

Abstract
Glycans play a key role as recognition elements in the communication of cells and other organisms. Thus, the analysis of carbohydrate–protein interactions has gained significant importance. In particular, nuclear magnetic resonance (NMR) techniques are considered powerful tools to detect relevant features in the interaction between sugars and their natural receptors. Here, we present the results obtained in the study on the molecular recognition of different mannose-containing glycans by Pisum sativum agglutinin. NMR experiments supported by Corcema-ST analysis, isothermal titration calorimetry (ITC) experiments, and molecular dynamics (MD) protocols have been successfully applied to unmask important binding features and especially to determine how a remote branching substituent significantly alters the binding mode of the sugar entity. These results highlight the key influence of common structural modifications in natural glycans on molecular recognition processes and underscore their importance for the development of biomedical applications.
Molecular recognition is at the heart of all processes in living beings. The understanding at the molecular level of how the interactions between biomolecules take place may provide new clues for the design and synthesis of novel entities able to modulate the corresponding events. In this context, glycoconjugates (glycoproteins, glycolipids, etc.) mediate a wide variety of actions critical for the development and function of a complex multicellular organism.1 The intrinsic variability of glycan structures enables sugars to encode specific information,2 which being recognized by natural receptors, can be translated into a specific biological process. As a consequence of this fine-tuned structure–function relationship, slight modifications on the structure of the glycan can influence their interaction with receptors and change the specific biological response.3
N-glycans are sophisticated oligosaccharides present in glycoproteins with a common core sugar sequence, Manα1–6(Manα1–3)Manβ1–4GlcNAcβ1–4GlcNAcβ1-Asn-X-Ser/Thr, to which additional monosaccharides are attached in various positions, giving rise to a wide variety of N-glycans.4 Frequently, these structural modifications, introduced by the action of different glycosidases and glycosyltransferases in the Golgi, have an impact on their binding features, thus triggering differentiating functions. In fact, nontemplate driven biosynthesis and a lack of glycosylation control mechanisms in the Golgi among other factors lead to a variety of different glycoforms for any given protein. Glycans can have an impact on circulatory half-life, protection against protease digestion, and sometimes protein function. However, detailed structural studies evaluating molecular recognition processes of these complex glycan entities are scarce and often limited to smaller fragments. Lectins are among the natural receptors that specifically recognize glycans. Beyond their biological roles as endogenous receptors, which are still unclear in many cases, plant and animal lectins are important tools for glycan detection and analysis, especially of N-glycans. Indeed, the technological improvements in different areas has resulted in the development of important lectin-based techniques (lectin blotting, lectin affinity chromatography ELLA, lectin arrays, 2D lectin-tandem electrophoresis), which have allowed a significant boost in the glycosciences field.5Pisum sativum agglutinin (PSA) is a plant lectin known to specifically bind to mannose (Man) and glucose (Glc) and that is especially used in different assays to detect α1,6-fucosylated N-glycans,6−8 for which it shows the highest affinity.9 α1,6-fucosylation is the main core modification of N-glycans in vertebrates and is increased in a number of cancers.10 The structural details for monosaccharide recognition (Man and Glc) by PSA have been elucidated by using X-ray diffraction.11,12 However, such detailed structural information is missing for larger N-glycans, including the key α1,6-fucosylated entities. In fact, the chemical nature of the glycosidic linkages and, especially the presence of 1–6 branches, endows a large flexibility to the molecule. This feature often precludes the crystallization of complex carbohydrates and/or impairs the detection of sufficient electron density for most of the glycan part in the X-ray diffraction analysis of large oligosaccharides. Moreover, the standard use of the corresponding fitting programs to deduce 3D sugar structures usually gives rise to incorrect geometries of the corresponding saccharide moieties.13 However, N-glycan processing including increased branching, the addition of core sugars, and branch “capping” often determines the function and recognition of glycoconjugates. These modifications not only determine the sugar content at the “peripheral” positions but also the overall shape of the glycan and become determining factors in the presentation of the glycans for their interaction with lectins. In our ongoing effort to analyze the impact of these structural modifications on N-glycan binding to natural receptors, we here report our molecular recognition study deciphering the binding features of PSA with several glycans depicted in Figure 1. Ligands 1 to 4, with gradually increasing structural complexity including branching, branch elongation, and α1,6-fucosylation provided a platform to study independently the effect of these structural features in binding to PSA.
Figure 1.

Sugars and glycans analyzed in this study.
Earlier X-ray diffraction data on the PSA/trimannoside complex (PDB code 1RIN)14 had shown only one Man residue to be located inside the lectin’s recognition site, with no electron density observed for the other two sugar units. We have therefore resorted to NMR methodologies since they are extremely useful to obtain a dynamic view of the binding event, which can complement X-ray diffraction data. NMR can also provide detailed structural information on the molecular recognition process in solution and help understand how specific modifications of the core of oligosaccharides might modulate the binding mode for the interaction with the receptors. These data, together with modeling protocols, have allowed us to correlate the increasing structural modifications with the differences observed in the binding modes and to explain the different levels of affinity. These findings show the existence of a particular binding mode for each glycan structure and highlight the importance of the overall structure of the oligosaccharide, including remote modifications, in the recognition by lectins.
Results and Discussion
The Binding Epitope for Small Oligosaccharides
Trimannoside 2 versus Mannoside 1
STD-NMR experiments15 were employed for studying the interaction between the oligosaccharides and pea lectin. STD experiments performed on a 1:30 molar ratio mixture of PSA and methyl α-d-mannopyranoside 1 (Figure 2) provided clear STD signals for all protons of 1, thus evidencing the existence of an interaction. The STD epitope mapping is in agreement with the carbohydrate recognition mode previously depicted from X-ray diffraction studies,12 in which H2 and H1 are more exposed to the solvent, while the rest of the protons are in tighter contact with the protein.
Figure 2.
