at mutational hot spots: Implications for the mechanism of double-strand cleavage. Proc Nat Acad Sci USA. 1990;87:8350–8354. doi: 10.1073/pnas.87.21.8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. The cytotoxicity of (−)-lomaiviticin A arises from induction of double-strand breaks in DNA. Nat Chem. 2014;6:504–510. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Siddiqi MA, Bothe E. Single- and double-strand break formation in DNA irradiated in aqueous solution: dependence on dose and OH• radical scavenger concentration. Radiat Res. 1987;112:449–463. [PubMed] [Google Scholar]
- 149.Frankenberg-Schwager M, Frankenberg D. DNA double-strand breaks: their repair and relationship to cell killing in yeast. Int J Radiat Biol. 1990;58:569–575. doi: 10.1080/09553009014551931. [DOI] [PubMed] [Google Scholar]
- 150.Eccles LJ, Lomax ME, O’Neill P. Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage. Nucleic Acids Res. 2010;38:1123–1134. doi: 10.1093/nar/gkp1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kozmin SG, Sedletska Y, Reynaud-Angelin A, Gasparutto D, Sage E. The formation of double-strand breaks at multiply damaged sites is driven by the kinetics of excision/ incision at base damage in eukaryotic cells. Nucleic Acids Res. 2009;37:1767–1777. doi: 10.1093/nar/gkp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Milligan JR, Ng JY, Wu CCL, Aguilera JA, Fahey RC, Ward JF. DNA repair by thiols in air shows two radicals make a double-strand break. Radiat Res. 1995;143:273–280. [PubMed] [Google Scholar]
- 153.Taverna Porro ML, Greenberg MM. Double-strand breaks from a radical commonly produced by DNA-damaging agents. Chem Res Toxicol. 2015;28:810–816. doi: 10.1021/acs.chemrestox.5b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dervan PB. Design of sequence-specific DNA-binding molecules. Science. 1986;232:464–471. doi: 10.1126/science.2421408. [DOI] [PubMed] [Google Scholar]
- 155.Giese B. Long-distance charge transport in DNA: the hopping mechanism. Acc Chem Res. 2000;33:631–636. doi: 10.1021/ar990040b. [DOI] [PubMed] [Google Scholar]
- 156.Giese B. Long-distance electron transfer through DNA. Annu Rev Biochem. 2002;71:51–70. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- 157.O’Neill MA, Barton JK. DNA-mediated charge transport chemistry and biology. Top Curr Chem. 2004;236:67–115. [Google Scholar]
- 158.Lewis FD, Letsinger RL, Wasielewski MR. Dynamics of photoinduced charge transfer and hole transport in synthetic DNA hairpins. Acc Chem Res. 2001;34:159–170. doi: 10.1021/ar0000197. [DOI] [PubMed] [Google Scholar]
- 159.Takada T, Fujitsuka M, Majima T. Single-molecule observation of DNA charge transfer. Proc Nat Acad Sci USA. 2007;104:11179–11183. doi: 10.1073/pnas.0700795104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Takada T, Kawai K, Fujitsaka M, Majima T. Rapid long-distance hole transfer through consecutive adenine sequence. J Am Chem Soc. 2007;128:11012–11013. doi: 10.1021/ja0641554. [DOI] [PubMed] [Google Scholar]
- 161.Genereux JC, Boal AK, Barton JK. DNA-mediated charge transport in redox sensing and signaling. J Am Chem Soc. 2010;132:891–905. doi: 10.1021/ja907669c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sontz PA, Muren NB, Barton JK. DNA charge transport for sensing and signaling. Acc Chem Res. 2012;45:1792–1800. doi: 10.1021/ar3001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Furst AL, Muren NB, Hill MG, Barton JK. Label-free electrochemical detection of human methyltransferase from tumors. Proc Nat Acad Sci USA. 2014;111:14985–14989. doi: 10.1073/pnas.1417351111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee PE, Demple B, Barton JK. DNA-mediated redox signaling for transcriptional activation of SoxR. Proc Nat Acad Sci USA. 2009;106:13164–13168. doi: 10.1073/pnas.0906429106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Joy A, Schuster GB. Long-range radical cation migration in DNA: Investigation of the mechanism. Chem Commun. 2005:2778–2784. doi: 10.1039/b500412h. [DOI] [PubMed] [Google Scholar]
