

INTRODUCTION
Chemotherapy-induced peripheral neuropathy (CIPN) is one of the most commonly and widely reported adverse side effects of cancer treatment. The overall incidence of CIPN ranges from 30%–80% in patients treated for cancer, depending on the chemotherapy regimens used and the duration of treatment [10; 36; 44]. The symptoms of CIPN include pain, numbness, tingling, and temperature sensitivity, and are normally present in a symmetric, distal, “stocking and glove” distribution [24]. The occurrence of CIPN can limit the chemotherapeutic dosage, delay additional treatment cycles, and even lead to early termination of treatment [30; 34]. Moreover, CIPN frequently persists or even worsens after completion of chemotherapy [43], thereby greatly reducing quality of life for cancer survivors. Despite the high prevalence and severity of CIPN, currently there are no effective FDA-approved drugs to prevent or reverse CIPN.
Cisplatin is a platinum-based chemotherapeutic that is commonly used for the treatment of solid tumors such as lung, ovarian, testis, bladder, and head and neck cancer [3]. Cisplatin treatment is associated with a high incidence of CIPN [12], and in rodents it induces mechanical allodynia, spontaneous pain, and numbness [2; 25; 35; 39]. Mechanisms that underlie development and maintenance of cisplatin-induced peripheral neuropathy are under active investigation. Chemotherapy-induced mitochondrial damage in peripheral sensory neurons is thought to play a major role in CIPN. Paclitaxel and oxaliplatin have been shown to cause mitochondrial swelling or vacuolization in cell bodies and axons of sensory neurons [4; 18; 23; 25; 52; 57]. In the peripheral nerves of paclitaxel-treated and oxaliplatin-treated rats, these mitochondrial morphological changes are associated with impaired Complex I-mediated and Complex II-mediated respiration and the production of ATP [57]. We recently showed that prevention of paclitaxel-induced mitochondrial morphological abnormalities by the compound pifithrin-μ also prevented paclitaxel-induced mechanical allodynia [25]. These findings further support a causal role of mitochondrial damage in the pathogenesis of CIPN. However, no effective treatment approach has been identified.
Histone deacetylase 6 (HDAC6) is a cytoplasmic class II histone deacetylase (HDAC) that, in contrast to the other HDACs, has a specificity for non-histone proteins, including α-tubulin[22]. HDAC6 regulates multiple intracellular processes, such as protein degradation, cell motility, and cell–cell interaction [22; 46; 56]. Importantly, HDAC6 has been implicated in the regulation of mitochondrial transport [22; 40; 56]. In vitro, HDAC6 inhibition increases α-tubulin acetylation and promotes mitochondrial transport in hippocampal neurons [14]. In vivo, it has been shown that an HDAC6 inhibitor increases α-tubulin acetylation in peripheral nerves and improves sensory-motor function in a mouse model of type 2 Charcot-Marie-Tooth disease [16]. In this model, the HDAC6 inhibitor also enhances mitochondrial transport as measured in dorsal root ganglia (DRG) explants [16].
We tested the hypothesis that inhibition of HDAC6 prevents and treats multiple symptoms of CIPN, including mechanical allodynia, numbness, and spontaneous pain by restoring mitochondrial function in peripheral nerve and dorsal root ganglia, and reversing the loss of intra-epidermal nerve fibers (IENFs) in mice treated with cisplatin.
MATERIALS AND METHODS
Animals
Adult male C57BL/6J mice of 8 – 10 weeks of age were obtained from Jackson Laboratories and housed at the Texas A&M Health Science Center Program for Animal Resources or The University of Texas MD Anderson Cancer Center animal facility in Houston, TX. Adult male Sprague Dawley rats were obtained from Harlan Laboratories and rat experiments were performed by M.D. Biosciences, Ness Ziona, Israel. Animals were housed on a regular 12-hour light/dark cycle and had free access to food and water. Animals were randomly assigned to treatment groups. All procedures were consistent with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and the Ethical Issues of the International Association for the Study of Pain [58] and were approved by the local Institution for Animal Care and Use Committee (IACUC).
HDAC enzyme assays
In vitro biochemical assays were performed as described previously [8]. Specifically, drugs were dissolved and diluted in assay buffer (50mM HEPES, pH 7.4, 100mM KCl, 0.001% Tween-20, 0.05% BSA, and 20μM Tris (2-carboxyethyl)phosphine to 6-fold the final concentration. HDAC enzymes (BPS Biosciences, San Diego, CA) were diluted to 1.5-fold of the final concentration in assay buffer and pre-incubated with tested compounds for 24 hours before the addition of the substrate. The amount of substrate (acetyl-lysine tripeptide substrate for HDAC1, HDAC2, HDAC3, and HDAC6 or trifluoroacetyl-lysine tripeptide for HDAC4, HDAC5, HDAC7, HDAC8, and HDAC9) used for each enzyme was equal to the Michaelis constant (Km), as determined by a titration curve. Substrate was diluted in assay buffer to 6-fold the final concentration with 0.3 μM sequencing grade trypsin (Sigma-Aldrich, St. Louis, MO). The substrate/trypsin mix was added to the enzyme/compound mix and the plate was shaken for 5 seconds and then placed into a SpectraMax M5 microtiter (Molecular Devices, Sunnyvale, CA) plate reader. The enzymatic reaction was monitored for release of 7-amino-4-methoxy-coumarin, after deacetylation of the lysine side chain in the peptide substrate, over 30 minutes and the linear rate of the reaction was calculated. The IC50 was determined using Graph Pad Prism and a 4-parameter curve fit.
In vivo pharmacokinetics for ACY-1083
For in vivo pharmacokinetic studies, mice were fasted overnight and i.p. injected with 5 mg/kg ACY-1083 dissolved in 10% dimethylacetamide (Sigma-Aldrich, St. Louis, MO) + 10% Solutol HS 15 (BASF, Houston, TX) in saline. Blood was collected by retro-orbital bleeding at 5 minutes, 15 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 8 hours, and 24 hours post injection. Plasma was obtained by centrifugation at 2,000 × g for 5 minutes at 4°C. Plasma compound level was analyzed using liquid chromatography-tandem mass spectrometry (Waters Corporation, Milford, MA), and was calculated from a standard curve of ACY-1083 in mouse plasma. Pharmacokinetic parameters were calculated using WinNonlin software.
Drug administration
Cisplatin (TEVA Pharmaceuticals, North Wales, PA) was diluted in sterile saline and administered i.p. at a daily dose of 2.3 mg/kg for 5 days followed by 5 days of rest and a second round of 5 doses [25; 39]. Paclitaxel (Tocris Biosciences, Bristol, United Kingdom) was diluted in sterile saline and administered i.p. at a dose of 6 mg/kg daily for 7 days and 12 mg/kg for 6 days. The HDAC6 inhibitor ACY-1083 (Acetylon Pharmaceuticals, Boston, MA) was dissolved in 20% 2-hydroxypropyl-B-cyclodextrin (Sigma-Aldrich, St. Louis, MO) + 0.5% hydroxypropyl methylcellulose (Spectrum Chemical, Gardena, CA) in water. Mice received i.p. injections of ACY-1083 at a dose of 3 or 10 mg/kg. Rats received ACY-1083 by oral gavage at a dose of 3 mg/kg twice per day. The HDAC inhibitor ACY-1215 (Acetylon Pharmaceuticals) [32; 42] was dissolved in 10% DMSO, 30% Propylene glycol and 60% PEG-300, and was administered to mice at 30 mg/kg via oral gavage. The dosing schedules are shown in Supplementary Figure 1.
Von Frey test
For mice, mechanical allodynia was measured using von Frey hairs (0.02, 0.07, 0.16, 0.4, 0.6, 1.0, and 1.4 g) (Stoelting, Wood Dale, Illinois, USA) starting with the application of the 0.16 g hair as we described previously [25; 39]. For rats, we used von Frey hairs up to 60 g to determine the minimal force needed to elicit a withdrawal response. A positive response was defined as a clear paw withdrawal or shaking [13]. Testing was done by an experimenter blinded to treatment groups.
Adhesive removal test
To examine numbness, we used a modification of the adhesive removal test [7] as we described [39]. Briefly, a round adhesive patch (3/16” Teeny Touch-Spots, USA Scientific INC., Ocala, FL) was placed on the plantar surface of the hind paws. The time it took for the mouse to display a behavioral response to the patch (eg, shaking or attempted removal) was recorded within a 15-minute testing time.
