

Abstract
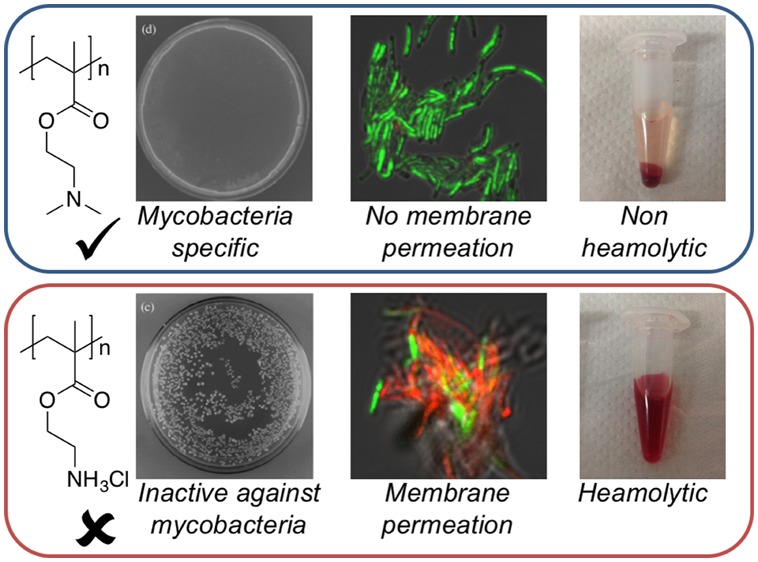
Antimicrobial resistance is a global healthcare problem with a dwindling arsenal of usable drugs. Tuberculosis, caused by Mycobacterium tuberculosis, requires long-term combination therapy and multi- and totally drug resistant strains have emerged. This study reports the antibacterial activity of cationic polymers against mycobacteria, which are distinguished from other Gram-positive bacteria by their unique cell wall comprising a covalently linked mycolic acid–arabinogalactan–peptidoglycan complex (mAGP), interspersed with additional complex lipids which helps them persist in their host. The present study finds that poly(dimethylaminoethyl methacrylate) has particularly potent antimycobacterial activity and high selectivity over two Gram-negative strains. Removal of the backbone methyl group (poly(dimethylaminoethyl acrylate)) decreased antimycobacterial activity, and poly(aminoethyl methacrylate) also had no activity against mycobacteria. Hemolysis assays revealed poly(dimethylaminoethyl methacrylate) did not disrupt red blood cell membranes. Interestingly, poly(dimethylaminoethyl methacrylate) was not found to permeabilize mycobacterial membranes, as judged by dye exclusion assays, suggesting the mode of action is not simple membrane disruption, supported by electron microscopy analysis. These results demonstrate that synthetic polycations, with the correctly tuned structure are useful tools against mycobacterial infections, for which new drugs are urgently required.
Introduction
The emergence and spread of antimicrobial resistance (AMR) is a rapidly growing, global, healthcare threat. There have been no new classes of antibiotics since 1987 with the pipeline of new antibiotics being scarce.1 Each year in the United States, at least 2 million people become infected with bacteria that are resistant to antibiotics and at least 23 000 people die each year as a direct result of these infections.2 Estimates of the cost of antibiotic-resistant infections in the United States alone are US$ 21 billion to US$ 34 billion3 needing a rethink of the approach to tackle this threat.4 New and innovative treatments and diagnostics are urgently required.5−9A re-emerging threat is multi- and totally drug resistant Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB). It is estimated that there are up to 2 billion latent infections globally (with London known as the TB capital of Europe10), and it is now found in wealthy countries where it had previously been eradicated. In 2014 alone, there were about 480 000 new cases of multidrug-resistant Mtb (MDR-TB). Extensively drug-resistant Mtb (XDR-TB) has been identified in 100 countries.11 The slow growth rate of Mtb and complex lipid-rich cell wall contribute to its persistence inside host organisms and there are few new candidate drugs against it.12,13 To counter the threat of AMR, new and innovative approaches to drug design are needed, which might not be limited to the “small molecule-single target” paradigm or strict adherence to Lipinski’s rules of small hydrophobic drugs.14 An interesting class of antimicrobials are cationic antimicrobial peptides (CAMPS).15−17 CAMPS are thought to function by interaction with the anionic bacterial cell membrane followed by cell wall permeabilization, or pore formation which ultimately leads to cell death, although the exact mechanisms are not fully understood and are under debate.18,19 Examples of CAMPS include the peptide Nisin which is widely used in the dairy industry as an antibiotic20 and Colistin a (polymyxin cyclic peptide with a hydrophobic tail) which is an antibiotic of last resort for infections such as Pseudomonas aeruginosa but its use is limited by its high toxicity. The biocompatibility of CAMPS is a key issue as they can also disrupt the anionic membrane of eukaryotic (mammalian) cells, particularly red blood cells (RBCs). Reduction of the net cationic charge mitigates toxicity but reduces activity meaning a precise balance must be maintained.21
In an effort to create synthetic mimics of antimicrobial peptides (SMAMPS), cationic polymers have emerged as an easy-access class of antibacterial materials which have been extensively reviewed.22−25 Synthetic polymers are particular appealing as their composition and architecture can be finely tuned using controlled (e.g., radical or ring-opening) polymerization methods and a huge range of monomers are available. These have been widely incorporated as biocidal coatings26 or as mediators of bacteria aggregation and signaling which can effect quorum sensing as well as killing.27 Fernandez-Trillo and co-workers recently demonstrated that polyionic complexes of an antibacterial polymer with an anionic peptide which upon exposure to elastase secreted by P. aeruginosa released the polymer leading to antibacterial activity, as a new route to targeted antimicrobials.28
The mechanism of the membrane disrupting polycations, is thought to make resistance less likely to develop as there is no single protein target. Cationic, second generation PAMAM (poly(amidoamine)) dendrimers have been reported to not induce resistance in penicillin resistant Escherichia coli and methicillin-resistant Staphylococcus aureus.29 In contrast, CAMP resistance through neutralization of anionic surface charges or changes in efflux pumps have been found, indicating that resistance is possible and that the mechanism of action varies between polycations.30,31 Ikeda et al. found that molecular weight of the polycations was crucial. Using cationic polyacrylates, it was found that a molecular weight of 5–10 kDa was optimal with shorter or longer polymers leading to a decrease in antimicrobial activity against S. aureus.32 Bacterial clustering triggered by polycations has recently been found to play a complex role in cell signaling beyond their cell lytic behavior also.33 Generally, Gram-negative bacterial membranes are harder to disrupt than Gram-positive due to the presence of an inner and outer membrane structure, which limits the action of simple polycations. However, Tew et al. have developed a range of facially amphiphilic polymers, which reproduce the cationic/lipophilic character of natural CAMPs such as magainins which are broad spectrum antimicrobials.34