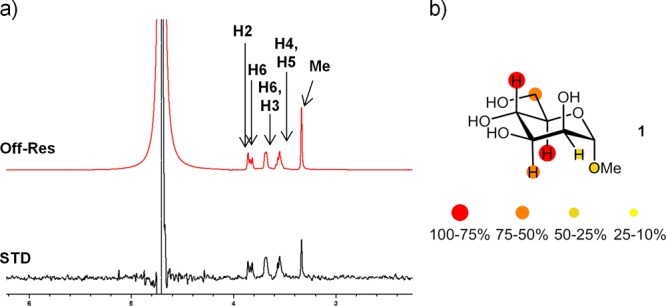
(a) 1H NMR reference spectrum (off-resonance frequency 100 ppm) and STD spectrum (on-resonance frequency 0.6 ppm) of a sample containing 0.9 mM of ligand 1 and 30 μM of PSA at 25 °C (600 MHz). (b) Relative STD-AF (amplification factor) for H2–H6 protons is indicated by color code.
We next investigated the binding with trimannose 2. This trisaccharide is a common structural motif of the core pentasaccharide of N-glycans, positioned in the center of the glycan structure. Although X-ray diffraction studies have been described for the complex between PSA and the methyl 3,6-di-O-(α-d-mannopyranosyl)-α-d-mannopyranose, only a single mannose residue could be resolved in the binding site.14 In our STD-NMR study, however, clear STD responses were observed for H1 and H2 protons of all three mannopyranose rings (Figure 3).
Figure 3.
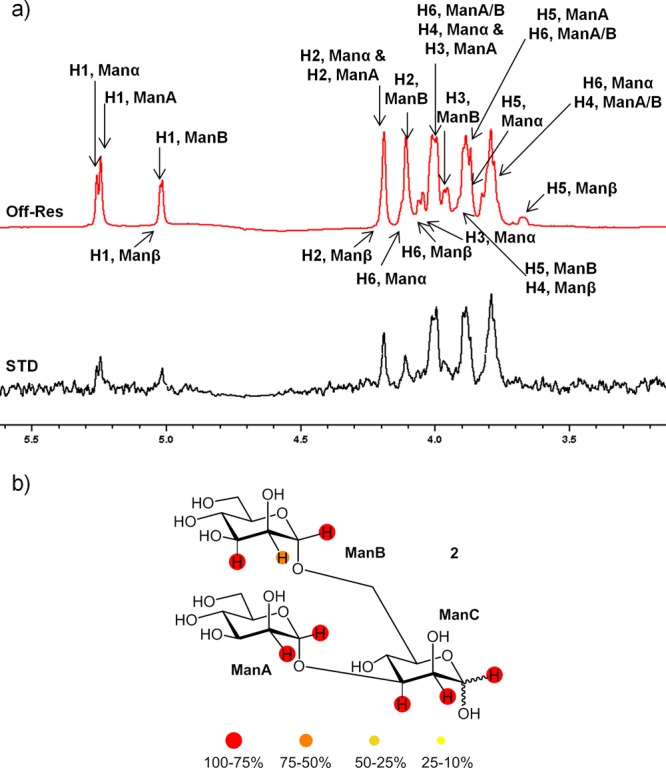
(a) 1H NMR reference spectrum (off-resonance frequency 100 ppm) and STD spectrum (on-resonance frequency 0.6 ppm) of a sample containing 1.2 mM of ligand 2 and 30 μM of PSA at 40 °C (600 MHz). (b) Relative STD-AF for nonoverlapped protons is indicated by color code.
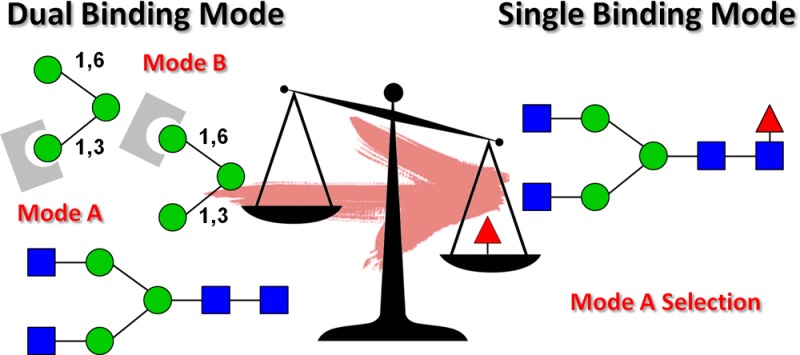
These nonoverlapping protons might be used as a fingerprint for detecting residues near the protein and indicate a similar proximity of ManA, ManB, and ManC residues to the pea lectin (Figure 3b). In order to rationalize these STD data, two binding poses were built using the crystal structure of PSA in complex with d-mannopyranose as a template. In pose A, the Manα-1,3 branch was docked onto the primary mannose binding site. In pose B, the alternative Manα-1,6 residue (Figure 4a) was docked. No steric clashes were found for any of the binding poses. Although binding mode A explained the STD responses observed for mannose A and mannose C residues, the observed STD signals for mannose B of the α-1,6 branch could not be accounted for. Conversely, the binding mode B places mannoses A and B near the protein, while mannose C is located away from the protein surface. A critical inspection of these structures clearly shows that a single binding mode is not enough to explain the STD results. The Corcema-ST16 analysis strongly suggested that only the simultaneous presence of two binding modes could account for the observed saturation profile in ligand 2 (Figure 4b). Strategies based on a combination of STD experiments and Corcema–ST calculations have been previously employed to demonstrate the dual binding character of different sugar-containing molecules.17
Figure 4.

(a) Binding pose A with mannopyranose A (1–3 linked) located in the mannose binding site (left) and binding pose B with mannopyranose B (1–6 linked) located in the mannose binding site (right). (b) Representation of the relative saturation of nonoverlapped protons of the ligand 2 for experimental value obtained after 2 s irradiation of PSA (▲, —, irradiation frequency 0.6 ppm); calculated values using Corcema-ST for binding mode geometry A (⧫, ---) and B (■, ···).