- 166.Turro NJ, Barton JK. Accelerated electron transfer between metal complexes mediated by DNA. Science. 1988:241. doi: 10.1126/science.241.4873.1645. [DOI] [PubMed] [Google Scholar]
- 167.Murphy CJ, Arkin MR, Jenkins Y, et al. Long-range photoinduced electron transfer through a DNA helix. Science. 1993;262:1025–1029. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 168.Turro NJ, Barton JK. Long-range photoinduced electron transfer through a DNA helix. Science. 1993;262:1025. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 169.Slinker JD, Muren NB, Renfrew SE, Barton JK. DNA charge transport over 34 nm. Nat Chem. 2011;3:228–233. doi: 10.1038/nchem.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Magliozzo RS, Peisach J, Ciriolo MR. Transfer RNA is cleaved by activated bleomycin. Mol Pharmacol. 1989;35:428–432. [PubMed] [Google Scholar]
- 171.Carter BJ, de Vroom E, Long EC, van der Marel GA, van Boom JH, Hecht SM. Site-specific cleavage of RNA by Fe(II)-bleomycin. Proc Natl Acad Sci USA. 1990;87:9373–9377. doi: 10.1073/pnas.87.23.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Abraham AT, Lin J-J, Newton DL, Rybak S, Hecht SM. RNA cleavage and inhibition of protein synthesis by bleomycin. Chem Biol. 2003;10:45–52. doi: 10.1016/s1074-5521(02)00306-x. [DOI] [PubMed] [Google Scholar]
- 173.Morgan MA, Hecht SM. Iron(II) bleomycin-mediated degradation of DNA–RNA heteroduplex. Biochemistry. 1994;33:10286. doi: 10.1021/bi00200a008. [DOI] [PubMed] [Google Scholar]
- 174.Strittmatter H, Dussy A, Schwitter U, Giese B. Differences between 4′-RNA and 4′-DNA radicals during anaerobic and aerobic strand cleavage. Angew Chem Int Ed. 1999;38:135–137. [Google Scholar]
- 175.Crich D, Mo X-S. Nucleotide C3′,4′-radical cations and the effect of a 2′-oxygen substituent. The DNA/RNA paradox. J Am Chem Soc. 1997;119:249–250. [Google Scholar]
- 176.Kappen LS, Goldberg IH, Liesch JM. Identification of thymidine-5′-aldehyde at DNA strand breaks induced by neocarzinostatin chromophore. Proc Natl Acad Sci USA. 1982;79:744–748. doi: 10.1073/pnas.79.3.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xu J, Zhen Y, Goldberg IH. C1027 chromophore, a potent new enediyne antitumor antibiotic, induces sequence-specific double-strand DNA cleavage. Biochemistry. 1994;33:5947–5954. doi: 10.1021/bi00185a036. [DOI] [PubMed] [Google Scholar]
- 178.Epstein JL, Zhang X, Doss GA, et al. Interplay of hydrogen abstraction and radical repair in the generation of single- and double-strand DNA damage by the esperamicins. J Am Chem Soc. 1997;119:6731–6738. [Google Scholar]
- 179.Xu Y, Xi Z, Zhen Y, Goldberg IH. A single binding mode of activated enediyne C1027 generates two types of double-stranded DNA lesions: deuterium iostope-induced shuttling between adjacent nucleotide target sites. Biochemistry. 1995;34:12451–12460. doi: 10.1021/bi00038a044. [DOI] [PubMed] [Google Scholar]
- 180.Meschwitz SM, Goldberg IH. Selective abstraction of 2H from C-5′ of thymidylate in an oligodeoxynucleotide by the radical center at C-6 of the diradical species of neocarzinostatin: chemical evidence for the structure of the activated drug-DNA complex. Proc Nat Acad Sci USA. 1991;88:3047–3051. doi: 10.1073/pnas.88.8.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pratviel G, Bernadou J, Meunier B. Carbon-hydrogen bonds of DNA sugar units as targets for chemical nucleases and drugs. Angew Chem Int Ed Engl. 1995;34:746–769. [Google Scholar]
- 182.Chin DH, Carr SA, Goldberg IH. Incorporation of 18O2 into thymidine 5′-aldehyde in neocarzinostatin chromophore-damaged DNA. J Biol Chem. 1984;259:9975–9978. [PubMed] [Google Scholar]
- 183.Pitié M, Pratviel G. Activation of DNA carbon, hydrogen bonds by metal complexes. Chem Rev. 2010;110:1018–1059. doi: 10.1021/cr900247m. [DOI] [PubMed] [Google Scholar]
- 184.Boussicault F, Kaloudis P, Caminal C, Mulazzani QG, Chatgilialoglu C. The fate of C5′ radicals of purine nucleosides under oxidative conditions. J Am Chem Soc. 2008;130:8377–8385. doi: 10.1021/ja800763j. [DOI] [PubMed] [Google Scholar]