Conditioned place preference test
Spontaneous pain was tested using a conditioning paradigm with retigabine (#R-100, Alomone Laboratory, Jerusalem, Israel) as the conditioned stimulus to briefly relieve pain (45–60 minutes), as originally described in a rat model [6; 53]. Briefly, mice were first allowed to freely explore the conditioned place preference apparatus, which consists of 2 chambers (18 × 20 cm, 1 dark, 1 light) connected by a 15 cm hallway (Stoelting, Wood Dale, IL), for 15 minutes. The time spent in the light chamber was recorded. During the conditioning phase, mice were first i.p. injected with saline and kept in the dark chamber for 20 minutes. 3 hours later, the analgesic retigabine was injected i.p. and the mice were placed in the light chamber for 20 minutes. Conditioning was done for 4 consecutive days. The mice were again allowed to freely explore the apparatus for 15 minutes without any injections. A mouse experiencing pain should show an increase in time spent in the light chamber that was paired with retigabine as compared to the pre-conditioning phase.
Immunofluorescence staining
For quantification of intra-epidermal nerve fibers (IENFs) in glabrous skin, biopsies from the plantar surface of the hind paws were collected and processed as described previously [25; 39]. 25 μm frozen sections were incubated with antibodies against the pan-neuronal marker PGP9.5 (rabbit; AbD Serotec, Oxford, United Kingdom) and Collagen IV (goat; Southern Biotech, Birmingham, AL) followed by Alexa-594 donkey anti-rabbit and Alexa-488 donkey anti-goat (Life Technologies, Carlsbad, CA). For assessing astrocyte activation, 6 μm frozen sections of lumbar spinal cord (L1–L6) were incubated with glial fibrillary acidic protein (GFAP) antibody (rabbit; Abcam, Cambridge, MA) followed by Alexa-594 goat anti-rabbit (Life Technologies) as described previously [55]. Images were captured using Leica SPE confocal microscope (Leica Microsystems, Buffalo Grove, IL). IENF density was determined as the total number of fibers that crossed the collagen-stained dermal-epidermal junction/length of epidermis (IENFs/mm). 5 random pictures were quantified from each mouse to get individual IENF counts, and 4 mice from each group were measured. GFAP staining intensity was quantified using Image J software for percent positive GFAP staining. Analysis was done by an experimenter blinded to treatment groups.
Mitochondrial bioenergetics analysis
Tibial nerves were placed into islet capture XF24 microplate (Seahorse Bioscience, North Billerica, MA) in XF media supplemented with 5.5 mM glucose, 0.5 mM sodium pyruvate, and 1 mM glutamine. Oxygen consumption rate (OCR) was measured with an XF24 Flux Analyzer (Seahorse Bioscience). Oligomycin (12 μM), carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP, 20 μM), and rotenone/antimycin A (20 μM each) (Sigma-Aldrich) were injected sequentially during the assay. An assay cycle of 3-minute mix, 3-minute wait, and 4-minute measure combination was repeated 4 times for baseline rates and after each port injection.
Lumbar DRGs were digested with 1.25% collagenase (Thermo Fisher Scientific, Waltham, MA) and 2% trypsin (Sigma-Aldrich), and dissociated by triturating in Ham’s F10 medium (Corning Inc., Corning, NY) with a fire-polished glass pipette [28; 29]. Dissociated neurons were layered on a 10 ml gradient of sterile 26% Percoll (GE Healthcare Life Sciences, Little Chalfont, United Kingdom) in PBS, and centrifuged at 800 × g for 20min at room temperature. The cell pellet was resuspended in Ham’s F10 medium supplemented with N2 supplement (Thermo Fisher Scientific) and cultured overnight in an XF24 plate. The cell maintenance medium was changed to XF media 1 hour prior to starting the assay. Oligomycin (2 μM), FCCP (4 μM) and rotenone/antimycin A (2 μM each), and a 3-time repeat of a 2-minute mix, 3-minute wait, and 2-minute measure assay cycle were used. OCR values were normalized to the total protein content of each well. Basal respiration, ATP-linked respiration, proton leak, and maximal respiratory capacity (MRC) were determined as described previously [9; 28]. Basal OCR was determined before any port injection. ATP-coupled OCR was determined by the magnitude of decrease after oligomycin injection, whereas proton leak was estimated from the residual OCR. MRC was assessed by addition of the protonophore FCCP. Non-mitochondrial respiration was determined by the addition of rotenone and antimycin A.
Western blot analysis
SK-N-BE2 cells were grown in 12-well plates to 70% confluence before treatment with ACY-1083 in DMSO for 5 hours. (The total DMSO is 0.1% for all treatments). The final ACY-1083 concentrations in well range from 3 nM to 10 μM. The cells were washed with cold PBS two times and scraped into cold RIPA denaturing buffer with protease inhibitors and PMSF. They were incubated on ice for 10 minutes, sonicated in a bioruptor (high setting), and spun down at 100,000 rpm for 5 minutes. The supernatant was used for Western blot analysis of acetylated α-tubulin (mouse monoclonal; Sigma-Aldrich) and Histone H2BK5ac (rabbit polyclonal; Active Motif, Carlsbad, CA).
Tibial nerves retrieved from the XF24 assay were used for Western blot analysis of acetylated α-tubulin (rabbit monoclonal; Abcam), α-tubulin (rabbit polyclonal; Cell Signaling Technology, Danvers, MA) and mitochondrial proteins cytochrome c oxidase subunit IV (Cox IV; mouse monoclonal; Life Technologies), succinate dehydrogenase complex subunit A (SDHA; rabbit polyclonal; Cell Signaling Technology) and voltage-dependent anion channel (VDAC; rabbit polyclonal; Cell Signaling Technology). Horseradish-peroxidase–conjugated anti-mouse, anti-rabbit, or anti-β-actin antibodies (Jackson Laboratory, Bar Harbor, ME) were used for visualizing the immunoreactivity using a chemiluminescence detection kit (GE Healthcare Life Sciences). Images were acquired using ImageQuant LAS 4000 (GE Healthcare Life Sciences) and densitometrically analyzed using Image J software.
Mitochondrial movement in cultured DRG neurons
DRG neurons acquired from Lonza (Lonza, Basel, Switzerland) were cultured for 5–7 days according to the supplier’s protocol. 24-hour prior to chemotherapy treatment, cells were infected with Bacmam 2.0 (ThermoFisher Scientific) to label mitochondria with GFP. Cells were treated with cisplatin (80 μM) +/− ACY-1083 (100 nM) for 3 hours. For live imaging, cells were maintained in CO2-independent Hybernate E media (BrainBits LLC, Springfield, IL) containing cisplatin and ACY-1083. Imaging was performed using Zeiss 3i system (Intelligent Imaging Innovations, Denver, CO) at 37°C. Time-lapse images were acquired every 2 seconds over a course of 2 minutes. Image analysis was performed using Fiji and Multiple Kymograph plugin.
Statistics
Data are expressed as mean ± SEM. Statistical analysis of the differences between groups was performed using one-way or two-way ANOVA with Tukey’s post-hoc analysis (GraphPad, San Diego, CA) as appropriate; P<0.05 was deemed as statistically significant.
RESULTS
HDAC6 inhibition prevents and reverses cisplatin-induced mechanical allodynia
To investigate whether inhibition of HDAC6 reverses existing CIPN, we used the highly selective HDAC6 inhibitor ACY-1083. ACY-1083 has negligible inhibitory activity against other enzymes. In vitro, ACY-1083 was shown to inhibit HDAC6 with an IC50 of 3 nM and was 260-fold more selective for HDAC6 than all other classes of HDAC isoforms tested, HDAC1-9 (Figure 1A). The selectivity of ACY-1083 for inhibiting HDAC6 activity was validated in SK-N-BE2 cells. SK-N-BE2 cells were treated with various concentrations of ACY-1083 for 5 hours, and analyzed for acetylation level of the known HDAC6 substrate, α-tubulin, and a non-HDAC6 substrate, histone. ACY-1083 induced a dose-dependent increase in α-tubulin acetylation starting at a concentration of 30 nM, whereas its effect on histone acetylation was only evident at 10 μM, indicating an over 300-fold selectivity of ACY-1083 towards HDAC6 (Figure 1B). Mice dosed with 5 mg/kg ACY-1083 by intraperitoneal (i.p.) injection have a maximum plasma concentration (Cmax) of 936 ng/mL, a half-life (T1/2) of 3.5 hours, and a biologically active plasma exposure of 8 hours after dosing (Figure 1C).
Figure 1. Structure, selectivity and pharmacokinetics of ACY-1083.