The stability of the two complexes was then analyzed by molecular dynamics (MD) simulations. Both complexes were conformationally stable along the MD simulations and preserved the intermolecular interactions reported for the PSA-D-mannopyranoside complex,9 where the key interactions take place almost exclusively with the mannopyranose ring placed at the primary binding site. Importantly, no further significant interactions for remaining residues in the ligand were found in any of the two complexes (see Supporting Information for additional details). In order to support this result, KD values for the interaction of PSA with both ligands were measured by isothermal titration calorimetry (ITC) experiments. Previous affinity studies18 had shown that the dissociation constant of methyl 3,6-di-O-(α-d-mannopyranosyl)-α-d-mannopyranoside from PSA is moderate, with a KD = 0.19 ± 0.07 mM, although three times stronger than for the α-methyl-mannopyranoside. As shown in Table 1, ligands 1 and 2 exhibited similar affinities, with nearly equal KD values in the micromolar range, suggesting that additional residues in ligand 2 do not play a key role in the binding event.14,19,20 Notably, Concanavalin A (ConA), a related mannose binding lectin, recognizes the trimannose 2 in a similar fashion as PSA, but it displays a unique binding mode with the α-1,6 branch in the primary binding site. In this case, additional contacts from the α-1,3 branch are present, which implies a 50-fold increase in the KD with respect to the monosaccharide.21 On the basis of the ITC results and MD findings, a comparable stability for the proposed binding modes A and B for PSA-trimannose 2 complex can be assumed. This fact also strongly suggests the presence of the dynamic equilibrium revealed by the STD-NMR experiments for the complex between PSA and ligand 2.
Table 1. Dissociation (KD) Constants Determined by ITC at 300.7K.
| ligand | KD (μM) |
|---|---|
| mannoside 1 | 806 ± 32 |
| trimannose 2 | 621 ± 11 |
The Binding Epitope for Larger Oligosaccharides
The binding features of monosaccharide 1 and trisaccharide 2 to PSA suggest that only the external Man residues strongly contribute to the binding event, although the central βMan residue also contributes marginally to the affinity. However, for entire N-glycans, the global structure of the glycan might be more relevant for their recognition by lectins.22 The initial results encouraged us to carry on the study with larger oligosaccharides. Therefore, we analyzed the binding of the heptasaccharide 3 and octasaccharide 4 to PSA. Glycan 3 could be considered as the simplest complex-type biantennary N-glycan with N-acetylglucosamine units attached to the N-glycan core pentasaccharide, whereas glycan 4, with an α-1,6 fucose residue, includes one of the most prominent core modifications in vertebrates, with key implications in diverse phenomena like prostate and liver cancer, chronic liver diseases, or immune response.23−25 STD-NMR experiments were then performed under similar conditions for both ligands in the presence of PSA (Figure 5).
Figure 5.

(a) STD results for heptasaccharide 3 and (b) octasaccharide 4 for experiments recorded at 25 and 40 °C, respectively. STD-AF for nonoverlapped protons was indicated. Samples contained 1.2 mM of ligand 3 and 4 and 30 μM of PSA (800 MHz).
The STD spectrum obtained for heptasaccharide 3 indicated STD response for H1 and H2 protons of Man residues A and B (3- and 6-arms, respectively). Moreover, although H1 of ManC could not be observed (below the HDO signal), STD for H2 of ManC was also evident. These results resembled those obtained for trimannose 2, with comparable STD responses for H1 and H2 protons of ManA, ManB, and ManC. Thus, analogous A and B binding poses to those deduced for trimannose 2 were built by docking/minization strategies for the PSA-3 complex (Figure 6). Low energy conformations were used for the heptasaccharide 3, previously built using standard molecular mechanics protocols.26
Figure 6.
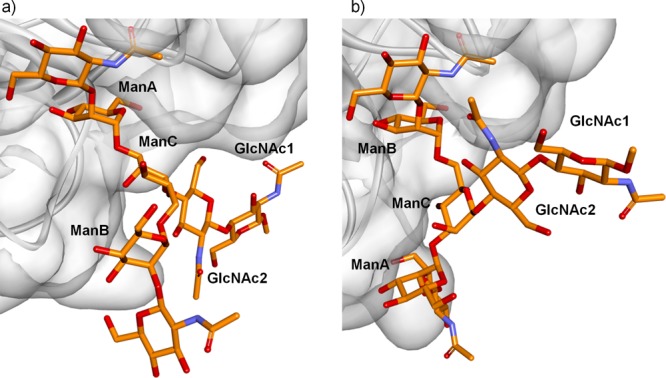
Binding poses proposed for the interaction between PSA and ligand 3. (a) A-type (3-arm) binding pose. (b) B-type (6-arm) binding pose.
Binding pose A (Figure 6a) locates ManA at the primary binding site, placing GlcNAc1 and GlcNAc2 residues closer to the protein, whereas the 6-arm is oriented toward the solvent. This binding mode explains the STD signals observed for ManA and ManC, but it is inconsistent with the STD response observed for ManB. In contrast, binding pose B (Figure 6b) locates ManB at the primary binding site, with the GlcNAc1 and GlcNAc2 residues exposed to the solvent. Herein, the 3-arm is positioned near the C loop of the protein. In this binding mode, ManC is located far away from the protein surface and would not show the observed STD response. Thus, the combination of the A- and B-type binding modes is again needed in order to explain the STD profile obtained for ligand 3, as depicted in Figure 6. Therefore, the dynamic equilibrium described for trimannose 2 seems also to occur for the biantennary glycan 3.27,28 The elongation of the core trimannoside at both arms with N-acetylglucosamine as well as the introduction of a chitobiose moiety at the reducing end provide new interactions with the protein. However, these additional interactions are present in both binding modes, and they do not significantly affect the dual binding character observed with PSA.