- 185.Chin DH, Kappen LS, Goldberg IH. 3′-Formyl phosphate-ended DNA: high-energy intermediate in antibiotic-induced DNA sugar damage. Proc Nat Acad Sci USA. 1987;84:7070–7074. doi: 10.1073/pnas.84.20.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kawabata H, Takeshita H, Fujiwara T, Sugiyama H, Matsuura T, Saito I. Chemistry of neocarzinostatin-mediated degradation of d(GCATGC). Mechanism of spontaneous thymine release. Tetrahedron Lett. 1989;30:4263–4266. [Google Scholar]
- 187.Kodama T, Greenberg MM. Preparation and analysis of oligonucleotides containing lesions resulting from C5′-oxidation. J Org Chem. 2005;70:9916–9924. doi: 10.1021/jo051666k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Weng L, Greenberg MM. Rapid histone-catalyzed DNA lesion excision and accompanying protein modification in nucleosomes and nucleosome core particles. J Am Chem Soc. 2015;137:11022–11031. doi: 10.1021/jacs.5b05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dhar S, Kodama T, Greenberg MM. Selective detection and quantification of oxidized abasic lesions in DNA. J Am Chem Soc. 2007;129:8702–8703. doi: 10.1021/ja073014e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Manetto A, Georganakis D, Leondiadis L, et al. Independent generation of C5′-nucleosidyl radicals in thymidine and 2′-deoxyguanosine. J Org Chem. 2007;72:3659–3666. doi: 10.1021/jo062518c. [DOI] [PubMed] [Google Scholar]
- 191.Chatgilialoglu C, Bazzanini R, Jimenez LB, Miranda MA. (5′S)- and (5′R)-5′, 8-cyclo-2′-deoxyguanosine: mechanistic insights on the 2′-deoxyguanosin-5′-yl radical cyclization. Chem Res Toxicol. 2007;20:1820–1824. doi: 10.1021/tx700282x. [DOI] [PubMed] [Google Scholar]
- 192.Chatgilialoglu C, Guerra M, Mulazzani QG. Model studies of DNA C5′ radicals. Selective generation and reactivity of 2′-deoxyadenosin-5′-yl radical. J Am Chem Soc. 2003;125:3839–3848. doi: 10.1021/ja029374d. [DOI] [PubMed] [Google Scholar]
- 193.You C, Dai X, Yuan B, et al. A quantitative assay for assessing the effects of DNA lesions on transcription. Nat Chem Biol. 2012;8:817–822. doi: 10.1038/nchembio.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jasti VP, Das RS, Hilton BA, Weerasooriya S, Zou Y, Basu AK. (5′-S)-8,5′-cyclo-2′-deoxyguanosine is a strong block to replication, a potent pol V-dependent mutagenic lesion, and is inefficiently repaired in Escherichia coli. Biochemistry. 2011;50:3862–3865. doi: 10.1021/bi2004944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Belmadoui N, Boussicault F, Guerra M, Ravanat JL, Chatgilialoglu C, Cadet J. Radiation-induced formation of purine 5′,8-cyclonucleosides in isolated and cellular DNA: high stereospecificity and modulating effect of oxygen. Org Biomol Chem. 2010;8:3211–3219. doi: 10.1039/c004531d. [DOI] [PubMed] [Google Scholar]
- 196.Wang Y. Bulky DNA lesions induced by reactive oxygen species. Chem Res Toxicol. 2008;21:276–281. doi: 10.1021/tx700411g. [DOI] [PubMed] [Google Scholar]
- 197.Shaik R, Ellis MW, Starr MJ, Amato NJ, Bryant-Friedrich AC. Photochemical generation of a C5′-uridinyl radical. ChemBioChem. 2015;16:2379–2384. doi: 10.1002/cbic.201500330. [DOI] [PubMed] [Google Scholar]
- 198.Wagner JR, Decarroz C, Berger M, Cadet J. Hydroxyl-radical-induced decomposition of 2′-deoxycytidine in aerated aqueous solutions. J Am Chem Soc. 1999;121:4101–4110. [Google Scholar]
- 199.Deeble DJ, von Sonntag C. Radiolysis of poly(U) in oxygenated solution. Int J Radiat Biol. 1986;49:927–936. doi: 10.1080/09553008514553161. [DOI] [PubMed] [Google Scholar]
- 200.Deeble DJ, Schulz D, Von Sonntag C. Reactions of OH radicals with poly(U) in deoxygenated solutions: sites of OH radical attack and the kinetics of base release. Int J Radiat Biol. 1986;49:915–926. doi: 10.1080/09553008514553151. [DOI] [PubMed] [Google Scholar]
- 201.Martini M, Termini J. Peroxy radical oxidation of thymidine. Chem Res Toxicol. 1997;10:234–241. doi: 10.1021/tx960154l. [DOI] [PubMed] [Google Scholar]
- 202.Wang W, Razskazovskii Y, Sevilla MD. Secondary radical attack on DNA nucleotides: reaction by addition to DNA bases and abstraction from sugars. Int J Radiat Biol. 1997;71:387–399. doi: 10.1080/095530097144003. [DOI] [PubMed] [Google Scholar]