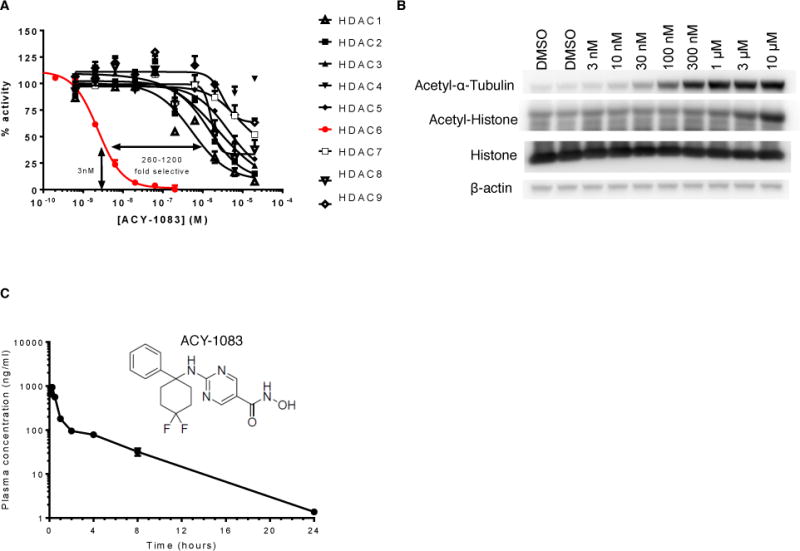
(A) The inhibitory effects of ACY-1083 on the enzymatic activities of HDAC1-HDAC9. The Y-axis represents the percentage of basal enzymatic activity corresponding to various concentrations of ACY-1083. The curve fit was generated by a 4-parameter, nonlinear regression in Graph Pad Prism. (B) The effects of various concentrations of ACY-1083 on the acetylation level of the HDAC6 substrate α-tubulin and on histone, which is not an HDAC6 substrate in SK-N-BE2 cells. (C) Structure and pharmacokinetics of ACY-1083. ACY-1083 is present at biologically active concentrations 8 hours after dosing in the plasma of mice received ACY-1083 i.p. (n=3/group).
To test the effect of ACY-1083 on chemotherapy-induced peripheral neuropathy, mice received two rounds of cisplatin for a cumulative dose of 23 mg/kg [25; 39]. Starting 3 days after the last dose of cisplatin, when mechanical allodynia had already developed, mice received 7 daily doses of ACY-1083 (i.p. 3 mg/kg or 10 mg/kg) or vehicle (Supplementary Figure 1). Treatment with 10 mg/kg ACY-1083 effectively relieved cisplatin-induced mechanical allodynia, whereas the 3 mg/kg dose did not (Figure 2A). The effect of ACY-1083 was first detected 24-hour after the first dose (Figure 2B). Treatment with 10 mg/kg ACY-1083 in the absence of cisplatin treatment did not alter mechanical sensitivity. In a separate set of experiments, we found that ACY-1083 was also capable of preventing the development of mechanical allodynia when administered 1 hour prior to each dose of cisplatin (Figure 2C). ACY-1083 also reversed paclitaxel-induced mechanical allodynia in rats received 13 daily injection of paclitaxel followed by two daily oral doses of 3 mg/kg ACY-1083 for 7 days (Figure 2D). The protective effect was still detectable 3 days after the last dose of ACY-1083, which was the last day tested. CIPN is associated with spinal cord astrocyte activation as evidenced by an increase in GFAP expression [38; 55]. Consistently, we detected an increase in GFAP expression in the dorsal horn of the spinal cord in cisplatin-treated mice. ACY-1083 treatment normalized GFAP expression to levels comparable to saline-treated animals (Supplement Figure 2). We did not detect changes in Iba-1 expression in response to cisplatin, indicating that spinal cord microglia were not activated (data not shown).
Figure 2. HDAC6 inhibition prevents and reverses cisplatin-induced mechanical allodynia.
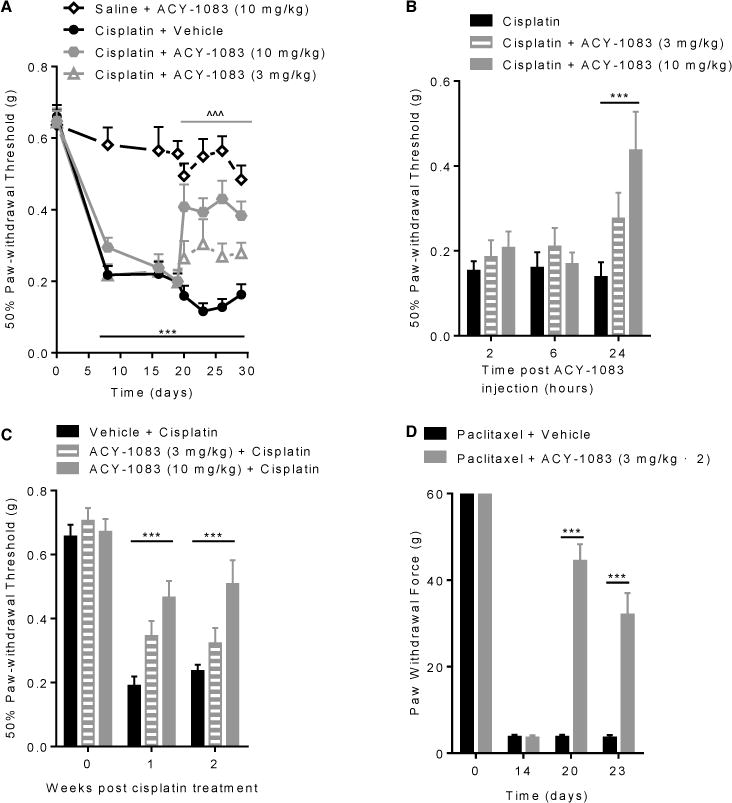
Mechanical allodynia was measured using von Frey hairs, and the 50% paw withdrawal threshold was calculated by the up–down method. (A) Mice were administered with 2 rounds of cisplatin treatment; 3 days after the last cisplatin dose, mice received ACY-1083 (10 mg/kg or 3 mg/kg) for 7 days. Two-way repeated-measure ANOVA revealed a main effect for time (P<0.01), group (P<0.01), and a group-by-time interaction (P<0.01). Tukey post-hoc analysis was used to determine differences between groups at specified time points: ***P<0.001 between cisplatin + vehicle vs. saline + vehicle; ^^^P<0.001 between cisplatin + ACY-1083 (10mg/kg) vs. cisplatin + vehicle. n=10–12/group. (B) Two-way repeated-measure ANOVA showed a main effect for time (P<0.05) and a group-by-time interaction (P<0.05). Tukey post-hoc analysis for cisplatin + ACY-1083 (10 mg/kg) vs. cisplatin + vehicle: ***P<0.001. n= 6–8/group. (C) Mice received cisplatin daily for 5 days. ACY-1083 (10 mg/kg or 3 mg/kg) was administered i.p. 1 hour prior to each cisplatin injection and for 2 days after the last cisplatin injection. Two-way repeated-measure ANOVA showed a main effect for time (P<0.0001), group (P<0.03), and a group-by-time interaction (P<0.03). Tukey post-hoc analysis for ACY-1083 (10 mg/kg) + cisplatin vs. vehicle + cisplatin: ***P<0.001. n= 6–8/group. (D) Rats were treated with paclitaxel for 13 days. Two daily doses of ACY-1083 (3 mg/kg) were given orally for 7 days. Mechanical allodynia were measured at baseline, post-paclitaxel treatment (day 14), after completion of ACY-1083 treatment (day 20) and 3 days after completion of ACY-1083 treatment (day 23). Two-way ANOVA with Tukey’s post-hoc analysis showed a significant treatment effects at day 20 and day 23: ***P<0.001. n= 11–12/group.
The beneficial effect on mechanical allodynia of 7 daily doses of 10 mg/kg ACY-1083 was maintained until 4 days after the last dose. Administration of a second round of 7 daily injections of ACY-1083 transiently reversed cisplatin-induced mechanical allodynia again (Figure 3A). Notably, when we administered ACY-1083 for 2 consecutive weeks starting 3 days after the last dose of cisplatin, we observed complete and sustained recovery from mechanical allodynia (Figure 3A). The beneficial effect of the prolonged regimen of ACY-1083 was maintained until at least 1 month after the last dose of ACY-1083 (experiment termination). It is of note that, at this time point, mice treated with cisplatin alone still displayed marked mechanical allodynia (Figure 3A).
Figure 3. Effect of repeated or prolonged ACY-1083 treatment on symptoms of cisplatin-induced peripheral neuropathy.