This particular behavior was not found for the fucosylated octasaccharide 4. Previous affinity chromatography studies had reported an increase in affinity to PSA for fucosylated mono- and biantennary N-glycans.7,29 Indeed, an interaction was measurable by STD-NMR experiments (Figure 5b). Although both terminal GlcNAc residues showed overlapping signals and were indistinguishable, we used the isolated protons of the ManA and -B as a fingerprint to differentiate between both arms. Fittingly, the STD signals for H1 and H2 protons of ManB, at the 6-arm, were significantly weaker than the STD signals for the same protons of ManA, at the 3-arm. Moreover, a relevant STD response was observed for the methyl protons of the fucose moiety. These results deviate from the data obtained for heptasaccharide 3 and strongly suggest the existence of a different binding mode. In contrast to ligand 3, the much weaker STD signals for ManB with respect to ManA in the STD spectrum of octasaccharide 4 points out the existence of a unique binding mode, where the 6-arm is exposed to the solvent. In this case, the observation of STD signals for residues located at opposite ends of the glycan (terminal GlcNAc and Fuc) suggests an extended binding mode with different residues playing a key role in the interaction. To shed light into these experimental findings, a docking/minimization-based approach for the complex between PSA and the fucosylated octasaccharide 4 was performed. The structure of the N-glycan was superimposed in the binding site using the more populated conformations found for the free state (according to the observed NOEs, as described in the experimental section). Among the different possibilities explored, binding pose A, locating ManA at the primary binding site, resulted to be the best one to match the STD results and did not show any steric clashes with the protein (Figure 7).
Figure 7.
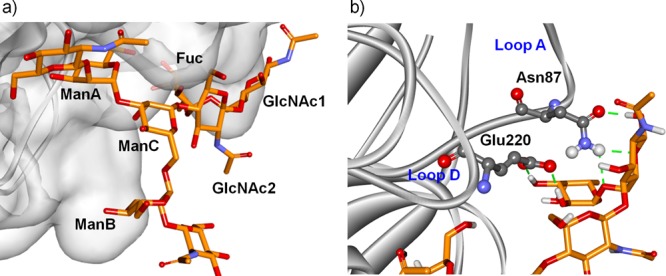
(a) Docking/minimization pose for type-A binding mode of the octasaccharide 4 in complex with PSA. (b) The expansion shows the close proximity of the fucose residue to the pea lectin surface. Protein neighboring residues involved in hydrogen bonds are highlighted.
This binding mode locates the 3-arm and ManC, as well as the fucose moiety, closer to the protein, whereas the 6-arm is exposed to the solvent. In fact, the corresponding 1,6-linked ManB displays much lower STD intensities. Type-B binding mode thus was excluded since it would locate the 6-arm near the protein and the fucose residue far away from the protein surface, which would be in contrast to the NMR observations. Thus, the seemingly remote α1,6-fucosylation drastically modifies the multiple dynamic binding modes observed for ligands 2 and 3 and results in only one productive binding mode for the fucosylated octasaccharide 4. Herein, only the A-type binding mode is selected.
Core fucosylation modulates the conformational space accessible to N-glycans,30 and as a consequence changes in glycan-receptor interactions might be induced. Thus, an in depth analysis of the conformational features of the octasaccharide 4 was undertaken. High-resolution NMR methods together with MD calculations revealed that the core fucose did not affect the dynamic behavior expected for octasaccharide 4, especially for the 6-arm. Indeed, biantennary N-glycans are known to exist as an ensemble of conformers, where flexibility exists around the Ψ and ω torsions of the α-1,6-linkage.31 Analysis of the 1H multiplicity, in the 1H–13C HSQC spectrum, of the signals for H6 and H6′ of residues ManC and GlcNAc1 was performed (Figure 8a). While only gg conformers seem to be present for the Fucα1 → 6GlcNAcβ linkage (two small 3JH,H), a medium value for the 3JH5H6′ coupling constant of residue ManC (ca. 6 Hz) indicates the presence of a conformational equilibrium between gg and gt conformations for the Manα1 → 6Manβ linkage.32 The dynamic behavior of the octasaccharide 4 was further analyzed over 100 ns MD-tar simulations.33 The simulation predicted the coexistence of gg and gt conformations for the Manα1 → 6Manβ linkage, while the contribution of other conformations was minor (Figure 8b,c).
Figure 8.

Conformational study of the octasaccharide 4. (a) J(H,H) coupling constant determination for the Fucα1 → 6GlcNAcβ and Manα1 → 6Manβ linkages. (b) Superimposition of 5-ns-averaged frames obtained from MD calculations. The minimum-energy conformations for the α-1,6 arm of the disaccharide fragment of 4 are depicted. (c) MD-tar results, indicating the relative population of the different conformers.
Thus, the NMR and MD results excluded the participation of conformational restrictions in glycan 4 compared to 3 as a consequence of the presence of core fucose, with both gg and gt conformers in equilibrium. Hence, the existence of a direct relationship between specific conformational features of ligand 4 and the changes observed in the interaction with PSA was ruled out.