Considerable effect has been placed on the development of cationic polymers for Gram-negative bacteria due to their widespread role in human disease. Gram-positive bacteria and the Mycobacterium genus, including Mtb, have been less studied in the context of polymer antimicrobials. The double versus single membrane cell wall structure between Gram-negative and Gram-positive (respectively) results in vastly different susceptibility to cationic polymers.35 The cell wall components of Gram-positive and mycobacteria equally display vastly different components, with mycobacteria having a distinct mAGP cell wall complex, rich in complex lipids and carbohydrates. Synthetic CAMPS with just 10 amino acids have been found to be active against Mtb at 10 μM concentrations36 as have naturally occurring anti-TB peptides from several kingdoms of life.37 Synergistic anti-Mtb activity was seen with CAMPS in combination with the front line drug rifampicin, potentially due to the CAMPS disrupting the membrane to promote drug uptake.38 It has also been found that antimicrobial peptides attach to teichoic acids in cell wall of Gram-positives (which are not present in mycobacteria) and not the anionic cell membrane supporting the concept that the design rules to target mycobacteria will be significantly different for that of Gram-negative and Gram-positive organisms.39 Tew and co-workers showed that facially amphiphilic SMAMPS were broad spectrum Gram-negative/positive active, but disruption of this amphiphilicity results in only maintaining activity against Gram-positives (S. aureus) again suggesting that a simplicity driven approach toward antituberculars might be possible.40
Considering the above observations, it would seem that the development of new synthetic materials that can selectively target mycobacteria is possible, especially as other Gram-positive strains seem particularly susceptible. This study reports a preliminary investigation into the use of RAFT-derived (reversible addition–fragmentation chain transfer) synthetic cationic polymers against mycobacteria, which may enable them to be used as a new tool for treating such infections. Poly(dimethylaminoethyl methacrylate) in particular is shown to have potent, and selective activity against mycobacteria relative to Gram-negative strains.
Experimental Section
Materials
4-Cyano-4-(phenylcarbonothioylthio)pentanoic acid (>97.0%), 4,4′-azobis(4-cyanovaleric acid) (≥98.0%), mesitylene (analytical standard), 2-mercaptoethanol (≥99.0%), tribasic potassium phosphate (reagent grade, ≥98.0%), carbon disulfide (≥99.0%), and benzyl bromide (98.0%) were purchased from Sigma-Aldrich. Monomers were purchased from Sigma-Aldrich and inhibitors removed by passing through a column of basic alumina prior to polymerization. RAFT agents were prepared as previously described.45
Physical and Analytical Methods
SEC analysis was performed using a Varian 390-LC MDS system equipped with a PL-AS RT/MT autosampler, a PL-gel 3 μm (50 × 7.5 mm) guard column, two PL-gel 5 μm (300 × 7.5 mm) mixed-D columns using DMF with 5 mM NH4BF4 at 50 °C as eluent at a flow rate of 1.0 mL.min–1. The SEC system was equipped with ultraviolet (UV)/visible (set at 280 and 461 nm) and differential refractive index (DRI) detectors. Narrow molecular weight PMMA standard (200–1.0 × 106 g mol–1) were used for calibration using a second order polynomial fit. NMR spectroscopy (1H, 13C) was conducted on a Bruker DPX-300, Bruker DRX-500 or Bruker AV III 600 spectrometer using deuterated chloroform or deuterated methanol as solvent. All chemical shifts are reported in ppm (δ) relative to the solvent used. FTIR spectra were acquired using a Bruker Vector 22 FTIR spectrometer with a Golden Gate diamond attenuated total reflection cell. A total of 64 scans were collected on samples in their native state. Microscopy was performed using a Zeiss LSM 880 microscope. SYTO-9 dye was imaged by excitation at 488 nm and emission at 530 nm for green fluorescence. Propidium iodide was imaged by excitation at 561 nm and emission at 646 nm for red fluorescence.
Synthetic Section
Polymerization of Dimethylaminoethyl Methacrylate
Dimethylaminoethyl methacrylate (2.00 g, 12.7 mmol), 2-cyano-2-propyl dodecyl trithiocarbonate (87.9 mg, 255 μmol), and azobis(isobutyronitrile) (4.18 mg, 25.5 μmol) were dissolved in dioxane (2 mL) in a glass vial containing a stir bar. The mixture was added to a dry ampule under a N2 atmosphere and degassed by three freeze–pump–thaw cycles. The reaction mixture was stirred and heated by an oil bath thermostated at 65 °C. After 6 h, the reaction mixture was opened to air and quenched in liquid nitrogen. An aliquot was removed and conversion determined by 1H NMR spectroscopic analysis. The product was purified by dialysis into deionized water (MWCO = 1000 g·mol–1). The solid was isolated by lyophilization to give a waxy, yellow solid. Conversion (NMR): 51.9%; Mn (theoretical): 4100 g·mol–1; Mn (SEC) 11 000 g·mol–1; Mw/Mn (SEC): 1.25.
Polymerization of Aminoethyl Methacrylate Hydrochloride
2-Aminoethyl methacrylate hydrochloride (0.83 g, 5.01 mmol), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (3.50 mg, 12.53 μmol) and 4,4′-azobis(4-cyanovaleric acid) (3.57 mg, 12.72 μmol) were dissolved in 0.6 mL of acetate buffer at pH 5.2 (produced using 0.27 mol·L–1 acetic acid and 0.73 mol·L–1 sodium acetate) in a 50 mL round bottomed flask, from a stock solution and subsequently diluted to 5 mL with the addition of further acetate buffer. The flask was purged with nitrogen for 45 min and placed in an oil bath at 70 °C. After 180 min the reaction was quenched using liquid nitrogen. Dialysis using acetate buffer (24 h, 5 water changes) and lyophilization were then used to purify the product. 1H NMR (D2O): δ 4.21 (br, 2H, −OCH2); δ 3.31 (br, 2H, −NH2CH2); δ 1.95 (br, 2H, backbone CH2); δ 0.83–1.36 (br, 3H, backbone CH3).