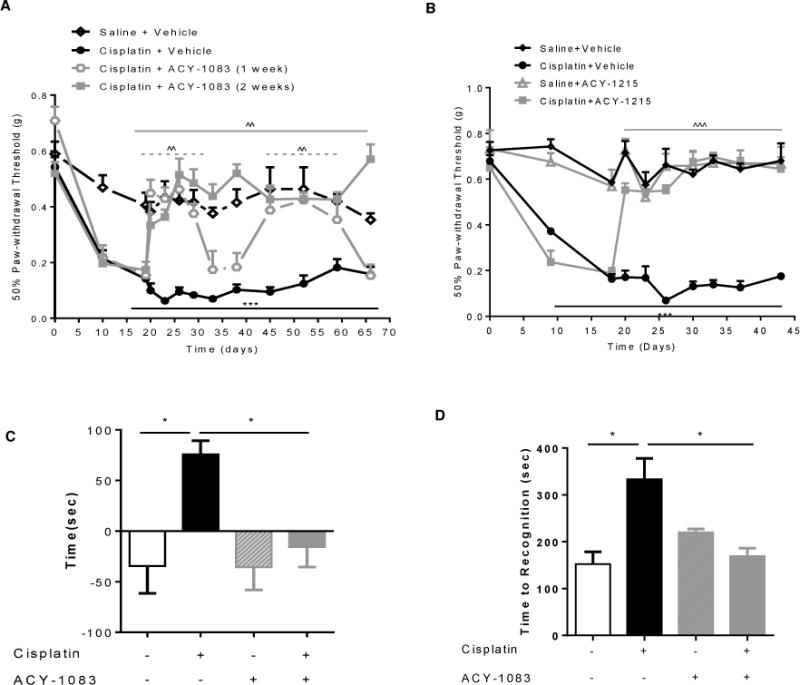
(A) Mice were administered with 2 rounds of cisplatin treatment; 3 days after the last cisplatin dose, mice received ACY-1083 (10 mg/kg) for 7 days followed by 7 days rest and then another 7 days of cisplatin treatment. Alternatively, mice received ACY-1083 (10 mg/kg) for 14 consecutive days. Mechanical allodynia was measured using von Frey hairs, and the 50% paw withdrawal threshold was calculated by the up–down method. Two-way repeated-measure ANOVA revealed a main effect for time (P<0.01), a group effect (P<0.01), and a group-by-time interaction (P<0.01). Tukey post-hoc analysis was used to determine differences between groups at specified time points. ***P<0.001 between cisplatin + vehicle vs. saline + vehicle; ^^P<0.01 between cisplatin + ACY-1083 vs. cisplatin + vehicle. n=6–14/group. (B) Mice were administered with 2 rounds of cisplatin treatment; 3 days after the last cisplatin dose, mice received ACY-1215 (30 mg/kg) orally for 14 days. Mechanical allodynia was measured using von Frey hairs, and the 50% paw withdrawal threshold was calculated by the up–down method. Two-way repeated-measure ANOVA revealed a main effect for time (P<0.01), a group effect (P<0.01), and a group-by-time interaction (P<0.01). Tukey post-hoc analysis was used to determine differences between groups at specified time points. ***P<0.001 between cisplatin + vehicle vs. saline + vehicle; ˆˆˆP<0.01 between cisplatin + ACY-1215 vs. cisplatin + vehicle. n=6–8/group. (C) Spontaneous pain was measured by the conditioned place preference test after 2 weeks of ACY-1083 treatment. The Y-axis indicates the change in time spent in light chamber. Two-way ANOVA revealed a signification interaction (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. n=6–8 mice/group. (D) Cisplatin-induced numbness was measured by the adhesive removal test. Mice were tested in week 5 for cisplatin-induced numbness. Statistical analysis using Two-way ANOVA revealed a significant interaction (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. n=4–10/group.
We also tested the HDAC6 inhibitor, ACY-1215 (Ricolinostat), which is currently in several clinical studies for the treatment of multiple myeloma and lymphoma. ACY-1215 is an orally bioavailable HDAC6 inhibitor that is less selective for HDAC6 than ACY-1083, but still shows over 10-fold selectivity against HDAC6 compared with Class I HDACs [32; 42]. Mice received 2 rounds of cisplatin treatment, and ACY-1215 was given orally at a daily dose of 30 mg/kg for two weeks, starting 3 days after the last dose of cisplatin. The results in Figure 3B demonstrate that daily administration of the less selective HDAC6 inhibitor ACY-1215 also effectively reversed cisplatin-induced mechanical allodynia. In line with what we observed for ACY-1083, the beneficial effect of ACY-1215 was still present one week after completion of treatment, which was the last time point tested.
HDAC6 inhibition reverses spontaneous pain and numbness induced by cisplatin
Patients with CIPN not only report allodynia, but also spontaneous pain and numbness [11; 24; 50]. Therefore, we investigated whether inhibition of HDAC6 would also reverse spontaneous pain and numbness in our mouse model. To measure spontaneous pain, we utilized a conditioned place preference test with the nerve blocker retigabine as the conditioning stimulus [53]. During the conditioning phase, mice received saline injections paired with exposure to a dark chamber and retigabine injections paired with exposure to a light chamber. The mice were allowed to freely explore both chambers before and after conditioning, and the change in time spent in the light chamber was recorded. The results in Figure 3C show that cisplatin-treated mice increased the time spent in the light chamber paired with retigabine, indicating ongoing pain. Saline-treated mice maintain a preference for the dark chamber. Mice that were treated with cisplatin followed by 2 weeks of daily ACY-1083 did not develop a preference for the retigabine-paired light chamber, indicating that these mice no longer experienced pain. There were no group differences in time spent in the light chamber before conditioning.
To assess the effect of ACY-1083 on cisplatin-induced numbness, we used the adhesive removal test that we recently validated for this purpose [39]. A small sticker was placed on the hind paw and the time until the mouse displays a behavioral response to the sticker was recorded. Consistent with our previous study, cisplatin treatment prolonged the time to respond in this test, indicating numbness [39]. Treatment with ACY-1083 starting after completion of the 2 rounds of cisplatin normalized the response time, indicating reversal of the numbness (Figure 3D).
HDAC6 inhibition induces α-tubulin acetylation in vivo
To control whether ACY-1083 functions as an inhibitor of HDAC6-mediated protein de-acetylation in vivo, we examined the effect of ACY-1083 on acetylation of α-tubulin [22]. Tibial nerves were collected from mice treated with cisplatin followed by ACY-1083 or vehicle treatment. Figure 4 shows that ACY-1083 treatment increased α-tubulin acetylation in mice treated with saline + ACY-1083 and in mice treated with cisplatin + ACY-1083. We did not detect changes in α-tubulin acetylation in mice treated with cisplatin alone.
Figure 4. Effect of ACY-1083 on α-tubulin acetylation.
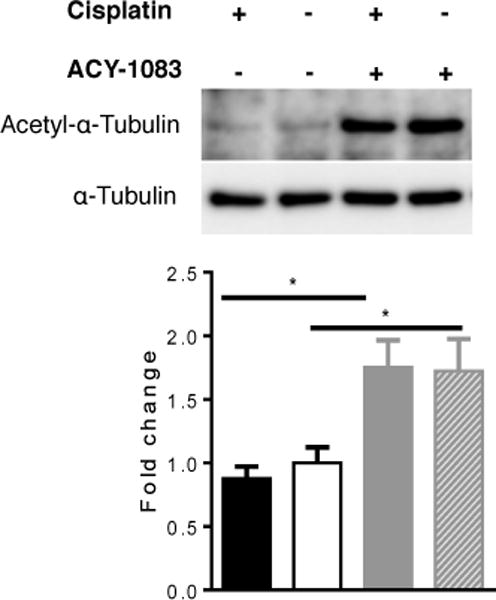
Acetylated α-tubulin levels in tibial nerves from mice that had received 2 rounds of cisplatin and 11 doses of ACY-1083 were assessed by Western blot analysis. ACY-1083 treatment induced α-tubulin acetylation. Two-way ANOVA revealed a significant treatment (ACY-1083) effect (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. n=4 mice/group.
Effect of cisplatin and ACY-1083 on peripheral nerve mitochondrial function
α-Tubulin acetylation status is a key regulator of neuronal mitochondrial transport [14]. Efficient mitochondrial transport towards distal nerve endings and sufficient energy production is essential for proper function of the peripheral nervous system [4]. Therefore, we tested if ACY-1083 impacts mitochondrial function in vivo. We assessed mitochondrial bioenergetics in tibial nerves and DRGs of mice treated with 2 cycles of cisplatin followed by 11 doses of ACY-1083. In tibial nerves, cisplatin significantly decreased mitochondrial respiration (Figure 5A); specifically, we detected decreases in basal respiration, ATP-linked respiration, proton leak and maximal respiration (Figure 5A). Notably, 11 days of treatment with ACY-1083 normalized all aspects of the cisplatin-induced decrease in mitochondrial respiration in the tibial nerve, indicating normalization of mitochondrial bioenergetics (Figure 5A). The non-mitochondrial respiration was not altered in either cisplatin-treated or ACY-1083-treated mice (measurements after rotenone/antimycin A administration). ACY-1083 treatment alone did not have any effect on mitochondrial bioenergetics.
Figure 5. ACY-1083 enhances mitochondrial bioenergetics and contents in the tibial nerve of cisplatin-treated mice.
