

Abstract
Directed evolution is a powerful tool for optimizing enzymes, and mutagenesis methods that improve enzyme library quality can significantly expedite the evolution process. Here, we report a simple method for targeted combinatorial codon mutagenesis (CCM). To demonstrate the utility of this method for protein engineering, CCM libraries were constructed for cytochrome P450BM3, pfu prolyl oligopeptidase, and the flavin-dependent halogenase RebH. 22–26 sites were targeted for codon mutagenesis in each of these enzymes, and libraries with a tunable average of 1–7 codon mutations per gene were generated. Each of these libraries provided improved enzymes for their respective transformations, which highlights the generality, simplicity, and tunability of CCM for targeted protein engineering.
Keywords: codon mutagenesis, directed evolution, halogenase, prolyl oligopeptidase, cytochrome P450
Directed evolution allows for systematic improvement of enzyme function with only minimal information regarding enzyme structure.1 A classical directed evolution campaign consists of iterative rounds of gene mutagenesis and screening for improved enzymes until a desired level of function is obtained (Fig. 1A-1).2 A variety of methods have been used to introduce mutations throughout enzymes, but error prone PCR is probably the simplest and most commonly used (Fig. 1A-2).2,3 Importantly, however, the low probability of mutating contiguous bases limits the range of mutations possible at each site, biases exist for mutation of individual bases,3 and a large fraction of libraries typically contains no mutations.
Figure 1.
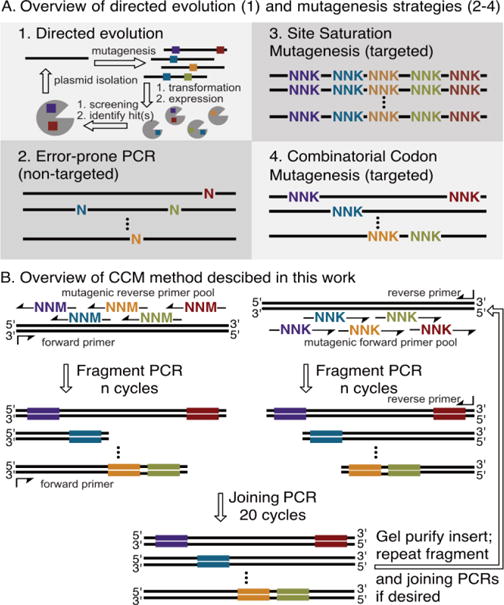
Overview of A) directed evolution and three common mutagenesis strategies; B) combinatorial codon mutagenesis involving iterative rounds of fragment PCR using mutagenic primer pools followed by joining PCR.
If available, sequence activity relationships or structural information can be used to identify sites within an enzyme that significantly impact or are predicted to impact its function. Ideally, libraries targeting these sites can be screened to rapidly improve enzyme function and decrease the effort associated with directed evolution.2 While predicting specific beneficial mutations remains challenging, degenerate codons can be used to examine different mutations at targeted sites. Both single and multiple site saturation mutagenesis can be conducted using synthetic oligonucleotides containing degenerate codons or a combination of codons to encode desired mutations at targeted sites within an enzyme (Fig. 1A-3).4,5
Beyond the fundamental difficulty of accurately predicting which sites to target, however, saturation mutagenesis efforts must also address a number of technical issues. Simultaneous mutation of several residues often results in unfolded peptides, so only small subsets of residues are usually targeted. This process can be iterated to cover a desired region (e.g. within an enzyme active site).6,7 Beneficial epistasis resulting from simultaneous mutation of sites in different sub-libraries will be missed by this approach, but the full diversity of sequences within each sub-library can be readily screened. Various bioinformatics tools have been developed and applied to design libraries comprised of discrete enzyme mutants that collectively sample diversity at large numbers of targeted sites.8–10 Library members must be individually cloned or synthesized, the latter of which, at least currently, remains cost prohibitive for most academic efforts. Simple methods for targeted mutagenesis of large numbers of sites within enzymes are therefore valuable tools for protein engineering efforts.