Polymerization of Dimethylaminoethyl Acrylate
Dimethylaminoethyl acrylate (1.00 g, 6.98 mmol), 2-(dodecylthiocarbonothioylthio)-2-methylpropanoic acid (25.47 g, 69.80 μmol) and 4,4′-azobis(4-cyanovaleric acid) (3.92 mg, 14.00 μmol) were dissolved in dioxane (4 mL) in a glass vial containing a stir bar. Mesitylene (200 μL) was added as an internal reference and the mixture stirred (5 min). An aliquot of this starting mixture was removed for 1H NMR spectroscopic analysis. The vial was fitted with a rubber septum and degassed by bubbling with nitrogen gas (30 min). The vial was then placed in an oil bath thermostated at 70 °C. After 6 h, the reaction mixture was opened to air and quenched in liquid nitrogen. An aliquot was removed and conversion determined by 1H NMR spectroscopic analysis. The product was purified three times by precipitation from tetrahydrofuran into hexane, isolated by centrifugation and dried under vacuum overnight to give a waxy, yellow solid. Conversion (NMR): 77.6%; Mn (theoretical): 11 000 g·mol–1; Mn (SEC) 6900 g·mol–1; Mw/Mn (SEC): 1.43.
Microbiology Section
Bacterial Strains and Growth Conditions
Mycobacterium smegmatis MC2155 was grown in either Tryptic Soy broth with the addition 0.05% Tween 80 (TSBT) or Middlebrook 7H9 media supplemented with 0.2% glycerol and 0.05% Tween 80. Escherichia coli Top10 and Pseudomonas putida KT4224 were grown in Luria–Bertani (LB) media.
Determination of Antibacterial Activities of Compounds 1–6
Minimum inhibitory concentrations (MICs) of the compounds were determined against M. smegmatis, P. putida, and E. coli. The bacteria were cultured to mid log phase and the inoculum standardized to 1 × 105 CFU·mL–1 before addition to a 96-well microtiter plate in which P1–6 were serially diluted 2-fold across the plate. Control wells containing culture controls and a reference drug were undertaken. In the case of M. smegmatis rifampicin was used, for E. coli ampicillin was used and for P. putida tetracycline was used. The plates were incubated at 37 °C for 72 h for M. smegmatis, at 37 °C for 18 h for E. coli and at 30 °C for 18 h for P. putida. Following this incubation period 25 μL resazurin was added (one resazurin tablet (VWR) in 30 mL sterile PBS) and left for a further incubation period (24 h for M. smegmatis, 4 h for E. coli and P. putida. The MIC values were determined as the lowest concentration of drug concentration that prevented the color change of resazurin (blue, no bacterial growth) to resorufin (pink, bacterial growth). The MIC values were determined in triplicate.
Minimal Bactericidal Concentration (MBC) Determination
Minimal bactericidal concentrations (MBCs) against replicating M. smegmatis were determined. M. smegmatis (inoculum of 105 CFU·mL–1) was cultured in the presence of P1–3 in a 96-well microtiter plate for 24 h at 37 °C. The highest concentrations of P1–3 were equivalent to a final concentration of 10 × MIC in the first wells and this was serially diluted 2-fold across the plate. Following incubation in the presence of compounds P1–3, 100 μL of the cells from each well were plated onto LB agar and incubated at 37 °C for 72 h. After 72 h the colonies on each plate were counted and the MBC determined to be the lowest concentration of the compound that resulted in the observation of no growth of M. smegmatis. The MBC values were determined in triplicate.
Time Kill Assays
Cultures of M. smegmatis (inoculum of 105 CFU·mL–1) were incubated with 2× MBC P3 (62.5 μg·mL–1) and 2 × MBC Rifampicin (6.25 μg·mL–1) in 7H9 broth at 37 °C. At defined intervals (0, 1, 2, 3, 4, 6, 24, and 48 h), 100 μL of each culture was plated onto LB agar containing no antibiotics and incubated at 37 °C for 2 days. Cell viability was assessed by determining CFU·mL–1 values. Each assay was performed in triplicate.
Confocal Microscopy
M. smegmatis was grown to late log phase before being harvested by centrifugation at 3300g for 15 min. The cell pellet was resuspended in 0.85% NaCl, then aliquoted and incubated with 0.5× MBC (15.625 μg·mL–1) and 2 × MBC (62.5 μg·mL–1) P3, 0.5 × MBC (62.5 μg·mL–1) and 2 × MBC (250 μg·mL–1) P4 for 15 min at room temperature. A single aliquot was used as a live cells control incubated with no drug and a further aliquot was used as a heat killed cells control, incubated at 80 °C for 30 min. After incubation, all samples were washed with 0.85% NaCl twice, with the suspension volume halved the second time. A Live/Dead BacLight bacterial viability kit (Invitrogen) was used to check cell viability. Briefly, Dye A (Syto-9) and Dye B (propidium iodide) were used in a 1:1 ratio and added to all samples before incubation for 15 min in the dark. Samples were then analyzed using a Zeiss LSM 880 confocal microscope to check for green (Ex 488 nm Em 520 nm) and red (Ex 561 nm, Em 646 nm) fluorescence.
Hemolysis Assay
Samples containing 250 μL ovine red blood cells (RBCs) and 250 μL of polymer solution (at indicated concentration) were incubated at 37 °C for 1 h. After centrifugation, 10 μL of the supernatant was added to 90 μL of PBS buffer in a 96 well plate. The absorbance was measured at 450 nm and compared against a PBS buffer and deionized water (to lyse cells) controls to determine the % hemolysis.