- 203.Chatgilialoglu C, D’Angelantonio M, Kciuk G, Bobrowski K. New insights into the reaction paths of hydroxyl radicals with 2′-deoxyguanosine. Chem Res Toxicol. 2011;24:2200–2206. doi: 10.1021/tx2003245. [DOI] [PubMed] [Google Scholar]
- 204.Kumar A, Pottiboyina V, Sevilla MD. Hydroxyl radical (OH•) reaction with guanine in an aqueous environment: a DFT study. J Phys Chem B. 2011;115:15129–15137. doi: 10.1021/jp208841q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chatgilialoglu C, D’Angelantonio M, Guerra M, Kaloudis P, Mulazzani QG. A reevaluation of the ambident reactivity of the guanine moiety towards hydroxyl radicals. Angew Chem Int Ed. 2009;48:2214–2217. doi: 10.1002/anie.200805372. [DOI] [PubMed] [Google Scholar]
- 206.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 207.Rokhlenko Y, Cadet J, Geacintov NE, Shafirovich V. Mechanistic aspects of hydration of guanine radical cations in DNA. J Am Chem Soc. 2014;136:5956–5962. doi: 10.1021/ja412471u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rokhlenko Y, Geacintov NE, Shafirovich V. Lifetimes and reaction pathways of guanine radical cations and neutral guanine radicals in an oligonucleotide in aqueous solutions. J Am Chem Soc. 2012;134:4955–4962. doi: 10.1021/ja212186w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bienvenu C, Wagner JR, Cadet J. Photosensitized oxidation of 5-methyl-2′-deoxycytidine by 2-methyl-1,4-naphthoquinone: characterization of 5-(hydroperoxymethyl)-2′-deoxycytidine and stable methyl group oxidation products. J Am Chem Soc. 1996;118:11406–11411. [Google Scholar]
- 210.Douki T, Cadet J. Modification of DNA bases by photosensitized one-electron oxidation. Int J Radiat Biol. 1999;75:571–581. doi: 10.1080/095530099140212. [DOI] [PubMed] [Google Scholar]
- 211.Wagner JR, van Lier JE, Johnston LJ. Quinone sensitized electron transfer photooxidation of nucleic acids: chemistry of thymine and thymidine radical cations in aqueous solution. Photochem Photobiol. 1990;52:333–343. doi: 10.1111/j.1751-1097.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 212.Adhikary A, Kumar A, Munafo SA, Khanduri D, Sevilla MD. Prototropic equilibria in DNA containing one-electron oxidized GC: Intra-duplex vs. Duplex to solvent deprotonation. Phys Chem Chem Phys. 2010;12:5353–5368. doi: 10.1039/b925496j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Al-Sheikhly M. The reactivity of adenyl and guanyl radicals towards oxygen. Radiat Phys Chem. 1994;44:297–301. [Google Scholar]
- 214.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: Induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 215.Dizdaroglu M, Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Rad Res. 2012;46:382–419. doi: 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- 216.Wagner JR, Cadet J. Oxidation reactions of cytosine DNA components by hydroxyl radical and one-electron oxidants in aerated aqueous solutions. Acc Chem Res. 2010;43:564–571. doi: 10.1021/ar9002637. [DOI] [PubMed] [Google Scholar]
- 217.Fleming AM, Muller JG, Dlouhy AC, Burrows CJ. Structural context effects in the oxidation of 8-oxo-7,8-dihydro-2′-deoxyguanosine to hydantoin products: Electrostatics, base stacking, and base pairing. J Am Chem Soc. 2012;134:15091–15102. doi: 10.1021/ja306077b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Alshykhly OR, Fleming AM, Burrows CJ. 5-carboxamido-5-formamido-2-iminohydantoin, in addition to 8-oxo-7,8-dihydroguanine, is the major product of the iron-fenton or X-ray radiation-induced oxidation of guanine under aerobic reducing conditions in nucleoside and DNA contexts. J Org Chem. 2015;80:6996–7007. doi: 10.1021/acs.joc.5b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Dizdaroglu M, Kirkali G, Jaruga P. Formamidopyrimidines in DNA: mechanisms of formation, repair, and biological effects. Free Radic Biol Med. 2008;45:1610–1621. doi: 10.1016/j.freeradbiomed.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 220.Earley LF, Minko IG, Christov PP, Rizzo CJ, Lloyd RS. Mutagenic spectra arising from replication bypass of the 2,6-diamino-4-hydroxy-N5-methyl formamidopyrimidine adduct in primate cells. Chem Res Toxicol. 