Combinatorial codon mutagenesis (CCM) methods can be used to replace, with tunable frequency, each codon within a set of targeted sites with a desired codon. Thus, diversity at several sites in a protein can be introduced into a library without the detrimental effects of simultaneous codon mutagenesis (Fig. 1A-4). Inspired by early work from Stemmer,11 Herman and Tawfik described a method in which a target gene is amplified using biotinylated primers, the PCR product is fragmented with DNaseI, and the fragments are assembled in a self-primed extension PCR using a set of mutagenic oligonucleotides. The assembled genes are enriched by capture on streptavidin-coated magnetic beads, which maintains diversity created in the assembly reaction by minimizing mis-priming and amplification of short products.12 Firnberg and Ostermeier later described a method for comprehensive codon mutagenesis that allowed for creation of libraries containing a single codon mutation per gene. To accomplish this, uracil-containing template DNA, thermostable ligase, phosphorylated primers, and other specialized reagents are required.13 In a very recent report, Jin described a method that uses phosphorylated mutagenic primers that simultaneously anneal to the template DNA and mutate contiguous nucleotide bases. Template DNA is linearly amplified with T4 DNA polymerase, and the resulting fragments are ligated using a thermostable T4 DNA ligase.14
While effective, these CCM methods are technically demanding and require the use of costly, specialized reagents. Splicing by overlap extension (SOE) PCR15 has been used to encode mixtures of wild-type and mutagenic codons at multiple targeted sites in enzymes without these complications, but tedious cloning procedures are required if large numbers of sites are targeted.16,17 Bloom recently reported a CCM method18 that incorporates elements SOE PCR and megaprimer PCR19 using mutagenic oligonucleotide primers to create random mutants of influenza nucleoprotein for experimentally determined evolutionary models (Figure 1B). Herein, we show that this method can be used to target a tunable number of codon mutations to a large number of sites in enzymes (Figure 1B). The utility of this method is illustrated via optimization of cytochrome P450BM3 (BM3), a prolyl oligopeptidase (POP), and the flavin-dependent halogenase RebH for different organic transformations.
Initial optimization of Bloom’s protocol for targeted CCM was conducted using BM3. This enzyme has been extensively engineered using a range of mutagenesis strategies, including error-prone PCR,20,21 targeted site saturation mutagenesis (SSM) on single sites, and CASTing/iterative saturation mutagenesis (ISM) on multiple sites22. We envisioned that CCM could be used to generate a single library that would target sites found to improve BM3 catalysis in these earlier efforts (Fig. 2C),16,22,23 allowing for rapid BM3 optimization with minimal cloning effort.
Figure 2.
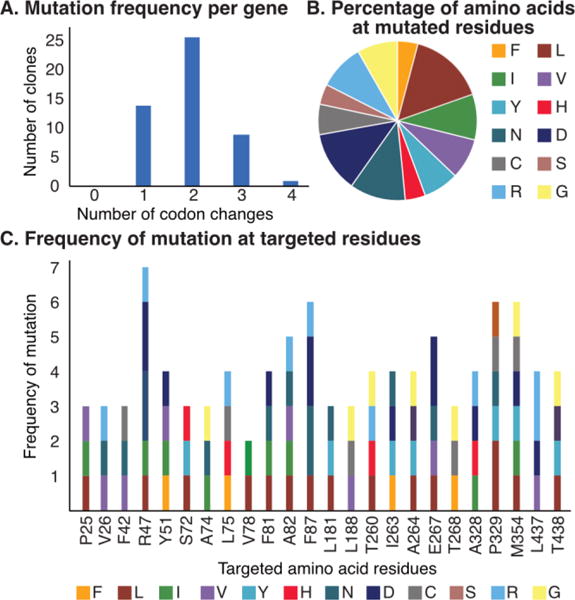
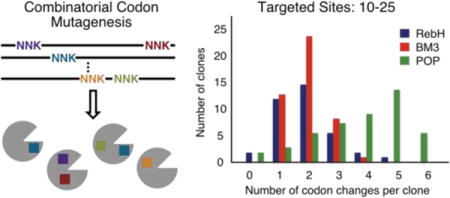
A) Codon changes per gene for 50 randomly selected clones from a BM3 CCM library. B) Percentage of the 12 possible amino acid residues at mutated sites. C) The 22 targeted sites are all mutated with similar efficiency.