Transmission Electron Microscopy (TEM)
Transmission electron microscopy (TEM) was performed as previously described.46 Briefly, M. smegmatis was cultured in TBST for 72 h at 37 °C, 200 rpm. The culture was then diluted 1/100 in fresh TBST media and P1 was added to the culture to give a final concentration of P1 of 0.5 × MIC99. The culture was incubated for 18 h at 37 °C, 200 rpm. The cells were then centrifuged at 2400g for 10 min. The cell pellets were resuspended in 1× PBS and centrifuged and resuspended in 1× PBS twice more. The cell pellets were then fixed and processed as described previously.46 Samples were fixed in 2% glutaraldehyde washed in 3× PBS, then fixed with 2% osmium tetroxide with ruthenium red, washed, dehydrated through an ethanol gradient, proplylene oxide then low viscocity epoxy resin. Polymerized at 60 °C overnight and sectioned on a UltracutE ultramicrotome at 80 nm, stained with 2% uranyl acetate and Reynolds Lead Citrate before imaging. TEM imaging was carried out on a JEOL 2200FS at 200 kV using a Gatan K2 summit camera.
Results and Discussion
To evaluate if cationic polymers are potential lead candidates against mycobacteria, with a unique cell wall distinct from that of Gram-positive and Gram-negative organisms, a small panel of cationic polymers was selected, based on commercially available monomers. To this end, DMAEMA (dimethylaminoethyl methacrylate), DMAEA (dimethylaminoethyl acrylate), and AEAM (aminoethyl acrylate) were chosen based on previous reports on their antimicrobial activity and the commercial availability of the starting monomers.41 To ensure control over polymer molecular weight and dispersity, RAFT polymerization was employed, Scheme 1, using the indicated chain transfer agents (CTAs).
Scheme 1. RAFT Polymerization to Generate Cationic Polymers.

DMAEMA and DMAEA were polymerized in dioxane, and AEMA in acetate buffer (pH 5.2) to ensure protonation of the primary amine, which could otherwise undergo side reactions with the RAFT agent/ester group.42 Following polymerization, the polymers were isolated by precipitation and dialyzed against Milli-Q water to remove excess monomer/solvent. The resulting polymers were characterized by SEC (size exclusion chromatography) and NMR spectroscopy, Figure 1 and Table 1. SEC analysis revealed monomodal distributions but higher than expected dispersity values, which could be attributed to column interactions, as is common for amino-containing polymers. The amine side chains could also lead to some reduction in the fidelity of the RAFT agent also, leading to broadening. Nonetheless, these are accep for the purposes of this study (vide infra) where a library of different chain lengths was the target. The SEC molecular weights were also larger than expected from conversion and the feed ratio, supporting the assumption of some column interactions. PAEMA is challenging to characterize by SEC, as we have previously reported meaning only an estimated Mn based on conversion and the [monomer]:[CTA] feed ratio.42
Figure 1.
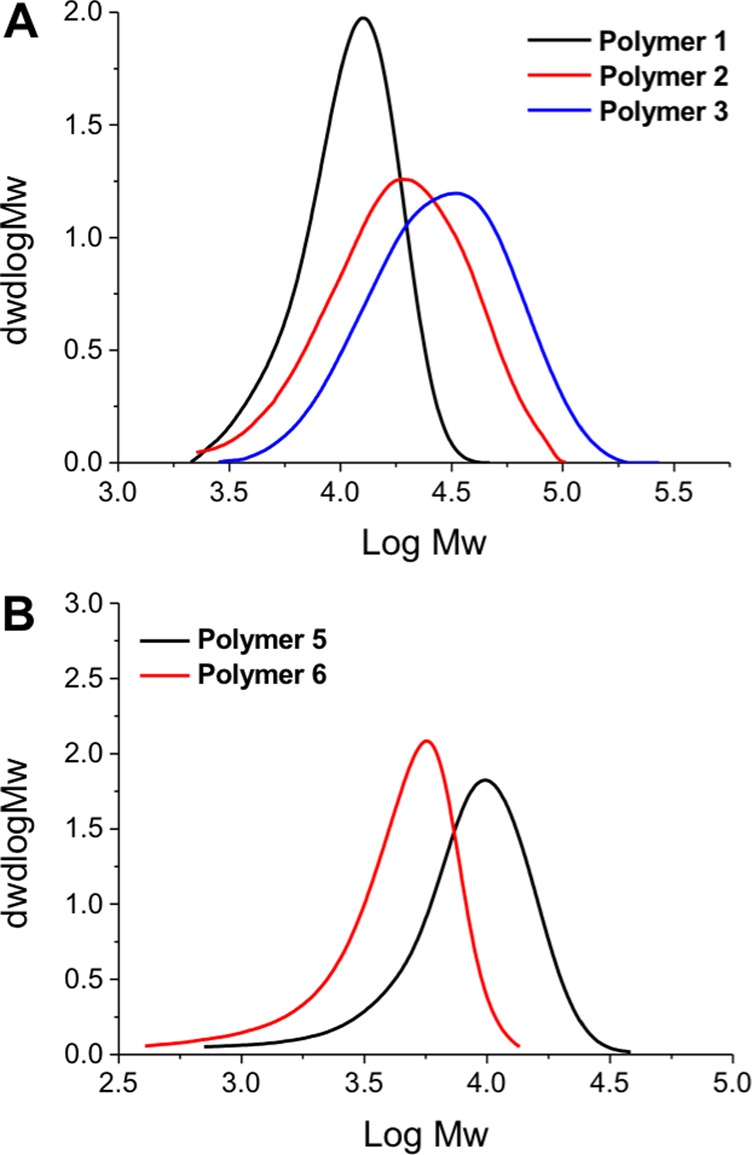
SEC analysis of polymers used in this study, as shown in Table 1.
Table 1. Cationic Polymers Synthesized by RAFT Polymerization.
| polymer | monomer | [M]:[CTA] | conversion (%)a | Mn(th) (g mol-1)b | Mn(SEC) (g mol-1) | Mw/Mnc |
|---|---|---|---|---|---|---|
| P1 | DMAEMA | 50 | 51.9 | 4500 | 11 000 | 1.25 |
| P2 | DMAEMA | 50 | 77.3 | 6100 | 13 900 | 1.61 |
| P3 | DMAEMA | 100 | 71.4 | 11 200 | 21 800 | 1.64 |
| P4 | AEMA | 100 | 71.4 | 11 200 | N/A | N/A |
| P5 | DMAEA | 100 | 77.6 | 11 000 | 6900 | 1.43 |
| P6 | DMAEA | 25 | 89.6 | 3200 | 3300 | 1.47 |
Determined by 1H NMR against an internal mesitylene standard.