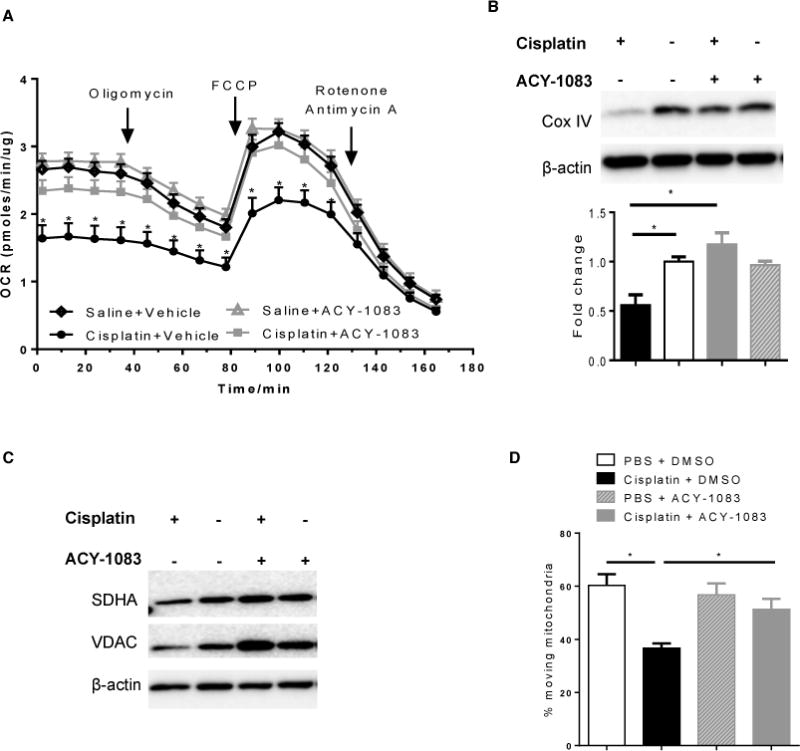
(A) Mitochondrial bioenergetics in tibial nerves from mice that had received 2 rounds of cisplatin treatment and 11 injections of ACY-1083. Two-way ANOVA revealed a signification interaction (P<0.05) for baseline respiration, ATP-coupled respiration, proton leak, and maximal respiratory capacity (MRC). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. n=6–8 mice/group. (B) Tibial nerves retrieved from the XF-analysis were used for Western blot analysis of mitochondrial marker protein Cox IV. Two-way ANOVA revealed a signification interaction (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. n=6–8 mice/group. (C) Tibial nerves from mice received 2 rounds of cisplatin treatment and 11 injections of ACY-1083 were used for Western blot analysis of additional mitochondrial marker protein SDHA and VDAC. (D) Mitochondrial transport was determined with rat DRG neuron cultures in vitro. The percent of moving mitochondria was determined and shown in the Y-axis. Two-way ANOVA revealed a signification interaction (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. n=3–4/group.
To determine whether the cisplatin-induced impairment of mitochondrial bioenergetics in tibial nerve and the normalization in response to ACY-1083 were associated with changes in mitochondrial content in the distal nerves, we performed Western blot analysis for the mitochondrial proteins Cox IV, SDHA and VDAC. The level of Cox IV in the tibial nerves of cisplatin-treated mice was significantly reduced when compared to saline-treated mice. Moreover, treatment with ACY-1083 normalized Cox IV levels in the tibial nerve (Figure 5B). Similar effects were observed when we assessed SDHA and VDAC (Figure 5C). These findings indicate that inhibition of HDAC6 restored cisplatin-induced reductions in mitochondrial bioenergetics, which is likely due to increased mitochondrial content in the distal nerve.
To further examine the effect of cisplatin and ACY-1083 on mitochondrial motility, primary cultures of rat DRG neurons with fluorescently labeled mitochondria were treated with cisplatin and ACY-1083 for 3 hours. Cisplatin treatment decreased the percentage of moving mitochondria when compared to vehicle-treated cultures. Co-administration of ACY-1083 prevented this cisplatin-induced decrease in mitochondrial transport (Figure 5D).
Effects of HDAC6 inhibition on mitochondrial function in DRG neurons
A change in mitochondrial bioenergetics in the distal peripheral nerves could also reflect a change in overall neuronal mitochondrial health. Therefore, we tested the mitochondrial bioenergetics in adult DRG neurons isolated from mice treated with cisplatin and ACY-1083. As expected, we detected an overall decrease in mitochondrial bioenergetics in cisplatin-treated mice, with significant reduction in maximal respiratory capacity (Figure 6A). However, at the time point when we observed a full recovery of mitochondrial function in the tibial nerves (Figure 5), we did not detect a similar recovery of the bioenergetic profile in the DRG neuronal cells (Figure 6A). Interestingly, 2 weeks after the last dose of ACY-1083, there was a significant recovery of mitochondrial bioenergetics in DRG neurons from mice treated with cisplatin + ACY-1083 as compared to mice received cisplatin treatment alone (Figure 6B). At this time point, cisplatin-treated mice still displayed marked pain, while the mice treated with ACY-1083 had fully recovered (Figure 3A). ACY-1083 treatment alone did not have any effect on mitochondrial bioenergetics in DRG neurons at either time point measured.
Figure 6. ACY-1083 enhances mitochondrial bioenergetics in DRG neurons of cisplatin-treated mice.
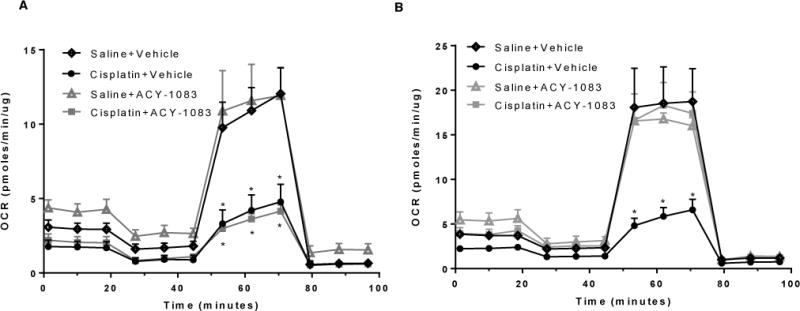
(A) Mitochondrial bioenergetics in DRG neurons from mice that had received 2 rounds of cisplatin treatment and 11 injections of ACY-1083. Two-way ANOVA revealed a signification difference for cisplatin + vehicle and cisplatin + ACY-1083 versus saline + vehicle and saline + ACY-1083 for maximal respiratory capacity: *P<0.05. (B) Mitochondrial bioenergetics in DRG neurons from mice received 2 rounds of cisplatin and two weeks of ACY-1083, tissues were taken two weeks after the last ACY-1083 treatment. Two-way ANOVA revealed a signification difference for cisplatin + vehicle versus all three other groups for maximal respiratory capacity: *P<0.05.
Effect of HDAC6 inhibition on intra-epidermal nerve fiber density
Cisplatin-treatment is known to reduce IENF density in the plantar surface of the paw [39]. It has been hypothesized that this reduction is due to mitochondrial damage, as these peripheral nerve fibers represent bioenergetically demanding regions [4]. Consistent with our previous report [39], cisplatin treatment reduced the IENF density. Remarkably, the prolonged regimen of ACY-1083 treatment completely reversed the cisplatin-induced loss of IENFs (Figure 7). In contrast, short-term treatment with ACY-1083 had no effect on IENF density (Supplementary Figure 3), indicating that prolonged treatment with ACY-1083 promoted restoration of IENF density rather than preventing the progression of IENF loss.
Figure 7. ACY-1083 reverses the loss of intra-epidermal nerve fibers in cisplatin-treated mice.
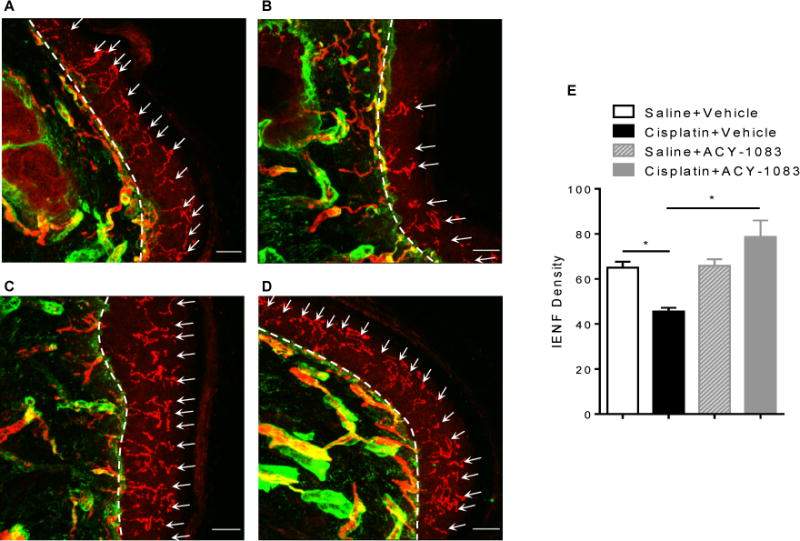
Paw biopsies were obtained from the hind paws of mice that received 2 cycles of cisplatin and 11 doses of ACY-1083. Tissues were stained with antibodies for IENFs (PGP9.5; red) and collagen (green). Representative images from saline + vehicle-treated mice: (A) saline + vehicle; (B) cisplatin + vehicle; (C) saline + ACY-1083; and (D) cisplatin + ACY-1083. The basement membrane is indicated by the dashed lines, the nerve fibers crossing the basement membrane are indicated by arrows. (E) Quantification of IENF density expressed as the number of nerve fibers crossing the basement membrane/length of the basement membrane (mm), scale bar = 20 μm, magnification 40×. Two-way ANOVA revealed a signification interaction (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. n=4 mice/group.