A library targeting 22 active site residues was therefore constructed using 22 forward and reverse primers that contained a degenerate NDT codon at the targeted residues. Standard primer design considerations and restriction digest cloning methods were used; no specialized (e.g. biotinylated, phosphorylated, etc.) primers are required. The mutagenic primers were pooled and used in fragment and joining PCRs in analogy to Bloom’s procedure (Fig. 1B).18 Amplification of the appropriately sized gene was observed, and the full-length genes were purified using agarose gel electrophoresis. The purified product was used as a template for a second round of fragment and joining PCRs, and the product was again purified and cloned into a pET vector using standard restriction methods. Fifty random clones from this library were sequenced, which revealed a mutation frequency of ~2 mutations per gene (Fig. 2A). All 50 random clones contained at least one mutation, which is not commonly observed in error-prone PCR libraries. Further analysis demonstrated that all targeted sites were mutated with similar efficiency (Fig. 2C) and all possible codon mutations were observed (Fig. 2B). In essence, the CCM library involves random mutagenesis of all 22 targeted sites with residues encoded by the NDT codon and little parent background (none observed in the 50 sequenced clones).
From this initial CCM library, 1,000 variants were screened for demethylation activity on hexyl methyl ether (HME) and benzyl methyl ether (BME). Various combinations of mutations at sites F87, T268, P239, and L437 improved demethylation activity on HME and BME compared to wild-type BM3 (Fig. S1). The variant with the highest increase in conversion of BME, 1DF (F87D and T286F, Fig. 3A), was further characterized by steady state kinetic analysis. Variant 1DF displayed a 1.4-fold improvement of KM and a 2.2-fold improvement of kcat compared to BM3 (Fig. 3C, Fig. S2), indicating that CCM can be used to identify new BM3 variants with improved catalytic efficiency.
Figure 3.
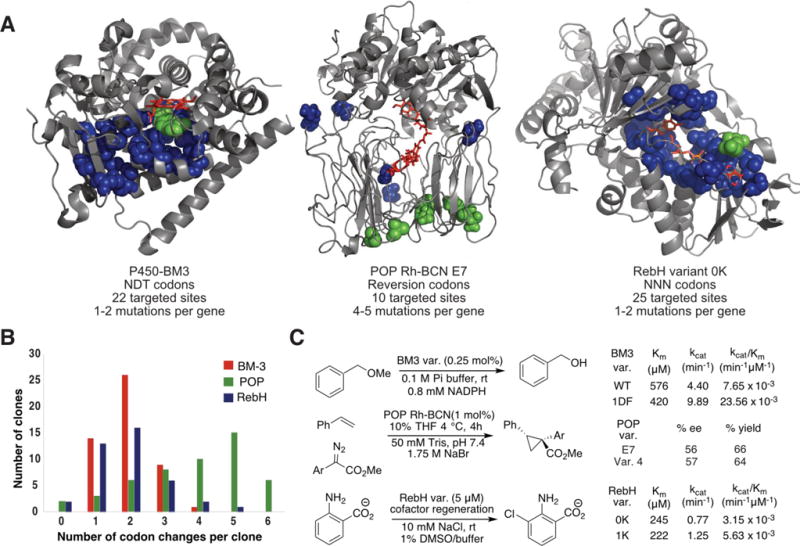
A) Crystal structures of RebH24 and BM325, and a homology model of POP26 containing a covalently-linked dirhodium cofactor27. Cofactors shown as red sticks. Targeted residues shown as blue and green spheres; green indicates mutations in optimized variants. B) Codon changes per gene for each library. C) Reactions catalyzed by CCM library members (Ar=4-methoxyphenyl).
We next sought to establish the generality of CCM for optimizing other enzymes and the extent to which mutation frequency during CCM can be tuned. The latter should vary as a function of fragmentation PCR cycle number and the number of rounds of fragmentation and joining PCRs conducted. Both of these variables were explored by generating libraries of POP, which our laboratory has explored as a scaffold for generating artificial metalloenzymes.27 The high stability of POP was expected to allow for evaluation of CCM libraries with several mutations in each library member.