In fact, the remote fucose residue plays a key role in the selection of the binding mode A, as deduced from the analysis of the PSA–ligand 4 complex. This binding mode places the fucose near the loops A and D, where several polar residues in the protein can engage in hydrogen bonds with the fucose residue (Figure 7b). In fact, a 20 ns MD simulation for the PSA–4 complex supported the participation of intermolecular hydrogen bonds between amino acids E220 and N87 of the lectin and Fuc and GlcNAc1 residues of ligand 4 (see Supporting Information). In particular, the hydrogen bond pattern involving the donor–acceptor pairs H3O(Fuc)–OE1(Glu220) and H2O(Fuc)–OE2(Glu220) remained stable during the entire simulation with occupancies of 99.6% and 82.9% and average bond distances of 2.62 and 2.71 Å, respectively. Additional contact is provided by the GlcNAc1 residue (O5/O6) and the amino group of Asn87. These interactions would account for the enhanced stability of the complex in the proposed A-type binding mode, as earlier proposed for other legume lectins.34,35 Accordingly, STD-NMR competition experiments unequivocally demonstrated the higher affinity of octasaccharide 4 to bind PSA. The STD intensity variations of the fucose methyl protons of octasaccharide 4 were monitored as a function of increased amounts of ligand 1 (Figure S1). Progressive reduction of STD intensity for the signals of 4 after the addition of ligand 1 strongly evidenced the competition between both compounds for the same binding site and allowed us to deduce that ligand 4 binds with much higher affinity than compound 1. In fact, a 1/4 ratio >5:1 was needed to produce a 50% reduction in the STD signal intensities of 4. Indeed, the analysis of the STD titration data indicated that ligand 4 is a strong binder with 6-fold higher affinity (KD = 140 μM) than ligand 1. These results are in complete accordance with the affinity chromatography data published for the interaction of PSA with fucosylated and nonfucosylated sugars6,7 and pointed to the existence of enlarged interactions between fucosylated ligand 4 and PSA. Thus, the presence of the remote fucose moiety in ligand 4 is responsible for the selection of one unique binding mode, with additional favorable interactions between the protein and the fucosylated ligand.
Conclusions
The large structural diversity of the glycome makes the detailed analysis of carbohydrate–lectin interactions a challenging task. Moreover, subtle differences in glycan structure may imply significant changes for binding events, demanding a more rigorous analysis of the process. STD-NMR spectroscopy supported by modeling protocols has shown the strength of NMR techniques to characterize epitope selection, as well as identify the relevant features in the interaction between sugars and their natural receptors. The combination of these techniques has been successfully applied for studying the molecular recognition of different mannose-containing structures by Pisum sativum agglutinin. A dual binding mode was found for 3,6-di-O-(α-d-mannopyranosyl)-α-d-mannopyranose that was also displayed by a complex biantennary N-glycan. These results go far beyond the previous X-ray crystallographic studies and provide a dynamic view of the binding event. Moreover, core fucosylation dramatically changed the binding mode. In this case, a single binding mode is selected, where additional interactions between the fucose residue and the protein exist. These results highlight the key influence of common structural modifications of glycans on molecular recognition processes and underscore their importance for the development of biomedical applications involving glycan-lectin recognition processes.
Methods
Lectins
PSA was purchased from Sigma–Aldrich and was used after two runs of dialysis in phosphate-buffered saline (pH = 7.2) solution.
Ligands
Compounds 1 and 2 were purchased from Sigma–Aldrich and Carbosynth, respectively. Compound 3 was synthesized as described previously.36 Synthesis of compound 4 based on a selective debenzylation of a protected precursor was described previously.29
NMR
The samples for saturation-transfer difference (STD) experiments were prepared in phosphate-buffered saline (20 mM PBS, NaCl 150 mM, pH = 7.2) using ligand/lectin ratios varying from 1:30 to 1:50 with a lectin concentration of 30 μM. The applied temperatures varied between 298 and 313 K. Molar ratio and temperature were optimized in each case. Representative experiments with significant STD responses are presented in the figures. In all cases, the on-resonance frequency was set at aliphatic regions (0.45–0.6 ppm) and the off-resonance frequency at 100 ppm. Protein saturation was achieved by using a series of 30 ms PC9 pulses with a total saturation time of the protein of 2 s with or without water suppression in a 600 or 800 MHz (cryo) spectrometer. A spin-lock filter (50 ms) was used to remove the NMR signals of the macromolecule.
Competition experiments were performed recording sequential STD experiments at a distinct concentration of the competitor (ligand 1) at 318 K with a constant protein concentration of 30 μM and constant octasaccharide 4 concentration of 1.2 mM. The interference caused by the competitor (ligand 1, Kd = 806 μM) was quantified by assessing the decrease of the STD intensity of the methyl protons of the fucose unit of octasaccharide 4. Thus, the KD value of octasaccharide 4 (KD = 140 μM) was determined from the IC50 value of the competitor by using the equation of Yung-Chi and Prusoff.37
Isothermal Titration Calorimetry
PSA was dissolved in buffer (20 mM PBS, NaCl 150 mM, pH = 7.2). The lectin was dialyzed overnight and centrifuged. Concentration was determined with an ND-1000 spectrophotometer (Nanodrop Technology) at 280 nm using an extinction coefficient of 40 910 as estimated by the ExPASy ProtParam tool. Pre-equilibrated solutions of 200 μM protein and 10 mM ligand were used for each assay. Titration was performed with a VP-ITC microcalorimeter at 27.7 °C with PSA in the cell and 6 μL injections of the carbohydrate ligand in the same buffer. Data were analyzed and fitted using MicroCal Origin 7 software.
Molecular Modeling (Ligands)
Initial geometries of ligands 2 and 3 were built using the carbohydrate builder module available in the GLYCAM web portal (Glycam Biomolecule Builder), www.glycam.org. The ligand structure was submitted to an energy minimization with a low gradient convergence threshold (0.05) in 1000 steps. The MM3 force field was employed, as integrated in the MAESTRO suite of programs. Distance (derived from NOESY spectra using the isolated spin-pair approximation) and J(H,H) coupling constant data were used to check the goodness of the structures modeled.