DISCUSSION
The high prevalence and incidence of CIPN, along with the lack of effective treatments, make identification of therapeutics and understanding of mechanistic drivers of CIPN vital for improving quality of life in cancer patients and survivors. In the current study, we demonstrated that pharmacological inhibition of HDAC6 with ACY-1083 effectively prevents cisplatin-induced mechanical allodynia in mice. The beneficial effect of HDAC6 inhibition is not limited to the cisplatin-model in mice, but extends to paclitaxel-induced mechanical allodynia in rats. Prolonged treatment with either the selective HDAC6 inhibitor ACY-1083 or the less selective HDAC6 inhibitor ACY-1215 reverses established cisplatin-induced mechanical allodynia. Notably, HDAC6 inhibition also reverses cisplatin-induced spontaneous pain and numbness. To our knowledge, this is the first study to identify a single agent capable of inducing full recovery from multiple symptoms of CIPN. The reversal of cisplatin-induced neuropathy by HDAC6 inhibition was associated with normalization of cisplatin-induced mitochondrial bioenergetic deficits in the peripheral nervous system and reduced mitochondrial content in the distal axons of tibial nerves, which was accompanied by restoration of IENF density.
Our results identify HDAC6 as a novel therapeutic target for treatment of existing cisplatin-induced peripheral neuropathy. The ability of HDAC6 inhibitors to reverse existing symptoms of CIPN is an important finding, as no FDA-approved therapeutics is available for treatment of established CIPN. In addition, the ability of the HDAC6 inhibitor ACY-1083 to prevent the primary dose-limiting toxicity of cisplatin would potentially allow oncologists to dose higher and/or longer and thus achieve better clinical outcomes for cancer treatment. Importantly, we also showed that the HDAC6 inhibitor ACY-1215, which is currently tested in clinical trials for combinational treatment of several cancers [32; 42], also reverses cisplatin-induced mechanical allodynia to a similar extent as ACY-1083. These findings provide important evidence for novel clinical applications of HDAC6 inhibitors as prevention and treatment of CIPN.
Bioenergetic deficits associated with neuronal mitochondrial dysfunction have been proposed as a driver for CIPN [4]. The symptoms of CIPN present in a distal-to-proximal “stocking and glove” distribution, with the longest peripheral axons having the highest susceptibility. This pattern of symptoms suggests that insufficient distribution of mitochondria to the distal peripheral axons might contribute to CIPN as well. Indeed, the results in our study implicate impaired mitochondrial transport as a contributing factor to cisplatin-induced peripheral neuropathy. In our in vitro model of DRG neuron cultures, cisplatin treatment reduced mitochondrial movement in the axons (Figure 5D). We did not directly measure mitochondrial transport in vivo. However, we show impaired mitochondrial bioenergetics and decreased mitochondrial content in the distal tibial nerves of cisplatin-treated mice (Figure 5A–C).
Inhibition of HDAC6 has been proposed to protect against neurological disorders by facilitating distribution of mitochondria throughout the neuronal network [16; 17; 19; 41]. Transport of mitochondria along microtubules is increased by acetylation of α-tubulin, which serves as a recognition signal for the anchoring of mitochondrial motor proteins [40]. HDAC6 is a well-known α-tubulin deacetylase, and inhibition of HDAC6 increases α-tubulin acetylation. Indeed, increased acetylation of α-tubulin by HDAC6 inhibition is associated with improved mitochondrial transport [14; 16; 22]. In our model of cisplatin-induced peripheral neuropathy, we show that inhibition of HDAC6 with ACY-1083 promoted α-tubulin acetylation (Figure 4). Corresponding with increased α-tubulin acetylation, we show that ACY-1083 prevented cisplatin-induced reduction in mitochondrial motility in DRG cultures (Figure 5D). More importantly, we show that 11 doses of ACY-1083 treatment improved mitochondrial bioenergetics and contents in the tibial nerves of cisplatin-treated mice (Figure 5A–C). Interestingly, at the time point when ACY-1083 treatment had normalized mitochondrial function in the distal nerves (day 11 after start ACY-1083), DRG mitochondrial bioenergetics were still impaired. However, at a later time point DRG mitochondrial function also normalized, indicating that HDAC6 may not only affect mitochondrial transport but also mitochondrial health (Figure 6). Thus it is of interest that HDAC6 inhibition has been shown to induce mitochondrial fusion/elongation and movement in neuronal cultures [20], and it has been proposed that elongated mitochondrial networks are more efficient at energy production [33; 45]. Although the exact mechanism of how HDAC6 inhibition enhances mitochondrial bioenergetics in our model is still to be determined, our data clearly shows that pharmacological inhibition of HDAC6 with ACY-1083 slowly reverses mitochondrial bioenergetic deficits in the peripheral nervous system. Our findings also indicate that in mice treated with cisplatin and ACY-1083 healthy mitochondria were preferentially transported towards distal nerve endings. The latter point is consistent with previous studies suggesting that the transport of mitochondria along axons is dependent on the mitochondrial membrane potential, and healthier mitochondria with higher membrane potential are preferentially transported anterogradely towards axonal terminals [15]. We therefore propose that HDAC6 inhibition reverses CIPN by improving mitochondrial health and by increasing α-tubulin acetylation and promoting the transport of healthy mitochondria to the peripheral endings of sensory neurons.
IENF loss has been suggested as the earliest sign of axonal pathology, as the IENFs represent bioenergetically active regions and are therefore highly susceptible to chemotherapy-induced mitotoxic insults [4]. Consistent with previous reports [27; 39], we show here that cisplatin treatment decreased IENF density in the hind paws. The data in supplemental figure 3 demonstrate that IENF loss was already present before the start of HDAC6 inhibitor treatment. Importantly, 11 doses of ACY-1083 treatment fully reversed cisplatin-induced IENF loss, indicating re-innervation (Figure 7) and restored distal-nerve mitochondrial bioenergetics (Figure 6). As both maintenance of peripheral nerve axons as well as re-innervation are highly energy-demanding process [5], the effect of ACY-1083 on IENFs is likely attributable to normalization of mitochondrial bioenergetics in the distal axons. Therefore, our data provide important evidence for the mitotoxic hypothesis of IENF loss in CIPN. Importantly, we show here that prolonged inhibition of HDAC6 with ACY-1083 reversed IENF loss and at the same time led to persistent recovery from multiple symptoms of CIPN. Clinically, IENF loss has been regarded as a reliable diagnostic tool for peripheral neuropathy [26], and our data indicate that restoration of IENF density could be used as a biomarker to evaluate drug efficacy and predict long-term recovery in CIPN patients.
Our findings strongly implicate increased α-tubulin acetylation and improved mitochondrial health and transport in the reversal of CIPN by HDAC6 inhibition. However, it is possible that ACY-1083 also affects other cellular processes. For example, HDAC6 inhibition has been implicated in redox regulation by increasing acetylation of peroxiredoxins-1 and -2, thereby increasing their reducing activity [37]. The anti-inflammatory effect of selective HDAC6 inhibition has also been shown to have beneficial effects in models of rheumatoid arthritis [47]. In addition, there is evidence that HDAC6 inhibition amplifies production of the anti-inflammatory cytokine interleukin-10, which is known to have pain-reducing effects [31; 48]. In this work we show that ACY-1083 treatment reversed cisplatin-induced astrocyte activation (Supplementary Figure 2), implicating a potential contribution of anti-inflammatory effects to the efficacy of ACY-1083. As both oxidative stress and inflammatory cascade activation have been implicated in the initiation and progression of CIPN [1; 49], inhibition of HDAC6 might also contribute to resolution of CIPN through targeting these pathways. In addition, spontaneous discharges in DRG neurons have been shown to contribute to CIPN in rodent models [51; 54]. Notably, it has been suggested that spontaneous discharges might be a consequence of mitotoxicity-induced energy deficiency [4]. Therefore, it is possible that the beneficial effects of HDAC6 inhibition include reversal of chemotherapy-induced neuronal spontaneous activity. Further studies will be needed to determine the involvement of other pathways in the protective effects of HDAC6 inhibition against CIPN.