Determined by the [M]:[CTA] ratio and conversion, assuming 100% CTA efficiency.
Determined by SEC against PMMA standards.
This focused library of polymers was then used to screen for antibacterial activity. Three representative strains of bacteria were chosen for this: Escherichia coli, a Gram-negative bacteria; Pseudomonas putida, which is a Gram-negative bacteria closely related to pathogenic pseudomonas strains; and Mycobacterium smegmatis, from the Mycobacterium genus, and an avirulent model organism for Mtb.12 The minimum inhibitory concentrations (MIC) of the polymer library was determined against all the bacteria using a resazurin microtiter viability assay. In short, the polymers were serially diluted, added to the bacterial cultures, and the concentration where growth was inhibited reported as the MIC99 value. All assays were carried out in triplicate and the results of this are shown in Table 2.
Table 2. Minimum Inhibitory Concentrations of Polymers P1–6a.
| polymer | M. smegmatis (Mycobacteria) (μg·mL–1) | P. putida (Gram-negative) (μg·mL–1) | E. coli (Gram-negative) (μg·mL–1) |
|---|---|---|---|
| P1 | 31.25 | 156.25 | 62.5 |
| P2 | 31.25 | >5000 | 250 |
| P3 | 31.25 | >5000 | 250 |
| P4 | 500 | 625 | 1000 |
| P5 | >5000 | >5000 | >5000 |
| P6 | 3125 | >5000 | >5000 |
| tetracycline | × | 156.25 | × |
| rifampicin | 6.25 | × | × |
| ampicillin | × | × | 3.125 |
All values from three repeats; × indicates the antibiotic was not used against that strain. Antibiotics chosen based on their front-line use against associated infections.
Due to the presence of the inner and outer cell membranes, Gram-negative bacteria are challenging to kill using polycations, requiring careful installation of hydrophobic groups to promote insertion and disruption of the inner/outer membranes, or in the case of PDMAEMA a reduction in pH to increase the net positive charge (the aim here was to test at physiological pH hence it was maintained at 7.4). Our results were in agreement with this hypothesis, with all polymers having higher MIC99 values against P. putida and E. coli than against M. smegmatis. In the case of P5 and P6, extremely high concentrations were needed (concentrations above 3125 μg·mL–1, Table 2) despite the only structural difference compared to P1–3 being the absence of the backbone methyl group (Scheme 1). These subtle effects highlight the challenge of designing potent macromolecular antimicrobials as each individual strain has subtly different membrane components that govern the initial interactions, but also may offer a route to selectivity. Of particular interest was that P1–3 were very active against M. smegmatis, with the MIC99 value only 5× higher than that of rifampicin (on a mass basis, more active on molar due to high MW of polymers), a front-line antibiotic used against mycobacteria. The observed potency is similar to what has been reported for other antimicrobial polymers against nonmycobacteria, such as degradable polycarbonates25 or amino-terminated dendrimers with in vivo activity against E. coli infections in mice.29 The activity of these polymers is somewhat surprisingly considering the complex mAGP complex that surrounds mycobacteria, which would be expected to reduce the electrostatic interactions between polycations and the anionic cell membrane components. Against M. smegmatis, there was no clear effect of molecular weight on activity, but against the Gram-negative organisms, the shortest (4500 g·mol–1) polymers (P1) showed increased activity (Table 2). This is in line with observations by Ikeda et al., who saw that molecular weight effects on antimicrobial polycations were most pronounced below 10 kg mol–1.32
Based on their selectivity toward M. smegmatis the PDMAEMAs (P1–3) were further tested to determine their minimum bactericidal concentration (MBC). Briefly, the bacteria were grown in the presence of serial dilutions of P1–3 starting from 10× MIC99 and after 24 h plated on solid agar media and counted. The minimum concentration required to kill all the bacteria was reported, Table 3. Pleasingly, these values were found to closely correlate with those from the resazurin MIC testing in liquid media, confirming the bactericidal activity of the polymers. There was some evidence that P1 (lowest Mn) had slightly higher activity than the longer P2 and P3 which agrees with the observations from the MIC99 results (from Table 2, above). P3 was also tested in a time-kill assay at 2× MBC and compared to the front-line drug rifampicin (also at 2× MBC), to determine the rate of antibacterial action against M. smegmatis, Figure 2. The polymer killed all the bacteria (down to the detection limit of the assay) within 6 h, but rifampicin took 48 h. This highlights the potent nature of PDMAEMA against mycobacteria, which functions by a mechanism distinct (vide infra) from front-line drugs, which are slow to act, as they disrupt physiological pathways, which are themselves slow due to the slow-growing nature of mycobacteria.
Table 3. Minimum Bactericidal Concentration of P1–3 against M. smegmatis.
| polymer | μg·mL–1 |
|---|---|
| P1 | 31.25 |
| P2 | 31.25–62.5 |
| P3 | 31.25–62.5 |
Figure 2.

Time-kill assay of P3 against M. smegmatis. Errors bars represent the standard deviation from n = 3.
Previous reports of PDMAEMA against Gram-positive bacteria (not mycobacteria) suggested that the mode of action involved membrane binding, followed by permeabilization of the cell membrane, enabling leakage of cytoplasmic contents.41 This is also a potential route to cytotoxicity through lysis of mammalian cell membranes. In particular, this is an issue for red blood cells (RBCs) a key clinical side-effect in the field of antimicrobial polymers/peptides. P2–4 were tested up to 5 mg·mL–1 at 37 °C against (ovine) RBCs (Figure 3). P2 and P3, which were active against M. smegmatis, showed very little hemolysis, below 2% even at the highest concentration tested as would be expected for this polymer.43 Higher concentrations of PDMAEMA (>100 μg·mL–1) and molecular weights above 20 kDa than used here have been reported to lead to RBC agglutination, but was not seen here.43 In contrast, P4 with a primary amine group showed significant hemolysis at all concentrations. These results show that the identified PDMAEMA are more selective toward mycobacteria and less hemolytic than the primary amine containing polymers. (It should be noted that this does not necessarily indicate a complete lack of cytotoxicity44).
Figure 3.