2013;26:1108–1114. doi: 10.1021/tx4001495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kamiya H. Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: Approaches using synthetic oligonucleotides and nucleotides. Nucleic Acids Res. 2003;31:517–531. doi: 10.1093/nar/gkg137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lemaire DGE, Bothe E, Schulte-Frohlinde D. Yields of radiation-induced main chain scission of poly U in aqueous solution: strand break formation via base radicals. Int J Radiat Biol. 1984;45:351–358. doi: 10.1080/09553008414550491. [DOI] [PubMed] [Google Scholar]
- 223.Hildenbrand K, Schulte-Frohlinde D. E.S.R. studies on the mechanism of hydroxyl radical-induced strand breakage of polyuridylic acid. Int J Radiat Biol. 1989;55:725–738. doi: 10.1080/09553008914550781. [DOI] [PubMed] [Google Scholar]
- 224.Milligan JR, Aguilera JA, Nguyen T-TD. Yield of DNA strand breaks after base oxidation of plasmid DNA. Radiat Res. 1999;151:334–342. [PubMed] [Google Scholar]
- 225.Jones GDD, O’Neill P. Kinetics of radiation-induced strand break formation in single-stranded pyrimidine polynucleotides in the presence and absence of oxygen; a time-resolved light-scattering study. Int J Radiat Biol. 1991;59:1127–1145. doi: 10.1080/09553009114551031. [DOI] [PubMed] [Google Scholar]
- 226.Jones GD, O’Neill P. The kinetics of radiation-induced strand breakage in polynucleotides in the presence of oxygen: a time-resolved light-scattering study. Int J Radiat Biol. 1990;57:1123–1139. doi: 10.1080/09553009014551241. [DOI] [PubMed] [Google Scholar]
- 227.Hildenbrand K, Behrens G, Schulte-Frohlinde D. Comparison of the reaction of OH and of SO4−· radicals with pyrimidine nucleosides. An electron spin resonance study in aqueous solution. J Chem Soc Perkin Trans II. 1989:283–289. [Google Scholar]
- 228.Catterall H, Davies MJ, Gilbert BC. An EPR study of the transfer of radical-induced damage from the base to sugar in nucleic acid components: Relevance to the occurrence of strand-breakage. J Chem Soc Perkin Trans II. 1992:1379–1385. [Google Scholar]
- 229.Wolf P, Jones GD, Candeias LP, O’Neill P. Induction of strand breaks in polyribonucleotides and DNA by the sulphate radical anion: role of electron loss centres as precursors of strand breakage. Int J Radiat Biol. 1993;64:7–18. doi: 10.1080/09553009314551061. [DOI] [PubMed] [Google Scholar]
- 230.Patrzyc HB, Dawidzik JB, Budzinski EE, Freund HG, Wilton JH, Box HC. Covalently linked tandem lesions in DNA. Radiat Res. 2012;178:538–542. doi: 10.1667/RR2915.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yuan B, Jiang Y, Wang Y, Wang Y. Efficient formation of the tandem thymine glycol/8-oxo-7,8-dihydroguanine lesion in isolated DNA and the mutagenic and cytotoxic properties of the tandem lesions in Escherichia coli cells. Chem Res Toxicol. 2009;23:11–19. doi: 10.1021/tx9004264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Douki T, Riviere J, Cadet J. DNA tandem lesions containing 8-oxo-7,8-dihydroguanine and formamido residues arise from intramolecular addition of thymine peroxyl radical to guanine. Chem Res Toxicol. 2002;15:445–454. doi: 10.1021/tx0155909. [DOI] [PubMed] [Google Scholar]
- 233.Tallman KA, Greenberg MM. Oxygen-dependent DNA damage amplification involving 5,6-dihydrothymidin-5-yl in a structurally minimal system. J Am Chem Soc. 2001;123:5181–5187. doi: 10.1021/ja010180s. [DOI] [PubMed] [Google Scholar]
- 234.Box HC, Patrzyc HB, Dawidzik JB, et al. Double base lesions in DNA X-irradiated in the presence or absence of oxygen. Radiat Res. 2000;153:442–446. doi: 10.1667/0033-7587(2000)153[0442:dblidx]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 235.Bourdat A-G, Douki T, Frelon S, Gasparutto D, Cadet J. Tandem base lesions are generated by hydroxyl radical within isolated DNA in aerated aqueous solution. J Am Chem Soc. 2000;122:4549–4556. [Google Scholar]