Time-Averaged Restrained (tar-MD) Simulations with Distance-Based Restraints
Tar-MD simulations were performed for ligand 4 using the AMBER12 package with GLYCAM_06h parameters. The tar-MD used a 12 Å octahedral box of explicit TIP3P waters. The starting geometries were generated from the carbohydrate builder module in the glycam webpage. Distance restraints derived from NOE (using the isolated spin-pair approximation) and J(H,H) coupling constant data were included as tar restraints. Two initial consecutive minimizations were performed involving (1) only the water molecules and (2) the whole system with a higher number of cycles, using the steepest descent algorithm. Then, the system was heated and equilibrated in two steps: (1) 20 ps of MD heating the whole system from 0 to 300 K (NVT ensemble, cutoff 10 Å), followed by (2) equilibration of the entire system over 100 ps at 300 K (NPT ensemble, cutoff 10 Å). The equilibrated structure was the starting point for the tar-MD simulation (100 ns) at constant temperature (300 K) and pressure (1 atm). Molecular dynamics simulations without constraints were recorded, using a NPT ensemble with periodic boundary conditions, a cutoff of 10 Å, and the particle mesh Ewald method. A total of 100 000 000 molecular dynamics steps were run with a time step of 1 fs per step. Coordinates and energy values were recorded every 1000 steps (2 ps) for a total of 100 000 MD models. A detailed analysis of the MD trajectory (for example, bond angle evaluation) was accomplished using the cpptraj module included in Amber-Tools 12 package. The ensemble of structures obtained from tar-MD simulation was in agreement with the experimental data (see Supporting Information for additional details).
Molecular Docking for PSA–Ligand Complexes
The initial binding poses for PSA–ligand complexes were built using the X-ray structures of PSA complexed with small mannose oligosaccharides (PDB codes: 1BQP and 1RIN). Manual docking of the structure of the N-glycan was performed by superimposition of the Man residue in the binding site of mannoside. The most populated conformations found for the N-glycan in the free state (according to a standard NOE/molecular modeling approach) were used.
Minimization of PSA–Ligand Complexes
The docked complex structure was submitted to a short molecular dynamics (MD) run, followed by energy minimization with a low gradient convergence threshold (0.05) in 1000–5000 steps. In all cases, the OPLS2005 force field38 was employed, as integrated in the MAESTRO (Schroedinger) suite of programs.39
Molecular Dynamics Simulations
Manually docked structures of complexes between PSA and mannotriose 2 in both binding modes were used as starting points for molecular dynamics (MD) simulations. The MD simulations were performed using the Amber12 program with the ff99SB force field parameters for protein and GLYCAM06h for the saccharides. Thereafter, the starting 3D geometries were placed into a 12 Å octaedral box of explicit TIP3P waters, and counterions were added to maintain electroneutrality. Two consecutive minimizations were performed involving (1) only the water molecules and ions and (2) the whole system with a higher number of cycles, using the steepest descent algorithm. The system was subjected to two rapid molecular dynamic simulations (heating and equilibration) before starting the real dynamic simulation: (1) 20 ps of MD heating the whole system from 0 to 300 K, using NVT ensemble and a cutoff of 10 Å, followed by (2) equilibration of the entire system over 100 ps at 300 K using NPT ensemble and a cutoff of 10 Å. A relaxation time of 2 ps was used in order to equilibrate the entire system in each step. The equilibrated structures were the starting points for the final MD simulations at constant temperature (300 K) and pressure (1 atm). Molecular dynamics simulations without constraints were recorded, using an NPT ensemble with periodic boundary conditions, a cutoff of 10 Å, and the particle mesh Ewald method. A total of 20 000 000 molecular dynamics steps were run with a time step of 1 fs per step. Coordinates and energy values were recorded every 1000 steps (2 ps) for a total of 20 000 MD models. A detailed analysis of each MD trajectory (for example, RMSD evaluation, hydrogen-bond) was accomplished using the cpptraj module included in Amber-Tools 12 package, and it is gathered in the Supporting Information.
Corcema-ST Calculation
Corcema-ST matlab scripts were applied to the modeled structures of the complexes, obtained after molecular dynamics calculations, between 1,3-α-1,6-α-d-mannotriose 2 with PSA. Average structures from MD simulations for both binding modes were selected and were analyzed by Corcema-ST. The input parameters used in the calculations were 2 s saturation time; amino acid in a radius of 10 Å around the ligand; direct irradiation on methyl groups of Ile, Leu, and Val (as an approximation to 0.6 ppm experimental irradiation frequency); experimental KD dissociation constants, 0.6 mM, experimental concentration conditions, 1.2 mM in the case of 2 and 0.03 mM in the case of PSA. A kon of 108 L mol–1 s–1 was used assuming a diffusion controlled kinetic model; correlation times of 0.5 and 30 ns for the ligand in the free and bound form, respectively, were estimated following an empirical approximation and considering a 55 kDa dimer form for the PSA.
Acknowledgments
We acknowledge funding by the Spanish Ministry of Economy and Competiveness, MINECO (CTQ2014-58779-R, CTQ2015-64597-C2-1P and -2P). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreements No. 642870 (ETN-Immunoshape), No. 642157 (ETN-Tollerant), and No. 675671 (ETN-GlycoVAX). A.A. and A.G. also thank MINECO for Ramón y Cajal and Juan de la Cierva contracts, respectively.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.6b01116.
Supporting figures of the analysis of the MD simulations, CORCEMA-ST calculations, STD spectra and corresponding quantifications tables, and STD competition experiments (PDF)
The authors declare no competing financial interest.