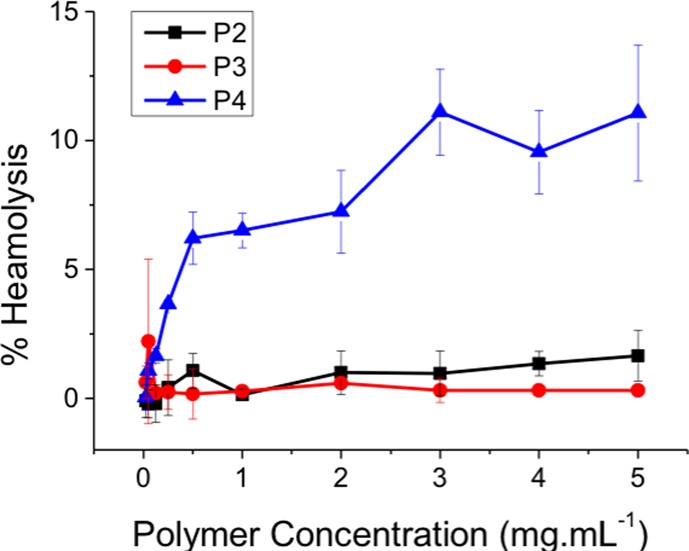
Assessment of hemolytic activity of polymers P2–4 after 1 h of exposure against ovine red blood cells. Error bars represent the standard deviation n = 3.
The lack of activity against RBCs even at high concentrations raises questions about the mechanism of action against bacteria. Tew and co-workers have extensively studied the mechanism of facially amphiphilic cations against Gram-negative bacteria, revealing a complex process which appears to involve the formation of pores in the membrane enabling leaking of cellular components.19 To probe the nature of the polymer–bacteria interaction the polymers were evaluated for their ability to permeabilize membranes using fluorescence microscopy in conjugation with membrane permeable/imperable dyes (“live/dead”), Figure 4. Control experiments of live M. smegmatis show green color due to the SYTO 9 dye associated with live (membrane intact) bacteria. Conversely, heat killed M. smegmatis (which does not lyse the bacteria) showed no green color but some were red, indicative of propidium iodide (dye) being able to cross damage cell membranes. Incubation of the M. smegmatis with P3 (PDMEAMA) for 30 min (as this effect would be direct and rapid and avoiding secondary interactions which could be mistaken for membrane damage and hence longer periods were not used, and guided by the kill-curve in Figure 2) both above (2× MIC) and below (0.5× MIC) MIC99 values resulted in green colored bacteria being obtained in both cases. As the assay does not actually probe if they are alive, but rather if the membrane is damaged, this result suggested that PDMAEMA does not immediately damage the cell membrane, in contrast to the expected mode of action of a polycation. P4 (PAEMA) with a primary amine side chain, resulted in red bacteria, due to extensive damage to the cell membrane, consistent with how other polycations function, and suggesting a unique mode of action between PDMAEMA and mycobacteria.
Figure 4.
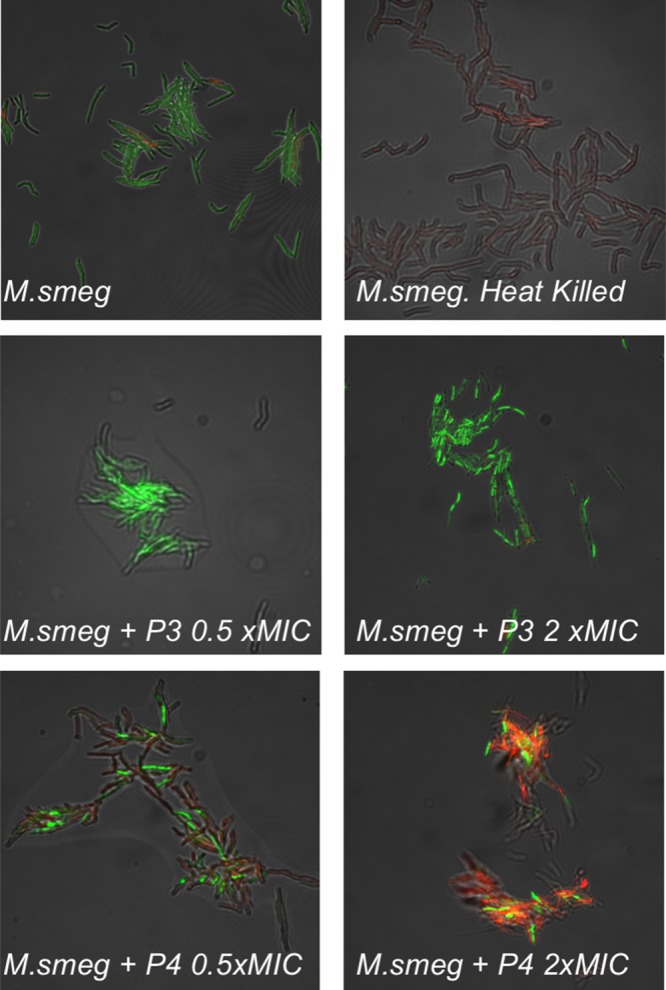
Fluorescence microscopy of M. smegmatis upon exposure to varying concentrations of P3 and P4. Green channel is SYTO-9 (nucleic acid stain), and red is propidium iodide (damage membrane stain). Each image is 85 × 85 μm2.
PDMAEMA (P1–3) was the most active polymer class against mycobacteria, with high selectivity toward these compared to Gram-negative bacteria and erythrocytes while also having relatively low membrane lytic activity. These assays do not, however, prove the lack of interaction/impact between the polymers and the bacterial membranes, just that they are intact and resistant to permeation. To visualize the impact on the membrane, transmission electron microscopy (TEM) was conducted on M. smegmatis, which was grown with and without the polymers at sublethal doses at 0.5 × MIC99, Figure 5. After incubation, the bacteria were fixed, stained and imaged. The wild type M. smegmatis without the polymer showed the expected cell morphology and membrane structure indicating it was intact when fixed. However, the M. smegmatis incubated with P1 clearly showed signs of distress, with significant signs of puckering strongly suggesting that the cell wall has been stressed. A possible explanation for this observation would be electrostatic interactions between the polymer and the unique mycobacterial cell wall lipids and/or anionic cell membranes. Nonetheless, these images clearly support a mechanism based on cell wall surface interactions of the polymer.