- 236.Sage E, Harrison L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat Res. 2011;711:123–133. doi: 10.1016/j.mrfmmm.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Saloua KS, Sonia G, Pierre C, Léon S, Darel HJ. The relative contributions of DNA strand breaks, base damage and clustered lesions to the loss of DNA functionality induced by ionizing radiation. Radiat Res. 2014;181:99–110. doi: 10.1667/RR13450.1. [DOI] [PubMed] [Google Scholar]
- 238.Cunniffe S, O’Neill P, Greenberg MM, Lomax ME. Reduced repair capacity of a DNA clustered damage site comprised of 8-oxo-7,8-dihydro-2′-deoxyguanosine and 2-deoxyribonolactone results in an increased mutagenic potential of these lesions. Mutat Res Fundam Mol Mech Mutagen. 2014;762:32–39. doi: 10.1016/j.mrfmmm.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Georgakilas A. Processing of DNA damage clusters in human cells: current status of knowledge. Mol Biosyst. 2008;4:30–35. doi: 10.1039/b713178j. [DOI] [PubMed] [Google Scholar]
- 240.Joseph J, Schuster GB. One-electron oxidation of DNA: reaction at thymine. Chem Comm. 2010;46:7872–7878. doi: 10.1039/c0cc02118k. [DOI] [PubMed] [Google Scholar]
- 241.Kanvah S, Schuster GB. One-electron oxidation of DNA: thymine versus guanine reactivity. Org Biomol Chem. 2010;8:1340–1343. doi: 10.1039/b922881k. [DOI] [PubMed] [Google Scholar]
- 242.Greenberg MM, Barvian MR, Cook GP, et al. DNA damage induced via 5,6-dihydrothymidin-5-yl in single-stranded oligonucleotides. J Am Chem Soc. 1997;119:1828–1839. [Google Scholar]
- 243.Barvian MR, Greenberg MM. Independent generation of 5,6-dihydrothymid-5-yl in single-stranded polythymidylate. O2 is necessary for strand scission. J Am Chem Soc. 1995;117:8291–8292. [Google Scholar]
- 244.Hong IS, Carter KN, Sato K, Greenberg MM. Characterization and mechanism of formation of tandem lesions in DNA by a nucleobase peroxyl radical. J Am Chem Soc. 2007;129:4089–4098. doi: 10.1021/ja0692276. [DOI] [PubMed] [Google Scholar]
- 245.Hong IS, Carter KN, Greenberg MM. Evidence for glycosidic bond rotation in a nucleobase peroxyl radical and its effect on tandem lesion formation. J Org Chem. 2004;69:6974–6978. doi: 10.1021/jo0492158. [DOI] [PubMed] [Google Scholar]
- 246.Carter KN, Greenberg MM. Independent generation and study of 5,6-dihydro-2′-deoxyuridin-6-yl, a member of the major family of reactive intermediates formed in DNA from the effects of gradiolysis. J Org Chem. 2003;68:4275–4280. doi: 10.1021/jo034038g. [DOI] [PubMed] [Google Scholar]
- 247.Carter KN, Greenberg MM. Tandem lesions are the major products resulting from a pyrimidine nucleobase radical. J Am Chem Soc. 2003;125:13376–13378. doi: 10.1021/ja036629u. [DOI] [PubMed] [Google Scholar]
- 248.San Pedro JMN, Greenberg MM. Photochemical generation and reactivity of the major hydroxyl radical adduct of thymidine. Org Lett. 2012;14:2866–2869. doi: 10.1021/ol301109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.San Pedro JMN, Greenberg MM. 5,6-dihydropyrimidine peroxyl radical reactivity in DNA. J Am Chem Soc. 2014;136:3928–3936. doi: 10.1021/ja412562p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zhang Q, Wang Y. The reactivity of the 5-hydroxy-5,6-dihydrothymidin-6-yl radical in oligodeoxyribonucleotides. Chem Res Toxicol. 2005;18:1897–1906. doi: 10.1021/tx050195u. [DOI] [PubMed] [Google Scholar]
- 251.Zhang Q, Wang Y. Independent generation of the 5-hydroxy-5,6-dihydrothymidin-6-yl radical and its reactivity in dinucleoside monophosphates. J Am Chem Soc. 2004;126:13287–13297. doi: 10.1021/ja048492t. [DOI] [PubMed] [Google Scholar]
- 252.Hwang J-T, Tallman KA, Greenberg MM. The reactivity of the 2-deoxyribonolactone lesion in single-stranded DNA and its implication in reaction mechanisms of DNA damage and repair. Nucleic Acids Res. 1999;27:3805–3810. doi: 10.1093/nar/27.19.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sato K, Greenberg MM. Selective detection of 2-deoxyribonolactone in DNA. J Am Chem Soc. 2005;127:2806–2807. doi: 10.1021/ja0426185. [DOI] [PubMed] [Google Scholar]
- 254.Miaskiewicz K, Miller J, Ornstein R, Osman R. Molecular dynamics simulations of the effects of ring-saturated thymine lesions on DNA structure. Biopolymers. 1995;35:113–124. doi: 10.1002/bip.360350112. [DOI] [PubMed] [Google Scholar]