A homology model of POP was used to identify 26 sites within β-propeller domain of this enzyme for mutagenesis.26 Oligonucleotides encoding NNN at the targeted residues were used, and three libraries were constructed using different numbers of fragmentation/joining rounds or fragmentation cycles. The expected increase in mutagenesis with increasing numbers of rounds or cycles was observed: three rounds of seven cycles, three rounds of fourteen cycles, and seven rounds of seven cycles provided three, five, and seven codon mutations/gene, respectively (Fig. S3). In all three libraries, all targeted sites were mutated with similar efficiencies (Fig. S4). 1,000 members of the second library, containing five codon mutations per gene, were screened for peptidase activity on the chromogenic substrate Z-Gly-Pro-(p-nitroaniline). 42% of the variants were active, indicating that at least this fraction of the library was folded (Fig. S5). While beyond the scope of this study, these CCM libraries will serve as a valuable source of scaffold proteins with extensive active site mutations for our artificial metalloenzyme efforts.28
We also envisioned that CCM could be used for deconvoluting or combining mutations accumulated throughout a directed evolution campaign. In deconvolution experiments, mutations in an improved enzyme are individually reverted to the wild-type residue to determine which mutations are necessary for improved function. When several beneficial mutations are discovered as separate hits in a single round of mutagenesis and screening, combining these mutations can be beneficial. Deconvolution and combination using CCM could be significantly faster and more efficient than individually cloning reversions or combinations of mutations.
A POP variant developed in our lab (E7) was chosen as a template to establish the utility of CCM for mutation deconvolution. E7 contains ten point mutations accumulated throughout a directed evolution campaign aimed at improving this enzyme as a scaffold for enantioselective cyclopropanation following covalent modification with a dirhodium cofactor (Fig. 3C, Fig. S6).26 Oligonucleotides for the reversions of the ten sites back to the parent POP enzyme (ZA2) residues were designed using standard primer design considerations. To construct this library, one round of fragmentation/joining with 17 fragmentation cycles was performed, which gave an average of ~4 mutations per gene. Approximately 200 clones from this library were expressed and bioconjugated with a synthetic dirhodium cofactor (Fig. S6). The resulting artificial metalloenzyme library was screened using a model cyclopropanation reaction, and several enzymes that provided the same conversion and enantioselectivity as E7 were observed (Fig. 4, Variants 1–5). When these clones were sequenced, residues at five sites (blue: 84, 99, 161, 166, and 301) were conserved while the remaining sites (green) were not, indicating that the latter were not required for the observed enantioselectivity of E7. Notably, Variant 4 contained all five reversions. Subsequent CCM aimed at further reducing the number of mutations required to maintain E7 activity using Variant 4 as a template provided no further consensus for unnecessary mutations.
Figure 4.

Targeted residues for five variants from a POP-E7 deconvolution library. Residues at targeted sites in POP-ZA2 and E7 are shown in grey. Residues shown in blue were targeted but not reverted; residues shown in green were reverted.
In a final test of the utility and generality of CCM for targeted protein engineering, we sought to explore mutagenesis of the FAD-binding site of RebH. Recent reports have shown that the catalytic efficiencies of several different enzymes can be improved via mutagenesis of residues in highly conserved redox cofactor binding sites.29,30 Library approaches to interrogate the effects of different mutations at each site within a cofactor binding site could provide a means to rapidly explore similar effects in other enzymes. The expected importance of most of these residues for catalytic activity, however, requires the ability to target degenerate codons to different sites with low frequency.
To explore this possibility, twenty-five residues that comprise the FAD-binding pocket of the RebH variant 0K (RebH-E461K)31 were selected, and a library targeting a frequency of 1–2 mutations per gene was constructed using one round of fragmentation/joining with 17 fragmentation cycles (Figure 3A–B). Sequencing random clones from this library revealed that, while mutation frequency per gene was easily tunable (Fig. S7), bias in the distribution of mutations was observed unlike the POP and BM3 libraries (Fig. 2C, S4). This could be due to the GC-rich nature of the RebH gene, and codon optimization of the gene could prove useful for removing this bias.
Despite a bias for certain sites, 1,000 variants from this library were screened by UPLC for activity on 2-aminobenzoic acid. As expected from the high conservation of residues in the FAD binding pocket of FDHs, a significant proportion of the library showed no activity. A few variants did, however, provide improved conversion relative to 0K. Sequencing and validating these hits using purified enzyme revealed that variant 1K (R231K) was found to increase conversion 1.7-fold. Kinetic characterization of 1K revealed an increase in kcat from 0.77 min−1 for that of 0K to 1.25 min−1 (Fig. S8). Remarkably, this variant also has a higher turnover number on the unnatural substrate 2-aminobenzoic acid than that of the wild-type RebH on its native substrate tryptophan (1.1 min−1).32
CCM libraries have been shown to aid directed evolution efforts by allowing rapid evaluation of mutations targeted to specific sites in enzymes with variable frequency.11,12,14 The CCM method outlined in this study, which is based on work by Bloom18, has proven similarly effective for optimizing three different enzymes, but it does not require the specialized primers or reagents used in other CCM methods.11,12,14 The procedure is essentially a variation of SOE PCR15 that can be performed using standard cloning procedures. Despite this simplicity, mutations encoded by degenerate codons or codon mixtures can be targeted to many non-contiguous sites (>20 demonstrated) in an enzyme to provide library members with a random, tunable distribution of codon mutations. We have shown that this method can be applied to deconvolution efforts, mutation of enzyme active sites, and conserved cofactor binding sites that are sensitive to mutagenesis. We expect that the simplicity and robustness of this method will lead to its application for a wide range of protein engineering efforts.