Importantly, potential therapeutics for CIPN should not interfere with the anti-tumor effects of chemotherapy. It is known that the HDAC inhibitor ACY-1215 has synergistic tumor-killing effects when combined with chemotherapy treatment in both preclinical and clinical settings [32; 42]. Another structurally related HDAC6 inhibitor (ACY-241) has also been shown to enhance the activity of paclitaxel in solid tumor models [21]. Despite the lower specificity of ACY-1215 towards HDAC6, our results show that oral administration of ACY-1215 effectively reverses cisplatin-induced mechanical allodynia to the same extent as ACY-1083 (Figure 3B). Currently, additional clinical trials for combinational therapy of ACY-1215 and paclitaxel or cisplatin for cancer treatment have been initiated or are in preparation and peripheral neuropathy is being monitored in one of them (https://clinicaltrials.gov/ct2/show/NCT02632071?term=ricolinostat&rank=3, https://clinicaltrials.gov/ct2/show/NCT02661815?term=ricolinostat&rank=6; https://clinicaltrials.gov/ct2/show/NCT02856568?term=ricolinostat&rank=2). Taken together previous data and our current findings indicate that addition of HDAC6 inhibitors to chemotherapeutic regimens may enhance the anti-tumor effects of chemotherapeutics while at the same time inhibiting CIPN.
CIPN represents an important challenge in cancer treatment due to the severity of symptoms and the lack of effective therapeutics for both prevention and treatment. In the current study, we show that HDAC6 inhibition completely reversed multiple symptoms of CIPN. The protective effect of HDAC6 inhibition is associated with increased α-tubulin acetylation, improved axonal mitochondrial bioenergetics, and enhanced IENF density. These findings provide important evidence that HDAC6 inhibitors are promising therapeutics for the prevention and treatment of CIPN. Our results also implicate IENF density as a reliable biomarker for clinical evaluation of drug efficacy in the treatment of CIPN.
Supplementary Material
Black X denotes cisplatin or paclitaxel treatment; gray X denotes ACY-1083 treatment.
Mice were treated with 2 rounds of cisplatin followed by 3 doses of 10 mg/kg ACY-1083 treatment starting at day 4 after completion of cisplatin treatment. Lumbar spinal cord was stained for the astrocyte activation marker GFAP. Representative examples of immunofluorescence: (A) saline + vehicle; (B) cisplatin + vehicle; (C) saline + ACY-1083; and (D) cisplatin + ACY-1083. (E) GFAP levels (area positive) in the spinal dorsal horn were quantified using Image J Analysis software, n=3–4 mice/group. Two-way ANOVA revealed a signification interaction (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. Scale bar = 20 μm; 40× magnification.
Paw biopsies were obtained from the hind paws of mice that received 2 cycles of cisplatin and 11 doses of ACY-1083. Tissues were stained with antibodies for IENFs (PGP9.5; red) and collagen (green). Representative images from saline + vehicle-treated mice: (A) saline + vehicle; (B) saline + ACY-1803; (C) cisplatin + vehicle; and (D) cisplatin + ACY-1083. The basement membrane is indicated by the dashed lines, the nerve fibers crossing the basement membrane are indicated by arrows. (E) Quantification of IENF density expressed as the number of nerve fibers crossing the basement membrane/length of the basement membrane (mm), scale bar = 20 μm, magnification 40×. Two-way ANOVA revealed significant differences between saline and cisplatin: *P<0.05. Tukey post-hoc analysis did not show a significant difference between cisplatin + vehicle vs. cisplatin +ACY-1083. n=4 mice per group.
Acknowledgments
This study is supported by funding from Acetylon Pharmaceuticals Inc. and grant CA1832736 from the National Institutes of Health. O.G, J.H.v.D. and M.J. are employed by, and have equity ownership in, Acetylon Pharmaceuticals Inc.. This study was supported by a research grant from Acetylon Pharmaceuticals to A.K. And C.J.H.
We thank Xiaojiao Huo, M. Sc., M.D. Anderson Cancer Center for technical support with tissue collection.
Financial Support: Acetylon Pharmaceuticals, NIH CA1832736
Footnotes
Conflict of Interest: O.G, J.H.v.D. and M.J. are employed by, and have equity ownership in, Acetylon Pharmaceuticals Inc..
References
- 1.Areti A, Yerra VG, Naidu V, Kumar A. Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014;2:289–295. doi: 10.1016/j.redox.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Authier N, Gillet JP, Fialip J, Eschalier A, Coudore F. An animal model of nociceptive peripheral neuropathy following repeated cisplatin injections. Exp Neurol. 2003;182(1):12–20. doi: 10.1016/s0014-4886(03)00003-7. [DOI] [PubMed] [Google Scholar]
- 3.Barabas K, Milner R, Lurie D, Adin C. Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol. 2008;6(1):1–18. doi: 10.1111/j.1476-5829.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 4.Bennett GJ, Doyle T, Salvemini D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol. 2014;10(6):326–336. doi: 10.1038/nrneurol.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett GJ, Liu GK, Xiao WH, Jin HW, Siau C. Terminal arbor degeneration–a novel lesion produced by the antineoplastic agent paclitaxel. Eur J Neurosci. 2011;33(9):1667–1676. doi: 10.1111/j.1460-9568.2011.07652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. 2003;460(2–3):109–116. doi: 10.1016/s0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- 7.Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, Freret T. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc. 2009;4(10):1560–1564. doi: 10.1038/nprot.2009.125. [DOI] [PubMed] [Google Scholar]
- 8.Bradner JE, Mak R, Tanguturi SK, Mazitschek R, Haggarty SJ, Ross K, Chang CY, Bosco J, West N, Morse E, Lin K, Shen JP, Kwiatkowski NP, Gheldof N, Dekker J, DeAngelo DJ, Carr SA, Schreiber SL, Golub TR, Ebert BL. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(28):12617–12622. doi: 10.1073/pnas.1006774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435(2):297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavaletti G, Alberti P, Frigeni B, Piatti M, Susani E. Chemotherapy-induced neuropathy. Curr Treat Options Neurol. 2011;13(2):180–190. doi: 10.1007/s11940-010-0108-3. [DOI] [PubMed] [Google Scholar]
- 11.Cavaletti G, Cornblath DR, Merkies IS, Postma TJ, Rossi E, Frigeni B, Alberti P, Bruna J, Velasco R, Argyriou AA, Kalofonos HP, Psimaras D, Ricard D, Pace A, Galie E, Briani C, Dalla Torre C, Faber CG, Lalisang RI, Boogerd W, Brandsma D, Koeppen S, Hense J, Storey D, Kerrigan S, Schenone A, Fabbri S, Valsecchi MG, Group CI-P The chemotherapy-induced peripheral neuropathy outcome measures standardization study: from consensus to the first validity and reliability findings. Ann Oncol. 2013;24(2):454–462. doi: 10.1093/annonc/mds329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol. 2010;6(12):657–666. doi: 10.1038/nrneurol.2010.160. [DOI] [PubMed] [Google Scholar]
- 13.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 14.Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5(5):e10848. doi: 10.1371/journal.pone.0010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox A, Blaikie A, MacEwen CJ, Jones D, Thompson K, Holding D, Sharma T, Miller S, Dobson S, Sanders R. Visual impairment in elderly patients with hip fracture: causes and associations. Eye (Lond) 2005;19(6):652–656. doi: 10.1038/sj.eye.6701610. [DOI] [PubMed] [Google Scholar]
- 16.d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- 17.Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27(13):3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122(3):245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ, De Vos KJ. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun. 2014;5:5245. doi: 10.1038/ncomms6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guedes-Dias P, de Proenca J, Soares TR, Leitao-Rocha A, Pinho BR, Duchen MR, Oliveira JM. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochimica et biophysica acta. 2015;1852(11):2484–2493. doi: 10.1016/j.bbadis.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Huang P, Almeciga-Pinto I, Jarpe M, van Duzer JH, Mazitschek R, Yang M, Jones SS, Quayle SN. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget. 2017;8(2):2694–2707. doi: 10.18632/oncotarget.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 23.Janes K, Doyle T, Bryant L, Esposito E, Cuzzocrea S, Ryerse J, Bennett GJ, Salvemini D. Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain. 2013;154(11):2432–2440. doi: 10.1016/j.pain.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JH, Dougherty PM, Abdi S. Basic science and clinical management of painful and non-painful chemotherapy-related neuropathy. Gynecol Oncol. 2015;136(3):453–459. doi: 10.1016/j.ygyno.2015.01.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krukowski K, Nijboer CH, Huo X, Kavelaars A, Heijnen CJ. Prevention of chemotherapy-induced peripheral neuropathy by the small-molecule inhibitor pifithrin-mu. Pain. 2015;156(11):2184–2192. doi: 10.1097/j.pain.0000000000000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lauria G, Lombardi R. Skin biopsy: a new tool for diagnosing peripheral neuropathy. BMJ. 2007;334(7604):1159–1162. doi: 10.1136/bmj.39192.488125.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauria G, Lombardi R, Borgna M, Penza P, Bianchi R, Savino C, Canta A, Nicolini G, Marmiroli P, Cavaletti G. Intraepidermal nerve fiber density in rat foot pad: neuropathologic-neurophysiologic correlation. J Peripher Nerv Syst. 2005;10(2):202–208. doi: 10.1111/j.1085-9489.2005.0010210.x. [DOI] [PubMed] [Google Scholar]