Dedication
Dedicated to Prof. M. Martín-Lomas on the occasion of his 75th anniversary
Supplementary Material
References
- Gabius H.-J. (2009) The Sugar Code: Fundamentals of Glycosciences, Wiley-VCH, Weinheim, Germany. [Google Scholar]
- Gabius H. J.; André S.; Jiménez-Barbero J.; Romero A.; Solís D. (2011) From lectin structure to functional glycomics: principles of the sugar code. Trends Biochem. Sci. 36, 298–313. 10.1016/j.tibs.2011.01.005. [DOI] [PubMed] [Google Scholar]
- André S.; Kozar T.; Schuberth R.; Unverzagt C.; Kojima S.; Gabius H.-J. (2007) Substitutions in the N-Glycan Core as Regulators of Biorecognition: The Case of Core-Fucose and Bisecting GlcNAc Moieties. Biochemistry 46, 6984–6995. 10.1021/bi7000467. [DOI] [PubMed] [Google Scholar]
- Lannoo N.; Van Damme E. J. M. (2015) Review/N-glycans: The making of a varied toolbox. Plant Sci. 239, 67–83. 10.1016/j.plantsci.2015.06.023. [DOI] [PubMed] [Google Scholar]
- Cummings R. D., and Etzler M. E. (2009) Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E., Eds.), 2nd ed., Chapter 45, Cold Spring Harbor Laboratory Press, New York. [PubMed] [Google Scholar]
- Yoshitake H.; Hashii N.; Kawasaki N.; Endo S.; Takamori K.; Hasegawa A.; Fujiwara H.; Araki A. (2015) Chemical Characterization of N-Linked Oligosaccharide As the Antigen Epitope Recognized by an Anti-Sperm Auto-Monoclonal Antibody, Ts4. PLoS One 10, e0133784. 10.1371/journal.pone.0133784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno H.; Nakamura-Tsuruta S.; Hirabayashi J. (2009) Comparative analysis of core-fucose-binding lectins from Lens culinaris and Pisum sativum using frontal affinity chromatography. Glycobiology 19, 527–536. 10.1093/glycob/cwp016. [DOI] [PubMed] [Google Scholar]
- Bierhuizen M. A.; Hansson M.; Odin P.; Debray H.; Öbrink B.; Van Dijk W. (1989) Structural assessment of the N-linked oligosaccharides of cell-CAM 105 by lectin-agarose affinity chromatography. Glycoconjugate J. 6, 195–208. 10.1007/BF01050648. [DOI] [PubMed] [Google Scholar]
- Kornfeld K.; Reitman M. L.; Kornfeld R. (1981) The carbohydrate-binding specificity of pea and lentil lectins. Fucose is an important determinant. J. Biol. Chem. 256, 6633–6640. [PubMed] [Google Scholar]
- Miyoshi E.; Moriwaki K.; Nakagawa T. (2008) Biological function of fucosylation in cancer biology. J. Biochem. 143, 725–729. 10.1093/jb/mvn011. [DOI] [PubMed] [Google Scholar]
- Pletnev V. Z.; Ruzheinikov S. N.; Tsygannik I. N.; Mikhailova Y. I.; Duax W.; Ghosh D.; Pangborn W. (1997) The structure of pea lectin-D-glucopyranose complex at a 1.9 A resolution. Russ. J. Bioorganic Chem. 23, 436–445. [Google Scholar]
- Ruzheinikov S. N.; Mikhailova Y. I.; Tsygannik I. N.; Pangborn W.; Duax W.; Pletnev V. Z. (1998) The structure of the pea lectin-D-mannopyranose complex at a 2.1 A resolution. Russ. J. Bioorganic Chem. 24, 277–279. [Google Scholar]
- Agirre J.; Iglesias-Fernández J.; Rovira C.; Davies G. J.; Wilson K. S.; Cowtan K. D. (2015) Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834. 10.1038/nsmb.3115. [DOI] [PubMed] [Google Scholar]
- Rini J. M.; Hardman K. D.; Einspahr H.; Suddath F. L.; Carver J. P. (1993) X-ray crystal structure of a pea lectin-trimannoside complex at 2.6 A resolution. J. Biol. Chem. 268, 10126–10132. [DOI] [PubMed] [Google Scholar]
- Meyer B.; Peters T. (2003) NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem., Int. Ed. 42, 864–890. 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- Krishna N. R.; Jayalakshmi V. (2007) Quantitative Analysis of STD-NMR Spectra of Reversibly Forming Ligand-Receptor Complexes. Top. Curr. Chem. 273, 15–54. 10.1007/128_2007_144. [DOI] [PubMed] [Google Scholar]
- Marcelo F.; Garcia-Martín F.; Matsushita T.; Sardinha J.; Coelho H.; Oude-Vrielink A.; Koller C.; André S.; Cabrita E. J.; Gabius H.-J.; Nishimura S.-I.; Jiménez-Barbero J.; Cañada F. J. (2014) Delineating binding modes of Gal/GalNAc and structural elements of the molecular recognition of tumor-associated mucin glycopeptides by the human macrophage galactose-type lectin. Chem. - Eur. J. 20, 16147–16155. 10.1002/chem.201404566. [DOI] [PubMed] [Google Scholar]
- Stubbs M. E.; Carver J. P.; Dunn R. J. (1986) Production of pea lectin in Escherichia coli. J. Biol. Chem. 261, 6141–6144. [PubMed] [Google Scholar]
- Loris R.; Van Walle I.; De Greve H.; Beeckmans S.; Deboeck F.; Wyns L.; Bouckaert J. (2004) Structural basis of oligomannose recognition by the Pterocarpus angolensis seed lectin. J. Mol. Biol. 335, 1227–1240. 10.1016/j.jmb.2003.11.043. [DOI] [PubMed] [Google Scholar]
- Naismith J. H.; Field R. A. (1996) Structural basis of trimannoside recognition by Concanavalin A. J. Biol. Chem. 271, 972–976. 10.1074/jbc.271.2.972. [DOI] [PubMed] [Google Scholar]
- Dam T. K.; Roy R.; Pagé D.; Brewer C. F. (2002) Thermodynamic binding parameters of individual epitopes of multivalent carbohydrates to Concanavalin A as determined by “reverse” isothermal titration microcalorimetry. Biochemistry 41, 1359–1363. 10.1021/bi015829k. [DOI] [PubMed] [Google Scholar]