Figure 5.

TEM analysis of M. smegmatis with and without P1. P1 applied at 10 μg·mL–1 (0.3× MIC99). Top three panels, scale bar = 1 μm. Bottom three panels, scale bar = 100 nm.
Conclusions
Here it is demonstrated that poly(dimethylaminoethyl methacrylate), PDMAEMA, has potent and selective antimycobacterial activity against a model strain for M. tuberculosis. Conversely, it was weakly active against two Gram-negative strains, indicating selectivity. Relatively low molecular weight PDMAEMA (11 kDa) was shown to have minimum inhibitory values as low as 30 μg·mL–1. An acrylate equivalent and polymers with primary amine side chains were found to be significantly less active. Mechanistic studies revealed that the PDMAEMA did not affect the integrity of the mycobacterial cell membrane, confirmed by fluorescent microscopy, indicating an intact membrane/cell wall, unlike primary amine-containing polymers, which despite having lower activity clearly disrupted the cell membranes. TEM analysis supported a mechanism where the PDMAEMA is binding to, and affecting the outer-cell components but without causing lysis. Finally, PDMAEMA was shown to be nonhemolytic to red blood cells, confirming that it has a unique mode of action in exerting its effect on mycobacteria as well as demonstrating some biocompatibility. The underlying reasons for the selectivity toward mycobacteria, compared to Gram-negative strains is not clear at this time but could be due to the unique mAGP complex layer surrounding mycobacteria, which are rich in anionic mycolic acids and interspersed with complex lipids that are not found in Gram-negative (or Gram-positive) organisms. Taken together, this shows that nontraditional antibiotics which target bacterial extracellular components, rather than traditional single drug/single protein drugs might offer a solution to the emergence of multidrug resistant mycobacteria and that (relatively) simple polycations could supplement the arsenal of antimycobacterial drugs, in particular against M. tuberculosis which is a devastating global disease.
Acknowledgments
Equipment used was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). The microscopy imaging facilities are funded by University of Warwick Advanced BioImaging Research Technology Platform and BBSRC ALERT14 award BB/M01228X/1. The Royal Society provided funds for the plate reader (Grant number RG120405). M.I.G. acknowledges the ERC for a starter grant (CRYOMAT 638661). E.F. has a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (Grant Number 104193/Z/14/Z). D.E.M. thanks the MOAC DTC for a Ph.D. studentship EP/F500378/1. D.P. and S.-J.R. thank the University of Warwick and Institute of Advanced Study for Early Career Fellowships. E.F. and M.I.G. acknowledge EPSRC (EP/M027503/1) for supporting JH with a Training Fellowship. E.F., M.I.G., and C.G. acknowledge the Antimicrobial Resistance Cross Council Initiative, support by BBSRC and MRC (MR/N006917/1).
The authors declare no competing financial interest.
References
- Wise R.; Carrs O.; Cassell G.; Fishman N.; Guidos R.; Levy S.; Powers J.; Norrby R.; Tillotson G.; Davies R.; Projan S.; Dawson M.; Monnet D.; Keogh-Brown M.; Hand K.; Garner S.; Findlay D.; Morel C.; Wise R.; Bax R.; Burke F.; Chopra I.; Czaplewski L.; Finch R.; Livermore D.; Piddock L. J. V.; White T. J. Antimicrob. Chemother. 2011, 66 (9), 1939–1940. 10.1093/jac/dkr261. [DOI] [PubMed] [Google Scholar]
- Centre for Disease Control; Antibiotic/Antimicrobial Resistance, https://www.cdc.gov/drugresistance/.
- World Economic Forum; Global Risks, 2013; http://reports.weforum.org/global-risks-2013/.
- Nathan C. Nature 2004, 431 (7011), 899–902. 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- O’Neil J.The Review on Antimicrobial Resistance; O’Neil Report.
- Richards S.-J.; Jones M. W.; Hunaban M.; Haddleton D. M.; Gibson M. I. Angew. Chem., Int. Ed. 2012, 51 (31), 7812–7816. 10.1002/anie.201202945. [DOI] [PubMed] [Google Scholar]
- Otten L.; Gibson M. I. RSC Adv. 2015, 5 (66), 53911–53914. 10.1039/C5RA08857G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten L.; Fullam E.; Gibson M. I. Mol. BioSyst. 2016, 12 (2), 341–344. 10.1039/C5MB00720H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D. I.; Hughes D. Nat. Rev. Microbiol. 2010, 8 (4), 260. 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- The Lancet; Elsevier, April 27, 2013; p 1431. [Google Scholar]
- World Health Organisation; AMR Factsheet 194, 2015. [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Möllmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; De Rossi E.; Belanova M.; Bobovska A.; Dianiskova P.; Kordulakova J.; Sala C.; Fullam E.; Schneider P.; McKinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. Science 2009, 324 (5928), 801–804. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neres J.; Pojer F.; Molteni E.; Chiarelli L. R.; Dhar N.; Boy-Röttger S.; Buroni S.; Fullam E.; Degiacomi G.; Lucarelli A. P.; Read R. J.; Zanoni G.; Edmondson D. E.; De Rossi E.; Pasca M. R.; McKinney J. D.; Dyson P. J.; Riccardi G.; Mattevi A.; Cole S. T.; Binda C. Sci. Transl. Med. 2012, 4 (150), 150ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. Nature 2012, 481 (7382), 455–456. 10.1038/481455a. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Nature 2002, 415 (6870), 389–395. 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- Seo M.