- 28.Ma J, Farmer KL, Pan P, Urban MJ, Zhao H, Blagg BS, Dobrowsky RT. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J Pharmacol Exp Ther. 2014;348(2):281–292. doi: 10.1124/jpet.113.210435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma J, Pan P, Anyika M, Blagg BS, Dobrowsky RT. Modulating Molecular Chaperones Improves Mitochondrial Bioenergetics and Decreases the Inflammatory Transcriptome in Diabetic Sensory Neurons. ACS Chem Neurosci. 2015;6(9):1637–1648. doi: 10.1021/acschemneuro.5b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mielke S, Sparreboom A, Mross K. Peripheral neuropathy: a persisting challenge in paclitaxel-based regimes. Eur J Cancer. 2006;42(1):24–30. doi: 10.1016/j.ejca.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 31.Milligan ED, Penzkover KR, Soderquist RG, Mahoney MJ. Spinal interleukin-10 therapy to treat peripheral neuropathic pain. Neuromodulation. 2012;15(6):520–526. doi: 10.1111/j.1525-1403.2012.00462.x. discussion 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mishima Y, Santo L, Eda H, Cirstea D, Nemani N, Yee AJ, O’Donnell E, Selig MK, Quayle SN, Arastu-Kapur S, Kirk C, Boise LH, Jones SS, Raje N. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br J Haematol. 2015;169(3):423–434. doi: 10.1111/bjh.13315. [DOI] [PubMed] [Google Scholar]
- 33.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. The Journal of cell biology. 2016;212(4):379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pachman DR, Barton DL, Watson JC, Loprinzi CL. Chemotherapy-induced peripheral neuropathy: prevention and treatment. Clin Pharmacol Ther. 2011;90(3):377–387. doi: 10.1038/clpt.2011.115. [DOI] [PubMed] [Google Scholar]
- 35.Park HJ, Stokes JA, Pirie E, Skahen J, Shtaerman Y, Yaksh TL. Persistent hyperalgesia in the cisplatin-treated mouse as defined by threshold measures, the conditioned place preference paradigm, and changes in dorsal root ganglia activated transcription factor 3: the effects of gabapentin, ketorolac, and etanercept. Anesth Analg. 2013;116(1):224–231. doi: 10.1213/ANE.0b013e31826e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park SB, Goldstein D, Krishnan AV, Lin CS, Friedlander ML, Cassidy J, Koltzenburg M, Kiernan MC. Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA Cancer J Clin. 2013;63(6):419–437. doi: 10.3322/caac.21204. [DOI] [PubMed] [Google Scholar]
- 37.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(28):9633–9638. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters CM, Jimenez-Andrade JM, Jonas BM, Sevcik MA, Koewler NJ, Ghilardi JR, Wong GY, Mantyh PW. Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp Neurol. 2007;203(1):42–54. doi: 10.1016/j.expneurol.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 39.Qi-Liang Mao-Ying AK, Karen Krukowski, Xiao-Jiao Huo, Theodore J Price, Cleeland Charles, Heijnen Cobi J. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLos One. 2014 Jun;9(6):e100701. doi: 10.1371/journal.pone.0100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16(21):2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds IJ, Malaiyandi LM, Coash M, Rintoul GL. Mitochondrial trafficking in neurons: a key variable in neurodegeneration? J Bioenerg Biomembr. 2004;36(4):283–286. doi: 10.1023/B:JOBB.0000041754.78313.c2. [DOI] [PubMed] [Google Scholar]
- 42.Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, Cirstea D, Rodig S, Eda H, Scullen T, Canavese M, Bradner J, Anderson KC, Jones SS, Raje N. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider BP, Hershman DL, Loprinzi C. Symptoms: Chemotherapy-Induced Peripheral Neuropathy. Adv Exp Med Biol. 2015;862:77–87. doi: 10.1007/978-3-319-16366-6_6. [DOI] [PubMed] [Google Scholar]
- 44.Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, Colvin LA, Fallon M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain. 2014;155(12):2461–2470. doi: 10.1016/j.pain.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 45.Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends in biochemical sciences. 2001;26(1):23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 46.Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18(6):291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Vishwakarma S, Iyer LR, Muley M, Singh PK, Shastry A, Saxena A, Kulathingal J, Vijaykanth G, Raghul J, Rajesh N, Rathinasamy S, Kachhadia V, Kilambi N, Rajgopal S, Balasubramanian G, Narayanan S. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int Immunopharmacol. 2013;16(1):72–78. doi: 10.1016/j.intimp.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Wang B, Rao YH, Inoue M, Hao R, Lai CH, Chen D, McDonald SL, Choi MC, Wang Q, Shinohara ML, Yao TP. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat Commun. 2014;5:3479. doi: 10.1038/ncomms4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang XM, Lehky TJ, Brell JM, Dorsey SG. Discovering cytokines as targets for chemotherapy-induced painful peripheral neuropathy. Cytokine. 2012;59(1):3–9. doi: 10.1016/j.cyto.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolf S, Barton D, Kottschade L, Grothey A, Loprinzi C. Chemotherapy-induced peripheral neuropathy: prevention and treatment strategies. Eur J Cancer. 2008;44(11):1507–1515. doi: 10.1016/j.ejca.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 51.Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: Abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain. 2008;135(3):262–270. doi: 10.1016/j.pain.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao WH, Zheng H, Zheng FY, Nuydens R, Meert TF, Bennett GJ. Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience. 2011;199:461–469. doi: 10.1016/j.neuroscience.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Q, Wu Z, Hadden JK, Odem MA, Zuo Y, Crook RJ, Frost JA, Walters ET. Persistent pain after spinal cord injury is maintained by primary afferent activity. J Neurosci. 2014;34(32):10765–10769. doi: 10.1523/JNEUROSCI.5316-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology. 2014;120(6):1463–1475. doi: 10.1097/ALN.0000000000000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Yoon SY, Zhang H, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of Paclitaxel-induced painful neuropathy. J Pain. 2012;13(3):293–303. doi: 10.1016/j.jpain.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22(5):1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng H, Xiao WH, Bennett GJ. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol. 2011;232(2):154–161. doi: 10.1016/j.expneurol.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16(2):109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Black X denotes cisplatin or paclitaxel treatment; gray X denotes ACY-1083 treatment.
Mice were treated with 2 rounds of cisplatin followed by 3 doses of 10 mg/kg ACY-1083 treatment starting at day 4 after completion of cisplatin treatment. Lumbar spinal cord was stained for the astrocyte activation marker GFAP. Representative examples of immunofluorescence: (A) saline + vehicle; (B) cisplatin + vehicle; (C) saline + ACY-1083; and (D) cisplatin + ACY-1083. (E) GFAP levels (area positive) in the spinal dorsal horn were quantified using Image J Analysis software, n=3–4 mice/group. Two-way ANOVA revealed a signification interaction (P<0.05). Tukey post-hoc analysis revealed significant differences between groups: *P<0.05. Scale bar = 20 μm; 40× magnification.
Paw biopsies were obtained from the hind paws of mice that received 2 cycles of cisplatin and 11 doses of ACY-1083. Tissues were stained with antibodies for IENFs (PGP9.5; red) and collagen (green). Representative images from saline + vehicle-treated mice: (A) saline + vehicle; (B) saline + ACY-1803; (C) cisplatin + vehicle; and (D) cisplatin + ACY-1083. The basement membrane is indicated by the dashed lines, the nerve fibers crossing the basement membrane are indicated by arrows. (E) Quantification of IENF density expressed as the number of nerve fibers crossing the basement membrane/length of the basement membrane (mm), scale bar = 20 μm, magnification 40×. Two-way ANOVA revealed significant differences between saline and cisplatin: *P<0.05. Tukey post-hoc analysis did not show a significant difference between cisplatin + vehicle vs. cisplatin +ACY-1083. n=4 mice per group.