- 255.Miaskiewicz K, Miller J, Osman R. Energetic basis for structural preferences in 5/6-hydroxy-5,6-dihydropyrimidines: products of ionizing and ultraviolet radiation action on DNA bases. Biochim Biophys Acta. 1994;1218:283–291. doi: 10.1016/0167-4781(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 256.Bergeron F, Auvré F, Radicella JP, Ravanat J-L. HO• radicals induce an unexpected high proportion of tandem lesion base lesions refractory to repair by DNA glycosylases. Proc Nat Acad Sci USA. 2010;107:5528–5533. doi: 10.1073/pnas.1000193107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Jovanovic SV, Jankovic I, Josimovic L. Electron-transfer reactions of alkyl peroxy radicals. J Am Chem Soc. 1992;114:9018–9021. [Google Scholar]
- 258.Yoshioka Y, Kitagawa Y, Takano Y, Yamaguchi K, Nakamura T, Saito I. Experimental and theoretical studies on the selectivity of GGG triplets toward one-electron oxidation in B-form DNA. J Am Chem Soc. 1999;121:8712–8719. [Google Scholar]
- 259.Sugiyama H, Saito I. Theoretical studies of GG-specific photocleavage of DNA via electron transfer: significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B-form DNA. J Am Chem Soc. 1996;118:7063–7068. [Google Scholar]
- 260.Saito I, Nakanura T, Nakatani K, Yoshioka Y, Yamaguchi K, Sugiyama H. Mapping of the hot spots for DNA damage by one-electron oxidation: Efficacy of GG doublets and GGG triplets as a trap in long-range hole migration. J Am Chem Soc. 1998;120:12686–12687. [Google Scholar]
- 261.Resendiz MJE, Pottiboyina V, Sevilla MD, Greenberg MM. Direct strand scission in double stranded RNA via a C5-pyrimidine radical. J Am Chem Soc. 2012;134:3917–3924. doi: 10.1021/ja300044e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Jacobs AC, Resendiz MJE, Greenberg MM. Product and mechanistic analysis of the reactivity of a C6-pyrimidine radical in RNA. J Am Chem Soc. 2011;133:5152–5159. doi: 10.1021/ja200317w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Jacobs AC, Resendiz MJE, Greenberg MM. Direct strand scission from a nucleobase radical in RNA. J Am Chem Soc. 2010;132:3668–3669. doi: 10.1021/ja100281x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Newman CA, Resendiz MJE, Sczepanski JT, Greenberg MM. Photochemical generation and reactivity of the 5,6-dihydrouridin-6-yl radical. J Org Chem. 2009;74:7007–7012. doi: 10.1021/jo9012805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Zhang Q, Wang Y. Independent generation of 5-(2′-deoxycytidinyl)methyl radical and the formation of a novel cross-link lesion between 5-methylcytosine and guanine. J Am Chem Soc. 2003;125:12795–12802. doi: 10.1021/ja034866r. [DOI] [PubMed] [Google Scholar]
- 266.Zhang Q, Wang Y. Generation of 5-(2′-deoxycytidyl)methyl radical and the formation of intrastrand cross-link lesions in oligodeoxyribonucleotides. Nucleic Acids Res. 2005;33:1593–1603. doi: 10.1093/nar/gki301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hong IS, Greenberg MM. Mild generation of 5-(2′-deoxyuridinyl)methyl radical from a phenyl selenide precursor. Org Lett. 2004;6:5011–5013. doi: 10.1021/ol047754t. [DOI] [PubMed] [Google Scholar]
- 268.Hong IS, Ding H, Greenberg MM. Oxygen independent DNA interstrand cross-link formation by a nucleotide radical. J Am Chem Soc. 2006;128:485–491. doi: 10.1021/ja0563657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Bellon S, Ravanat J-L, Gasparutto D, Cadet J. Cross-linked thyminepurine base tandem lesions: synthesis, characterization, and measurement in γ-irradiated isolated DNA. Chem Res Toxicol. 2002;15:598–606. doi: 10.1021/tx015594d. [DOI] [PubMed] [Google Scholar]
- 270.Anderson AS, Hwang JT, Greenberg MM. Independent generation and reactivity of 2′-deoxy-5-methyleneuridin-5-yl, a significant reactive intermediate produced from thymidine as a result of oxidative stress. J Org Chem. 2000;65:4648–4654. doi: 10.1021/jo000271s. [DOI] [PubMed] [Google Scholar]