Supplementary Material
Acknowledgments
This work was supported by the NIH (1R01GM115665) and the U.S. Army Research Laboratory and the U. S. Army Research Office under contract/grant number 62247-LS. MCA was supported by an NSF predoctoral fellowship (DGE-1144082), a University of Chicago Department of Chemistry Helen Sellei-Beretvas Fellowship, and an ARCS Scholar Award. We would like to thank Prof. Jesse Bloom for helpful discussion and insight.
Footnotes
A full description of materials used and experimental details are provided in the Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat Rev Genet. 2015;16:379–394. doi: 10.1038/nrg3927. [DOI] [PubMed] [Google Scholar]
- 3.Cirino PC, Mayer KM, Umeno D. Generating mutant libraries using error-prone PCR. Methods Mol Biol. 2003;231:3–9. doi: 10.1385/1-59259-395-X:3. [DOI] [PubMed] [Google Scholar]
- 4.Siloto RMP, Weselake RJ. Site saturation mutagenesis Methods and applications in protein engineering. Biocatal Agric Biotechnol. 2012;1:181–189. [Google Scholar]
- 5.Denard CA, Ren H, Zhao H. Improving and repurposing biocatalysts via directed evolution. Curr Opin Biotechnol. 2015;25:55–64. doi: 10.1016/j.cbpa.2014.12.036. [DOI] [PubMed] [Google Scholar]
- 6.Reetz MT, Carballeira JD. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc. 2007;2:891–903. doi: 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- 7.Reetz MT, Wang LW, Bocola M. Directed evolution of enantioselective enzymes: iterative cycles of CASTing for probing protein-sequence space. Angew Chem Int Ed. 2006;45:1236–1241. doi: 10.1002/anie.200502746. [DOI] [PubMed] [Google Scholar]
- 8.Lalonde J. Highly engineered biocatalysts for efficient small molecule pharmaceutical synthesis. Curr Opin Biotechnol. 2016;42:152–158. doi: 10.1016/j.copbio.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Midelfort KS, Kumar R, Han S, Karmilowicz MJ, McConnell K, Gehlhaar DK, Mistry A, Chang JS, Anderson M, Villalobos A, Minshull J, Govindarajan S, Wong JW. Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng Des Sel. 2012;26:25–33. doi: 10.1093/protein/gzs065. [DOI] [PubMed] [Google Scholar]
- 10.Govindarajan S, Mannervik B, Silverman JA, Wright K, Regitsky D, Hegazy U, Purcell TJ, Welch M, Minshull J, Gustafsson C. Mapping of Amino Acid Substitutions Conferring Herbicide Resistance in Wheat Glutathione Transferase. ACS Synth Biol. 2015;4:221–227. doi: 10.1021/sb500242x. [DOI] [PubMed] [Google Scholar]
- 11.Stemmer WP. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc Natl Acad Sci. 1994;91:10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman A, Tawfik DS. Incorporating Synthetic Oligonucleotides via Gene Reassembly (ISOR): a versatile tool for generating targeted libraries. Protein Eng Des Sel. 2007;20:219–226. doi: 10.1093/protein/gzm014. [DOI] [PubMed] [Google Scholar]
- 13.Firnberg E, Ostermeier M. PFunkel: Efficient, Expansive, User-Defined Mutagenesis. In: Jones DD, editor. PLoS ONE. Vol. 7. 2012. pp. e52031–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin P, Kang Z, Zhang J, Zhang L, Du G, Chen J. Combinatorial Evolution of Enzymes and Synthetic Pathways Using One-Step PCR. ACS Synth Biol. 2016;5:259–268. doi: 10.1021/acssynbio.5b00240. [DOI] [PubMed] [Google Scholar]