- Ardá A.; Blasco P.; Varon Silva D.; Schubert V.; André S.; Bruix M.; Cañada F. J.; Gabius H.-J.; Unverzagt C.; Jiménez-Barbero J. (2013) Molecular recognition of complex-type biantennary N-glycans by protein receptors: a three-dimensional view on epitope selection by NMR. J. Am. Chem. Soc. 135, 2667–2675. 10.1021/ja3104928. [DOI] [PubMed] [Google Scholar]
- Saldova R.; Fan Y.; Fitzpatrick J. M.; Watson R. W. G.; Rudd P. M. (2011) Core fucosylation and alpha2–3 sialylation in serum N-glycome is significantly increased in prostate cancer comparing to benign prostate hyperplasia. Glycobiology 21, 195–205. 10.1093/glycob/cwq147. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Fukuda T.; Isaji T.; Lu J.; Im S.; Hang Q.; Gu W.; Hou S.; Ohtsubo K.; Gu J. (2015) Loss of α1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 29, 3217–3227. 10.1096/fj.15-270710. [DOI] [PubMed] [Google Scholar]
- Li W.; Yu R.; Ma B.; Yang Y.; Jiao X.; Liu Y.; Cao H.; Dong W.; Liu L.; Ma K.; Fukuda T.; Liu Q.; Ma T.; Wang Z.; Gu J.; Zhang J.; Taniguchi N. (2015) Core fucosylation of IgG B cell receptor is required for antigen recognition and antibody production. J. Immunol. 194, 2596–2606. 10.4049/jimmunol.1402678. [DOI] [PubMed] [Google Scholar]
- Xiong X.; Chen Z.; Cossins B. P.; Xu Z.; Shao Q.; Ding K.; Zhu W.; Shi J. (2015) Force fields and scoring functions for carbohydrate simulation. Carbohydr. Res. 401, 73–81. 10.1016/j.carres.2014.10.028. [DOI] [PubMed] [Google Scholar]
- Bourne Y.; Rouge P.; Cambillau C. (1992) X-ray structure of a biantennary octasaccharide-lectin complex refined at 2.3 Å resolution. J. Biol. Chem. 267, 197–203. [PubMed] [Google Scholar]
- Buts L.; Garcia-Pino A.; Imberty A.; Amiot N.; Boons G.-J.; Beeckmans S.; Versées W.; Wyns L.; Loris R. (2006) Structural basis for the recognition of complex-type biantennary oligosaccharides by Pterocarpus angolensis lectin. FEBS J. 273, 2407–2420. 10.1111/j.1742-4658.2006.05248.x. [DOI] [PubMed] [Google Scholar]
- Recently, binding between octasaccharide 4 and an algal lectin has been investigated, and a key role of the α1,6-fucose residue in the binding was elucidated. See:Do Nascimento A. S. F.; Serna S.; Beloqui A.; Arda A.; Sampaio A. H.; Walcher J.; Ott D.; Unverzagt C.; Reichardt N.-C.; Jimenez-Barbero J.; Nascimento K. S.; Imberty A.; Cavada B. S.; Varrot A. (2015) Algal lectin binding to core (α1–6) fucosylated N-glycans: Structural basis for specificity and production of recombinant protein. Glycobiology 25, 607–616. 10.1093/glycob/cwv002. [DOI] [PubMed] [Google Scholar]
- Nishima W.; Miyashita N.; Yamaguchi Y.; Sugita Y.; Re S. (2012) Effect of bisecting GlcNAc and core fucosylation on conformational properties of biantennary complex-type N-glycans in solution. J. Phys. Chem. B 116, 8504–8512. 10.1021/jp212550z. [DOI] [PubMed] [Google Scholar]
- Canales A.; Mallagaray A.; Pérez-Castells J.; Boos I.; Unverzagt C.; André S.; Gabius H.-J.; Cañada F. J.; Jiménez-Barbero J. (2013) Breaking pseudo-symmetry in multiantennary complex N-glycans using lanthanide-binding tags and NMR pseudo-contact shifts. Angew. Chem., Int. Ed. 52, 13789–13793. 10.1002/anie.201307845. [DOI] [PubMed] [Google Scholar]
- Patel D. S.; Pendrill R.; Mallajosyula S. S.; Widmalm G.; MacKerell A. D. (2014) Conformational properties of α- or β-(1→6)-linked oligosaccharides: hamiltonian replica exchange MD Simulations and NMR experiments. J. Phys. Chem. B 118, 2851–2871. 10.1021/jp412051v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; McCallum S. A.; Xie J.; Nieto L.; Corzana F.; Jiménez-Barbero J.; Chen M.; Liu J.; Linhardt R. J. (2008) Solution structures of chemoenzymatically synthesized heparin and its precursors. J. Am. Chem. Soc. 130, 12998–13007. 10.1021/ja8026345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y.; Mazurier J.; Legrand D.; Rouge P.; Montreuil J.; Spik G.; Cambillau C. (1994) Structures of a legume lectin complexed with the human lactotransferrin N2 fragment, and with an isolated biantennary glycopeptide: role of the fucose moiety. Structure 2, 209–219. 10.1016/S0969-2126(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Sokolowski T.; Peters T.; Pérez S.; Imberty A. (1997) Conformational analysis of biantennary glycans and molecular modeling of their complexes with lentil lectin. J. Mol. Graphics Modell. 15, 37–42. 10.1016/S1093-3263(97)00011-9. [DOI] [PubMed] [Google Scholar]
- Serna S.; Etxebarria J.; Ruiz N.; Martin-Lomas M.; Reichardt N.-C. (2010) Construction of N-glycan microarrays by using modular synthesis and on-chip nanoscale enzymatic glycosylation. Chem. - Eur. J. 16, 13163–1317. 10.1002/chem.201001295. [DOI] [PubMed] [Google Scholar]
- Yung-Chi C.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. J. J. (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105, 6474–6487. 10.1021/jp003919d. [DOI] [Google Scholar]
- Maestro, A Powerful, All-Purpose Molecular Modeling Environment, version 10.2, Schroedinger, LLC, New York, 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.