- 271.Hong IS, Greenberg MM. Efficient DNA interstrand cross-link formation from a nucleotide radical. J Am Chem Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 272.Ding H, Majumdar A, Tolman JR, Greenberg MM. Multinuclear NMR and kinetic analysis of DNA interstrand cross-link formation. J Am Chem Soc. 2008;130:17981–17987. doi: 10.1021/ja807845n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ghosh A, Joy A, Schuster GB, Douki T, Cadet J. Selective one-electron oxidation of duplex DNA oligomers: reaction at thymines. Org Biomol Chem. 2008;6:916–928. doi: 10.1039/b717437c. [DOI] [PubMed] [Google Scholar]
- 274.Joy A, Ghosh AK, Schuster GB. One-electron oxidation of DNA oligomers that lack guanine: reaction and strand cleavage at remote thymines by long-distance radical cation hopping. J Am Chem Soc. 2006;128:5346–5347. doi: 10.1021/ja058758b. [DOI] [PubMed] [Google Scholar]
- 275.Veldhuyzen WF, Shallop AJ, Jones RA, Rokita SE. Thermodynamic versus kinetic products of DNA alkylation as modeled by reaction of deoxyadenosine. J Am Chem Soc. 2001;123:11126–11132. doi: 10.1021/ja011686d. [DOI] [PubMed] [Google Scholar]
- 276.Veldhuyzen WF, Parde P, Rokita SE. A transient product of DNA alkylation can be stabilized by binding localization. J Am Chem Soc. 2003;125:14005–14013. doi: 10.1021/ja036943o. [DOI] [PubMed] [Google Scholar]
- 277.Hong IS, Greenberg MM. DNA interstrand cross-link formation initiated by reaction between singlet oxygen and a modified nucleotide. J Am Chem Soc. 2005;127:10510–10511. doi: 10.1021/ja053493m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Hong IS, Ding H, Greenberg MM. Radiosensitization by a modified nucleotide that produces DNA interstrand cross-links under hypoxic conditions. J Am Chem Soc. 2006;128:2230–2231. doi: 10.1021/ja058290c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Peng X, Hong IS, Li H, Seidman MM, Greenberg MM. Interstrand cross-link formation in duplex and triplex DNA by modified pyrimidines. J Am Chem Soc. 2008;130:10299–10306. doi: 10.1021/ja802177u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Peng X, Greenberg MM. Facile SNP detection using bifunctional, cross-linking oligonucleotide probes. Nucleic Acids Res. 2008;36:e31. doi: 10.1093/nar/gkn052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Weng L, Zhou C, Greenberg MM. Probing interactions between lysine residues in histone tails and nucleosomal DNA via product and kinetic analysis. ACS Chem Biol. 2015;10:622–630. doi: 10.1021/cb500737y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Weng L, Horvat SM, Schiesser CH, Greenberg MM. Deconvoluting the reactivity of two intermediates formed from modified pyrimidines. Org Lett. 2013;15:3618–3621. doi: 10.1021/ol401472m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Lin G, Li L. Oxidation and reduction of the 5-(2′-deoxyuridinyl)methyl radical. Angew Chem Int Ed. 2013;52:5594–5598. doi: 10.1002/anie.201209454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Vrantza D, Kaloudis P, Leondiadis L, et al. Modification of guanine with photolabile n-hydroxypyridine-2(1H)-thione: Monomer synthesis, oligonucleotide elaboration, and photochemical studies. Helv Chim Acta. 2006;89:2371–2386. [Google Scholar]
- 285.Kaloudis P, Paris C, Vrantza D, et al. Photolabile n-hydroxypyrid-2(1H)-one derivatives of guanine nucleosides: a new method for independent guanine radical generation. Org Biomol Chem. 2009;7:4965–4972. doi: 10.1039/b909138f. [DOI] [PubMed] [Google Scholar]
- 286.Kuttappan-Nair V, Samson-Thibault F, Wagner JR. Generation of 2′-deoxyadenosine N6-aminyl radicals from the photolysis of phenylhydrazone derivatives. Chem Res Toxicol. 2010;23:48–54. doi: 10.1021/tx900268r. [DOI] [PubMed] [Google Scholar]
- 287.Davis WB, Bjorklund CC, Deline M. Probing the effects of DNA-protein interactions on DNA hole transport: the N-terminal histone tails modulate the distribution of oxidative damage and chemical lesions in the nucleosome core particle. Biochemistry. 2012;51:3129–3142. doi: 10.1021/bi201734c. [DOI] [PubMed] [Google Scholar]
- 288.Bjorklund CC, Davis WB. Stable DNA-protein cross-links are products of DNA charge transport in a nucleosome core particle. Biochemistry. 2007;46:10745–10755. doi: 10.1021/bi700475b. [DOI] [PubMed] [Google Scholar]