- 15.Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2007;2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 16.Lewis JC, Mantovani SM, Fu Y, Snow CD, Komor RS, Wong CH, Arnold FH. Combinatorial Alanine Substitution Enables Rapid Optimization of Cytochrome P450BM3 for Selective Hydroxylation of Large Substrates. ChemBioChem. 2010;11:2502–2505. doi: 10.1002/cbic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison KL, Weiss GA. Combinatorial alanine-scanning. Curr Opin Biotechnol. 2001;5:302–307. doi: 10.1016/s1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 18.Bloom JD. An experimentally determined evolutionary model dramatically improves phylogenetic fit. Mol Biol Evol. 2014;31:1956–1978. doi: 10.1093/molbev/msu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brøns-Poulsen J, Petersen NE, Hørder M, Kristiansen K. An improved PCR-based method for site directed mutagenesis using megaprimers. Mol Cell Probes. 1998;12:345–348. doi: 10.1006/mcpr.1998.0187. [DOI] [PubMed] [Google Scholar]
- 20.van Vugt-Lussenburg BMA, Stjernschantz E, Lastdrager J, Oostenbrink C, Vermeulen PE, Commandeur JNM. Identification of Critical Residues in Novel Drug Metabolizing Mutants of Cytochrome P450 BM3 Using Random Mutagenesis. J Med Chem. 2007;50:455–461. doi: 10.1021/jm0609061. [DOI] [PubMed] [Google Scholar]
- 21.Lewis JC, Bastian S, Bennett CS, Fu Y, Mitsuda Y, Chen MM, Greenberg WA, Wong CH, Arnold FH. Chemoenzymatic elaboration of monosaccharides using engineered cytochrome P450BM3 demethylases. Proc Natl Acad Sci. 2009;106:16550–16555. doi: 10.1073/pnas.0908954106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kille S, Zilly FE, Acevedo JP, Reetz MT. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat Chem. 2011;3:738–743. doi: 10.1038/nchem.1113. [DOI] [PubMed] [Google Scholar]
- 23.Agudo R, Roiban GD, Reetz MT. Achieving Regio- and Enantioselectivity of P450-Catalyzed Oxidative CH Activation of Small Functionalized Molecules by Structure-Guided Directed Evolution. ChemBioChem. 2012;13:1465–1473. doi: 10.1002/cbic.201200244. [DOI] [PubMed] [Google Scholar]
- 24.Bitto E, Huang Y, Bingman CA, Singh S, Thorson JS, Phillips GN., Jr The structure of flavin-dependent tryptophan 7-halogenase RebH. Proteins. 2007;70:289–293. doi: 10.1002/prot.21627. [DOI] [PubMed] [Google Scholar]
- 25.Haines DC, Tomchick DR, Machius M, Peterson JA. Pivotal Role of Water in the Mechanism of P450BM-3 †. Biochemistry. 2001;40:13456–13465. doi: 10.1021/bi011197q. [DOI] [PubMed] [Google Scholar]
- 26.Harris MN. Kinetic and Mechanistic Studies of Prolyl Oligopeptidase from the Hyperthermophile Pyrococcus furiosus. J Biol Chem. 2001;276:19310–19317. doi: 10.1074/jbc.M010489200. [DOI] [PubMed] [Google Scholar]
- 27.Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC. Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat Commun. 2015;6:7789. doi: 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis JC. Artificial Metalloenzymes and Metallopeptide Catalysts for Organic Synthesis. ACS Catal. 2013;3:2954–2975. [Google Scholar]
- 29.Cahn JKB, Baumschlager A, Brinkmann-Chen S, Arnold FH. Mutations in adenine-binding pockets enhance catalytic properties of NAD(P)H-dependent enzymes. Protein Eng Des Sel. 2015:gzv057–8. doi: 10.1093/protein/gzv057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osborne R, Mitchell V, HTWE KYN, Zhang X, Milczek EM, Moore JC. Novel 450-BM3 Variants with Improved Activity. US Patent Office; [Google Scholar]
- 31.Andorfer MC, Grob JE, Hajdin CE, Chael J, Siuti P, Lilly J, Tan KL, Lewis JC. Understanding Flavin-Dependent Halogenase Reactivity via Substrate Activity Profiling. doi: 10.1021/acscatal.6b02707. In Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Payne JT, Andorfer MC, Lewis JC. Regioselective Arene Halogenation using the FAD-Dependent Halogenase RebH. Angew Chem Int Ed. 2013;52:5271–5274. doi: 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.