-D.; Won H.-S.; Kim J.-H.; Mishig-Ochir T.; Lee B.-J. Molecules 2012, 17 (10), 12276–12286. 10.3390/molecules171012276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon Y. J.; Romanowski E. G.; McDermott A. M. Curr. Eye Res. 2005, 30 (7), 505–515. 10.1080/02713680590968637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogden K. A. Nat. Rev. Microbiol. 2005, 3 (3), 238–250. 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- Yang L.; Gordon V. D.; Trinkle D. R.; Schmidt N. W.; Davis M. A.; DeVries C.; Som A.; Cronan J. E.; Tew G. N.; Wong G. C. L. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (52), 20595–20600. 10.1073/pnas.0806456105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas L.; Clarkson M.; Delves-Broughton J.. Natural food antimicrobial systems; Naidu A.., Ed.; CRC Press, 2000. [Google Scholar]
- Lopez A. I.; Reins R. Y.; McDermott A. M.; Trautner B. W.; Cai C. Mol. BioSyst. 2009, 5 (10), 1148–1156. 10.1039/b904746h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenawy E.-R.; Worley S. D.; Broughton R. Biomacromolecules 2007, 8 (5), 1359–1384. 10.1021/bm061150q. [DOI] [PubMed] [Google Scholar]
- Jain A.; Duvvuri L. S.; Farah S.; Beyth N.; Domb A. J.; Khan W. Adv. Healthcare Mater. 2014, 3 (12), 1969–1985. 10.1002/adhm.201400418. [DOI] [PubMed] [Google Scholar]
- Hasan J.; Crawford R. J.; Ivanova E. P. Trends Biotechnol. 2013, 31 (5), 295–304. 10.1016/j.tibtech.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Chin W.; Yang C.; Ng V. W. L.; Huang Y.; Cheng J.; Tong Y. W.; Coady D. J.; Fan W.; Hedrick J. L.; Yang Y. Y. Macromolecules 2013, 46, 8797–8807. 10.1021/ma4019685. [DOI] [Google Scholar]
- Siedenbiedel F.; Tiller J. C. Polymers (Basel, Switz.) 2012, 4 (4), 46–71. 10.3390/polym4010046. [DOI] [Google Scholar]
- Lui L. T.; Xue X.; Sui C.; Brown A.; Pritchard D. I.; Halliday N.; Winzer K.; Howdle S. M.; Fernandez-Trillo F.; Krasnogor N.; Alexander C. Nat. Chem. 2013, 5 (12), 1058–1065. 10.1038/nchem.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insua I.; Liamas E.; Zhang Z.; Peacock A. F. A.; Krachler A. M.; Fernandez-Trillo F. Polym. Chem. 2016, 7 (15), 2684–2690. 10.1039/C6PY00146G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X.; Chen X.; Mao X.; Hou Z.; Zhou Y.; Bai H.; Meng J.; Da F.; Sang G.; Wang Y.; Luo X. AAPS J. 2013, 15 (1), 132–142. 10.1208/s12248-012-9416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Cha D. J.; Lai Y.; Villaruz A. E.; Sturdevant D. E.; Otto M. Mol. Microbiol. 2007, 66 (5), 1136–1147. 10.1111/j.1365-2958.2007.05986.x. [DOI] [PubMed] [Google Scholar]
- Tzeng Y.-L.; Ambrose K. D.; Zughaier S.; Zhou X.; Miller Y. K.; Shafer W. M.; Stephens D. S. J. Bacteriol. 2005, 187 (15), 5387–5396. 10.1128/JB.187.15.5387-5396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T.; Yamaguchi H.; Tazuke S. Antimicrob. Agents Chemother. 1984, 26 (2), 139–144. 10.1128/AAC.26.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louzao I.; Sui C.; Winzer K.; Fernandez-Trillo F.; Alexander C. Eur. J. Pharm. Biopharm. 2015, 95 (Pt A), 47–62. 10.1016/j.ejpb.2015.05.026. [DOI] [PubMed] [Google Scholar]
- Tew G. N.; Liu D.; Chen B.; Doerksen R. J.; Kaplan J.; Carroll P. J.; Klein M. L.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (8), 5110–5114. 10.1073/pnas.082046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienkamp K.; Kumar K.-N.; Som A.; Nüsslein K.; Tew G. N. Chem. - Eur. J. 2009, 15 (43), 11710–11714. 10.1002/chem.200802558. [DOI] [PubMed] [Google Scholar]
- Ramón-García S.; Mikut R.; Ng C.; Ruden S.; Volkmer R.; Reischl M.; Hilpert K.; Thompson C. J. Antimicrob. Agents Chemother. 2013, 57 (5), 2295–2303. 10.1128/AAC.00175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedinzadeh M.; Gaeini M.; Sardari S. J. Antimicrob. Chemother. 2015, 70 (5), 1285–1289. 10.1093/jac/dku570. [DOI] [PubMed] [Google Scholar]
- Khara J. S.; Wang Y.; Ke X.-Y.; Liu S.; Newton S. M.; Langford P. R.; Yang Y. Y.; Ee P. L. R. Biomaterials 2014, 35 (6), 2032–2038. 10.1016/j.biomaterials.2013.11.035. [DOI] [PubMed] [Google Scholar]
- Scott M. G.; Gold M. R.; Hancock R. E. Infect. Immun. 1999, 67 (12), 6445–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker H. D.; Cankaya A.; Scott R. W.; Tew G. N. ACS Med. Chem. Lett. 2013, 4 (5), 481–485. 10.1021/ml300307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlinson L.-A. B.; Ryan S. M.; Mantovani G.; Syrett J. A.; Haddleton D. M.; Brayden D. J. Biomacromolecules 2010, 11 (2), 443–453. 10.1021/bm901166y. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Lilliman M.; Spain S. G.; Gibson M. I. Biomater. Sci. 2014, 2 (12), 1787–1795. 10.1039/C4BM00153B. [DOI] [PubMed] [Google Scholar]
- Cerda-Cristerna B. I.; Flores H.; Pozos-Guillén A.; Pérez E.; Sevrin C.; Grandfils C. J. Controlled Release 2011, 153 (3), 269–277. 10.1016/j.jconrel.2011.04.016. [DOI] [PubMed] [Google Scholar]
- Rawlinson L.-A. B.; O’Brien P. J.; Brayden D. J. J. Controlled Release 2010, 146 (1), 84–92. 10.1016/j.jconrel.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Phillips D. J.; Wilde M.; Greco F.; Gibson M. I. Biomacromolecules 2015, 16 (10), 3256–3264. 10.1021/acs.biomac.5b00929. [DOI] [PubMed] [Google Scholar]
- Etienne G.; Villeneuve C.; Billman-Jacobe H.; Astarie-Dequeker C.; Dupont M. A.; Daffé M. Microbiology 2002, 148 (10), 3089–3100. 10.1099/00221287-148-10-3089. [DOI] [PubMed] [Google Scholar]